

Abstract
PURPOSE
Fibroblast growth factor receptor (FGFR) 2 alterations, present in 5%-15% of intrahepatic cholangiocarcinomas (IHC), are targets of FGFR-directed therapies. Acquired resistance is common among patients who respond. Biopsies at the time of acquired resistance to targeted agents may not always be feasible and may not capture the genetic heterogeneity that could exist within a patient. We studied circulating tumor DNA (ctDNA) as a less invasive means of potentially identifying genomic mechanisms of resistance to FGFR-targeted therapies.
MATERIALS AND METHODS
Serial blood samples were collected from eight patients with FGFR-altered cholangiocarcinoma for ctDNA isolation and next-generation sequencing (NGS) throughout treatment and at resistance to anti-FGFR–targeted therapy. ctDNA was sequenced using a custom ultra-deep coverage NGS panel, incorporating dual index primers and unique molecular barcodes to enable high-sensitivity mutation detection.
RESULTS
Thirty-one acquired mutations in FGFR2, 30/31 located in the kinase domain, were identified at resistance in six of eight patients with detectable ctDNA. Up to 13 independent FGFR2 mutations were detected per patient, indicative of striking genomic concordance among resistant subclones.
CONCLUSION
ctDNA could be an effective means to longitudinally monitor for acquired resistance in FGFR2-altered IHC. The numerous acquired genetic alterations in FGFR2 suggest frequent polyclonal mechanisms of resistance that cannot be detected from single-site tissue biopsies.
BACKGROUND
The fibroblast growth factor receptor (FGFR) is a therapeutic target in intrahepatic cholangiocarcinoma (IHC) and bladder cancer, among others. Up to 20% of urothelial cancers have alterations in FGFR, and the pan-FGFR inhibitor erdafitinib was recently approved for patients with bladder cancer harboring alterations in FGFR2 and FGFR3.1 FGFR2 fusions are present in 5%-15% of IHCs with fusion of the 5’ end of FGFR with the intact kinase domain to a diverse range of 3’ fusion partners including BICC1, KIAA1217, and others.2-4 The pan-FGFR inhibitor pemigatinib was recently US Food and Drug Administration (FDA)–approved for treatment of cholangiocarcinoma harboring an FGFR2 fusion, and clinical trials have evaluated the safety and efficacy of several other ATP-competitive pan-FGFR inhibitors, including Debio1347 and infigratinib, and the irreversible FGFR-inhibitor TAS-120.5-8 Consistent with nearly all targeted therapies, responses to FGFR-targeted therapies are short-lived with a progression-free survival of 5.8 months noted with infigratinib and 6.9 months with pemigatinib.5,6 Identification of acquired genomic mutations in this population is important to facilitate drug development and overcome resistance. Historically, tissue acquisition through repeat biopsies has been required to identify mechanisms of resistance; however, single-site biopsies may under-represent the complexity of a patient’s full clinical and biological presentation because of tumor heterogeneity.
CONTEXT
Key Objective
To what extent do polyclonal mechanisms of acquired resistance contribute to cancer progression in patients with fibroblast growth factor receptor (FGFR) 2–altered cholangiocarcinoma receiving FGFR-targeted therapy?
Knowledge Generated
For eight patients with FGFR2-altered cholangiocarcinoma receiving FGFR-targeted therapies, 31 acquired genetic alterations were identified in FGFR2 in six patients with detectable circulating tumor DNA.
Relevance
Upon treatment with FGFR-targeted therapy, polyclonal resistance is widespread and convergent on the kinase domain of FGFR2. Circulating tumor DNA analysis provides a sensitive and less invasive means to identify polyclonal mechanisms of resistance.
Circulating tumor DNA (ctDNA) has emerged as a noninvasive approach to monitor disease and longitudinally characterize tumor evolution. ctDNA can be used in the baseline evaluation of patients with biliary tract cancers, having recently shown to reveal detectable FGFR2 fusions in 2% of 139 samples from patients with IHC.9 Moreover, the analysis of ctDNA collected at progression on targeted therapy can enable the detection of acquired resistance mutations that emerge during treatment. Goyal et al10 used ctDNA, in combination with postprogression biopsies and rapid autopsy specimens, to identify 11 novel FGFR2 mutations at drug resistance in three patients treated on the phase II study of infigratinib.5,10 This same team subsequently evaluated the acquired resistance mutations in ctDNA from patients receiving TAS-120 after progression on the pan-FGFR inhibitors infigratinib and Debio1347, identifying shared and unique acquired resistance mutations in these patients depending on the drug received.11 More broadly within GI malignancies, in four patients with biliary cancers, Parikh et al12 observed a complex and often heterogeneous path to resistance to targeted therapies by comparing ctDNA with tumor biopsy. Motivated by these results, we reasoned that polyclonal acquired resistance may be a general feature of GI cancers when treated with targeted therapies and sought to more comprehensively characterize the prevalence and spectrum of acquired resistance mutations in the largest series to date of patients with FGFR-altered IHC.
MATERIALS AND METHODS
Patient Selection
Patients with IHC received treatment at Memorial Sloan Kettering (MSK) Cancer Center. Next-generation sequencing (NGS) of tumor tissue to identify potentially targetable alterations was completed. Eligible patients with FGFR2-altered IHC were identified using either a commercial NGS assay or MSK-IMPACT, an in-house FDA-authorized custom panel of up to 410 cancer genes, which included FGFR2. Patients were enrolled on Institutional Review Board (IRB)-approved clinical trials for treatment with FGFR2-targeted investigational agents with either Debio1347 or infigratinib.5,7 Eight patients were also consented to IRB-approved biospecimen protocols, allowing collection of additional blood samples. Patients underwent serial blood draws obtained longitudinally from before starting treatment, throughout treatment, and at the time of resistance.
ctDNA Analysis
Cell-free DNA (cfDNA) was extracted from blood plasma (QIAsymphony DSP Circulating DNA Kit, Qiagen) and sequenced using a custom, ultra-deep coverage NGS panel, MSK-ACCESS.13 The assay encompasses selected exons of 129 genes and introns of 10 genes harboring recurrent break points, including FGFR2, and uses duplex unique molecular indexes (UMIs) and dual index barcodes to minimize background sequencing errors and cross-contamination. Probes targeting all protein-coding exons and relevant introns of FGFR2 were spiked into the capture reaction at an equimolar ratio to ensure complete coverage. Sequencing data were analyzed using a custom bioinformatics pipeline that collapses UMI-tagged replicate read-pairs into consensus sequences and realigns the error-suppressed consensus reads. Somatic mutations were called using VarDict (v1.5.1),14 requiring at least three consensus reads with representation from both strands of the original cfDNA duplex. Mutations detected in each single time point were genotyped at the corresponding genomic position in all other plasma samples analyzed from the same patient. Variants present in a pool of unmatched plasmas from healthy donors were removed as systematic artifacts. Plasma cfDNAs were sequenced to an average depth of 23,901 × raw coverage (2,228 × unique coverage). All mutations and genomic rearrangements were manually reviewed (M.F.B. and J.P.).
RESULTS
Patient Characteristics
Of eight patients, baseline NGS tumor profiling of the primary tumor revealed that one patient’s tumor had an FGFR2 amplification, and the remaining seven patients’ tumors had FGFR2 fusions with diverse fusion partners (Table 1). Based on the presence of potentially targetable alterations, patients were enrolled in a clinical trial with a pan-FGFR inhibitor infigratinib (n = 7) or Debio1347 (n = 1). Two of eight patients experienced a partial response, 5 of 8 experienced stable disease, and 1 of 8 patient discontinued study treatment before undergoing follow-up imaging. Mean time on treatment was 5.4 months with a range of 0.8 to 10.6 months (Table 1). We observed no significant differences in the tumor genomic landscapes of patients with partial responses versus stable disease.
TABLE 1.
FGFR2 Alterations Present at Baseline and at Resistance in Each Patient Treated With an Investigational FGFR-Targeted Drug

Detection of Polyclonal Acquired Mutations in the FGFR2 Kinase Domain
Patients had serial blood samples taken throughout treatment and at disease progression while on study. Baseline structural alterations in FGFR2 were detected in plasma ctDNA in 7 of 8 patients, with allele fractions (AFs) of rearrangements ranging from 0.001 to 0.143 at progression (median 0.019). In 6 of 7 patients with detectable FGFR2 alterations, acquired genetic mutations were detected in FGFR2 that were not present in prior cfDNA samples. The remaining patient harbored an FGFR2-VCL fusion and remained on treatment for only 2.8 months. A total of 31 acquired mutations (all single-nucleotide substitutions) were identified at 12 different residues in FGFR2, with up to four distinct amino acid substitutions observed at the same site (Table 1).
Five of six patients harbored at least two independent FGFR2 mutations in ctDNA at progression, with as many as 13 mutations detected per patient (Fig 1A). When possible, mutations detected in the same patient were phased and confirmed to occur on distinct ctDNA molecules, suggesting convergent evolutionary paths to resistance in different subclones, as previously reported.10 In one patient with an FGFR2-KIAA1217 fusion, all three possible base substitutions were detected at the same genomic position, resulting in three different amino acid substitutions: p.V564I (AF = 0.015), p.V564L (AF = 0.034), and p.V564F (AF = 0.003). In patients with multiple blood samples analyzed during FGFR inhibitor therapy, resistance mutations could be detected prior to radiographic progression coincident with the re-emergence of the baseline FGFR2 fusion (Fig 1B). All but one of the 31 mutations detected in FGFR2 occurred in the kinase domain, further implicating a functional role for these mutations and evolutionary convergence of independent polyclonal resistance mechanisms (Figs 2A and 2B).
FIG 1.
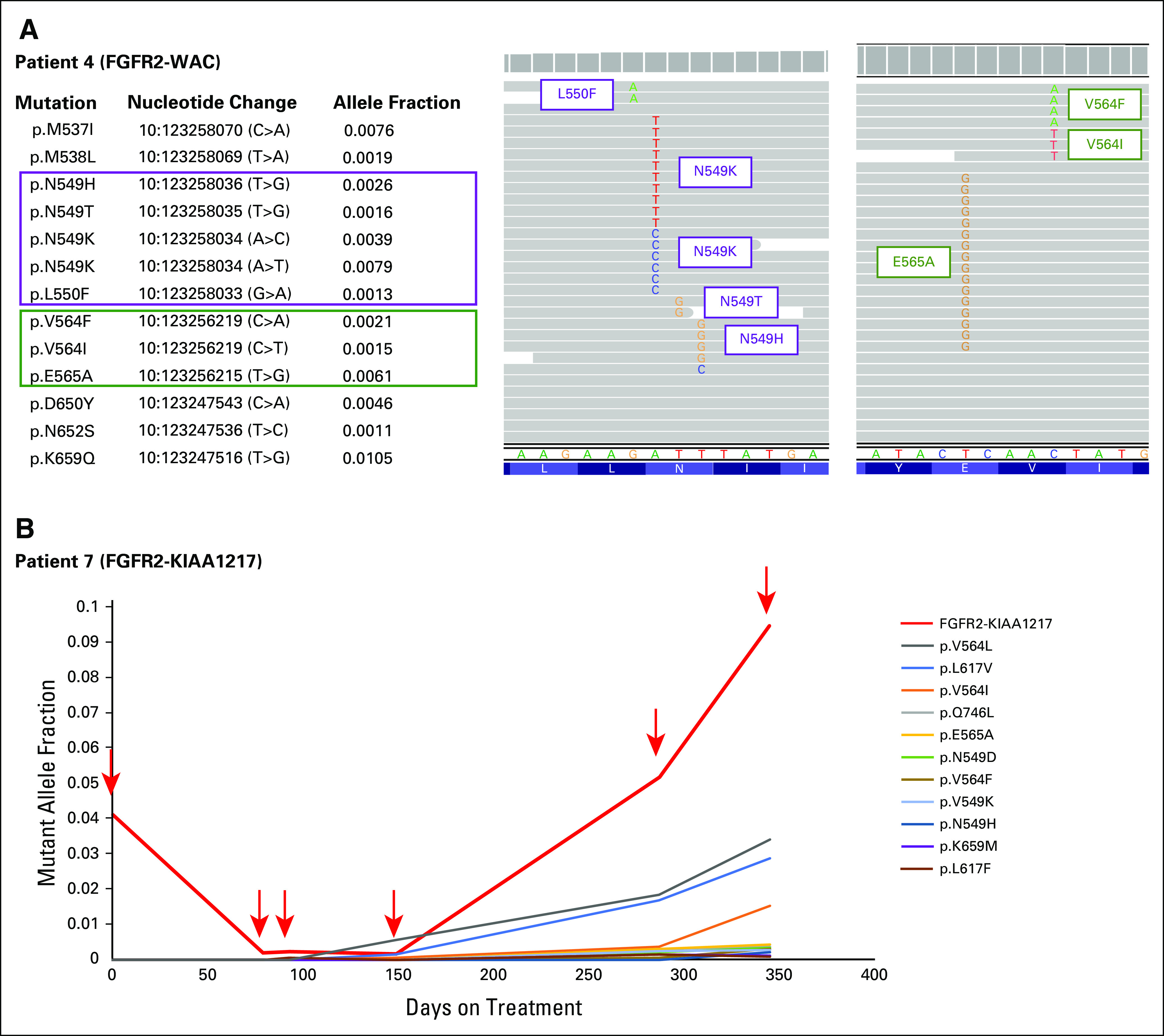
(A) Fibroblast growth factor receptor (FGFR) 2 acquired resistance mutations from a tumor harboring FGFR2-WAC fusion at baseline treated with an FGFR-targeted drug. Acquired mutations are present on distinct cfDNA molecules. (B) Development of FGFR2 acquired resistance mutations and re-emergence of FGFR2-KIAA1217 fusion, both detected prior to radiographic progression.
FIG 2.
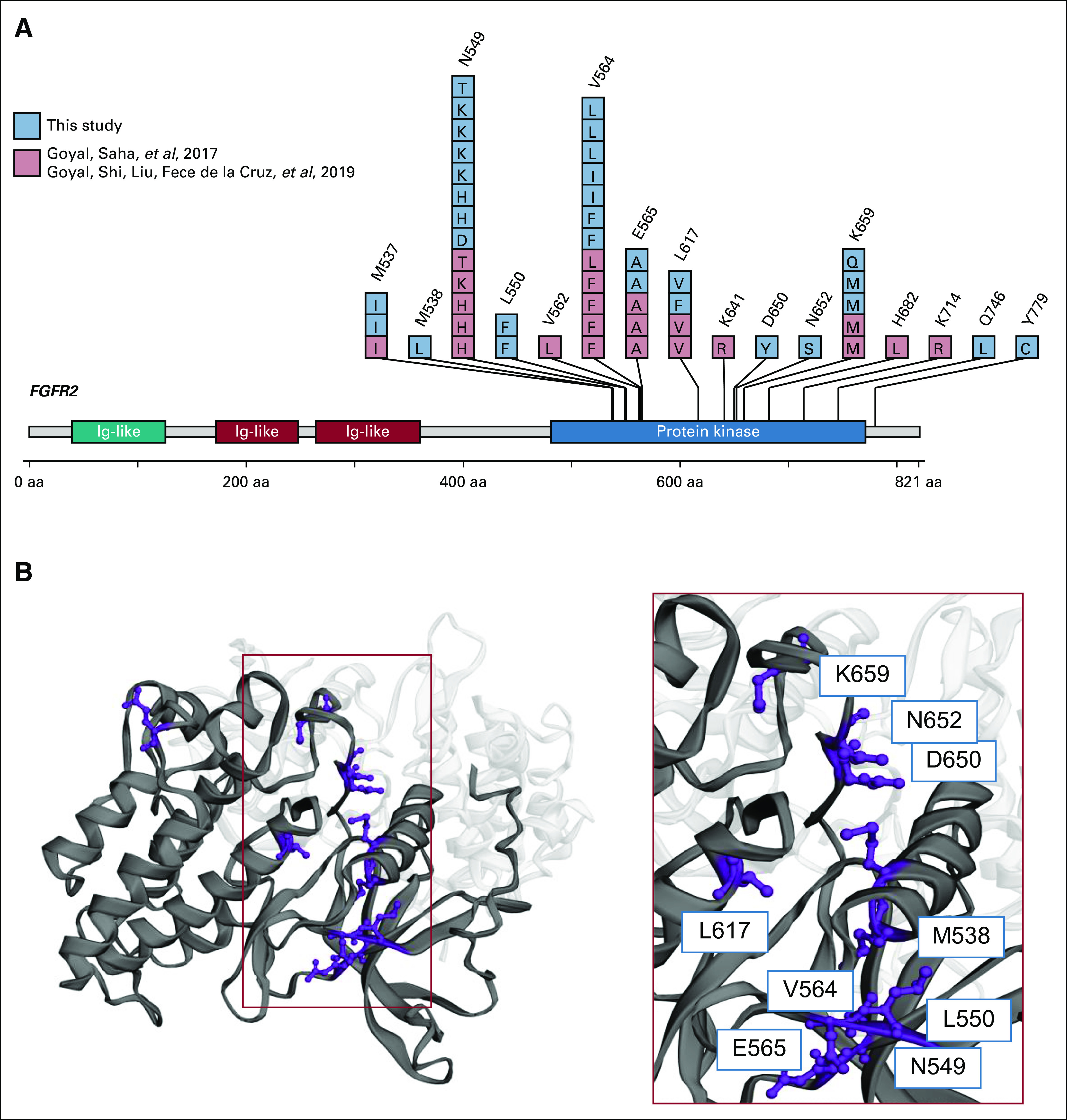
(A) Acquired resistance mutations identified from the six patients presented here and previously reported by Goyal et al,10 Cancer Discovery 2017, and Goyal et al,11 Cancer Discovery 2019. (B) Clustering of acquired resistance fibroblast growth factor receptor (FGFR) 2 mutations in the kinase domain from patients with FGFR-altered cholangiocarcinomas.
DISCUSSION
We report the identification of multiple novel FGFR2 alterations using a noninvasive approach to monitoring for acquired resistance in the largest series to date of patients with FGFR-altered IHC receiving targeted therapy. We identified that 6 of 8 patients had tumors that acquired mutations in FGFR2 in response to treatment with FGFR-targeted therapy, which were detectable in plasma ctDNA. Although the mutations identified here include the previously described gatekeeper mutation at FGFR2 V564 and mutations at the triad of residues N549, E565, and K741 that function as a molecular brake to FGFR activation, the diversity of FGFR2 alterations and multiple new FGFR2 mutations observed, even within individual patients, suggests an even more complex heterogeneity than previously described within each patient’s cancer contributing to resistance to FGFR-targeted therapy and convergence of these resistance mechanisms to the FGFR2 kinase domain (Fig 2A). Although these findings highlight the challenge in treatment of these patients, identification of novel acquired resistance mutations also provides targets for drug development, combination strategies, and sequential approaches of FGFR-targeted therapies. As noted by Goyal et al,11 Debio1347, infigratinib, and TAS-120 have unique resistance and sensitivity patterns to well-known acquired resistance mutations. The findings reported here provide further opportunities to sequence targeted treatments to overcome therapeutic resistance and also provide opportunities for further drug development to better target FGFR and its downstream effectors.
This study is exploratory and limited by sample size, variability in drugs and NGS assays used, and variable time points that were drawn. Serial biopsies and/or rapid autopsy analysis could also help further contextualize the heterogeneous tumor response and development of acquired resistance. Although ctDNA provides a less-invasive means of monitoring response and resistance, ctDNA analysis is limited by the quantity of tumor DNA shed into blood and the amount of blood that can be safely drawn. Since shedding of tumor DNA varies across patients and even within patients at different time points throughout treatment, negative results are challenging to interpret. Thus, even more mutations may exist below the assay limit of detection of approximately 0.1% variant allele frequency. Since MSK-ACCESS is also a targeted sequencing assay, this analysis may be insufficient to comprehensively investigate additional bypass mechanisms of resistance.
Nevertheless, our results clearly show that acquired FGFR2 mutations are highly prevalent at progression on FGFR inhibitor therapy and that genomic convergence of multiple independent mutations in the same is common. With the recent approval by the US Food and Drug Administration of pemigatinib, an FGFR inhibitor, as the first targeted treatment for cholangiocarcinoma, our results will inform strategies to monitor drug response and guide drug development efforts to combat acquired resistance.
In conclusion, ctDNA provides a noninvasive means to monitor for acquired resistance in IHC and captures the polyclonal mechanisms of resistance not easily identified from single-site biopsies.
PRIOR PRESENTATION
Presented in poster at ASCO Annual Meeting 2019, Chicago, IL, May 31-June 4.
SUPPORT
Supported in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Cycle for Survival, and the National Cancer Institute P30-CA008748.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Anna M. Varghese
Consulting or Advisory Role: Roche
Research Funding: Lilly, Verastem, BioMed Valley Discoveries, Bristol-Myers Squibb, Silenseed, Illumina
Travel, Accommodations, Expenses: Roche
Juber Patel
Research Funding: GRAIL
Patents, Royalties, Other Intellectual Property: I am an inventor on a pending patent application “Systems and Methods for Detecting Cancer via cfDNA Screening”
Yelena Y. Janjigian
Stock and Other Ownership Interests: Rgenix
Consulting or Advisory Role: Pfizer, Merck, Bristol-Myers Squibb, Merck Serono, Daiichi Sankyo, Ono Pharmaceutical, Michael J. Hennessy Associates, Jounce Therapeutics, Rgenix, Bayer, Imugene, AstraZeneca, Lilly, Paradigm Medical Communications LLC, Zymeworks, Seattle Genetics, PER (Physician's Education Resource), Merck Sharpe and Dohme Corp., MIL PeerView
Research Funding: Boehringer Ingelheim, Bayer, Lilly, Amgen, Roche, Genentech, Rgenix, Bristol-Myers Squibb, Merck
Gopakumar Iyer
Consulting or Advisory Role: Bayer, Janssen, Mirati Therapeutics
Research Funding: Mirati Therapeutics, Novartis, Debiopharm Group, Bayer
Brian Houck-Loomis
Patents, Royalties, Other Intellectual Property: BioLegend
James J. Harding
Consulting or Advisory Role: Bristol-Myers Squibb, CytomX Therapeutics, Lilly, Eisai, Imvax, Merck, Exelixis, Zymeworks, Adaptimmune
Research Funding: Bristol-Myers Squibb, Pfizer, Lilly, Novartis, Incyte, Calithera Biosciences, Polaris, Yiviva, Debiopharm Group, Zymeworks, Boehringer Ingelheim
Eileen M. O’Reilly
Consulting or Advisory Role: Merck, Agios, AstraZeneca, Bayer, BeiGene, Berry Genomics, Celgene, CytomX Therapeutics, Debiopharm Group, Eisai, Exelixis/Ipsen, Flatiron Health, Incyte, Janssen, LAM Therapeutics, Lilly, Loxo, Genentech/Roche, MINA, QED, RedHill Biopharma, Sillajen, SOBI, Yiviva, Autem, Gilead Sciences, Ipsen, Silenseed, Therabionics, twoXAR, Vector
Research Funding: AstraZeneca/MedImmune, Acta Biologica, Bristol-Myers Squibb, Celgene, Genentech, Halozyme, Halozyme, Roche, Silenseed
Ghassan K. Abou-Alfa
Consulting or Advisory Role: Celgene, Silenseed, Sillajen, Gilead Sciences, Agios, Bayer, Eisai, Ipsen, Merck Serono, AstraZeneca, CytomX Therapeutics, BeiGene, Genoscience Pharma, LAM Therapeutics, Lilly, Loxo, Mina, QED, RedHill Biopharma, SOBI, twoXAR, Yiviva, Flatiron Health, Roche/Genentech, Autem, Berry Genomics, Gilead Sciences, Incyte, TheraBionic, Vector
Research Funding: Bayer, Exelixis, CASI Pharmaceuticals, AstraZeneca, Bristol-Myers Squibb, Incyte, Agios, Polaris, Puma Biotechnology, QED
Travel, Accommodations, Expenses: Polaris
Maeve A. Lowery
Consulting or Advisory Role: Agios, Roche/Genentech
Travel, Accommodations, Expenses: Ipsen
Michael F. Berger
Consulting or Advisory Role: Roche
Research Funding: Grail
Patents, Royalties, Other Intellectual Property: Provisional patent pending for “Systems and Methods for Detecting Cancer via cfDNA Screening”
No other potential conflicts of interest were reported.
AUTHOR CONTRIBUTIONS
Conception and design: Anna M. Varghese, Ghassan K. Abou-Alfa, Maeve A. Lowery, Michael F. Berger
Administrative support: S. Duygu Selcuklu
Collection and assembly of data: Anna M. Varghese, Yelena Y. Janjigian, Fanli Meng, S. Duygu Selcuklu, Brian Houck-Loomis, James J. Harding, Ghassan K. Abou-Alfa, Maeve A. Lowery, Michael F. Berger
Data analysis and interpretation: Anna M. Varghese, Juber Patel, Yelena Y. Janjigian, Gopakumar Iyer, James J. Harding, Eileen M. O’Reilly, Ghassan K. Abou-Alfa, Michael F. Berger
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
REFERENCES
- 1.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma N Engl J Med 381338–3482019 [DOI] [PubMed] [Google Scholar]
- 2.Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention Clin Cancer Res 244154–41612018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma Hum Pathol 451630–16382014 [DOI] [PubMed] [Google Scholar]
- 4.Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing Oncologist 19235–2422014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma J Clin Oncol 36276–2822018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020 doi: 10.1016/S1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voss MH, Hierro C, Heist RS, et al. A phase I, open-label, multicenter, dose-escalation study of the oral selective FGFR inhibitor Debio 1347 in patients with advanced solid tumors harboring FGFR gene alterations Clin Cancer Res 252699–27072019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meric-Bernstam F, Arkenau H, Tran B, et al. O-00—Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors Ann Oncol 29v100–v1102018. (suppl 5) [Google Scholar]
- 9.Mody K, Kasi PM, Yang J, et al. Circulating tumor DNA profiling of advanced biliary tract Cancers 1–92019 [DOI] [PubMed] [Google Scholar]
- 10.Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma Cancer Discov 7252–2632017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion–positive intrahepatic cholangiocarcinoma Cancer Discov 91064–10792019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers Nat Med 251415–14212019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cocco E, Schram AM, Kulick A, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation Nat Med 251422–14272019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108-e108. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]