

PURPOSE
We hypothesized that circulating tumor DNA (ctDNA) molecular residual disease (MRD) analysis without prior mutational knowledge could be performed after neoadjuvant chemotherapy to assess oligometastatic colorectal cancer (CRC) treated surgically with curative intent. We also investigated urine as an alternative analyte for ctDNA MRD detection in this nongenitourinary setting.
PATIENTS AND METHODS
We applied AVENIO targeted next-generation sequencing to plasma, tumor, and urine samples acquired on the day of curative-intent surgery from 24 prospectively enrolled patients with oligometastatic CRC. Age-related clonal hematopoiesis was accounted for by removing variants also present in white blood cells. Plasma and urine ctDNA MRD were correlated with tumor cells detected in the surgical specimen, and adjuvant treatment strategies were proposed based on ctDNA-inferred tumor mutational burden (iTMB) and targetable alterations.
RESULTS
Seventy-one percent of patients were treated with neoadjuvant chemotherapy. Tumor-naive plasma ctDNA analysis detected MRD at a median level of 0.62% with 95% sensitivity and 100% specificity, and 94% and 77% sensitivity when only considering patients treated with neoadjuvant chemotherapy and putative driver mutations, respectively. In urine, ctDNA MRD detection specificity remained high at 100%, but sensitivity decreased to 64% with median levels being 11-fold lower than in plasma (P < .0001). Personalized ctDNA MRD oncogenomic analysis revealed 81% of patients might have been candidates for adjuvant immunotherapy based on high iTMB or targeted therapy based on actionable PIK3CA mutations.
CONCLUSION
Tumor-naive plasma ctDNA analysis can sensitively and specifically detect MRD in patients with oligometastatic CRC after neoadjuvant chemotherapy. Urine-based ctDNA MRD detection is also feasible; however, it is less sensitive than plasma because of significantly lower levels. Oligometastatic patients with detectable MRD may benefit from additional personalized treatment based on ctDNA-derived oncogenomic profiling.
INTRODUCTION
Colorectal cancer (CRC) is the second most common cause of cancer death worldwide.1 Although patients with localized or locally advanced disease typically have a high rate of long-term survival, those with distant metastatic disease have a 5-year survival rate of only 14%.2 Still, with advances in radiologic imaging and localized and systemic treatment modalities, some of these patients with oligometastatic disease can now be treated with curative intent to achieve survival outcomes typical of their more localized counterparts.3-13 The majority of these patients, however, relapse,3,10 exposing an unmet need for dependable biomarkers to optimally select patients for potentially curative treatment in this oligometastatic disease setting and to precisely monitor their treatment responses.
CONTEXT
Key Objective
Can plasma and urine circulating tumor DNA (ctDNA) analysis detect molecular residual disease (MRD) in patients with oligometastatic colorectal cancer (CRC) after neoadjuvant chemotherapy on the day of curative-intent surgery and inform adjuvant treatment personalization?
Knowledge Generated
Ultra-deep targeted sequencing of plasma cell-free DNA, without prior knowledge of tumor mutations, detected ctDNA MRD with high sensitivity (95%) and specificity (100%) in patients with oligometastatic CRC undergoing curative-intent surgery. Urine ctDNA MRD was also detected in this nongenitourinary cancer type, albeit with reduced sensitivity (64%). ctDNA MRD oncogenomic analysis enabled tumor mutational burden inference and targetable mutation identification, which could help personalize adjuvant systemic therapy in the future.
Relevance
We demonstrate that ctDNA analysis can detect MRD from plasma and urine in oligometastatic CRC, and that further oncogenomic analysis could inform adjuvant treatment strategies including immunotherapy and targeted systemic therapy.
Plasma circulating tumor DNA (ctDNA) has been shown to be capable of detecting molecular residual disease (MRD) in several cancer types including CRC.14-19 However, these applications of plasma ctDNA MRD detection after curative-intent treatment have focused exclusively on nonmetastatic disease. Oligometastatic cancer, arguably, represents a greater clinical conundrum and potentially a more useful avenue for the application of ctDNA MRD technology. Additionally, all published ctDNA MRD detection methodologies require that the hypothesis space be limited to tumor-specific mutations by initially sequencing tumor or pretreatment plasma (tumor-informed approach).16 This is a practical limitation as patients are often seen in a different practice setting for their initial diagnosis than their specialized treatment for oligometastatic disease.20 Finally, despite its potential logistical advantages, urine-based ctDNA MRD analysis has not been investigated before for nongenitourinary (GU) malignancies. Therefore, we strive here to fill the following unmet clinical needs: (1) plasma ctDNA MRD detection without prior knowledge of tumor mutations (tumor-naive approach), (2) plasma ctDNA MRD detection in the oligometastatic setting, and (3) investigation of urine-based ctDNA MRD detection for this non-GU malignancy.
There is also significant debate regarding the administration and timing of systemic therapy for oligometastatic CRC.5,11,13,21-23 In the absence of reliable biomarkers, it is challenging to decide between the different options of surgery alone, surgery plus chemotherapy, and the timing of chemotherapy when offered (neoadjuvant v adjuvant v both). Additionally, more modern systemic modalities such as immunotherapy and targeted inhibitors have not yet been used in the oligometastatic setting after curative-intent local treatment. ctDNA MRD analysis could help fill these clinical gaps by providing oncogenomic data to help guide the timing, strength, regimentation, and type of systemic therapy, thus optimizing clinical decision making for oligometastatic disease in a more personalized manner.
In this prospective cohort study, we used the AVENIO platform, which is based on Cancer Personalized Profiling by deep Sequencing (CAPP-Seq)24 with integrated digital error suppression (iDES),25 for tumor-naive plasma ctDNA MRD detection and mutational analysis. We aimed to determine whether ctDNA analysis using this ultra-sensitive method can reliably detect MRD in patients with oligometastatic CRC on the day of curative-intent surgery. We also investigated urine as an alternative cell-free DNA (cf-DNA) analyte for ctDNA MRD detection. Finally, we addressed the hypothesis that ctDNA MRD oncogenomic analysis can help guide precision adjuvant treatment approaches such as targeted inhibitors or immunotherapy in a personalized fashion for patients with oligometastatic CRC.
PATIENTS AND METHODS
We prospectively enrolled patients with oligometastatic CRC between September 2018 and November 2019 onto this single-institution prospective cohort study assessing ctDNA as a liquid biopsy biomarker for solid tumor response to therapy (Data Supplement). Patients with oligometastatic CRC had blood and urine samples collected on the day of surgery preoperatively, with tissue collected shortly thereafter at the time of curative-intent resection. All samples were collected with informed consent and institutional review board approval in accordance with the Declaration of Helsinki. Following DNA extraction, all samples were processed using the Roche AVENIO next-generation sequencing (NGS) assay with the Surveillance hybrid-capture panel (Data Supplement), and then sequenced on an Illumina HiSeq 4000. All tumor tissue was measured and examined by a board-certified surgical pathologist who was blinded to the ctDNA and tumor genomic data. Patients continued clinical follow-up as per the standard-of-care with the managing clinicians blinded to the ctDNA and tumor genomic data. This study was approved by the Washington University School of Medicine institutional review board (protocol numbers 201107221 and 201903142). See the Data Supplement for methodologic details.
RESULTS
Patient Characteristics and Samples Collected
We profiled a total of 82 plasma, peripheral blood mononuclear cell (PBMC), tumor tissue, and urine samples acquired on the day of curative-intent surgery from 24 prospectively enrolled patients with oligometastatic CRC (Data Supplement). Median age was 57 years, and the median study follow-up time was 12 months. Most patients (71%) were treated with neoadjuvant chemotherapy before surgery and had oligometastasis to the liver (92%). Fourteen patients (58%) had metachronous and the remainder had synchronous oligometastatic disease.
Ultra-deep NGS Detected ctDNA MRD and Clonal Hematopoiesis in Plasma
We sequenced and analyzed plasma cfDNA collected on the day of curative-intent surgery. For ultra-deep targeted sequencing, we used the Roche AVENIO Surveillance panel, which spans 198 kb and encompasses 197 genes frequently mutated in lung cancer and CRC (Data Supplement). Indicative of high performance, 95% of patients with CRC in The Cancer Genome Atlas (TCGA)26,27 had mutations detectable within the genomic space covered by our targeted sequencing panel (Data Supplement), with a median of six single-nucleotide variants (SNVs) detected per patient (Data Supplement). In our cohort, we detected 152 somatic mutations in plasma cfDNA from 24 patients, which ranged in variant allele frequency (VAF) between 0.02% and 38.31% (Fig 1A).
FIG 1.
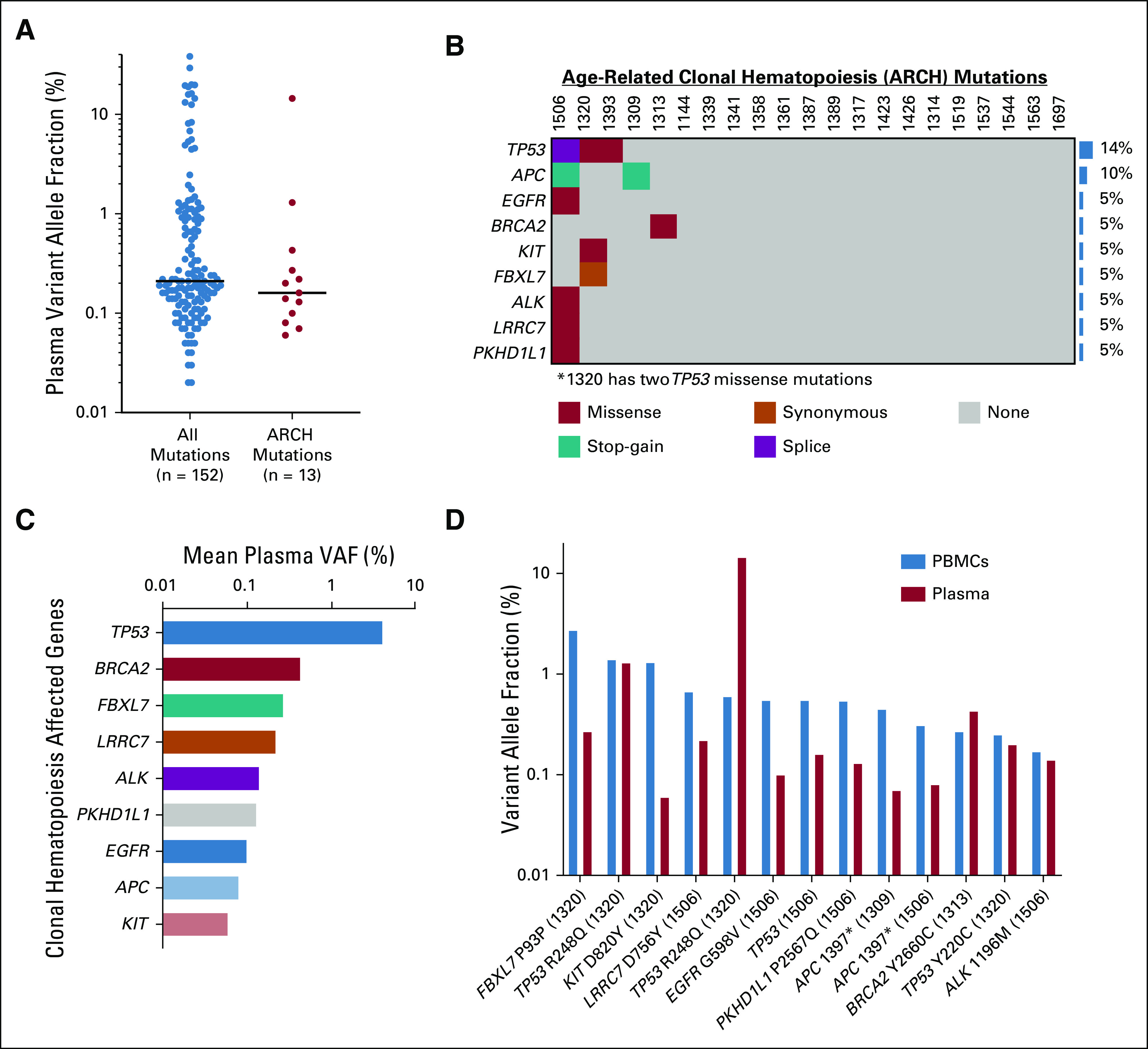
PBMC-derived mutations in plasma cell-free DNA. (A) Plasma VAFs for all detected somatic mutations (n = 152) and those also detected in PBMCs, referred to as ARCH mutations (n = 13). Horizontal black bars indicate median values. (B) Comutation plot illustrating these 13 ARCH mutations among 21 patients with detectable plasma ctDNA. Each column represents the day-of-surgery blood draw data from a single patient. Colors represent mutation type, and CH rate at the gene level is depicted by the bar graph to the right. (C) Genes in the NGS panel affected by clonal hematopoiesis with associated mean VAFs detected in plasma cfDNA on the day of surgery. (D) Comparison of VAFs in plasma cfDNA for each CH variant detected on the day of surgery with VAFs of the same variants detected in PBMCs. Each variant has a column representing its VAF in PBMCs (blue) and in plasma cfDNA (red). Some variants were detected in different patients with different VAFs, so the patient number in which each CH variant was detected is depicted in parentheses at the bottom of the graph. Patient 1506's TP53 mutation is a splice variant, as such there is no amino acid change listed. ARCH, age-related clonal hematopoiesis; cfDNA, cell-free DNA; CH, clonal hematopoiesis; ctDNA, circulating tumor DNA; MRD, molecular residual disease; NGS, next-generation sequencing; PBMC, peripheral blood mononuclear cell; VAF, variant allele fraction.
For each patient in our cohort, we also sequenced matched PBMCs to an average unique depth of 4,197×. Notably, we observed that age-related clonal hematopoiesis (ARCH),28-32 also known as clonal hematopoiesis of indeterminate potential (CHIP), accounted for 13 of the 152 (9%) somatic mutations we detected in plasma cfDNA (Fig 1A; Data Supplement), which would have confounded plasma ctDNA quantitation in 21% of cases in our cohort (Fig 1B). All 13 of these mutations were indexed within the catalog of somatic mutations in cancer (COSMIC),33 highlighting the challenge of excluding them from our plasma ctDNA results without dedicated high-depth PBMC sequencing. The gene most commonly mutated in both PBMCs and plasma was TP53 (Fig 1B), with these mutations also having a higher mean VAF in cfDNA than other genetic mutations shared between PBMCs and plasma (Fig 1C). The median VAF of PBMC-derived variants detected in plasma was 0.16%, lower than in matched PBMCs in 85% of cases (0.55% v 0.16%, P = .005; Fig 1D; Data Supplement). These data suggest that ARCH variants can confound plasma ctDNA results when employing ultra-deep sequencing but can be adequately accounted for by sequencing matched PBMCs to high depth.
We next analyzed ctDNA MRD within the plasma cfDNA compartment on the day of surgery without any prior knowledge of tumor mutations (tumor-naive approach). To do this, we performed ultra-deep sequencing with the AVENIO Surveillance panel of plasma cfDNA, achieving an average deduplicated sequencing depth of 8,372×. We then applied the AVENIO variant-caller, which is based on iDES-enhanced CAPP-Seq,24,25 to detect single-nucleotide variants. Following mutational calling, we removed variants also detected in PBMCs (Fig 1; Data Supplement), thus filtering out those resulting from ARCH or other PBMC-specific somatic variants. The AVENIO ctDNA variant-caller also bioinformatically filters out nonreference bases present in databases of healthy individuals.34 As such, both potential ARCH and single-nucleotide polymorphisms were accounted for.
We detected plasma ctDNA MRD in 21 of 22 (95%) patients with residual disease detected by surgical pathology. We detected on average seven mutations per patient with a median ctDNA level of 0.62% (Fig 2A; Data Supplement), approximately 6-fold lower than previously observed in advanced-stage CRC,35 and consistent with what we previously observed for localized lung cancer after curative-intent treatment.15 Among the mutations we detected in plasma ctDNA were nonsynonymous mutations in the candidate driver genes TP53, APC, KRAS, PIK3CA, FBXW7, SOX9, and NRAS (Fig 2A; Data Supplement). We also observed a significant correlation between tumor size and ctDNA levels in plasma (Fig 2B). Similar to ctDNA MRD studies in lung cancer,15,36 most mutations that we identified were not drivers, but rather appeared to be passenger mutations that were either silent or without known functional impact in CRC (Fig 2C; Data Supplement). When these nondriver mutations were discounted, the plasma ctDNA detection rate in our cohort decreased significantly from 95% to 77% (P = .04; Fig 3A), demonstrating the importance of including both mutation types to achieve high sensitivity.
FIG 2.

Plasma ctDNA analysis in patients with oligometastatic colorectal cancer. (A) Comutation plot based on day-of-surgery plasma ctDNA analysis of patients with oligometastatic CRC. Each column represents data from a single patient with detectable ctDNA (n = 21). Mean variant allele fraction in cfDNA (ctDNA %) is shown in the top bar graph, with the median indicated by a dashed line. The mutation rate at the gene level is depicted by the bar graph to the right. Nonsynonymous mutations in candidate driver genes are shown in descending order of prevalence in the comutation plot. The number of other (likely passenger) mutations detected is indicated in the bottom heat map. (B) Correlation of plasma ctDNA levels (mean VAF) with tumor size depicted as the SLD. (C) Pie chart showing the number and type of mutations detected in plasma cfDNA from all patients on the day of surgery. Variants are stratified based on the number of candidate driver mutations, other mutations, CH in candidate driver genes, and CH in other genes detected in plasma cfDNA. cfDNA, cell-free DNA; CH, clonal hematopoiesis; cm, centimeters; CRC, colorectal cancer; ctDNA, circulating tumor DNA; SLD, sum of longest diameters; VAF, variant allele fraction.
FIG 3.
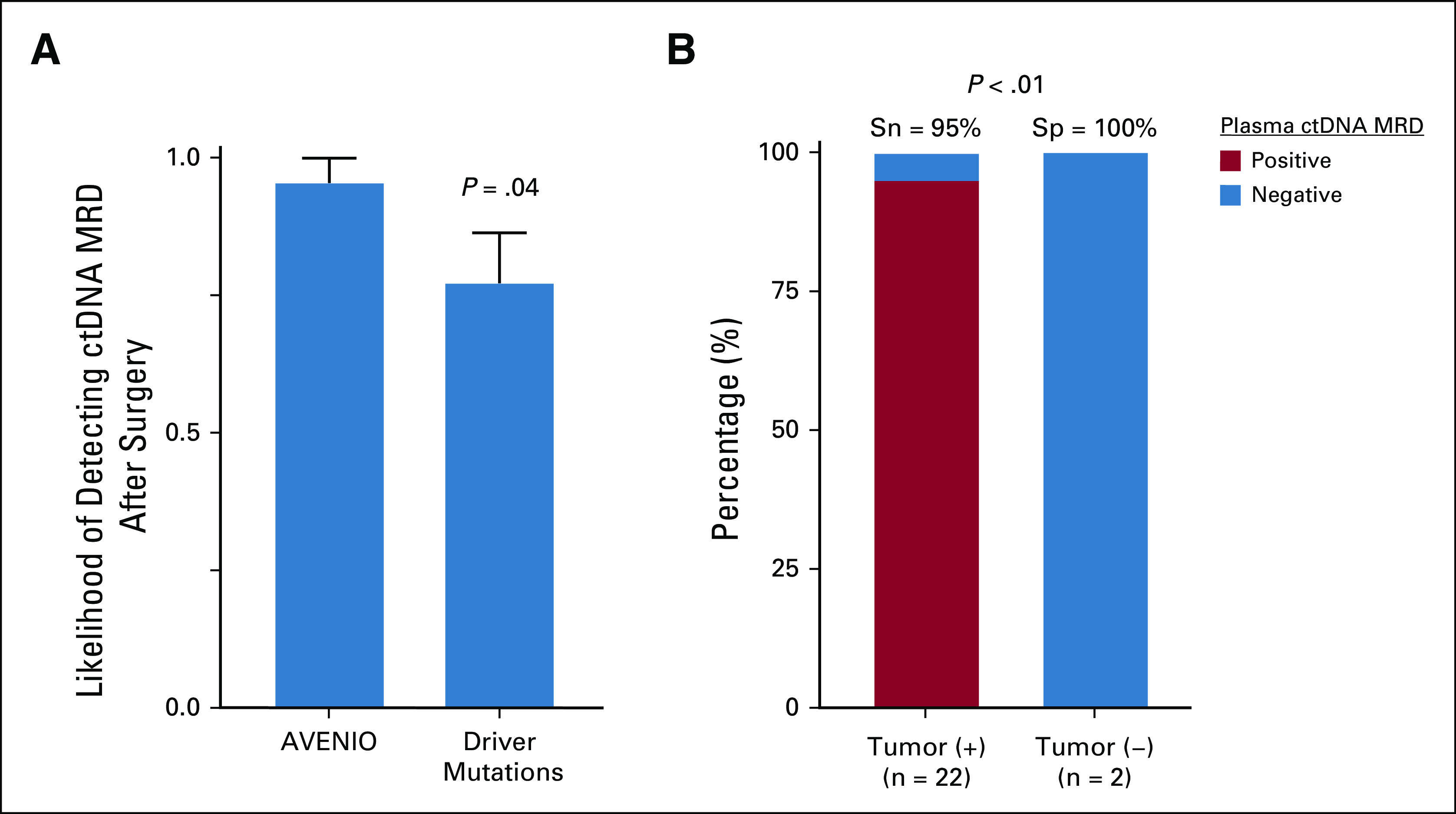
Plasma ctDNA MRD detection predicts residual disease in the surgical specimen. (A) Likelihood of detecting plasma ctDNA MRD on the day of surgery by tracking all mutations within the AVENIO target panel space or tracking only driver mutations (mean ± SEM). This figure illustrates data from the 22 patients with detectable tumor cells in the surgical specimen. P value was calculated by Student t-test. (B) Stacked bar plot depicting the sensitivity and specificity of tumor-naive plasma ctDNA MRD detection on the day of surgery according to the presence of viable tumor cells in the surgical specimen. P value was calculated by Fisher's exact test. ctDNA, circulating tumor DNA; MRD, molecular residual disease; Sn, sensitivity; Sp, specificity; Tumor (−), no viable tumor cells; Tumor (+), tumor cells present.
Among patients with tumor cells present in their surgical resection specimens, 95% had detectable plasma ctDNA MRD (Fig 3B; Data Supplement). Considering only those treated with neoadjuvant chemotherapy with persistent tumor cells detected by surgical pathology, 94% had detectable plasma ctDNA MRD prior to metastasectomy (Data Supplement). Among patients with undetectable plasma ctDNA MRD, one had < 10% tumor cells detected in his surgical specimen, whereas the others had no pathologic evidence of residual disease (Data Supplement).
Targeted NGS Detected ctDNA in Urine at Lower Levels Than in Plasma
Next, we asked whether urine-based analysis could also detect oligometastatic CRC ctDNA MRD. To enrich for plasma-derived cfDNA in urine, we applied a chromatography-based protocol using Q-Sepharose to isolate urine cfDNA,37,38 followed by a custom magnetic bead-based protocol to enrich for 70-450 base-pair fragments (Data Supplement). We sequenced the resulting enriched urine cfDNA using the AVENIO Surveillance panel to an ultra-high average deduplicated depth of 5,765×, which enabled detection of ctDNA MRD from the urine of 14 patients, with a median detectable ctDNA level of 0.05%, 11-fold lower than in plasma (P < .0001; Fig 4A; Data Supplement). We furthermore found that cfDNA fragments containing mutations were shorter in urine with an average size of 150.1 bp, compared to 180.2 bp in plasma (P < .0001; Data Supplement).
FIG 4.

Urine-based ctDNA MRD detection in oligometastatic CRC. (A) Comparison between detectable ctDNA MRD levels in plasma (n = 21) and urine (n = 14) from the day of curative-intent surgery for oligometastatic CRC. Each dot represents ctDNA mean VAF data from a single patient. Horizontal black bars represent median values. P value was calculated by the Mann-Whitney U test. (B) Comutation plot for 14 patients with detectable urine ctDNA MRD. (C) Stacked bar plot depicting the sensitivity and specificity of urine-based ctDNA MRD detection on the day of surgery according to the presence of viable tumor cells in the surgical specimen. (D) Comutation plot illustrating mutations in urine cfDNA that are in common with those in matched PBMCs on the day of surgery. For both B and D, each column represents data from a single patient with IDs in the row above the plot, colors represent mutational type, and gene-level mutation rates are depicted by bar graphs to the right. cfDNA, cell-free DNA; CRC, colorectal cancer; ctDNA, circulating tumor DNA; MRD, molecular residual disease; PBMCs, peripheral blood mononuclear cells; Sn, Sensitivity; Sp, Specificity; Tumor (−), no viable tumor cells; Tumor (+), tumor cells present; VAF, variant allele fraction.
A total of 44 mutations identified in tumor or plasma were also detected in urine on the day of surgery (Fig 4B; Data Supplement). The average number of SNVs detected in urine was 3-fold lower than in plasma (P = .03; Data Supplement). Using residual tumor cells in the surgical sample as our gold standard, the sensitivity of urine-based ctDNA MRD detection was 64%, whereas the specificity was 100% (Fig 4C). We also observed that 9% of mutations in urine cfDNA, all involving the TP53 gene, were detectable in PBMCs, consistent with a hematopoietic origin (Fig 4D; Data Supplement). Thus, ctDNA MRD can be measured from urine, with a subset shared with PBMCs; however, levels and sensitivity were lower than from plasma.
Targeted NGS in Tumor Tissue Compared With Plasma and Urine
Ten patients had their liver metastases available for analysis using the Roche AVENIO Surveillance panel, with 64% of identified genetic alterations also detected in matched plasma cfDNA samples (Data Supplement). Tumoral SNVs detected in both tissue and plasma had significantly higher VAFs when compared with SNVs solely detected in tumor tissue (P = .0002; Data Supplement). As expected, the mutant allele fractions of detected variants were significantly higher in tumor tissue when compared with plasma or urine cfDNA (P = .0007 and P < .0001, respectively; Data Supplement).
Plasma ctDNA MRD as an Oncogenomic Biomarker for Treatment Personalization
Immune checkpoint blockade has shown efficacy in the metastatic CRC non–curative-intent setting,39 and pembrolizumab was recently approved by the US Food and Drug Administration for patients with unresectable or metastatic solid tumor malignancies with high tumor mutational burden (TMB) (≥ 10 nonsynonymous mutations per megabase).40,41 We and others also previously showed that TMB can be inferred from hybrid-capture plasma cfDNA analysis in patients with non–small-cell lung cancer.15,42 To determine if similar methodology can be applied to CRC, we derived an equation relating nonsynonymous mutational burden in the AVENIO targeted sequencing space to exome-wide TMB using CRC data from TCGA26,27 (Fig 5A).
FIG 5.
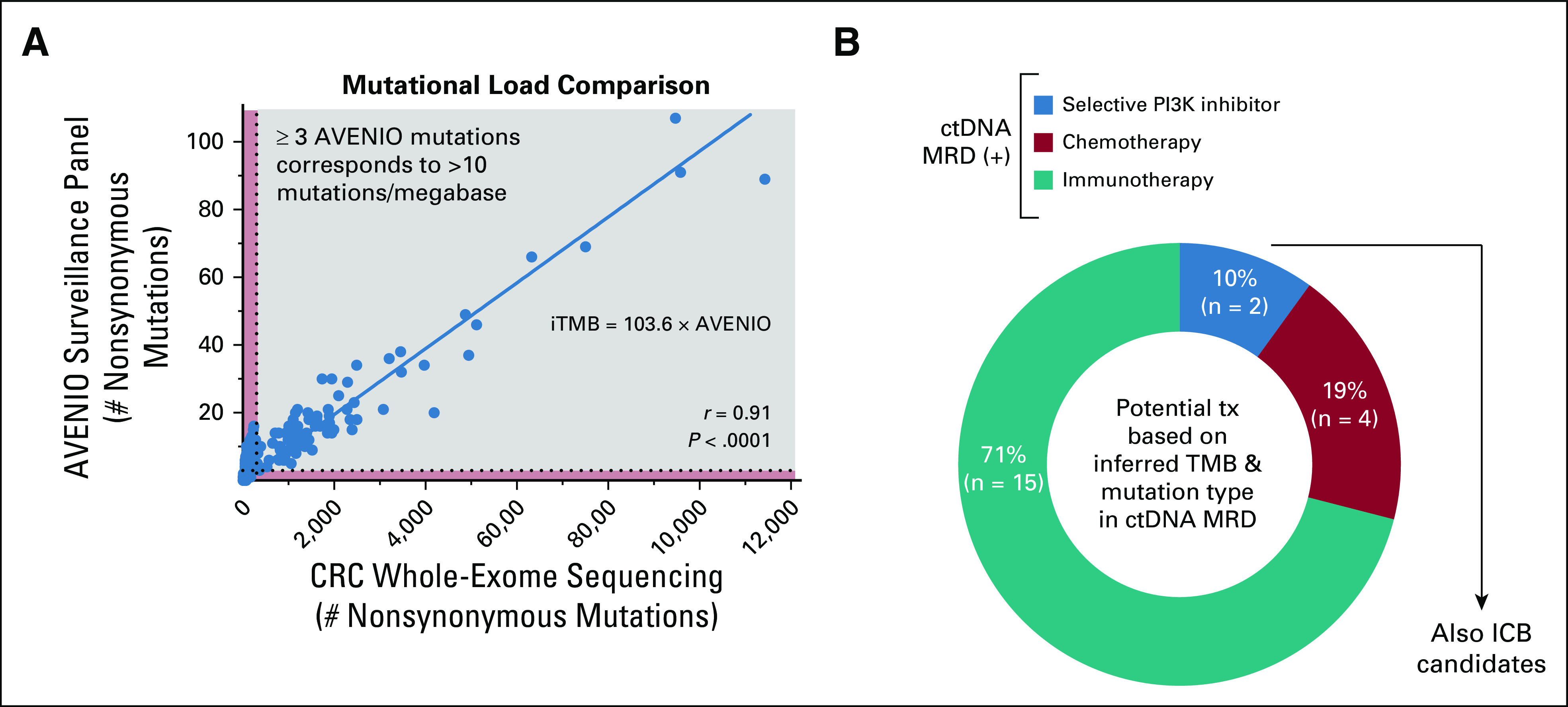
Plasma ctDNA MRD as a biomarker for adjuvant treatment personalization. (A) Comparison of mutational load between CRC WES and the AVENIO surveillance panel. Mutational data were obtained from 528 patients with CRC profiled by WES by TCGA. Linear regression was performed, with the shown equation indicating the inferred exome-wide TMB (iTMB) based on the number of nonsynonymous mutations detected within the surveillance panel. Specifically, > 3 AVENIO surveillance panel nonsynonymous mutations correspond to iTMB > 300 or > 10 nonsynonymous mutations per megabase in the exome. (B) Potential personalized adjuvant treatment strategies based on oncogenomic data for patients in this cohort with detectable plasma ctDNA MRD. Patients with targetable PIK3CA mutations would be offered selective PI3K inhibitors, those with high iTMB would be offered immune checkpoint blockade, and the remainder would be offered chemotherapy. In our cohort, patients with targetable PIK3CA mutations would also be ICB candidates based on high iTMB. CRC, colorectal cancer; ctDNA, circulating tumor DNA; ICB, immune checkpoint blockade; iTMB, inferred tumor mutational burden; MRD, molecular residual disease; TCGA, The Cancer Genome Atlas; WES, whole-exome sequencing.
Applying this equation to our cohort, we determined that 81% of plasma ctDNA MRD-positive patients had inferred tumor mutational burden (iTMB) levels exceeding 300 nonsynonymous variants (> 10 mutations per megabase; Data Supplement). Plasma iTMB demonstrated reasonable concordance with tumor tissue and urine cfDNA-based analysis too (Data Supplement) and could have potentially been applied to patients in our cohort to identify candidates for adjuvant immune checkpoint blockade (Fig 5B; Data Supplement).
We next queried our cohort for potentially actionable mutations in plasma ctDNA by reviewing the Clinical Interpretation of Variants in Cancer (CIViC)43 database (Data Supplement). This revealed that 10% of patients with detectable plasma ctDNA MRD also had a targetable PIK3CA mutation (Data Supplement).
Applying both of these oncogenomic analysis methodologies, we hypothesize that 10% of ctDNA MRD-positive patients in our cohort could have been candidates for selective PI3K inhibition or early immunotherapy, 71% might have been candidates for early immune checkpoint blockade, and the remaining 19% would have been candidates for further chemotherapy (Fig 5B). This has the potential to open the door to personalized precision adjuvant systemic therapy for patients with oligometastatic CRC.
Plasma ctDNA MRD as a Potentially Prognostic Biomarker
Our study was underpowered for survival analysis and the median follow-up time was only 12 months; however, as part of an exploratory analysis, we assess disease-free survival (DFS) for patients based on ctDNA MRD status. Although no statistically significant differences in DFS were noted, there was a trend toward shorter DFS for patients with detectable ctDNA MRD before metastasectomy. The median DFS was 9 months for patients with positive ctDNA MRD and was not reached for those with negative ctDNA MRD (hazard ratio, 3.4; 95% CI, 0.92 to 12.3; P = .2; Data Supplement).
DISCUSSION
Using a prospective study design, our data show that MRD can be reliably detected through tumor-naive plasma ctDNA analysis in patients with oligometastatic CRC. In addition to overcoming the practical limitation of having access to biopsy tissue, a tumor-naive approach may better account for geographic tumor heterogeneity.17,36 Although ctDNA MRD has been measured in several tumor types after curative-intent treatment,15-19 to our knowledge, this is the first study applying ctDNA MRD analysis to oligometastatic disease and without any prior knowledge of tumor mutations.
Given the prevalence of ARCH reported in other plasma ctDNA studies,29,34,44,45 we also sequenced matched PBMCs to an average unique depth > 4,000×. Notably, the sequencing panel we used does not include the canonical ARCH genes DNMT3A, TET2, or ASXL128-32; however, it was recently validated for ARCH/CHIP analysis by Yaung et al. Similar to Yaung et al34 who observed BRCA2 as the second most commonly mutated gene in their CHIP analysis, we also observed PBMC mutations in noncanonical ARCH genes. These mutations present in both PBMCs and plasma, and also indexed in COSMIC,33 would have been counted as ctDNA variants in the absence of high-depth PBMC sequencing. This finding emphasizes the importance of PBMC deep-sequencing to discriminate between ctDNA MRD mutations and confounding PBMC-derived variants.
Despite a recurrence risk of over 50% after surgical resection,3,10 the role for adjuvant systemic therapy in patients with oligometastatic CRC remains controversial.5,11,13,21-23 Our data suggest that ctDNA MRD oncogenomic analysis could potentially help clarify decision making regarding adjuvant treatment. We inferred tumor mutational burden from ctDNA results and queried potentially targetable mutations in the CIViC43 database. In this way, we showed that 10% of patients with detectable plasma ctDNA MRD in our study could have been candidates for selective PI3K inhibition, and 81% might have been candidates for adjuvant immune checkpoint blockade. This treatment paradigm, guiding adjuvant therapy based on oncogenomic ctDNA MRD features, will need to be tested prospectively in a clinical trial. Future studies may include ctDNA measurement before and after metastasectomy and randomly assign patients to receive additional treatment following resection according to ctDNA MRD detection and oncogenomic analysis.
We also identified a strong correlation between undetectable ctDNA MRD and pathologic complete response to neoadjuvant chemotherapy. If validated, future studies should use ctDNA MRD after neoadjuvant treatment to inform the optimal duration and regimentation of neoadjuvant chemotherapy and further optimize surgical timing. Future studies should also expand upon ours by measuring ctDNA at multiple timepoints during neoadjuvant treatment, and correlate ctDNA dynamics to therapeutic response. In this way, it may be possible to predict responses to neoadjuvant chemotherapy even earlier through ctDNA analysis, for example, by using the continuous individualized risk index.46 Furthermore, it is important that the ctDNA technology we used here be investigated in the postoperative setting to better inform the selection of adjuvant treatment in oligometastatic CRC.
Unique to previous ctDNA MRD studies in non-GU malignancies, we also explored urine as a biofluid analyte. Although we achieved ultra-high unique sequencing depths, the detection sensitivity of ctDNA MRD from urine was lower than from plasma, likely related to the 11-fold lower ctDNA levels that we observed. Interestingly, the rate of shared mutations with PBMCs relative to the total number of mutations detected in urine was similar to plasma, suggesting that PBMC-derived variants can also be detected in urine. Overall, the feasibility of urine-based ctDNA MRD detection for a non-GU cancer type is novel. However, our data suggest that urine is an inferior biofluid compared to plasma for ctDNA MRD detection in patients with oligometastatic CRC.
Limitations of our study include a modestly sized cohort with ctDNA assessment at a single timepoint and the inability to sequence paired tumor tissue for all patients. The recruited cohort was also heterogeneous, including patients with metachronous and synchronous oligometastatic disease; most patients received neoadjuvant chemotherapy but some did not. Still, we believe it was important to allow this heterogeneity as oligometastatic CRC is intrinsically a heterogeneous entity in terms of its presentation and treatment approaches.5,8,12,13 Although our study was underpowered for survival analysis, we observed a trend toward worse DFS in patients with detectable ctDNA MRD. This finding needs to be validated in larger studies. Finally, we observed 64% mutational concordance between tumor tissue and plasma cfDNA, which is consistent with prior studies of CRC and other cancer types.16,37,47-49
In conclusion, we used a prospective study design to demonstrate that ctDNA is a promising biomarker for MRD detection on the day of surgery in patients with oligometastatic CRC. Our results have strong clinical implications, and in the future could enable clinicians to personalize treatment paradigms for oligometastatic disease based on oncogenomic features derived from cell-free DNA.
ACKNOWLEDGMENT
We are grateful to the patients and their families for study participation. We also thank A. Newman for providing critical feedback on the manuscript. B. Pellini acknowledges the Washington University School of Medicine R25 STRENGTH Program (R25CA190190; Principal Investigator, Ramaswamy Govindan) for protected research time. Images from Servier Medical Art were used to create the Data Supplement. Images from Biorender.com were used to create the Data Supplement.
DISCLAIMER
The views expressed in the submitted article are the authors' views and not the official position of Washington University.
SUPPORT
Supported by the National Cancer Institute of the National Institutes of Health under award number K08CA238711 (A.A.C.), the NCI P50CA196510 Career Enhancement Program (A.A.C., W.G.H.), the Cancer Research Foundation Young Investigator Award (A.A.C.), the V Foundation V Scholar Award (A.A.C.), the American Surgical Association Foundation Fellowship (R.C.F.), the American Cancer Society Institutional Review Grant (R.C.F.), the Society of Surgical Oncology James Ewing Foundation Clinical Investigator Award (R.C.F.), the Sidney Kimmel Translational Science Scholar Award (R.C.F.), and the David Riebel Cancer Research Fund (R.C.F).
B.P., N.P., and W.F. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Bruna Pellini, Aadel A. Chaudhuri
Financial support: Aadel A. Chaudhuri
Administrative support: Aadel A. Chaudhuri
Provision of study materials or patients: Jacqueline Mudd, Lauren E. Henke, Matthew G. Mutch, Katrina S. Pedersen, Ryan C. Fields, Aadel A. Chaudhuri
Collection and assembly of data: Bruna Pellini, Nadja Pejovic, Peter K. Harris, Abul Usmani, Jacqueline Mudd, Marvin Petty, Ashla Singh, Katrina S. Pedersen, Benjamin R. Tan, William G. Hawkins, Ryan C. Fields, Aadel A. Chaudhuri
Data analysis and interpretation: Bruna Pellini, Wenjia Feng, Noah Earland, Peter K. Harris, Jeffrey J. Szymanski, Faridi Qaium, Yuqiu Jiang, Christopher A. Maher, Lauren E. Henke, Haeseong Park, Matthew A. Ciorba, Matthew G. Mutch, Katrina S. Pedersen, Ryan C. Fields, Aadel A. Chaudhuri
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Bruna Pellini
Honoraria: Bio Ascend
Consulting or Advisory Role: Guidepoint
Research Funding: Bristol-Myers Squibb
Noah Earland
Stock and Other Ownership Interests: Illumina, Inc, NASDAQ: ILMN
Faridi Qaium
Stock and Other Ownership Interests: Centene
Yuqiu Jiang
Employment: Roche
Stock and Other Ownership Interests: Roche
Travel, Accommodations, Expenses: Roche
Lauren E. Henke
Honoraria: ViewRay, Varian Medical Systems
Speakers' Bureau: ViewRay, Varian Medical Systems
Research Funding: ViewRay, Varian Medical Systems
Travel, Accommodations, Expenses: ViewRay
Haeseong Park
Research Funding: Amgen, AstraZeneca, Bayer, BeiGene, Bristol-Myers Squibb, Daiichi Sankyo, Lilly, EMD Serono, Gilead Sciences, Incyte, Macrogenics, MedImmune, Medivation, Merck, Millennium, Novartis, Pfizer, Puma Biotechnology, Regeneron, Taiho Pharmaceutical, Vertex, Ambrx, GlaxoSmithKline, Array BioPharma, Genentech, Oncologie, Turning Point Therapeutics, Xencor, BJ Bioscience, Five Prime Therapeutics, Roche, ImmuneOncia Therapeutics, Immunomedics, Mirati Therapeutics, PsiOxus Therapeutics, Synermore Biologics, TopAlliance Biosciences, Vedata Biosciences, Aprea Therapeutics, Gossamer Bio, Jounce Therapeutics, MabSpace Biosciences, Seattle Genetics
Travel, Accommodations, Expenses: Bayer, Daiichi Sankyo, Vedanta Biosciences
Matthew A. Ciorba
Consulting or Advisory Role: Pfizer
Speakers' Bureau: Takeda
Research Funding: Incyte
Hyun Kim
Honoraria: Varian Medical Systems, ViewRay
Consulting or Advisory Role: Varian Medical Systems
Research Funding: Varian Medical Systems, ViewRay
Travel, Accommodations, Expenses: Varian Medical Systems, ViewRay
Katrina S. Pedersen
Consulting or Advisory Role: Bayer, Array BioPharma, Pfizer
Speakers' Bureau: Clinical Care Options
Research Funding: Merck, AbbVie, Roche/Genentech, Bristol-Myers Squibb, Pfizer, Daiichi Sankyo/Lilly, Nouscom
Travel, Accommodations, Expenses: Array BioPharma, BeiGene, Nouscom
(OPTIONAL) Open Payments Link: https://openpaymentsdata.cms.gov/physician/876456
Benjamin R. Tan
Research Funding: Roche/Genentech, Eisai, Exelixis, Merck Serono, Tyrogenex, Sillajen, Boehringer Ingelheim, Bristol-Myers Squibb, AstraZeneca
William G. Hawkins
Patents, Royalties, Other Intellectual Property: Member of board of directors for Accuronix Therapuetics
Aadel A. Chaudhuri
Stock and Other Ownership Interests: Geneoscopy, Droplet Biosciences
Honoraria: Foundation Medicine, Roche
Consulting or Advisory Role: Geneoscopy, Roche, Fenix Group International, Tempus, DeciBio, Guidepoint
Patents, Royalties, Other Intellectual Property: US Patent No. US8685727B2
Travel, Accommodations, Expenses: Roche, Foundation Medicine
Other Relationship: Roche
No other potential conflicts of interest were reported.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. : Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394-424, 2018 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A: Cancer statistics, 2019. CA Cancer J Clin 69:7–34, 2019 [DOI] [PubMed] [Google Scholar]
- 3.Abdalla EK, Vauthey JN, Ellis LM, et al. : Recurrence and outcomes following hepatic resection, radiofrequency ablation, and combined resection/ablation for colorectal liver metastases. Ann Surg 239:818-825, 2004; discussion 825-827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adam R, Avisar E, Ariche A, et al. : Five-year survival following hepatic resection after neoadjuvant therapy for nonresectable colorectal [liver] metastases. Ann Surg Oncol 8:347-353, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Misiakos EP, Karidis NP, Kouraklis G: Current treatment for colorectal liver metastases. World J Gastroenterol 17:4067-4075, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okumura T, Boku N, Hishida T, et al. : Surgical outcome and prognostic stratification for pulmonary metastasis from colorectal cancer. Ann Thorac Surg 104:979-987, 2017 [DOI] [PubMed] [Google Scholar]
- 7.Robertson DJ, Stukel TA, Gottlieb DJ, et al. : Survival after hepatic resection of colorectal cancer metastases: A national experience. Cancer 115:752-759, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weichselbaum RR, Hellman S: Oligometastases revisited. Nat Rev Clin Oncol 8:378-382, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Agolli L, Bracci S, Nicosia L, et al. : Lung metastases treated with stereotactic ablative radiation therapy in oligometastatic colorectal cancer patients: Outcomes and prognostic factors after long-term follow-up. Clin Colorectal Cancer 16:58-64, 2017 [DOI] [PubMed] [Google Scholar]
- 10.Chen F, Sakai H, Miyahara R, et al. : Repeat resection of pulmonary metastasis is beneficial for patients with colorectal carcinoma. World J Surg 34:2373-2378, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Salah S, Watanabe K, Park JS, et al. : Repeated resection of colorectal cancer pulmonary oligometastases: Pooled analysis and prognostic assessment. Ann Surg Oncol 20:1955-1961, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Salah S, Watanabe K, Welter S, et al. : Colorectal cancer pulmonary oligometastases: Pooled analysis and construction of a clinical lung metastasectomy prognostic model. Ann Oncol 23:2649-2655, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Mitin T, Enestvedt CK, Thomas CR Jr: Management of oligometastatic rectal cancer: Is liver first? J Gastrointest Oncol 6:201-207, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azad TD, Chaudhuri AA, Fang P, et al. : Circulating tumor DNA analysis for detection of minimal residual disease after chemoradiotherapy for localized esophageal cancer. Gastroenterology 158:494-505.e6, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. : Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 7:1394-1403, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chin RI, Chen K, Usmani A, et al. : Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol Diagn Ther 23:311-331, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corcoran RB, Chabner BA: Application of cell-free DNA analysis to cancer treatment. N Engl J Med 379:1754-1765, 2018 [DOI] [PubMed] [Google Scholar]
- 18.Tie J, Cohen JD, Wang Y, et al. : Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 68:663-671, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tie J, Wang Y, Tomasetti C, et al. : Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 8:346ra92, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raman V, Adam MA, Turner MC, et al. : Disparity of colon cancer outcomes in rural America: Making the case to travel the extra mile. J Gastrointest Surg 23:2285-2293, 2019 [DOI] [PubMed] [Google Scholar]
- 21.Nordlinger B, Sorbye H, Glimelius B, et al. : Perioperative chemotherapy with FOLFOX4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC Intergroup Trial 40983): A randomised controlled trial. Lancet 371:1007-1016, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nordlinger B, Sorbye H, Glimelius B, et al. : Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): Long-term results of a randomised, controlled, phase 3 trial. Lancet Oncol 14:1208-1215, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Reddy SK, Zorzi D, Lum YW, et al. : Timing of multimodality therapy for resectable synchronous colorectal liver metastases: A retrospective multi-institutional analysis. Ann Surg Oncol 16:1809-1819, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Newman AM, Bratman SV, To J, et al. : An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20:548-554, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman AM, Lovejoy AF, Klass DM, et al. : Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34:547-555, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Network : Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330-337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Sethi NS, Hinoue T, et al. : Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 33:721-735.e8, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genovese G, Kahler AK, Handsaker RE, et al. : Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371:2477-2487, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Y, Ulrich BC, Supplee J, et al. : False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res 24:4437-4443, 2018 [DOI] [PubMed] [Google Scholar]
- 30.Jaiswal S, Fontanillas P, Flannick J, et al. : Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371:2488-2498, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanton C, Venn O, Aravanis A, et al. : Prevalence of clonal hematopoiesis of indeterminate potential (CHIP) measured by an ultra-sensitive sequencing assay: Exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. J Clin Oncol 36, 2018. (suppl; abstr 12003) [Google Scholar]
- 32.Xie M, Lu C, Wang J, et al. : Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20:1472-1478, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tate JG, Bamford S, Jubb HC, et al. : COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res 47:D941-D947, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaung SJ, Fuhlbrück F, Peterson M, et al. : Clonal hematopoiesis in late-stage non–small-cell lung cancer and its impact on targeted panel next-generation sequencing. JCO Precision Oncol 4:1271-1279, 2020 [DOI] [PubMed] [Google Scholar]
- 35.Zill OA, Banks KC, Fairclough SR, et al. : The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res 24:3528-3538, 2018 [DOI] [PubMed] [Google Scholar]
- 36.Abbosh C, Birkbak NJ, Wilson GA, et al. : Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545:446-451, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudley JC, Schroers-Martin J, Lazzareschi DV, et al. : Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov 9:500-509, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shekhtman EM, Anne K, Melkonyan HS, et al. : Optimization of transrenal DNA analysis: Detection of fetal DNA in maternal urine. Clin Chem 55:723-729, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Ganesh K, Stadler ZK, Cercek A, et al. : Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 16:361-375, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marabelle A, Fakih M, Lopez J, et al. : Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 21:1353-1365, 2020 [DOI] [PubMed] [Google Scholar]
- 41.Subbiah V, Solit DB, Chan TA, et al. : The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥10: A decision centered on empowering patients and their physicians. Ann Oncol 31:1115-1118, 2020 [DOI] [PubMed] [Google Scholar]
- 42.Gandara DR, Paul SM, Kowanetz M, et al. : Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 24:1441-1448, 2018 [DOI] [PubMed] [Google Scholar]
- 43.Griffith M, Spies NC, Krysiak K, et al. : CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 49:170-174, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chabon JJ, Hamilton EG, Kurtz DM, et al. : Integrating genomic features for non-invasive early lung cancer detection. Nature 580:245-251, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leal A, van Grieken NCT, Palsgrove DN, et al. : White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat Commun 11:525, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtz DM, Scherer F, Jin MC, et al. : Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol 36:2845-2853, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung JH, Pavlick D, Hartmaier R, et al. : Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann Oncol 28:2866-2873, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Adams HP, Yao L, et al. : Concordance of genomic alterations by next-generation sequencing in tumor tissue versus cell-free DNA in stage I-IV non-small cell lung cancer. J Mol Diagn 22:228-235, 2020 [DOI] [PubMed] [Google Scholar]
- 49.Schrock AB, Welsh A, Chung JH, et al. : Hybrid capture-based genomic profiling of circulating tumor DNA from patients with advanced non-small cell lung cancer. J Thorac Oncol 14:255-264, 2019 [DOI] [PubMed] [Google Scholar]