

Abstract
Electrons added to TiO2 and other semiconductors often occupy trap states, whose reactivity can determine the catalytic and stoichiometric chemistry of the material. We previously showed that reduced aqueous colloidal TiO2 nanoparticles have two distinct classes of thermally-equilibrated trapped electrons, termed Red/e− and Blue/e−. Presented here are parallel optical and electron paramagnetic resonance (EPR) kinetic studies of the reactivity of these electrons with solution-based oxidants. Optical stopped-flow measurements monitoring reactions of TiO2/e− with sub-stoichiometric oxidants showed a surprising pattern: an initial fast (seconds) decrease in TiO2/e− absorbance followed by a secondary, slow (minutes) increase in the broad TiO2/e− optical feature. Analysis revealed that the fast decrease is due to the preferential oxidation of the Red/e− trap states, and the slow increase results from re-equilibration of electrons from Blue to Red states. This kinetic model was confirmed by freeze-quench EPR measurements. Quantitative analysis of the kinetic data demonstrated that Red/e− react ~5 times faster than Blue/e− with the nitroxyl radical oxidant, 4-MeO-TEMPO. Similar reactivity patterns were also observed in oxidations of TiO2/e− by O2, which like 4-MeO-TEMPO is a proton-coupled electron transfer (PCET) oxidant, and by the pure electron transfer (ET) oxidant KI3. This suggests that the faster intrinsic reactivity of one trap state over another on the seconds–minutes timescale is likely a general feature of reduced TiO2 reactivity. This differential trap state reactivity is likely to influence the performance of TiO2 in photochemical/electrochemical devices, and it suggests an opportunity for tuning catalysis.
Graphical Abstract
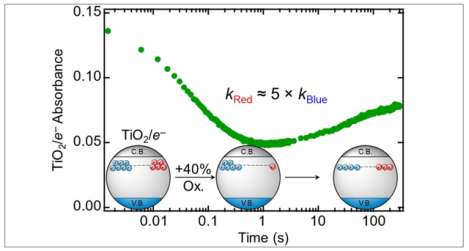
Introduction
Metal oxide semiconductors have long garnered interest across the physical sciences due to their attractive chemical, photochemical, electrochemical, optical, and electronic properties. One important feature is their ability to store and transport multiple charge carriers (redox equivalents). Understanding the chemical reactivity of such charge carriers in nanoscale metal oxides is crucial in their applications in renewable energy technologies and catalysis.1 In-depth spectroscopic studies of reduced titanium dioxide (TiO2) have identified various shallow and deep electron traps at diverse defect sites, including surface and interstitial Ti(III) centers and oxygen atom vacancies.2–8 The relaxation of photo-generated delocalized conduction band electrons to trap states occurs very quickly, typically on the fs–ns timescale.8–11 Much slower relaxations can occur up to tens of ms, attributed to proton involvement.12 As such, trapped electrons play an important role in the function of almost all TiO2-containing devices.8, 13–15
Presented here are kinetic studies of thermally equilibrated electron trap states in colloidal aqueous TiO2 nanoparticles (NPs). We examine the chemical reactivity of the reduced NPs on the seconds-to-minutes timescale more typical of catalysis. Our results are consistent with previous kinetic studies of colloidal TiO2 by many groups, by many different techniques.16–30 Such studies are complementary to the much more common work using transient, ultrafast kinetics by transient spectroscopies or under continuous UV illumination.31
The current studies build on our prior discovery that the occupied trap states in these particular reduced colloidal TiO2 NPs fall into two distinct types. On the basis of their optical spectra (Figure 1), we termed these trap states Red/e− and Blue/e−.7 The Red/e− and Blue/e− trap states are generated via UV-photochemical reduction and are stable under an inert atmosphere. The EPR spectra of the Red/e− and Blue/e− indicated that these trap states are structurally distinct and have different symmetries (Figure 1).7 These states are in thermal equilibrium, with the Red/e− higher in enthalpy (ΔH°) by 3.0 ± 0.6 kcal/mol (130 ± 30 meV), based on their relative ratio as a function of temperature.7 Surprisingly, re-equilibrations of the electrons between the Red/e− and Blue/e− traps upon changes in temperature were slow, occurring over at least several minutes. While the Red/e− states have a higher ΔH°, at equilibrium the Red/e− and Blue/e− states have the same free energies (ΔG).
Figure 1:

(A) A TiO2/e− EPR spectrum (dashed trace) with simulated rhombic (Blue/e−) and axial (Red/e−) EPR components. (B) The molar absorptivity (ε) of the TiO2/e− optical spectrum (dashed trace) with simulated Red/e− and Blue/e− component peaks. Thermally equilibrated TiO2/e− colloids under our general conditions (Table-SI 8.1) typically contain Red/e− and Blue/e− electrons in a roughly 40:60 ratio. The simulated spectra were derived by fitting TiO2/e− EPR and optical spectra with both Red/e− and Blue/e− components as described in Peper et. al.7
We show here that Red/e− and Blue/e− react at different rates with solution-based oxidants. The reaction of Red/e− with the nitroxyl radical 4-methoxy-2,2,6,6-tetramethyl-1-piperidinyloxyl (4-MeO-TEMPO) occurs ~5 times more quickly than the reaction of Blue/e−. Such differential trap-state reactivity can be observed because of the unusually simple Red/e− and Blue/e− system. While the electronic landscapes of TiO2 and other semiconductor materials are typically more complex, containing a variety of reduced trap states,4–10, 32–33 the faster reactivity of one trap state over another is likely a common feature of NP reaction chemistry. For example, these results help to explain the typical ill-defined, multi-exponential behavior observed in kinetic studies of TiO24, 16, 34 and other nanomaterials.30, 35–38 These results show the importance of trap state populations in the performance of TiO2 materials.31 They also raise questions about the connections and contrasts between fast-timescale photoinduced processes and slower-timescale, equilibrated and thermochemically-driven chemical reactions of trap states.
Methods
Anatase titanium dioxide nanoparticles (diameter = 4±1 nm) were synthesized via hydrolysis of titanium tetrachloride (TiCl4) as previously described, see Supporting Information (SI)-Sections 1.3–1.5.7, 16 The resulting white solid TiO2 product was re-suspended in neutral 18 MΩ cm water (without ligands) to give a transparent colloid (3g solid product L−1) with pH 2.2–2.4, due to residual HCl from the synthesis. The colloid concentrations were roughly 30 μM in nanoparticles, with each NP containing ~1,000 Ti atoms.
Photochemical reduction of the colloidal NP suspension in the presence of 0.1–0.15 M methanol resulted in a blue coloration of the colloid and the appearance of EPR signals (SI-Sections 2.1 and 4.1).16 As previously reported, the optical and EPR signals of the reduced nanoparticles (TiO2/e−) result from two structurally distinct trap states. The Red/e− states have a longer-wavelength, a higher intensity optical absorbance (λmax = 740 nm) and a broad axial EPR signal, while the Blue/e− states have a low-intensity, blue-shifted absorbance (λmax = 470 nm) and a rhombic EPR signal (Figure 1).7 Room temperature TiO2/e− samples were handled inside a N2 glovebox or with gas tight syringes, and colloids exposed to air were kept at temperatures < −78 °C. The reduced colloids were stable under both conditions. The extent of colloid reductions, evaluated in terms of the concentration of electrons, [e−] (mol e− L−1), were determined via standard optical titrations with 4-MeO-TEMPO as previously described (SI-Section 2.2).7, 17 Photolysis of TiO2 colloids (30 μM NPs) for 5 hours typically gave 1.0–1.5 mM e−.
The time course of TiO2/e− oxidations by various quantities of 4-MeO-TEMPO, potassium triiodide (KI3) or O2 were followed by optical spectroscopy via stopped-flow techniques (SI-Section 3.1–3.3). Additionally, the reaction of TiO2/e− and 4-MeO-TEMPO was followed by freeze-quench EPR spectroscopy (SI-Section 4.2). In both the stopped-flow and freeze-quench experiments, gas-tight syringes were used to mix equal volumes of TiO2/e− colloid and a pH-matched solution of the oxidant. The typical experimental conditions were pH 2.2–2.4, 15 μM TiO2 nanoparticles, and 0.05–0.075 M methanol, where concentrations given refer to the solutions after mixing. For each specific experiment presented, these and other details are given in Table-SI 8.1.
Results
I. 100% Oxidation of TiO2/e–
The addition of excess 4-MeO-TEMPO to photochemically reduced TiO2 results in the disappearance of the blue TiO2/e− color and the appearance of new diamagnetic 1H NMR signals, consistent with the formation of protonated hydroxylamine 4-MeO-TEMPO-H(H+) (Figure-SI 2.2–2.3). The full oxidation of TiO2/e− (0.55 mM e−) by just over one equivalent of 4-MeO-TEMPO (0.57 mM) occurs in less than 2 seconds as indicated by the disappearance of the broad TiO2/e− optical signal in Figure 2. The change in TiO2/e− absorbance with oxidation is not uniform across the spectrum, resulting in a subtle blueshift in the TiO2/e− optical density, from λmax = ~600 to ~460 nm (black markers, Figure 2). We will return to this blueshift in Section I of the Discussion below.
Figure 2:

Optical spectra monitoring the disappearance of TiO2/e− (0.55 mM e−) absorbance upon mixing with 4-MeO-TEMPO (0.57 mM). Black markers highlight the blueshift in the λmax of the broad TiO2/e− absorbance band.
II. Sub-stoichiometric Oxidation of TiO2/e−
Reactions of photochemically reduced NPs with sub-stoichiometric oxidants were monitored from 1.5 ms to 300 s using an optical stopped-flow instrument (SI-Section 3). These measurements revealed two distinct and surprising optical trends upon addition of 0.4 oxidizing equivalents of 4-MeO-TEMPO to TiO2/e− (0.49 mM e−), Figure 3A. The initial TiO2/e− absorbance (Ainit) decreased quickly, within ~2 seconds, to a minimum value (Amin) before increasing slowly over several minutes to a final value (Afin). The fast and slow changes in absorbance intensity are respectively coupled to blue- and red-shifts in the broad TiO2/e− absorbance peak. The same qualitative optical trends were observed in reactions with other sub-stoichiometric oxidants, including KI3 and O2 (Figure 3B and SI-Sections 3.5, 3.6). Based on the optical spectra in Figure 1, these changes suggest initial depletion of the more absorbing Red/e− followed by slow re-establishment of the Red/e− – Blue/e− equilibrium, as discussed in more detail below.
Figure 3:
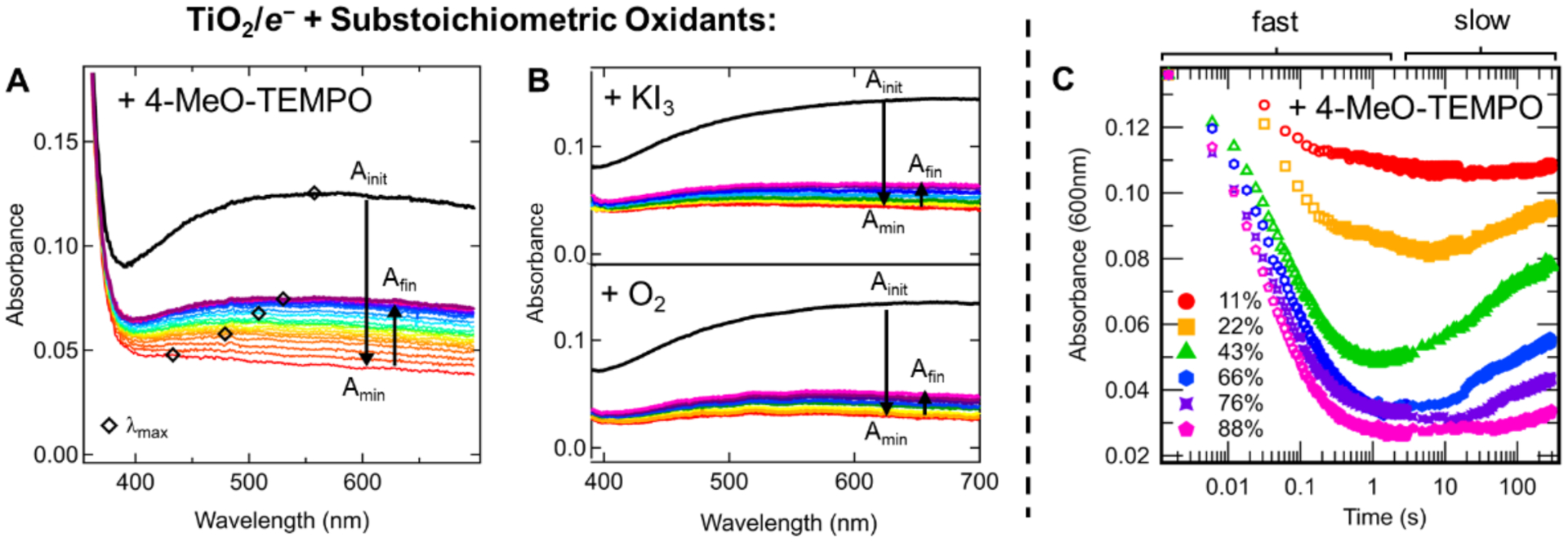
(A) Optical spectra monitoring the oxidation of TiO2/e− (0.49 mM e−) by 0.4 oxidizing equivalents of 4-MeO-TEMPO (0.19 mM). Black markers highlight the redshift in λmax in the secondary, slow absorbance increase from Amin to Afin. (B) Optical spectra following reactions of TiO2/e− (0.50 mM e−) with (top) 0.50 oxidizing equivalents KI3 (0.125 mM I2) and (bottom) ~0.50 oxidizing equivalents of O2 (0.065 mM O2). (C) Kinetic traces: Absorbance at 600 nm of a TiO2/e− colloid (0.50 mM e−) as a function of time after mixing with various oxidizing equivalents of 4-MeO-TEMPO. Absorbance is plotted versus a log time scale to show details of both the fast and slow absorbance changes.
The sub-stoichiometric reactivity of TiO2/e− with 4-MeO-TEMPO was broadly evaluated. The fast and slow reaction phases, and their distinct timescales, are seen clearly in the full kinetic traces following TiO2/e− (0.50 mM e−) absorbance upon reaction with various quantities of sub-stoichiometric 4-MeO-TEMPO, (Figure 3C and SI-Section 3.4). The magnitudes of the fast, slow, and total absorbance changes (ΔAfast, ΔAslow, and ΔAtot = Afin - Ainit) as a function of %TiO2/e− oxidation are discussed below.
In order to further probe the sub-stoichiometric oxidation of TiO2/e−, the time course of the reaction with 4-MeO-TEMPO was followed by EPR spectroscopy using freeze-quench techniques. A colloid of photochemically reduced TiO2 NPs (0.69 mM e−) was mixed with 0.46 oxidizing equivalents 4-MeO-TEMPO (0.32 mM) and allowed to rest (i.e., react) for 0.1 – 600 s before being shot into liquid nitrogen. The frozen samples were maintained at T ≤ −78 °C and prepared for EPR measurements at 10 K (SI-Section 4.2). The EPR spectra collected after different resting times are shown in Figure 4. The actual reaction quench times are somewhat longer than the resting times indicated in the Figure 4 legend, because water droplets freeze relatively slowly in liquid nitrogen (the Leidenfrost phenomenon).39 We crudely estimate the average sample freezing time of 1–2 seconds (SI-Section 4.4).
Figure 4:
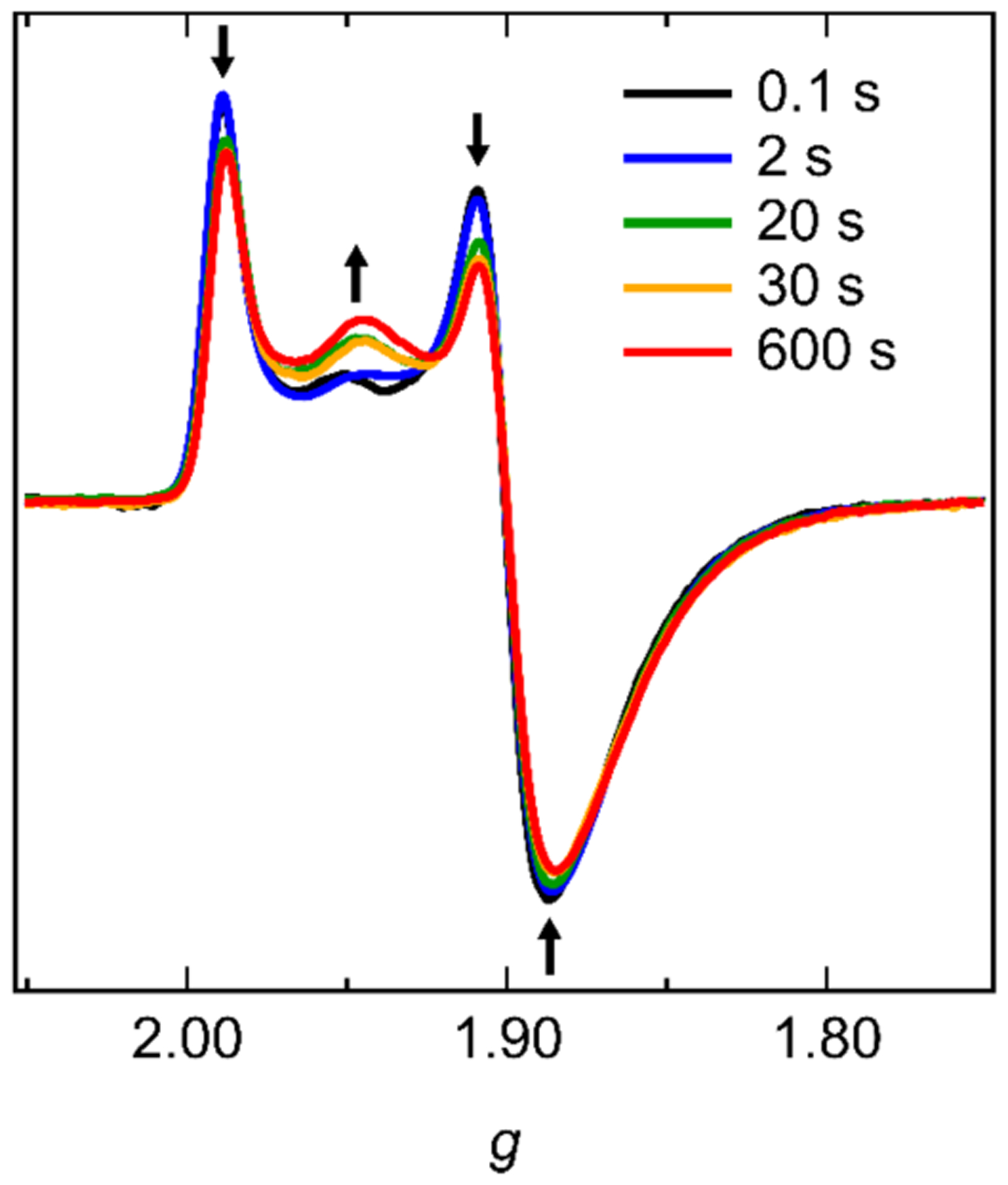
EPR spectra collected after mixing TiO2/e− (0.69 mM e−) and 0.46 oxidizing equivalents 4-MeO-TEMPO (0.32 mM) via freeze-quench techniques. The arrows denote spectral trends as a function of resting time. The resting times indicated in the legend underestimate of the actual reaction quench times due to slow freezing (see text and SI-Section 4.4). All spectra are baseline corrected and normalized to total spin density, SI-Section 4.3.
The EPR signals are consistent with Ti(III)-based electrons previously observed in this (and other) reduced TiO2 systems.5–7 The change in the freeze-quench spectra as a function of resting time qualitatively shows a shift in spin density from the rhombic signal, characteristic of Blue/e−, to the axial signal of Red/e−. The EPR signal of unreduced 4-MeO-TEMPO radical is completely absent from the 2 s spectrum, and only a very small 4-MeO-TEMPO signal (<1% of the total spin density) is observed in the baseline of the 0.1 s spectrum (Figure-SI 4.3). The absence of 4-MeO-TEMPO in these early EPR spectra is consistent with the optical experiment, which shows the complete oxidation of TiO2/e− by 1 equivalent 4-MeO-TEMPO in less than 2 seconds (Figure 2). These data confirm that the oxidation of TiO2/e− by 4-MeO-TEMPO is fast, occurring in a couple seconds (SI-Section 4.4).
Because the reaction of 4-MeO-TEMPO and TiO2/e− is already complete by the first timepoint of the freeze-quench experiment, the EPR spectra in Figure 4 (nominally 0.1 – 600 s) represent colloids containing the same concentration of electrons, the initial [TiO2/e−] minus 0.46 oxidizing equivalents 4-MeO-TEMPO. The spectral differences between the freeze-quench samples therefore correspond to changes in the electronic environments of the remaining trapped electrons.
The freeze-quench EPR spectra were well-simulated with a rigid two-state parameter set consisting of an axial and a rhombic signal, Figure 5A.40 The ratio of the axial to rhombic signals for each freeze-quench spectrum was optimized via least squares fitting, with the g and gStrain values held constant (SI-Section 4.5). The axial and rhombic percentages of total spin density in the simulation of each freeze-quench spectrum are plotted as a function of resting time in Figure 5B. The spectrum collected 0.1 s after the sub-stoichiometric oxidation of TiO2/e− by 4-MeO-TEMPO is only 20% axial. This %axial is the lowest we have ever observed compared to equilibrated TiO2/e− samples under these conditions (SI-Section 4.1). The spectra obtained over the ten minutes following the reaction showed an increase in the axial component from 20 to 33% of total spin density (Figure 5B).
Figure 5:

(A) The experimental (black) and simulated (green) EPR spectra collected 20 s after mixing TiO2/e− (0.69 mM e−) and 0.46 oxidizing equivalents 4-MeO-TEMPO (0.32 mM). The weighted axial and rhombic simulation components are included (dashed traces). Axial signal: g = 1.9376, 1.8289 and gStrain = 0.0474, 0.0462, and rhombic signal: g =1.987, 1.8966, 1.8667 and gStrain = 0.0152, 0.0185, 0.0384. (B) The optimized %axial (Red/e−) and %rhombic (Blue/e−) of total spin density as a function of the nominal resting time (see text).
The TiO2/e− EPR spectra are much broader than those of molecules, requiring large gStrain parameters in the simulation. This broadening is likely due to both the collection of random orientations present in our frozen solutions, and to a distribution of spin signals arising from a range of similar, but not identical, electronic trap states.7 Because of this inhomogeneity, the analysis of the freeze-quench EPR data with a rigid two-state model can only be semi-quantitative and the simulation parameters presented in Figure 5A are not unique (i.e. multiple combinations of parameters appear to fit the spectra well). We present three additional simulations of the freeze-quench EPR spectra, performed by two independent researchers, in SI-Section 4.5. Importantly, the four independent simulations (SI-Figures 4.4–4.5) give the same 12±2% increase in %Red/e− over the time-course of the freeze-quench experiment (SI-Figure 4.6). This consistency across simulations provides strong support that the shift in electron density depicted in Figure 5B is an accurate representation of the post-oxidation re-equilibration of the remaining electrons from Blue/e− (rhombic) to Red/e− (axial) trap states.
Discussion
TiO2 NPs are good reducing agents and react within seconds with both proton-coupled electron transfer oxidants (the TEMPO nitroxyl radical and O2) and the I3– oxidant that just accepts electrons. Our observations of rapid reactions of colloidal TiO2 with excess oxidant are consistent with previous kinetic studies.16–30 However, we are not aware of prior studies reporting relative rates of different trap states in solution reactions of this kind.
The treatment of reduced TiO2 NPs with sub-stoichiometric oxidant gives rise to unusual observations: the TiO2/e− absorbance rapidly decays and then rises partially back to its original value (Figure 3). The freeze-quench EPR experiments (Figure 4) connect this unusual observation to the distinct Red/e− and Blue/e− trap states previously identified in this colloidal TiO2/e− system.7 In this Discussion, we show that both the EPR and optical kinetic experiments demonstrate the preferential oxidation of Red/e− over Blue/e− in the fast sub-stoichiometric oxidation of TiO2/e− (Scheme 1, ΔAfast). This kinetic disparity disrupts the Red/e− and Blue/e− trap state equilibrium, leading to the slower second reaction phase—the re-equilibration of electrons from Blue/e− to Red/e− states (Scheme 1, ΔAslow).
Scheme 1:

Proposed reaction scheme for the oxidation of TiO2/e− by 50% 4-MeO-TEMPO. The initial fast reaction primarily of the Red/e− with 4-MeO-TEMPO (ΔAfast) is followed by the slow re-equilibration of Red/e− and Blue/e− (ΔAslow).
In an effort to further understand the different kinetic reactivity of the Red/e− and Blue/e− trapped states, we have applied a traditionally molecular, physical-organic relative rates approach. Applying this type of methodology to NP reactions is typically challenging due to the existence of multiple, inhomogeneous distributions of trap states and complex kinetic behavior (typically multi-exponential under pseudo-first-order conditions). Therefore, this Discussion is divided into three sections, with increasing complexity and quantitation. Section I brings together the direct spectroscopic evidence supporting the two-phase reaction depicted in Scheme 1. Next, Section II builds a simplistic, qualitative kinetic model that reproduces the unusual optical trends of sub-stoichiometric TiO2/e− oxidation. Finally, Section III presents a quantitative estimate of the relative and absolute rate constants for the Red/e− and Blue/e− oxidation. Together, these different levels of analysis strongly support the two-phase reaction depicted in Scheme 1.
I. Spectroscopic Evidence for Scheme 1
Addition of ≥1 equivalent of 4-MeO-TEMPO to TiO2/e− resulted in the complete loss of the TiO2/e− optical absorption within 2 seconds. However, the absorbance decay was not uniform across the TiO2/e− spectrum (Figure 2). This was an initial indication that the two classes of trapped electrons (Red/e− and Blue/e−) differ in kinetic reactivity. If the Red/e−:Blue/e− ratio of trapped electrons were maintained throughout the oxidation reaction, the TiO2/e− absorption feature would have decreased uniformly across the optical spectrum. Instead, longer wavelengths decayed at an increased rate, resulting in a blueshift in λmax. This indicates that Red/e− (λmax = 740 nm) react more quickly than Blue/e− (λmax = 470 nm) with 4-MeO-TEMPO.
Addition of sub-stoichiometric amounts of oxidant resulted in a rapid initial ~2 second drop in absorbance (ΔAfast) that mirrored the results of the 100% oxidation reaction (Figure 3). However, in the sub-stoichiometric oxidations, ΔAfast showed a larger than anticipated absorbance decay based on the amount of oxidant added. For instance, in the 40% oxidation by 4-MeO-TEMPO, ΔAfast corresponded to a loss of nearly 70% of Ainit (Figure 3A). The large absorbance change per %oxidation (ΔAfast/Δ[e−] ≡ εfast) indicated the preferential oxidation of Red/e− over Blue/e−. This is because Red/e− have a higher molar absorptivity than the Blue/e− (εRed = 630 M−1cm−1 > εBlue = 85 M−1cm−1 at 600 nm, Figure 1B). Following this initial phase, the slower (minutes) absorbance recovery occurred with a redshift in the optical spectrum of the TiO2/e− (Figure 3A). This indicates an increase in the Red/e− population during the second kinetic phase.
Together these results are consistent with the reactivity depicted in Scheme 1. The thermodynamic equilibrium of Red/e− and Blue/e− trap states is disturbed upon fast oxidation of TiO2/e−, due to the higher kinetic reactivity of Red/e− with 4-MeO-TEMPO (Scheme 1, ΔAfast). The fast oxidation is followed by the slow re-equilibration of electrons from Blue to Red trap states (Scheme 1, ΔAslow).
The parallel freeze-quench EPR spectra that followed the time course of the sub-stoichiometric oxidation of TiO2/e− (Figure 4) provide strong and independent support for this description of the reaction. First, the freeze-quench data confirm that the oxidation of TiO2/e− by 4-MeO-TEMPO is fast under these reaction conditions. Since 4-MeO-TEMPO radical is readily observed by EPR spectroscopy, the almost complete depletion of the radical in the earliest freeze-quench spectrum confirms the reduction of 4-MeO-TEMPO is finished in less than a few seconds (Scheme 1, ΔAfast, SI-Section 4.4).
Second, the freeze quench data confirm the faster oxidation of Red/e− over Blue/e− by 4-MeO-TEMPO. In our previous study, Red/e− and Blue/e− were correlated with the respective axial and rhombic EPR signals (Figure 1A).7 The spectra of early-time free-quench samples show low axial:rhombic ratios (low Red/e− populations), relative to the longer equilibrated TiO2/e− colloids, particularly the 600 s spectrum. This validates the higher kinetic reactivity of Red/e− relative to Blue/e− (Scheme 1, ΔAfast).
Finally, the freeze-quench EPR spectra collected over the slow, second kinetic phase of the reaction shows a shift in the EPR spin density from the rhombic to the axial signal as a function of resting time (Figure 5B). These spectra confirm that the secondary optical changes result from the slow equilibration of electrons from Blue/e− to Red/e− states (Scheme 1, ΔAslow).
II. Qualitative kinetic model of Red/e− and Blue/e− oxidation
A deeper analysis of the optical and EPR studies offers two important conclusions about the kinetics of Red/e− and Blue/e− reactivity with 4-MeO-TEMPO. First, these data sets inform the distinct timescales of the two reactions shown in Scheme 1. As per the discussion above, the transition from the fast to the slow phase of the reaction (from TiO2/e− oxidation to trap state equilibration) occurs at roughly the time of the minimum optical absorbance in the stopped-flow experiments (). For the reaction with 4-MeO-TEMPO, occurs ~2 s after mixing, while the secondary slow absorbance increase, resolving at , lasts for several minutes (Figure 3C). Thus, as indicated in the qualitative model in Scheme 2, the oxidation of TiO2/e− within a few seconds (eqs 1 and 2), and the trap state re-equilibration over minutes (eq 3), occur on substantially different timescales (eq 4).
Scheme 2:
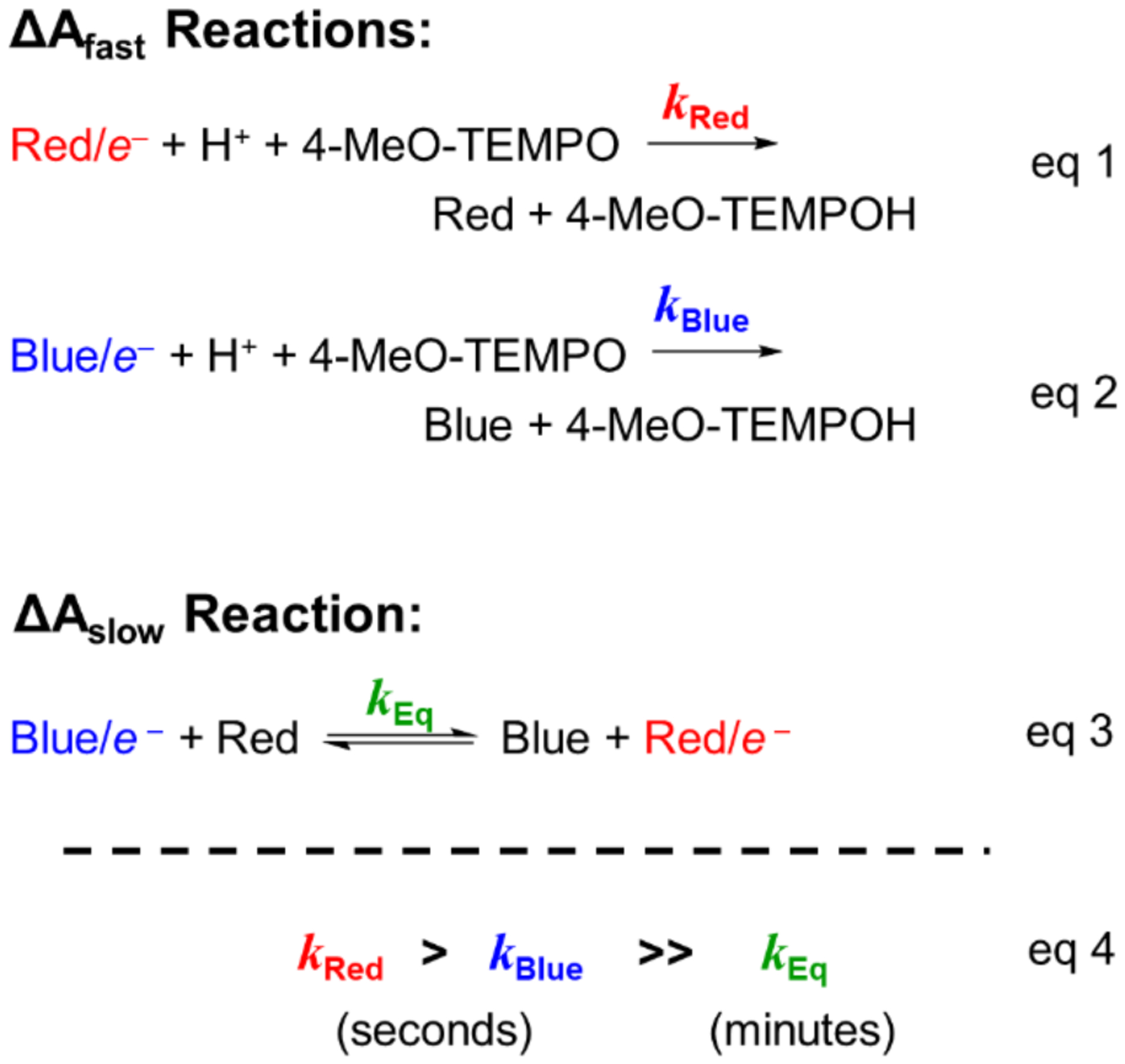
Simplified kinetic model of Red/e− and Blue/e− reactivity with 4-MeO-TEMPO.
Second, we can conclude that the initial, fast absorbance decay (ΔAfast, Figure 3C) is due to the reaction of both Red/e− and Blue/e− with 4-MeO-TEMPO (eqs 1 and 2). This is clear from the optical data following the reaction of TiO2/e− with ≥1 equivalent oxidant, which shows complete reaction of all the electrons within 2 s of mixing (Figure 2). Photochemically reduced TiO2/e− colloids under these conditions typically have ~40% Red/e− and ~60% Blue/e−. If only the Red/e− were reactive, the oxidation of TiO2/e− by excess 4-MeO-TEMPO would proceed rapidly only to ~40% completion, and then continue at the much slower, minute timescale of the trap state equilibration reaction (eq 3). Instead, we observe oxidation of both Red/e− and Blue/e−, within a couple of seconds, before significant trap state equilibration occurs (eq 4). While the Red/e− react faster than the Blue/e−, these results indicate that both trap states are oxidized within seconds by 4-MeO-TEMPO. Both the Red/e− and the Blue/e− react with 4-MeO-TEMPO much faster than they equilibrate with each other.
These conclusions have implications for the nature of the Red/e− and Blue/e− trap states and their reactions. One could imagine, for instance, that the slower reactions of Blue/e− could be the result of their being more buried in the nanoparticles, while the Red/e− are more at the NP surface. Then the reaction of Blue/e− with 4-MeO-TEMPO could be slower because they require diffusion through the NP or perhaps detrapping of the electron into conduction band states. However, we feel that a diffusive or detrapping process would likely also lead to equilibration of the two trap states (since electron trapping in such materials is usually rapid on these timescales8–12). Yet the reactions of both states are roughly two orders of magnitude faster than their equilibration. This suggests that both the Red/e− and the Blue/e− are close enough to the NP surface to react with 4-MeO-TEMPO directly. Perhaps this is because these NPs appear to be irregular in shape, with both crystalline (anatase) and amorphous regions, such that all of the trap states are close to the surface.
The generality of this model is demonstrated in the reactions of TiO2/e− with two additional oxidants, KI3 and O2 (SI-Sections 3.5–3.7). TiO2/e− suspensions are quickly and completely oxidized by stoichiometric amounts of these oxidants, as seen with 4-MeO-TEMPO (Scheme-SI 3.1). Stopped-flow studies show that these reactions with the three oxidants occur at different rates. Time traces for fully stoichiometric oxidations are shown in SI-Figure 3.7. Figures 3B and 6 above compare sub-stochiometric reactions of the oxidants under similar conditions: TiO2/e− suspensions with 0.50 mM e− plus ~0.5 oxidizing equivalents of each oxidant. The differences in reaction rates are indicated by the differences in the initial, ΔAfast phase of these reactions in Figure 6. The two-electron reduction of KI3 occurs in ~0.2 s; the proton-coupled reduction of 4-MeO-TEMPO is about an order of magnitude slower (~2 s) and the multi-proton, multi-electron reduction of O2 still slower (~10 s) (SI-Sections 3.5 and 3.6).
Figure 6:

Kinetic traces monitoring the sub-stoichiometric oxidation of TiO2/e− (0.50 mM e−) by (A) 0.50 oxidizing equivalents KI3 (0.125 mM), (B) 0.48 oxidizing equivalents of 4-MeO-TEMPO (0.24 mM), and (C) ~0.50 oxidizing equivalents of O2 (0.065 mM). The fuchsia arrows highlight the timescales of the ΔAfast phase of the sub-stoichiometric reactions.
Despite the differences between KI3, 4-MeO-TEMPO and O2 – in their oxidation rates and their electron transfer (ET) vs. proton-coupled electron transfer (PCET) oxidation nature – the same reactivity pattern outlined in Schemes 1 and 2 is observed for all three reagents. The optical absorbance initially drops rapidly and then partially recovers to a higher level, just as seen in oxidation by 4-MeO-TEMPO (Figure 3). The data imply that the KI3 and O2 oxidations of TiO2/e−, including both the Red/e− and the Blue/e−, are much faster than trap state equilibration. These results indicate that the faster reactivity of one trap state over another is likely a general feature of TiO2/e− reactivity.
The relative rates of kRed, kBlue, and kEq outlined in Scheme 2 were used to evaluate the changes in the fast and slow phases of the sub-stoichiometric reactions as a function of %TiO2/e− oxidation. In the six reactions of Figure 3C ranging from 11–88% TiO2/e− oxidation, the magnitude of the slow absorbance increase, ΔAslow, becomes larger from 11–43% oxidation and then diminishes from 43–88% oxidation. The initial increase and then decrease in the magnitude of ΔAslow as a function of %oxidation is easier to see in Figure 7B where the ΔA for each reaction phase is plotted as a function of %TiO2/e− oxidation. For reference, the magnitudes of the three arrows overlaying the 43% oxidation trace in Figure 7A (reprinted from Figure 3C) correspond to the 3 points plotted at 43% TiO2/e− oxidation in Figure 7B. Figure 7B also shows that the dependence of ΔAfast on %oxidation gets more shallow as %oxidation increases, as seen by the change in slope. Interestingly, the shifts (or inflections) of the ΔAfast and ΔAslow trends in Figure 7B are centered around ~40% oxidation. This is the %Red/e− in the initial TiO2/e− colloid prior to reaction (%Red/e−init).
Figure 7:

(A) TiO2/e− absorbance as a function of time after addition of 0.43 oxidizing equivalents of 4-MeO-TEMPO (from Figure 3C). The magnitudes of the three overlaying arrows (ΔAfast, ΔAslow, and ΔAtot) correspond to the 3 points (respectively, fuchsia, yellow, and black) plotted at 43% TiO2/e− oxidation in panel B. (B) The ΔAfast, ΔAslow, and ΔAtot of the six kinetic traces reported in Figure 3C plotted versus %TiO2/e− oxidation and [4-MeO-TEMPO]. Error bars signify ±1σ between multiple kinetic experiments. (C) ΔAfast, ΔAslow, and ΔAtot for simulated reactions starting from a theoretical colloid with a 40:60 Red/e−:Blue/e− ratio and adding 0–100% oxidant. In the simulations the relative rates of the kinetic model (eq 4) were approximated as kRed ≫ kBlue ≫ kEq. The full details of the kinetic simulations are reported in SI-Section 5.
In order to elucidate the origins of these striking experimental trends, the reactions presented in Figure 3C and Figure 7B were simulated starting from a colloid with a 40:60 Red/e− :Blue/e− ratio and adding 0–100% oxidant. For simplicity, the relative rates in this simulation were approximated as kRed ≫ kBlue ≫ kEq, such that all of the Red/e− were oxidized before any of the Blue/e−, and equilibration occurred only after all of the oxidant was consumed (SI-Section 5). The ΔAfast, ΔAslow, and ΔAtot of the simulated sub-stoichiometric oxidations plotted in Figure 7C very nicely capture the experimental trends. This strong agreement supports the approach of the qualitative kinetic model and confirms that the relative rates of eq 4 are the origin of the optical trends overserved in the sub-stoichiometric oxidations of TiO2/e−.
The qualitative kinetic model provides insights into both reaction phases. The initial, fast decrease in TiO2/e− absorbance (ΔAfast in Figure 7) tracks the oxidation of TiO2-based electrons by 4-MeO-TEMPO. During this oxidation, the Red/e−:Blue/e− ratio decreases because the electrons in the Red/e− trap states are oxidized faster (kRed ≫ kBlue). When less than ~40% 4-MeO-TEMPO was added, the oxidized electrons come predominantly from the Red/e−. In this region, ΔAfast is large and has a steep negative dependence on %TiO2/e− oxidation because of the higher absorbance of the Red/e− (Figure 7B,C; εRed = 7.4 × εBlue at 600 nm). Once %TiO2/e− oxidation exceeds the initial ~40% of Red/e−, then the Blue/e− states are also oxidized. Because of the lower absorbance of Blue/e−, the slope of ΔAfast vs. %TiO2/e− oxidation plot becomes shallower.
The simulation thus predicts a sharp change in the slope of ΔAfast when %oxidation = %Red/e−init (Figure 7C, fuchsia) because this is the point where the transition occurs between oxidation of only Red/e− (steep slope ≅ −εRed) to include Blue/e− (shallow slope ≅ −εBlue). The experimental data in Figure 7B show a more gradual transition from steep to shallow slopes. This shows that the oxidation of Blue/e− is partially competitive with Red/e−, as discussed in the next section.
The secondary, slow absorbance increase (ΔAslow in Figure 7A and B) results from the re-equilibration of electrons from Blue to Red trap states following the fast oxidation reaction. The magnitude of ΔAslow is largest (i) when the Red/e−:Blue/e− equilibrium position (40:60) is most perturbed during the oxidation reaction, and (ii) when the largest amount of Blue/e− remain after oxidation to re-equilibrate. As %TiO2/e− oxidation increases, the perturbation of the Red/e−:Blue/e− equilibrium increases, but less of the initial [TiO2/e−] remain for reequilibration. These opposing contributions give rise to the initial increase and then decrease in the magnitude of ΔAslow as a function of %oxidation in Figure 7B and C (yellow triangles). In the simulations, the maximum in the ΔAslow trend in Figure 7C occurs exactly at the equivalence of %oxidation and %Red/e−init (40%, Figure 7C), where all the Red/e− are oxidized and the maximum amount Blue/e− remains to reequilibrate. In the experimental data (Figure 7B), the maximum in ΔAslow occurs when %oxidation is slightly greater than %Red/e−init, because of the competitive oxidation of Blue/e−.
In summary, a qualitative simulation assuming that the TiO2/e− oxidations occur in three distinct phases, kRed ≫ kBlue ≫ kEq, captures the striking trends in the experimental absorbance changes (Figure 7B and C). Both the inflection in the ΔAfast as a function of %TiO2/e− oxidation by 4-MeO-TEMPO and the peak in the ΔAslow are reproduced. The same pattern of reactivity was observed using KI3 or O2 as the oxidant (Figure 6). The success of qualitative model confirms the two phases of the reaction (Scheme 2) and sets the foundation for the more rigorous quantitative analysis in the following section.
III. Quantitative Analysis of Red/e− and Blue/e− Reactivity During ΔAfast
The prior section showed both (i) that that the reaction of 4-MeO-TEMPO with Red/e− is faster than with Blue/e−, and (ii) that these rates must be somewhat competitive. The relative rates, kRed:kBlue, were estimated by evaluating the 20% TiO2/e− oxidation reaction as a pseudo-competition experiment. This reaction decreased the TiO2/e− concentration from its initial 0.5 mM e− to 0.4 mM (Scheme 3 and SI Section 6). Measured absorbances at 600 nm gave the individual Red/e− and Blue/e− concentrations before and after the reaction (using Ainit at , Amin at and εRed and εBlue;7 eqs 5 and 6). Note that in this system, the observed composite epsilon of TiO2/e− (εobs(t)) is not constant but changes based on the Red/e−:Blue/e− ratio (SI-Figure 2.4). This analysis showed that 70% of the 0.2 equivalents of 4-MeO-TEMPO were reduced by Red/e− and 30% were reduced by Blue/e−. Assuming two independent simple second-order reactions (eqs 1, 2), the [Red/e−] and [Blue/e−] concentrations at were calculated for various rate constant ratios kRed:kBlue. Comparisons of the predicted and experimental concentrations indicated that kRed is 8 ± 3 times faster than kBlue (SI-Section 6).
Scheme 3: [Red/e−] and [Blue/e−] before and after the ΔAfast phase of TiO2/e− oxidation by 0.2 equivalents of 4-MeO-TEMPO.a.
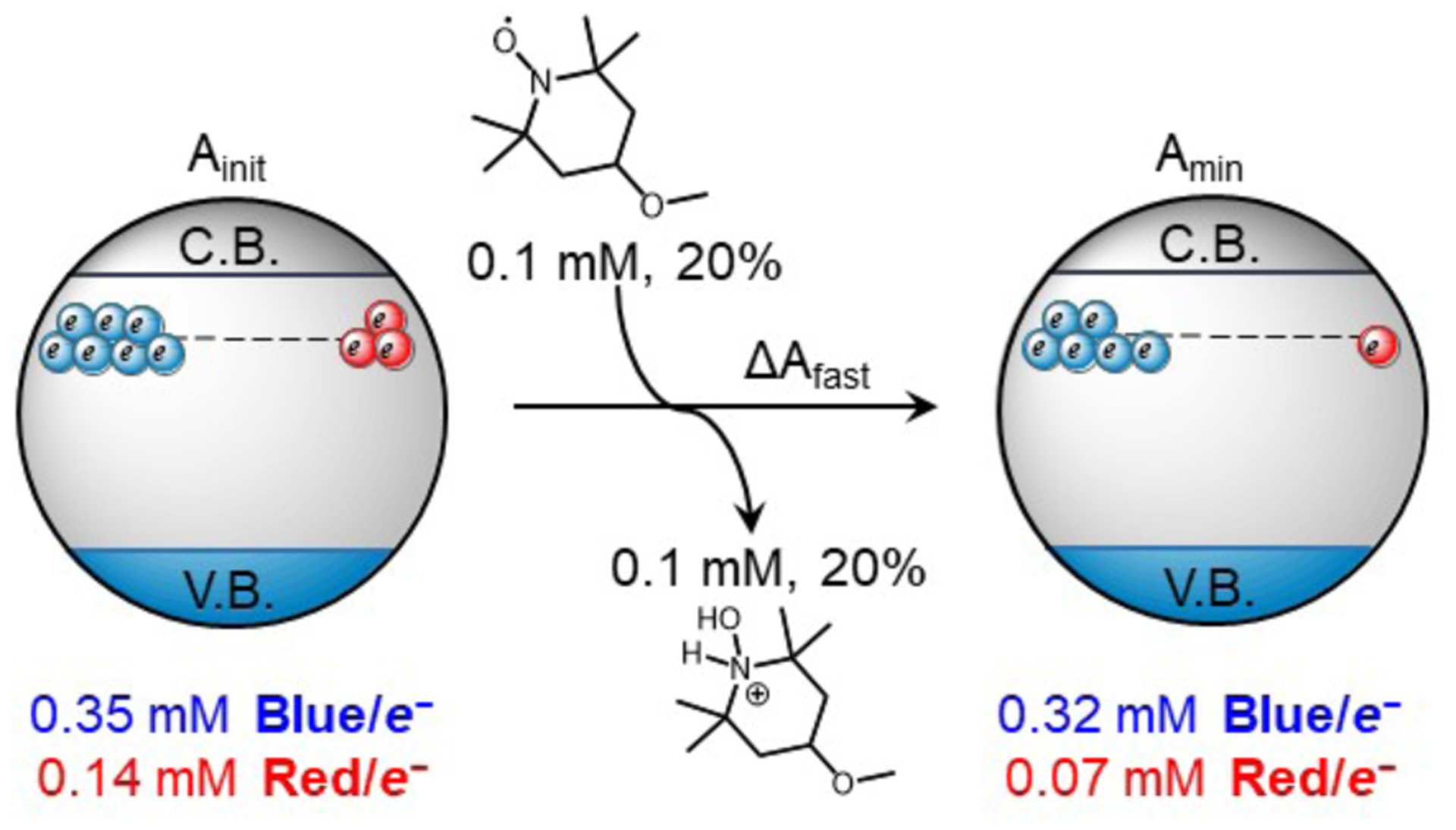
a Kinetic trace in SI-Figure 6.1. [Red/e−] and [Blue/e−] determined from Ainit and Amin at 600 nm, [TiO2/e−]init, and εRed and εBlue.7
| eq 5 |
| eq 6 |
In order to estimate the absolute values of kRed and kBlue, a more quantitative kinetic model was developed. The same assumption of independent, second order processes gave the rate law in eq 7 for the consumption of 4-MeO-TEMPO (Scheme 4). Based on the respective assignments of and as the beginning and end of the 4-MeO-TEMPO reduction reaction, [4-MeO-TEMPO]t = [4-MeO-TEMPO]init at and 0 mM at . However, because the concentration of 4-MeO-TEMPO and εobs(t) change throughout the TiO2/e− oxidation reaction, the [4-MeO-TEMPO]t at timepoints (t) between and cannot be uniquely determined. Thus, traditional kinetic modeling of the optical spectra over time is not straightforward with this system. Therefore, three different methods were used to estimate the time course of [4-MeO-TEMPO]t throughout the sub-stoichiometric TiO2/e− oxidation reactions: The percent conversion was estimated (1) using A600 as a simplistic metric for reaction progress, (2) assuming a second order disappearance of 4-MeO-TEMPO, and (3) assuming two competitive second order processes consuming 4-MeO-TEMPO. With the three different methods, [TiO2/e−]t was calculated by assuming mass balance and was used (in combination with eq 5 and 6) to extract [Red/e−]t and [Blue/e−]t. For further details of these three fitting methods see SI-Section 7.1.
Scheme 4: Quantitative kinetic model of Red/e− and Blue/e− reactivity with 4-MeO-TEMPOb.

bEqs 1 and 2 are also in Scheme 2 above. Because we believe that the protons required for the reactions are associated with the TiO2-based trapped electrons,17 they are not included in our rate law.
The concentration profiles of [4-MeO-TEMPO]t, [Red/e−]t, and [Blue/e−]t from each method were globally fit to the rate law in eq 7 to give optimized rate constants kRed and kBlue (Figure-SI 7.3–7.5). The A600nm resulting from the modeled [Red/e−]t, and [Blue/e−]t versus time fits well to the experimental data highlighting the accuracy of this model (Figure 8). The resulting rate constants were consistent across all three methods and gave kRed ≈ 1 × 105 M−1s−1 and kBlue ≈ 2 × 104 M−1s−1 (Table-SI 7.1). These are only estimated rate constants because the Red/e− and Blue/e− likely contain distributions of similar but non-identical trapped electrons, so the assumption of simple second order kinetics in eq 7 for each is an approximation. The extracted kRed/kBlue suggest that Red/e− react ~5 times faster than Blue/e− with 4-MeO-TEMPO, in good agreement with the results of the pseudo-competition experiment described above.
Figure 8:
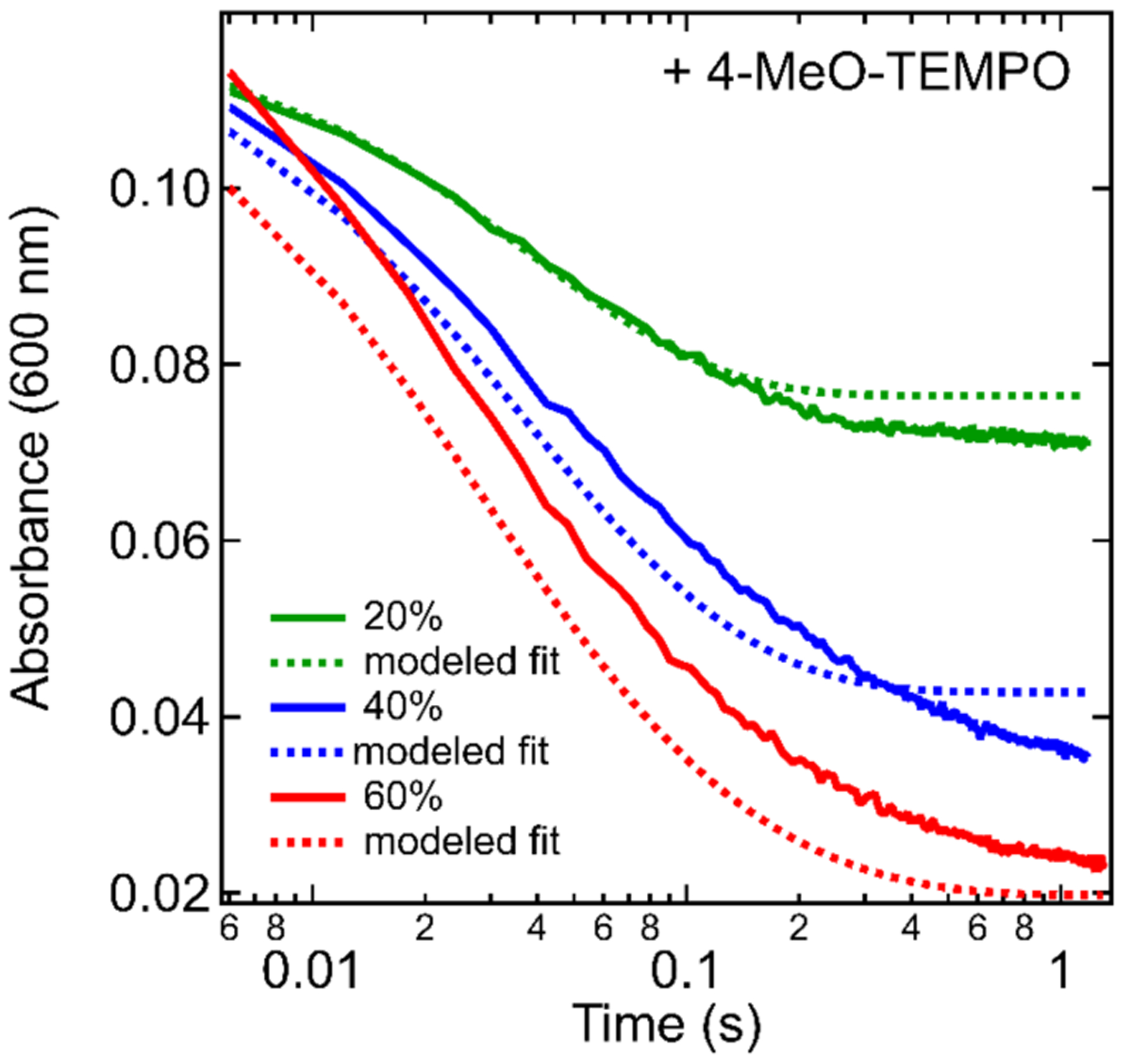
Experimental (solid lines) and modeled (dashed lines, method 3) A600nm vs. time plots for the ΔAfast portion of reactions of TiO2/e− (0.49 mM e−) with 0.2 (green), 0.4 (blue), and 0.6 (red) oxidizing equivalents of 4-MeO-TEMPO. (SI-Section 7.1).
Conclusions:
The oxidation kinetics of two distinct classes of trapped electrons, termed Red/e− and Blue/e−, in aqueous, colloidal TiO2/e− nanoparticles are presented here. Stopped-flow optical and freeze-quench EPR experiments demonstrate that the Red/e− react faster than Blue/e− with solution-based oxidant 4-MeO-TEMPO. Qualitative and semi-quantitative models confirm this kinetic behavior, with kRed ≅ 5 × kBlue ≫ kEq, with kRed ≈ 1 × 105 M−1s−1 and kBlue ≈ 2 × 104 M−1s−1. The disparity in reactivity between the trap states is the origin of the very peculiar “decrease then increase” trend in the optical absorbance of TiO2/e− during sub-stoichiometric reactions. This is due to fast oxidation of primarily Red/e− being followed by slow re-equilibration of electrons to the thermodynamic Red/e−:Blue/e− equilibrium. The same optical trends were observed in sub-stoichiometric reactions with two additional oxidants, KI3 and O2. This commonality is surprising because these oxidants are very different—KI3 is a simple electron only oxidant, 4-MeO-TEMPO is a proton-coupled electron transfer (PCET) oxidant41, and O2 reduction involves a many reaction steps.16, 42 The more rapid reactivity of the Red/e− with all three oxidants suggests that there are intrinsic differences in the reactivity of the Red/e− and Blue/e− trap states.
Implications and Outlook:
The preferential kinetic reactivity of one trap state over another observed here is likely a common feature in most TiO2 systems,4–10, 32–33, 43 and perhaps for many other semiconductor materials. Differences in trap state occupancy and reactivity seem likely to contribute to the complex kinetics commonly observed for reactions of colloidal NPs.16, 34–35
The reactivity difference between Red/e− and Blue/e− is common to a pure ET oxidant (KI3) and single- or multi-PCET reagents (4-MeO-TEMPO or O2). This could perhaps be due to different physical locations of the two states, perhaps with the Blue/e− requiring a diffusive or detrapping step to react. However, this explanation seems inconsistent with the reactions with oxidants being much faster than trap-state equilibration. Previous temperature-equilibration studies showed the Red/e− to be higher in enthalpy than the Blue/e−, by 3.0 ± 0.6 kcal mol−1 (130 ± 30 meV).7 While this is could provide an explanation of the higher reactivity of Blue/e−, ET and PCET reactivity should follow free energy differences, which could not be estimated due to the unknown concentrations of unoccupied traps. In general, the thermochemistry of nanoscale reactions is challenging to precisely define, especially when electron transfer is coupled to proton transfer as is the case here.44–45 Efforts to understand this chemical reactivity are underway, by more precisely defining the structures and stoichiometries of the trap states, and the nature and energetics of the rate limiting steps.
The millisecond timescale of the reactivity reported is of the order of typical turnover frequencies for electrocatalysis and photo(electro)chemical catalysis. Therefore, this study raises the tantalizing possibility that a future ability to intentionally tune trap states could be useful in catalytic applications.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Professors Andrew Borovik and Jenny Yang and their labs, especially Victoria Oswald, Justin Lee, Caitlin Hanna, and Bianca Ceballos (all UC Irvine), for use of their EPR spectrometer, optical spectrometer, and glove box as well as their support of the experiments conducted at UCI. We acknowledge Professor Ira Weinstock and Mrs. Sapir Avnaim (Ben Gurion University of the Negev) for helpful discussions. We acknowledge the Yale Chemical and Biophysical Instrumentation Center (CBIC) for use of their instrumentation and facilities. We further thank Gary Brudvig (Yale University) and Boris Dzikovski (ACERT Center at Cornell University) for help with EPR.
This work was supported primarily by (i) the Center for Light Energy Activated Redox Processes (LEAP), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DESC0001059; (ii) by US-Israel Binational Science Foundation Grant BSF grant 2016123; and (iii) by U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DE-SC0021298. The kinetic modeling by ACB was supported in part by an NIH postdoctoral fellowship F32GM129890 and in part by the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. The EPR freeze-quench studies were done collaboratively by JLP and NEG (while supported by LEAP), with MJF, who was supported by a National Sciences and Engineering Research Council of Canada (NSERC) Postgraduate Scholarship and University of California, Irvine funds, and with MTG who was supported by the National Institutes of Health, NIH (R01-GM101390). ACERT is supported by the National Institutes of Health Grants NIH/NIGMS P41GM103521.
Footnotes
SUPPORTING INFORMATION
General experimental consideration, synthesis and characterization of TiO2 nanoparticles, complete simulations of EPR data, and kinetics modeling of stopped-flow data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Ma Y; Wang X; Jia Y; Chen X; Han H; Li C, Titanium Dioxide-Based Nanomaterials for Photocatalytic Fuel Generations. Chem. Rev 2014, 114, 9987–10043. [DOI] [PubMed] [Google Scholar]
- 2.Rex RE; Knorr FJ; McHale JL, Surface Traps of TiO2 Nanosheets and Nanoparticles as Illuminated by Spectroelectrochemical Photoluminescence. J. Phys. Chem. C 2014, 118, 16831–16841. [Google Scholar]
- 3.Berger T; Sterrer M; Diwald O; Knözinger E; Panayotov D; Thompson TL; Yates JT, Light-Induced Charge Separation in Anatase TiO2 Particles. J. Phys. Chem. B 2005, 109, 6061–6068. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK; Wakim FG; Addiss RR, Photoelectronic Processes in Rutile. Phys. Rev 1969, 184, 979–988. [Google Scholar]
- 5.Howe RF; Graetzel M, EPR Observation of Trapped Electrons in Colloidal TiO2. J. Phys. Chem 1985, 89, 4495–4499. [Google Scholar]
- 6.Rajh T; Nedeljkovic JM; Chen LX; Poluektov O; Thurnauer MC, Improving Optical and Charge Separation Properties of Nanocrystalline TiO2 by Surface Modification with Vitamin C. J. Phys. Chem. B 1999, 103, 3515–3519. [Google Scholar]
- 7.Peper JL; Vinyard DJ; Brudvig GW; Mayer JM, Slow Equilibration between Spectroscopically Distinct Trap States in Reduced TiO2 Nanoparticles. J. Am. Chem. Soc 2017, 139, 2868–2871. [DOI] [PubMed] [Google Scholar]
- 8.Kohtani S; Kawashima A; Miyabe H, Reactivity of Trapped and Accumulated Electrons in Titanium Dioxide Photocatalysis. Catalysts 2017, 7, 303. [Google Scholar]
- 9.Schneider J; Matsuoka M; Takeuchi M; Zhang J; Horiuchi Y; Anpo M; Bahnemann DW, Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev 2014, 114, 9919–9986. [DOI] [PubMed] [Google Scholar]
- 10.Ke S-C; Wang T-C; Wong M-S; Gopal NO, Low Temperature Kinetics and Energetics of the Electron and Hole Traps in Irradiated TiO2 Nanoparticles as Revealed by EPR Spectroscopy. J. Phys. Chem. B 2006, 110, 11628–11634. [DOI] [PubMed] [Google Scholar]
- 11.Leytner S; Hupp JT, Evaluation of the Energetics of Electron Trap States at the Nanocrystalline Titanium Dioxide/Aqueous Solution Interface Via Time-Resolved Photoacoustic Spectroscopy. Chem. Phys. Lett 2000, 330, 231–236. [Google Scholar]
- 12.McCool NS; Swierk JR; Nemes CT; Saunders TP; Schmuttenmaer CA; Mallouk TE, Proton-Induced Trap States, Injection and Recombination Dynamics in Water-Splitting Dye-Sensitized Photoelectrochemical Cells. ACS Appl. Mater. Interfaces 2016, 8, 16727–16735. [DOI] [PubMed] [Google Scholar]
- 13.Konezny SJ; Richter C; Snoeberger RC; Parent AR; Brudvig GW; Schmuttenmaer CA; Batista VS, Fluctuation-Induced Tunneling Conductivity in Nanoporous TiO2 Thin Films. J. Phys. Chem. Lett 2011, 2, 1931–1936. [Google Scholar]
- 14.Halverson AF; Zhu K; Erslev PT; Kim JY; Neale NR; Frank AJ, Perturbation of the Electron Transport Mechanism by Proton Intercalation in Nanoporous TiO2 Films. Nano Lett. 2012, 12, 2112–2116. [DOI] [PubMed] [Google Scholar]
- 15.Swierk JR; McCool NS; Saunders TP; Barber GD; Mallouk TE, Effects of Electron Trapping and Protonation on the Efficiency of Water-Splitting Dye-Sensitized Solar Cells. J. Am. Chem. Soc 2014, 136, 10974–10982. [DOI] [PubMed] [Google Scholar]
- 16.Mohamed HH; Mendive CB; Dillert R; Bahnemann DW, Kinetic and Mechanistic Investigations of Multielectron Transfer Reactions Induced by Stored Electrons in TiO2 Nanoparticles: A Stopped Flow Study. J. Phys. Chem. A 2011, 115, 2139–2147. [DOI] [PubMed] [Google Scholar]
- 17.Schrauben JN; Hayoun R; Valdez CN; Braten M; Fridley L; Mayer JM, Titanium and Zinc Oxide Nanoparticles Are Proton-Coupled Electron Transfer Agents. Science 2012, 336, 1298–1301. [DOI] [PubMed] [Google Scholar]
- 18.Chen C; Zhao W; Lei P; Zhao J; Serpone N, Photosensitized Degradation of Dyes in Polyoxometalate Solutions Versus TiO2 Dispersions under Visible-Light Irradiation: Mechanistic Implications. Chem. Eur. J 2004, 10, 1956–1965. [DOI] [PubMed] [Google Scholar]
- 19.Du Y; Rabani J, The Measure of TiO2 Photocatalytic Efficiency and the Comparison of Different Photocatalytic Titania. J. Phys. Chem. B 2003, 107, 11970–11978. [Google Scholar]
- 20.Gao R; Stark J; Bahnemann DW; Rabani J, Quantum yields of hydroxyl radicals in illuminated TiO2 nanocrystallite layers. J. Photochem. Photobiol. A 2002, 148, 387–391. [Google Scholar]
- 21.Knorr FJ; Mercado CC; McHale JL, Trap-State Distributions and Carrier Transport in Pure and Mixed-Phase TiO2: Influence of Contacting Solvent and Interphasial Electron Transfer. J. Phys. Chem. C 2008, 112, 12786–12794. [Google Scholar]
- 22.Leytner S; Hupp JT, Evaluation of the energetics of electron trap states at the nanocrystalline titanium dioxide/aqueous solution interface via time-resolved photoacoustic spectroscopy. Chem. Phys. Lett 2000, 330, 231–236. [Google Scholar]
- 23.Liu Y; Jennings JR; Zakeeruddin SM; Grätzel M; Wang Q, Heterogeneous Electron Transfer from Dye-Sensitized Nanocrystalline TiO2 to [Co(bpy)3]3+: Insights Gained from Impedance Spectroscopy. J. Am. Chem. Soc 2013, 135, 3939–3952. [DOI] [PubMed] [Google Scholar]
- 24.Matylitsky VV; Lenz MO; Wachtveitl J, Observation of pH-Dependent Back-Electron-Transfer Dynamics in Alizarin/TiO2 Adsorbates: Importance of Trap States. J. Phys. Chem. B 2006, 110, 8372–8379. [DOI] [PubMed] [Google Scholar]
- 25.Meichtry JM; Dillert R; Bahnemann DW; Litter MI, Application of the Stopped Flow Technique to the TiO2-Heterogeneous Photocatalysis of Hexavalent Chromium in Aqueous Suspensions: Comparison with O2 and H2O2 as Electron Acceptors. Langmuir 2015, 31, 6229–6236. [DOI] [PubMed] [Google Scholar]
- 26.Moser J; Punchihewa S; Infelta PP; Graetzel M, Surface complexation of colloidal semiconductors strongly enhances interfacial electron-transfer rates. Langmuir 1991, 7, 3012–3018. [Google Scholar]
- 27.Safrany A; Gao R; Rabani J, Optical Properties and Reactions of Radiation Induced TiO2 Electrons in Aqueous Colloid Solutions. J. Phys. Chem. B 2000, 104, 5848–5853. [Google Scholar]
- 28.Serpone N; Lawless D; Khairutdinov R, Size Effects on the Photophysical Properties of Colloidal Anatase TiO2 Particles: Size Quantization versus Direct Transitions in This Indirect Semiconductor? J. Phys. Chem 1995, 99, 16646–16654. [Google Scholar]
- 29.Szczepankiewicz SH; Moss JA; Hoffmann MR, Slow Surface Charge Trapping Kinetics on Irradiated TiO2. J. Phys. Chem. B 2002, 106, 2922–2927. [Google Scholar]
- 30.Weng Y-X; Wang Y-Q; Asbury JB; Ghosh HN; Lian T, Back Electron Transfer from TiO2 Nanoparticles to FeIII(CN)63−: Origin of Non-Single-Exponential and Particle Size Independent Dynamics. J. Phys. Chem. B 2000, 104, 93–104. [Google Scholar]
- 31.Liu B; Zhao X; Yu J; Parkin IP; Fujishima A; Nakata K, Intrinsic Intermediate Gap States of TiO2 Materials and their Roles in Charge Carrier Kinetics. J. Photochem. Photobiol. C 2019, 39, 1–57. [Google Scholar]
- 32.Mandal D; Hamann TW, Charge Distribution in Nanostructured TiO2 Photoanode Determined by Quantitative Analysis of the Band Edge Unpinning. ACS Appl. Mater. Interfaces 2016, 8, 419–424. [DOI] [PubMed] [Google Scholar]
- 33.Morrison SR, Electrochemistry at Semiconductor and Oxidized Metal Electrodes. 1 ed.; Springer US: 1980. [Google Scholar]
- 34.Wilker MB; Shinopoulos KE; Brown KA; Mulder DW; King PW; Dukovic G, Electron Transfer Kinetics in CdS Nanorod–[FeFe]-Hydrogenase Complexes and Implications for Photochemical H2 Generation. J. Am. Chem. Soc 2014, 136, 4316–4324. [DOI] [PubMed] [Google Scholar]
- 35.Braten MN; Gamelin DR; Mayer JM, Reaction Dynamics of Proton-Coupled Electron Transfer from Reduced ZnO Nanocrystals. ACS Nano 2015, 9, 10258–10267. [DOI] [PubMed] [Google Scholar]
- 36.Wong NZ; Ogata AF; Wustholz KL, Dispersive Electron-Transfer Kinetics from Single Molecules on TiO2 Nanoparticle Films. J. Phys. Chem. C 2013, 117, 21075–21085. [Google Scholar]
- 37.Tsui EY; Carroll GM; Miller B; Marchioro A; Gamelin DR, Extremely Slow Spontaneous Electron Trapping in Photodoped n-Type CdSe Nanocrystals. Chem. Mater 2017, 29, 3754–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Utterback JK; Wilker MB; Mulder DW; King PW; Eaves JD; Dukovic G, Quantum Efficiency of Charge Transfer Competing against Nonexponential Processes: The Case of Electron Transfer from CdS Nanorods to Hydrogenase. J. Phys. Chem. C 2019, 123, 886–896. [Google Scholar]
- 39.Song YS; Adler D; Xu F; Kayaalp E; Nureddin A; Anchan RM; Maas RL; Demirci U, Vitrification and levitation of a liquid droplet on liquid nitrogen. Proc. Natl. Acad. Sci 2010, 107, 4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoll S; Schweiger A, EasySpin A Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- 41.Warren JJ; Tronic TA; Mayer JM, Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheng H; Ji H; Ma W; Chen C; Zhao J, Direct Four-Electron Reduction of O2 to H2O on TiO2 Surfaces by Pendant Proton Relay. Angew. Chem. Int. Ed 2013, 52, 9686–9690. [DOI] [PubMed] [Google Scholar]
- 43.Macdonald IR; Rhydderch S; Holt E; Grant N; Storey JMD; Howe RF, EPR Studies of Electron and Hole Trapping in Titania Photocatalysts. Catal. Today 2012, 182, 39–45. [Google Scholar]
- 44.Peper JL; Mayer JM, Manifesto on the Thermochemistry of Nanoscale Redox Reactions for Energy Conversion. ACS Energy Lett. 2019, 866–872. [Google Scholar]
- 45.Peper JL Studies of Titanium Dioxide Nanoparticles: Thermodynamics and Reactivity. Yale University, 2019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.