

PURPOSE
Tumor tissue from metastatic castration-resistant prostate cancer (mCRPC) harbors frequent copy number variations (CNVs) in the PTEN-PI3K-AKT pathway. However, identifying CNVs in plasma cell–free DNA (cfDNA) has proven to be challenging. With emerging data supporting Akt inhibition in PTEN-deficient mCRPC, we profiled PTEN-PI3K-AKT pathway aberrations in patients with mCRPC using a novel cfDNA assay optimized for CNV detection.
METHODS
A next-generation sequencing–based cfDNA assay was used to profile 231 patients with mCRPC from two independent cohorts (Australian, n = 78; United States, n = 153). PTEN-PI3K-AKT pathway genomic aberrations were correlated with clinical outcomes, including progression-free survival and overall survival (OS).
RESULTS
PTEN loss and PIK3CA gain were detected in 37% (85 of 231) and 17% (39 of 231) of patients, respectively. Poorer outcomes were observed in patients with PTEN-PI3K-AKT pathway aberrations, including those with dual PTEN loss and PIK3CA gain (hazard ratio 2.3, 95% CI 1.2 to 4.4). Cumulative CNV burden in the PTEN-PI3K-AKT and androgen receptor (AR) pathways was associated with significantly worse clinical outcomes (0 v 1 v ≥ 2 CNVs in Australian cohort: median OS 33.5 v 17.2 v 9.7 months, P < .001; 0 v 1 v ≥ 2 CNVs in US cohort: median OS 35.5 v 14.3 v 9.2 months, P < .001). Notably, 21% (31 of 146) of PTEN-neutral patients harbored alternative PTEN-PI3K-AKT pathway aberrations.
CONCLUSION
PTEN-PI3K-AKT pathway CNVs were readily detected using our cfDNA assay, with the prevalence of PTEN loss comparable with tissue-based studies. Additional PTEN-PI3K-AKT pathway aberrations were found in one fifth of PTEN-neutral cases. Concurrent CNVs in the PTEN-PI3K-AKT and AR pathways portended poor survival, and identifying this high-risk patient subset for dual AR/Akt inhibition may optimize precision treatment with Akt inhibitors in mCRPC.
INTRODUCTION
The last decade has seen an expansion in life-prolonging therapeutics for metastatic castration-resistant prostate cancer (mCRPC), including androgen receptor (AR) pathway inhibitors (ARPIs), the taxane chemotherapeutic agent cabazitaxel, and inhibitors of poly(ADP–ribose) polymerase.1-5 Although the development of drugs targeting phosphatidylinositol-3-kinase (PI3K) signaling has long been investigated across a range of solid tumors,6,7 recent findings from the IPATential-150 study of abiraterone and pan-Akt inhibitor ipatasertib underscore the potential of co-targeting these pathways in prostate cancer.8,9 Ipatasertib significantly improved radiographic progression-free survival (PFS); however, of note, the benefit was only observed in the context of PTEN loss.
CONTEXT
Key Objective
Can we robustly detect and determine the prognostic utility of blood-based genomic aberrations including copy number variations (CNVs) in the PTEN-PI3K-AKT pathway in metastatic castration-resistant prostate cancer (mCRPC)?
Knowledge Generated
PTEN-PI3K-AKT pathway aberrations were readily detected in plasma cell–free DNA (cfDNA) from 231 patients with mCRPC in two independent cohorts, including PTEN loss in 37%. CNVs in the PTEN-PI3K-AKT pathway were associated with significantly worse clinical outcomes, especially when combined with AR gain. Additional PTEN-PI3K-AKT pathway aberrations were detected in approximately one fifth of PTEN-neutral patients.
Relevance
PTEN-PI3K-AKT pathway CNVs were readily detected using a validated cfDNA liquid biopsy assay, with the prevalence of PTEN loss comparable with tissue-based studies. Plasma cfDNA profiling may facilitate and optimize patient selection for targeted treatment with Akt inhibitors in mCRPC.
Large-scale tissue sequencing efforts have demonstrated frequent dysregulation of the PTEN-PI3K-AKT pathway in mCRPC, most commonly with PTEN tumor suppressor loss (40%-60%) and less frequently by amplification or gain-of-function mutations in PIK3CA, PIK3CB, and AKT1 (< 10% each).10-20 The challenges of acquiring high-quality metastatic tissue for molecular analysis and recognition that disease evolution may not be adequately reflected in archival tissue have led to the emergence of liquid biopsies as an alternative path toward biomarker discovery in advanced prostate cancer.21 However, cell-free DNA (cfDNA) studies have consistently shown lower frequency of copy number variations (CNVs) compared with tissue cohorts, representative of challenges in ploidy assessment in environments with low circulating tumor DNA (ctDNA) fraction and high tumor heterogeneity.22,23 This is arguably no more apparent than with PTEN loss, where prevalence may be as low as 10%-15% in cfDNA studies after accounting for samples omitted because of low ctDNA fraction.24-27 Given that metastatic prostate cancer is characterized by recurrent gains and deletions,11-17 blood-based assays capable of accurate CNV status assessment are urgently needed.
Recent liquid biopsy studies have implicated PTEN-PI3K-AKT pathway aberrations in driving disease progression and treatment resistance in mCRPC,27-29 although not all are in agreement.18,30 Furthermore, the individual contribution of PTEN-PI3K-AKT pathway members besides PTEN is less well-defined. Accurate identification of PTEN-PI3K-AKT pathway aberrations in ctDNA and understanding their interaction with more established biomarkers including AR gain24,31-35 hold important clinical implications, particularly in light of the results from IPATential-150.8,9
Herein, we employ a targeted, high-sensitivity, next-generation sequencing (NGS) cfDNA assay to profile the PTEN-PI3K-AKT pathway in two independent mCRPC cohorts totaling 231 patients, correlating genomic aberrations with longitudinal clinical outcomes. We show that plasma cfDNA can be used to determine copy number status in mCRPC and may be a highly valuable tool for identifying patients suitable for precision treatment with PTEN-PI3K-AKT pathway inhibitors.
METHODS
Patient Cohorts
This multi-institutional prospective biomarker study collected plasma samples from 231 patients across two independent cohorts in Australia and the United States. Study approval was acquired from each institution's human research ethics committee, with all participants providing written informed consent before sample collection. In both cohorts, peripheral blood was collected immediately before commencing systemic therapy and processed as described in the Data Supplement.
The Australian cohort comprised 78 patients with mCRPC commencing systemic therapy at two tertiary institutions (Monash Health and Chris O'Brien Lifehouse) between September 2016 and April 2019. Forty-nine (63%) patients commenced ARPI therapy (abiraterone or enzalutamide) and 29 (37%) patients commenced taxane chemotherapy (docetaxel or cabazitaxel). Baseline clinical characteristics and previous systemic treatment exposure are presented in the Data Supplement. The median follow-up time for nondeceased patients was 28.0 months.
The US cohort comprised 153 patients from the Mayo Clinic enrolled between September 2009 and March 2014 with either biochemically or radiographically progressive mCRPC. Although genomic features of this cohort have been partially characterized,29,36 the analyses undertaken here represent unique hypotheses using a targeted and sensitive NGS panel that have not been explored before. Baseline clinical characteristics are presented in the Data Supplement. The median follow-up time for nondeceased patients was 80.7 months.
Targeted cfDNA Sequencing
All plasma samples underwent cfDNA sequencing (Predicine Inc., Hayward, CA) from a single tube (10 mL) of peripheral blood.29,37 Relevant to this study, the assay targets all coding regions of PTEN, PIK3CA, and AR and selected hotspot regions of AKT1, mTOR, PPP2R1A, STK11, TSC1, and TSC2 (see the Data Supplement for the full gene list). Orthogonal validation of CNVs with low-pass whole genome sequencing (LP-WGS) was performed on a subset of Australian cohort samples (n = 46). LP-WGS data were analyzed using ichorCNA software,38 which estimates plasma CNVs using a hidden Markov model. A detailed description of DNA extraction, sequencing, analytical validation, and bioinformatics analysis (somatic mutation identification, CNV detection and ctDNA fraction estimation, and ichorCNA tool settings) is presented in the Data Supplement.
Clinical Outcomes and Statistical Analysis
Kaplan-Meier survival estimates (log-rank test) and multivariable Cox regression models were used to assess the association between PTEN-PI3K-AKT pathway aberrations and clinical outcomes, including PFS (time from treatment commencement to first confirmed prostate-specific antigen progression, clinical or radiographic progression, or death from prostate cancer) and overall survival (OS; time from treatment commencement until death from any cause). Where an event had not occurred at time of data analysis, survival outcomes were right censored at the date of last patient contact. Statistical significance was defined as P < .05.
Study Oversight and Responsibilities
Samples were obtained as part of noninterventional biomarker studies. Study investigators were involved in initial sample acquisition, sample processing, and clinical data collection. Predicine Inc. were responsible for sequencing and bioinformatic analysis of cfDNA samples only and were not otherwise involved in study design, subject accrual, clinical data collection, or analysis of clinico-genomic associations.
RESULTS
Analytical Validation of a cfDNA-Based Assay Capable of Detecting CNV Alterations in Advanced Prostate Cancer
Given the challenges of accurate copy number estimation in liquid biopsy specimens, development of a novel NGS-based assay capable of enhanced detection of plasma CNVs, including PTEN loss, is a key priority. The assay used in this study employs a custom targeted panel-based approach, in combination with proprietary operation chemistry and analysis algorithms. Hybrid capture probes targeting single-nucleotide polymorphisms in the introns both upstream and downstream of relevant genes are used to capture additional copy number information.
Multiple steps were taken for analytical validation (see the Data Supplement for full details). Briefly, copy number loss performance was assessed using tumor- or normal-matched cancer cell lines as true positive and true negative samples. Extracted DNA was titrated to seven tumor fraction levels ranging from 5% to 75%. Copy number gain performance was evaluated using commercial ctDNA reference material at three tumor fraction levels ranging from 0.25% to 0.5%. The assay limit of detection was determined to be 1.75 copies for copy number loss and 2.23 copies for copy number gain (Fig 1A). Assay specificity was evaluated in 24 healthy donors and determined to be > 99%. Assay precision (both repeatability and within-lab reproducibility) was assessed by measuring variation between 18 sets of the same sample aliquots. The intra-run and intermediate-run precision was measured to be 97.8% (95% CI, 86.5% to 99.9%) and 99.6% (95% CI, 97.5% to 100%), respectively.
FIG 1.

Analytical and orthogonal validation of cell-free DNA (cfDNA) assay. A, Analytical sensitivity of Predicine cfDNA assay for detection of CNVs. B, Genomic landscape of PTEN-PI3K-AKT and AR pathway aberrations identified in cfDNA in Australian (top) and US (bottom) cohorts. C, Copy number plots for seven patients with discordant CNV status in the Australian cohort. Interrogation of the PTEN locus (top three panels) demonstrated that LP-WGS missed two patients with focal loss (AU-027 and AU-051) and was unable to accurately assess PTEN CNV status in one patient with adjacent copy number loss and copy number neutral segments (AU-072). Interrogation of the PIK3CA locus (middle panel) demonstrated that LP-WGS missed one patient with focal gain (AU-034). Interrogation of the AR locus (bottom three panels) demonstrated that LP-WGS missed two patients with low-level AR gain (AU-005 and AU-055) and one patient with focal AR gain (AU-027). D, Copy number correlation between targeted panel and LP-WGS for PTEN loss, PIK3CA gain, and AR gain in the Australian cohort. Abbreviations: AR, androgen receptor; ARPI, AR pathway inhibitor; cfDNA, cell–free DNA; CNV, copy number variation; LP-WGS, low-pass whole-genome sequencing; mCRPC, metastatic castration-resistant prostate cancer; PPV, positive predictive value; QC, quality control; Sn, sensitivity.
Detection, Validation, and Clinical Associations of PTEN-PI3K-AKT Pathway Aberrations
Having demonstrated that this cfDNA assay is capable of reliably detecting CNVs, we then applied it to determine PTEN and PIK3CA copy number status in our two independent mCRPC cohorts. Sequencing metrics for the Australian and US cohorts are provided in the Data Supplement. The workflow for the final 231 patients with analyzable cfDNA sequencing and clinical outcome data are shown in Appendix Fig A1.
PTEN loss was observed in 37% (85 of 231) of patients (Fig 1B). In a subset of Australian cohort patients with additional plasma available, LP-WGS confirmed targeted panel-detected PTEN loss in 90% (28 of 31) of patients (Data Supplement). Discordance was attributed to either focal copy number loss (n = 2) or failure of LP-WGS to accurately assess PTEN CNV status in adjacent copy number loss and copy number neutral segments (n = 1) (Fig 1C). There was evidence of high correlation for overall copy number as assessed by targeted panel and LP-WGS (R = 0.85, Fig 1D).
PTEN loss was strongly associated with shorter PFS and OS in the Australian cohort. When adjusting for baseline clinicopathologic prognostic factors including ctDNA fraction (Data Supplement), this association remained significant in multivariable analysis (Table 1, Fig 2A, and Data Supplement). A similar association was confirmed in the US cohort with respect to OS (Table 1, Fig 2B, and Data Supplement). Association with PFS in the US cohort was not performed as patient enrollment was before the widespread availability of ARPIs. Of note, in the 146 PTEN-copy number neutral patients in this study, 31 (21%) patients exhibited alternate PTEN-PI3K-AKT pathway aberrations (Fig 1B), most commonly PIK3CA mutation and/or gain (n = 21, 68%) and PTEN mutation (n = 7, 23%). In addition, none of the patients with mutations in AKT1 (n = 5) or mTOR (n = 2) across either cohort harbored concurrent PTEN loss and/or mutation.
TABLE 1.
Cox Proportional Hazards Analysis of Clinical Outcomes Based on PI3K/AR Pathway Aberrations in Australian and US Cohorts

FIG 2.
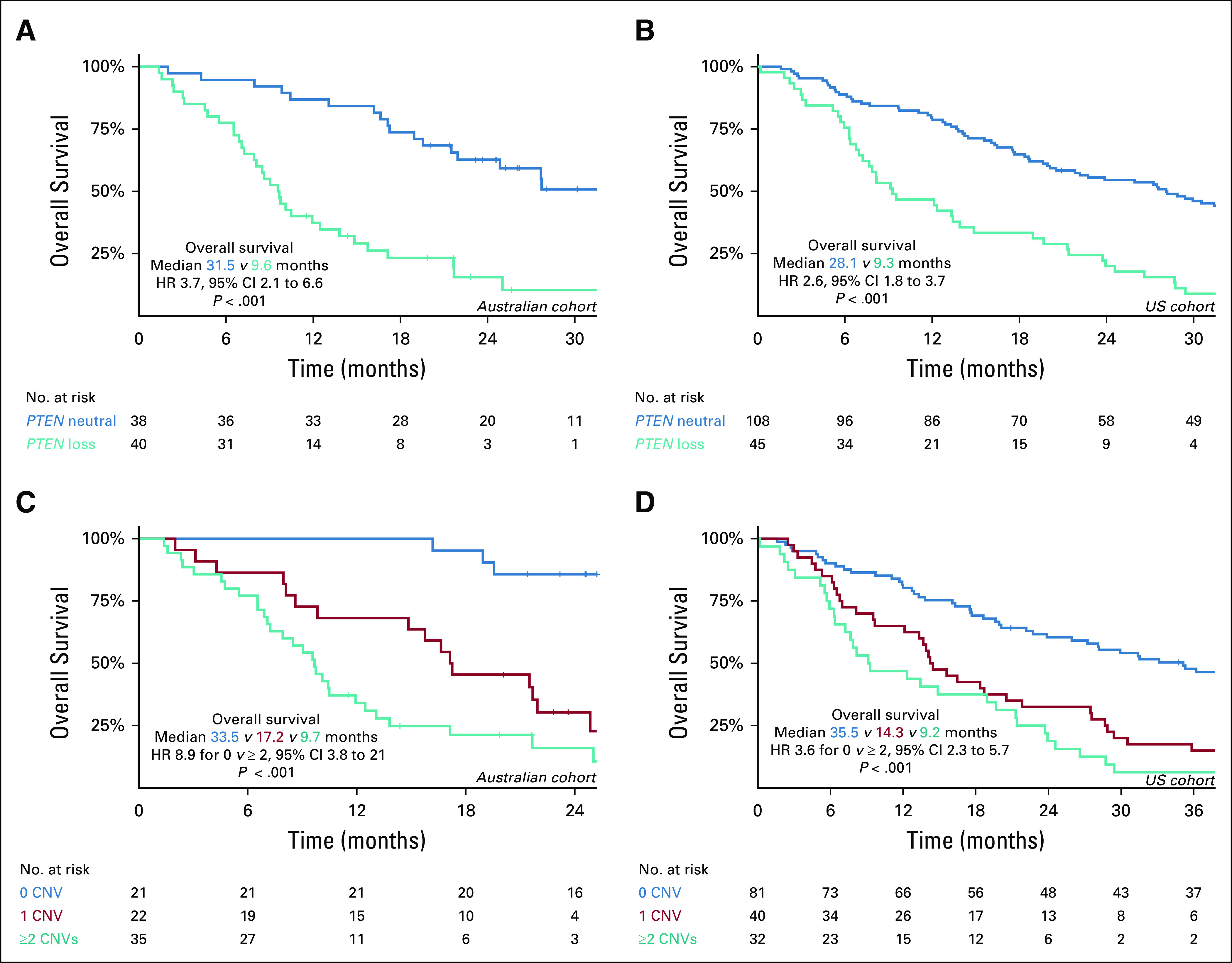
(A and B) Kaplan-Meier curves of overall survival according to PTEN copy number status and (C and D) cumulative CNVs in PTEN-PI3K-AKT and AR pathways. (A) PTEN CNV status in Australian cohort. (B) PTEN CNV status in US cohort. (C) Cumulative CNVs in Australian cohort. (D) Cumulative CNVs in US cohort. AR, androgen receptor; CNV, copy number variation; HR, hazard ratio.
PIK3CA gain was observed in 17% (39 of 231) of patients (Fig 1B). LP-WGS confirmed targeted panel-detected PIK3CA gain in 84% (16 of 19) of Australian cohort patients with additional available plasma, with one patient determined to be PIK3CA neutral and two patients with indeterminate LP-WGS results (Data Supplement). Examination of one patient with discordant CNV status showed evidence of focal copy number gain (Fig 1C). Strong correlation was again noted between targeted panel- and LP-WGS–assessed PIK3CA copy number (Fig 1D, R = 0.80). PIK3CA gain was also independently associated with poor survival outcomes in the Australian cohort, but not the US cohort (Table 1 and Appendix Fig A2).
In the Australian cohort, mutations were most frequently observed in PIK3CA (13 of 78, 17%). PTEN mutations were uncommon at 6% (5 of 78), with AKT1 and mTOR mutations rare (single case each). In the US cohort, PIK3CA mutations were again the most common, albeit at a lower prevalence than the Australian cohort at 10% (15 of 153). PTEN, AKT1, and mTOR mutations were observed in < 5% of patients. Given the low frequency of certain PTEN-PI3K-AKT pathway mutations (eg, AKT1 and mTOR) in both cohorts, correlation with clinical outcomes was restricted to genes mutated in at least five patient samples. In contrast to CNVs, mutations in PTEN-PI3K-AKT pathway genes alone did not significantly correlate with clinical outcomes (Table 1). A full list of PTEN-PI3K-AKT pathway CNVs and mutations for the Australian and US cohort can be found in the Data Supplement.
Cumulative CNVs in the PTEN-PI3K-AKT and AR Pathways Independently Confer Poor Prognosis
We next sought to analyze the clinical impact of cumulative CNVs. Given reciprocal regulation between the PTEN-PI3K-AKT and AR pathways,39-41 we included AR in analysis of clinico-genomic correlations. AR mutations were present in 19% (45 of 231) of patients (see the Data Supplement for the list of AR mutations in Australian and US cohorts, respectively). In the Australian cohort, AR mutations did not correlate with any clinical outcomes. By comparison, AR mutations in the US cohort were associated with shorter OS in univariable analysis (hazard ratio [HR] 1.7; 95% CI, 1.1 to 2.7; P = .02), but not in multivariable analysis (Table 1).
AR gain was present in 42% (96 of 231) of patients (Fig 1B). Of patients who underwent LP-WGS validation in the Australian cohort, 22 of 25 patients (88%) with AR gain were validated with LP-WGS, with excellent correlation between targeted panel and LP-WGS copy number (Fig 1D, R = 0.98). Examination of LP-WGS plots for the three discordant patients revealed low level AR gains (n = 2) and a focal copy number gain (n = 1) (Fig 1C). With respect to clinical outcomes, AR gain was associated with shorter PFS and OS in univariable and multivariable analyses in both cohorts (Table 1 and Appendix Fig A2).
Next, we hypothesized that cumulative CNVs in the PTEN-PI3K-AKT and AR pathways may predict for worse prognosis. To investigate this, we categorized patients based on the total number of CNVs detected at baseline. Considering PTEN loss, PIK3CA gain, and AR gain collectively across both cohorts, 0/1/≥ 2 CNVs were observed in 102 (44%), 62 (27%), and 67 (29%) samples. Cumulative CNVs in the PTEN-PI3K-AKT and AR pathways were significantly associated with OS in both the Australian (median OS for 0 v ≥ 2 CNVs: 33.5 months v 9.7 months, P < .001, log-rank test; Fig 2C) and US cohort (median OS for 0 v ≥ 2 CNVs: 35.5 months v 9.2 months, P < .001, log-rank test; Fig 2D). Critically, this relationship persisted when multivariable analysis was performed on each cohort separately (Table 1) and in an exploratory subgroup analysis based on ctDNA fraction (above and below 2%) (Appendix Fig A3). Analysis of outcomes by treatment subgroup in the Australian cohort demonstrated that findings were most apparent, as expected, in ARPI-treated patients (Data Supplement).
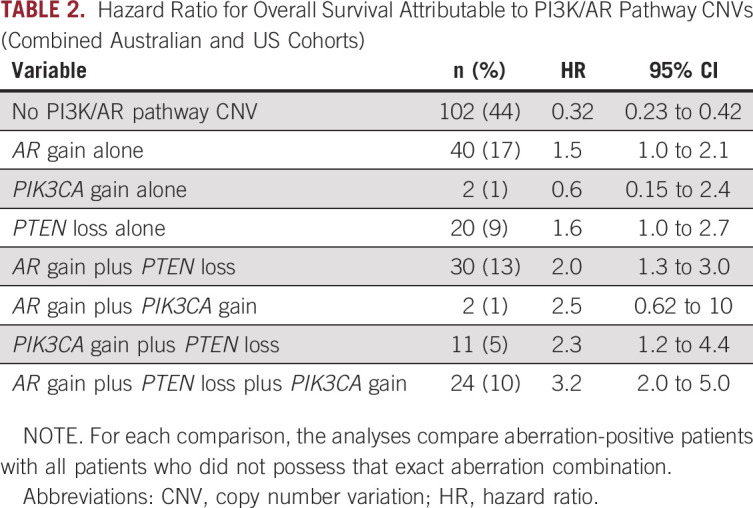
PIK3CA Gain May Further Potentiate PTEN Loss and Contribute to Worse Clinical Outcomes
To understand the extent to which different CNVs were contributing to poor outcomes, exploratory analyses of various CNV combinations were performed. Given the multiple testing, formal statistical comparison was not undertaken between these subgroups. Although isolated PTEN loss was associated with less favorable outcomes (median OS 11 months v 21 months; HR 1.6; 95% CI, 1.0 to 2.7; log-rank test), the negative impact of PIK3CA gain was most apparent when concurrently present with PTEN loss (median OS 7.2 months v 21 months; HR 2.3; 95% CI, 1.2 to 4.4; log-rank test) and with combined PTEN/PIK3CA/AR CNV (median OS 8.5 months v 21 months; HR 3.2; 95% CI, 2.0 to 5.0; log-rank test) (Table 2). To determine if treatment exposure affected the prevalence of dual PTEN-PI3K-AKT pathway CNV activation, we compared rates of prior systemic therapy in patients with PTEN loss without PIK3CA gain (n = 50) with patients with PTEN loss and PIK3CA gain (n = 35). There was no significant difference with respect to prior ARPI therapy (yes v no: 9 of 16 [56%] v 26 of 69 [38%], P = .3, chi-square test), taxane chemotherapy (20 of 51 [39%] v 15 of 34 [44%], P = 1.0, chi-square test), or any systemic therapy (25 of 59 [42%] v 10 of 26 [38%], P = .9, chi-square test).
TABLE 2.
Hazard Ratio for Overall Survival Attributable to PI3K/AR Pathway CNVs (Combined Australian and US Cohorts)
DISCUSSION
The PTEN-PI3K-AKT pathway is among the most frequently dysregulated oncogenic pathways in mCRPC, second only to the AR.12,13,16,17 With an emerging role for targeting the PTEN-PI3K-AKT axis, developing minimally invasive assays for profiling this pathway has clear clinical relevance for patients with mCRPC. CNVs in the PTEN-PI3K-AKT pathway are of particular importance, best reflected by the prognostic42 and predictive9 significance of PTEN loss. However, detection of PTEN loss in cfDNA has historically been challenging in low ctDNA purity and/or high tumor heterogeneity environments,22,23 accounting for suboptimal concordance with tissue-derived CNVs.43,44 Having undergone robust analytical and orthogonal validation with both reference material and cell-line titration samples, the cfDNA assay used in this study is capable of detecting copy number loss events with high sensitivity and specificity.
Applying this novel assay to two independent prospective clinical cohorts totaling 231 patients with mCRPC, we observed enhanced detection of PTEN loss (detected in 37% of patients with mCRPC compared with 10%-15% in previous cfDNA studies).24-27 We also validated a high proportion of panel-detected PTEN and PIK3CA CNVs using orthogonal validation with LP-WGS. Critically, where cfDNA PTEN copy number status was discordant between the sequencing methods, we showed that the targeted panel-based approach may possess greater sensitivity for detecting focal regions of copy number loss. This raises the appealing prospect that liquid biopsy panel–based assays such as the one described here could complement PTEN tissue testing to identify candidates for targeted treatment including ipatasertib and capivasertib45 or other PTEN-PI3K-AKT pathway inhibitors.
Our findings support the growing body of preclinical and clinical evidence pointing to significant interactions between the PTEN-PI3K-AKT and AR pathways that contribute to deleterious outcomes.39-41 Early attempts to target the PTEN-PI3K-AKT pathway with PI3K and Akt inhibitor monotherapy in PTEN-deficient mCRPC were met with disappointing results, with the reasons apparent with increased appreciation of negative feedback regulation between the two pathways.39,40 Acknowledging this relationship, the phase III IPATential-150 trial co-targeted PTEN-PI3K-AKT and AR pathways using abiraterone and the pan-Akt inhibitor ipatasertib, resulting in significant radiographic PFS improvement in PTEN-null tumors over abiraterone monotherapy.8,9 It remains to be seen if co-targeting these pathways results in cfDNA clearance of associated aberrations, for which the development of a sensitive and specific assay would be of utility. Furthermore, although this data signals renewed interest in PI3K/Akt targeting in mCRPC, additional aberrations beyond PTEN loss can activate PTEN-PI3K-AKT signaling and warrant exploration.
One such candidate may be PIK3CA gain. When PIK3CA gain was observed concurrently with PTEN loss, patients appeared to have worse outcomes than those with PTEN loss alone, suggesting that PIK3CA gain potentiates PTEN loss in prostate cancer progression. Traditionally, preclinical data in PTEN-deficient malignancies, including prostate cancer, have supported p110β (encoded by PIK3CB) as the primary PI3K isoform involved in tumorigenesis.46-48 However, dependent on the tissue type and pathology, the p110α catalytic subunit (encoded by PIK3CA) may in fact be of equal or greater importance in driving disease outcomes and responsiveness to PTEN-PI3K-AKT pathway inhibition.49-51 Pearson et al52 further illustrated a key role of PIK3CA in concert with PTEN. Using genetically engineered mouse models of prostate cancer, they demonstrated that a double hit culminating in concurrent PTEN loss and PIK3CA mutation is nonredundant and may instead cooperate to prime the PTEN-PI3K-AKT pathway for rapid progression to invasive carcinoma, greater tumor burden, and de novo castration resistance.52
Although statistically significant, we note that the absolute improvement in median radiographic PFS with ipatasertib in IPATential-150 was reasonably modest at 2 months.9 Therefore, much work remains to improve patient selection and thereby enhance the clinical utility of ipatasertib. Our data demonstrate the synergistic role of dual CNVs in the PTEN-PI3K-AKT pathway, and additionally, the impact of cumulative CNV burden in the PTEN-PI3K-AKT and AR pathways. It is plausible that these high-risk patients with dual PTEN-PI3K-AKT pathway with or without AR pathway activation might have tumors that are primed to best respond to co-targeting of PTEN-PI3K-AKT and AR signaling. This warrants further prospective evaluation in clinical trial cohorts to determine if a sensitive and specific cfDNA-based assay can optimize a rational targeted therapeutic decision for using agents such as ipatasertib.
It is also noteworthy that just more than one fifth of PTEN-neutral patients in our study had aberrations in other PTEN-PI3K-AKT pathway partners, with nearly 70% of this subset harboring either PIK3CA gain or mutation. Furthermore, AKT1 and mTOR mutations were exclusively seen in PTEN-neutral patients, with the former phenomenon having previously been observed in other studies.30,53 Identifying these patients with alternative drivers of PI3K signaling should be prioritized as they may derive clinical benefit from therapeutics targeting the PTEN-PI3K-AKT pathway. Recognizing that PTEN deletion is an early truncal event11,54,55 and PIK3CA aberrations emerge in response to systemic treatment resistance,55,56 molecular testing on primary tissue alone may underestimate the true extent of patients who exhibit PTEN-PI3K-AKT pathway activation because of non-PTEN deletion mechanisms. Thus, cfDNA assays of the nature described here will become of increasing importance by potentially broadening the clinical application of PI3K/Akt inhibitors.57,58
We acknowledge certain limitations in our study, including the need for larger and more homogenous data sets in future studies. Likewise, although our analysis was limited to genomic aberrations, multianalyte approaches incorporating transcriptomic, proteomic, and circulating tumor cell enumeration biomarkers have been shown to provide additional predictive value59-65 and would be of clear interest in follow-on studies.
In conclusion, multiple novel inhibitors of the PTEN-PI3K-AKT pathway are currently in late-phase development. With heterogeneity of response, strategies to correctly identify tumors with oncogenic addition to the PTEN-PI3K-AKT pathway are vital to optimizing treatment selection. Using a validated cfDNA liquid biopsy assay, we robustly identified PTEN loss in 37% of patients with mCRPC across two independent cohorts, similar to the prevalence observed in tumor tissue. We also demonstrated the negative prognostic potential of PTEN-PI3K-AKT with or without AR pathway CNVs in both cohorts. Importantly, we found that approximately one fifth of PTEN-neutral patients had other activating aberrations in the PTEN-PI3K-AKT pathway. Altogether, our data support the use of liquid biopsy assays to ascertain PTEN and PIK3CA copy number status in mCRPC and possess key clinical relevance given the recent findings of the IPATential-150 clinical trial. Future studies should examine the predictive biomarker potential of CNV quantification in clinical trials and investigate dynamic changes in the PTEN-PI3K-AKT and AR pathways that arise in the context of potent systemic therapy.
Appendix
FIG A1.

Workflow for assessable blood samples in the Australian and US cohorts. cfDNA, cell-free DNA; mCRPC, metastatic castration-resistant prostate cancer; NGS QC, next-generation sequencing quality control.
FIG A2.
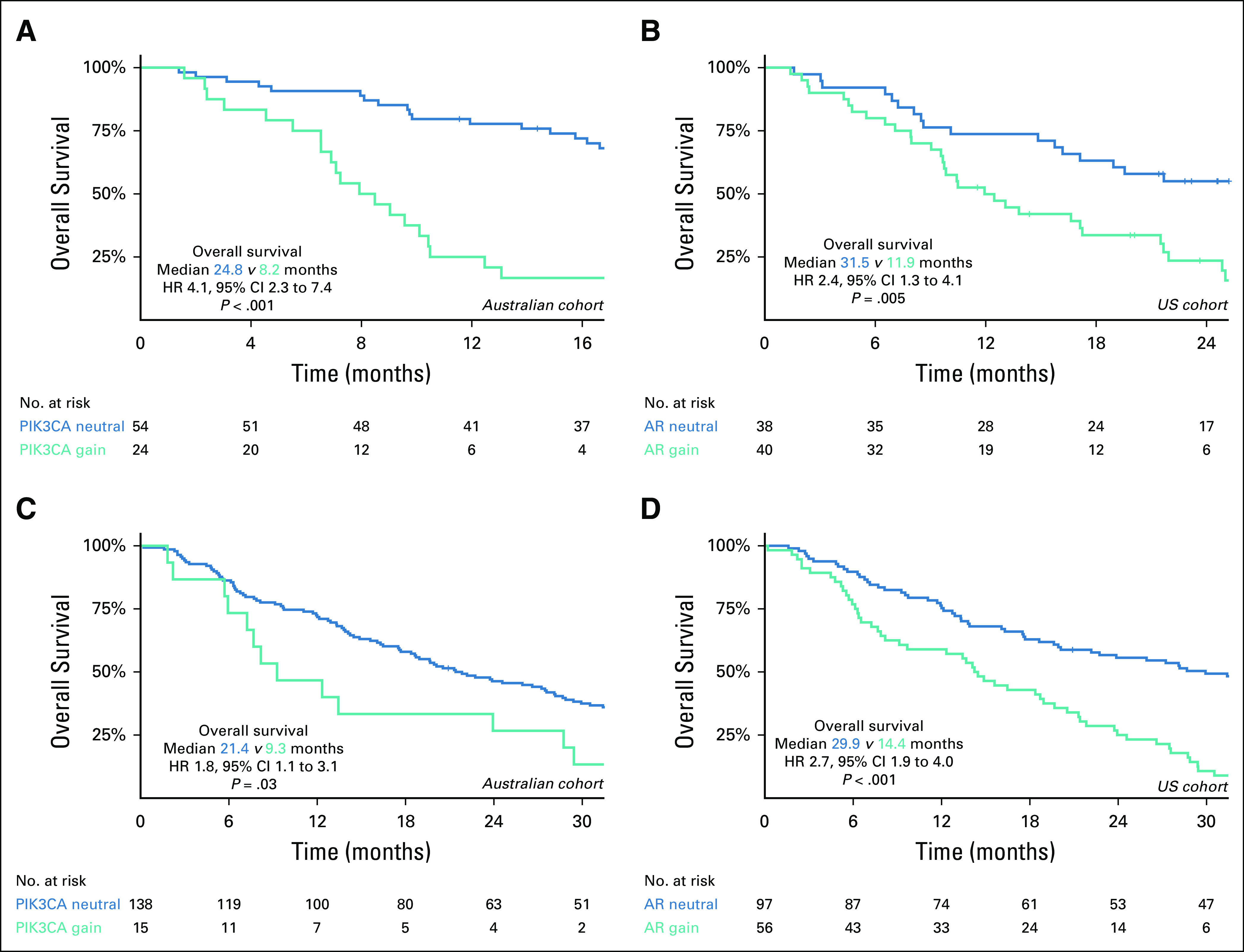
Kaplan-Meier curve of overall survival (OS) according to PIK3CA and AR copy number variation status. Kaplan-Meier analysis of OS according to the presence of (A) PIK3CA gain in Australian cohort, (B) AR gain in Australian cohort, (C) PIK3CA gain in US cohort, and (D) AR gain in US cohort. HR, hazard ratio.
FIG A3.
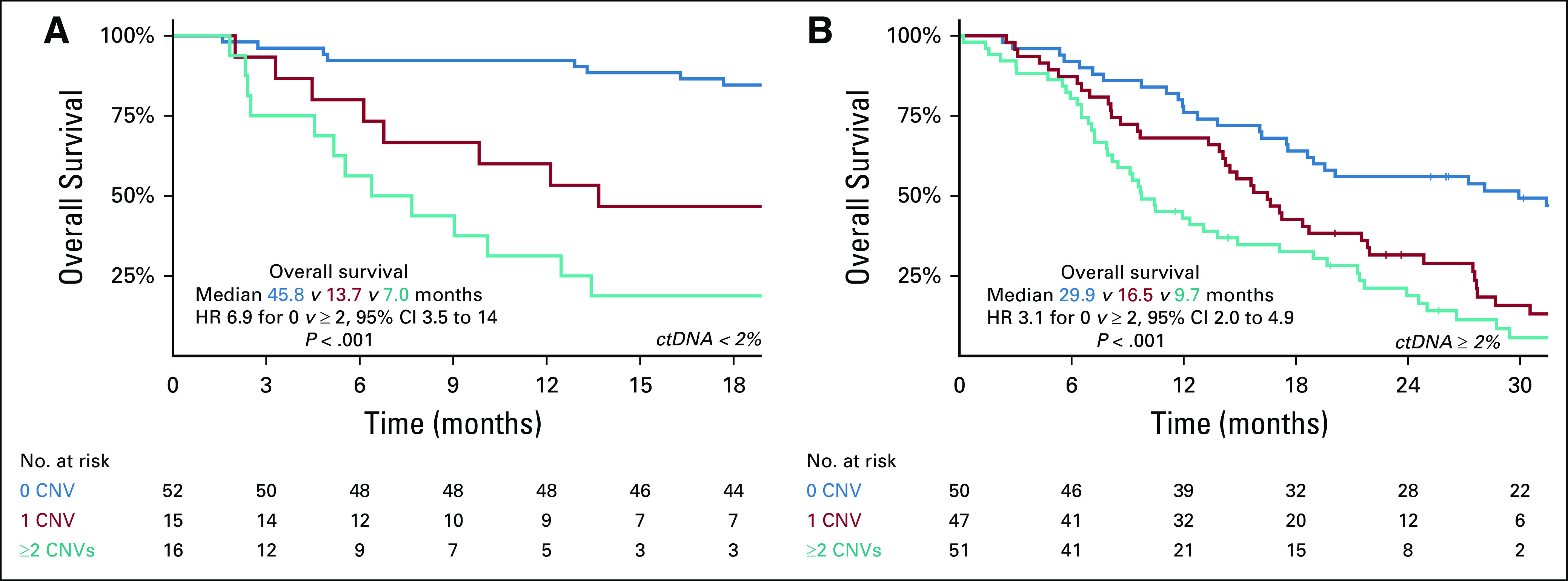
Kaplan-Meier curve of overall survival (OS) according to cumulative CNVs in PTEN-PI3K-AKT and AR pathways in combined Australian and US cohorts analyzed by ctDNA fraction. Kaplan-Meier analysis of OS according to cumulative CNVs for patients (A) below 2% ctDNA fraction and (B) above or equal to 2% ctDNA fraction. AR, androgen receptor; CNV, copy number variation; ctDNA, circulating tumor DNA; HR, hazard ratio.
Edmond M. Kwan
Honoraria: Janssen, Ipsen, Astellas Pharma, Research Review
Consulting or Advisory Role: Astellas Pharma, Janssen, Ipsen
Research Funding: Astellas Pharma, AstraZeneca
Travel, Accommodations, Expenses: Astellas Pharma, Pfizer, Ipsen, Roche
Siavash Foroughi
Stock and Other Ownership Interests: CSL Limited, Oncosil, Mesoblast
Lisa-Jane K. Graham
Patents, Royalties, Other Intellectual Property: I am listed in a “Rewards to Inventors” Scheme with the Institute of Cancer Research London Enterprise Unit as part of a research project I was involved in working at the Institute of Cancer Research London (in collaboration with Astex Therapeutics (now Astex Pharmaceuticals)) to develop novel small molecule inhibitors of PKB (AKT) from 2003-2005. The research was taken over by AstraZeneca in 2005. Payments are made to all inventors in the scheme when milestones are met, and paid by the ICR London Enterprise Unit
Kate Mahon
Research Funding: Mundipharma
Winston Tan
Honoraria: Medscape, Onclive
Xiaohong Wang
Employment: Predicine
Kemin Zhou
Employment: Denovo Biopharma, Predicine Inc.
Jianjun Yu
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Pan Du
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Lisa G. Horvath
Employment: Connected Medical Solutions
Leadership: Connected Medical Solutions
Stock and Other Ownership Interests: Connected Medical Solutions, Imagion Biosystems
Consulting or Advisory Role: Imagion Biosystems
Research Funding: Astellas Pharma
Travel, Accommodations, Expenses: Astellas Pharma, Janssen-Cilag, Pfizer
Shidong Jia
Employment: Predicine, Genentech
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Patents, Royalties, Other Intellectual Property: Liquid Biopsy Technology
Manish Kohli
Honoraria: Advanced Accelerator Applications
Travel, Accommodations, Expenses: Celgene
Arun A. Azad
Honoraria: Janssen, Astellas Pharma, Novartis, Tolmar, Amgen, Pfizer, Bayer, Telix Pharmaceuticals, Bristol-Myers Squibb, Merck Serono, AstraZeneca, Sanofi, Ipsen, Merck Sharp & Dohme, Noxopharm
Consulting or Advisory Role: Astellas Pharma, Novartis, Janssen, Sanofi, AstraZeneca, Pfizer, Bristol-Myers Squibb, Tolmar, Telix Pharmaceuticals, Merck Sharp & Dohme, Bayer, Ipsen, Merck Serono, Amgen, Noxopharm
Speakers' Bureau: Astellas Pharma, Novartis, Amgen, Bayer, Janssen, Ipsen, Bristol-Myers Squibb, Merck Serono
Research Funding: Astellas Pharma, Merck Serono, Novartis, Pfizer, Bristol-Myers Squibb, Sanofi, AstraZeneca, GlaxoSmithKline, Aptevo Therapeutics, MedImmune, Bionomics, Synthorx, AstraZeneca, Astellas Pharma, Ipsen, Merck Serono
Travel, Accommodations, Expenses: Astellas Pharma, Sanofi, Merck Serono, Amgen, Janssen, Tolmar, Pfizer
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented, in part, at the Annual Society of Clinical Oncology Genitourinary Cancers Symposium Virtual Meeting, February 11-13, 2021.
SUPPORT
E.M.K.: NHMRC Postgraduate Scholarship, Monash University Postgraduate Publications Award. H.F.: Australian Government Research Training Program (RTP) Scholarship, Monash University Postgraduate Publications Award. K.M.: Movember/Prostate Cancer Foundation of Australia Clinical Scientist Fellow. L.G.H.: Astellas Investigator-Initiated Grant, Cancer Institute NSW Translational Program Grant, Twin Towns services Community Foundation Grant. M.K.: National Institute of Health—RO1-CA212097. A.A.A.: NHMRC Project Grant (GNT1098647), Victorian Cancer Agency Clinical Research Fellowship (CRF14009), Astellas Investigator-Initiated Grant.
E.M.K., C.D., and H.F. share co-first authorship. S.J., M.K., and A.A.A. share co-senior authorship.
AUTHOR CONTRIBUTIONS
Conception and design: Edmond M. Kwan, Lisa G. Horvath, Manish Kohli, Arun A. Azad
Administrative support: Maria M. Docanto, Nicole Ng, Lisa G. Horvath
Provision of study materials or patients: Edmond M. Kwan, Nicole Ng, Kate Mahon, Winston Tan, Zhixin Zhao, Lisa G. Horvath, Shidong Jia, Manish Kohli, Arun A. Azad
Collection and assembly of data: Edmond M. Kwan, Heidi Fettke, Maria M. Docanto, Patricia Bukczynska, Nicole Ng, Siavash Foroughi, Lisa-Jane K. Graham, Kate Mahon, Winston Tan, Xiaohong Wang, Zhixin Zhao, Tiantian Zheng, Kemin Zhou, Manish Kohli, Arun A. Azad
Data analysis and interpretation: Edmond M. Kwan, Chao Dai, Heidi Fettke, Christine Hauser, Winston Tan, Jianjun Yu, Pan Du, Shidong Jia, Manish Kohli, Arun A. Azad
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Edmond M. Kwan
Honoraria: Janssen, Ipsen, Astellas Pharma, Research Review
Consulting or Advisory Role: Astellas Pharma, Janssen, Ipsen
Research Funding: Astellas Pharma, AstraZeneca
Travel, Accommodations, Expenses: Astellas Pharma, Pfizer, Ipsen, Roche
Siavash Foroughi
Stock and Other Ownership Interests: CSL Limited, Oncosil, Mesoblast
Lisa-Jane K. Graham
Patents, Royalties, Other Intellectual Property: I am listed in a “Rewards to Inventors” Scheme with the Institute of Cancer Research London Enterprise Unit as part of a research project I was involved in working at the Institute of Cancer Research London (in collaboration with Astex Therapeutics (now Astex Pharmaceuticals)) to develop novel small molecule inhibitors of PKB (AKT) from 2003-2005. The research was taken over by AstraZeneca in 2005. Payments are made to all inventors in the scheme when milestones are met, and paid by the ICR London Enterprise Unit
Kate Mahon
Research Funding: Mundipharma
Winston Tan
Honoraria: Medscape, Onclive
Xiaohong Wang
Employment: Predicine
Kemin Zhou
Employment: Denovo Biopharma, Predicine Inc.
Jianjun Yu
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Pan Du
Employment: Predicine
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Lisa G. Horvath
Employment: Connected Medical Solutions
Leadership: Connected Medical Solutions
Stock and Other Ownership Interests: Connected Medical Solutions, Imagion Biosystems
Consulting or Advisory Role: Imagion Biosystems
Research Funding: Astellas Pharma
Travel, Accommodations, Expenses: Astellas Pharma, Janssen-Cilag, Pfizer
Shidong Jia
Employment: Predicine, Genentech
Leadership: Predicine
Stock and Other Ownership Interests: Predicine
Patents, Royalties, Other Intellectual Property: Liquid Biopsy Technology
Manish Kohli
Honoraria: Advanced Accelerator Applications
Travel, Accommodations, Expenses: Celgene
Arun A. Azad
Honoraria: Janssen, Astellas Pharma, Novartis, Tolmar, Amgen, Pfizer, Bayer, Telix Pharmaceuticals, Bristol-Myers Squibb, Merck Serono, AstraZeneca, Sanofi, Ipsen, Merck Sharp & Dohme, Noxopharm
Consulting or Advisory Role: Astellas Pharma, Novartis, Janssen, Sanofi, AstraZeneca, Pfizer, Bristol-Myers Squibb, Tolmar, Telix Pharmaceuticals, Merck Sharp & Dohme, Bayer, Ipsen, Merck Serono, Amgen, Noxopharm
Speakers' Bureau: Astellas Pharma, Novartis, Amgen, Bayer, Janssen, Ipsen, Bristol-Myers Squibb, Merck Serono
Research Funding: Astellas Pharma, Merck Serono, Novartis, Pfizer, Bristol-Myers Squibb, Sanofi, AstraZeneca, GlaxoSmithKline, Aptevo Therapeutics, MedImmune, Bionomics, Synthorx, AstraZeneca, Astellas Pharma, Ipsen, Merck Serono
Travel, Accommodations, Expenses: Astellas Pharma, Sanofi, Merck Serono, Amgen, Janssen, Tolmar, Pfizer
No other potential conflicts of interest were reported.
REFERENCES
- 1.Sumanasuriya S, De Bono J: Treatment of advanced prostate cancer—A review of current therapies and future promise. Cold Spring Harb Perspect Med 8:a030635, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nuhn P, De Bono JS, Fizazi K, et al. : Update on systemic prostate cancer therapies: Management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur Urol 75:88-99, 2019 [DOI] [PubMed] [Google Scholar]
- 3.de Wit R, de Bono J, Sternberg CN, et al. : Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med 381:2506-2518, 2019 [DOI] [PubMed] [Google Scholar]
- 4.de Bono J, Mateo J, Fizazi K, et al. : Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 382:2091-2102, 2020 [DOI] [PubMed] [Google Scholar]
- 5.Abida W, Patnaik A, Campbell D, et al. : Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol 38:3763-3772, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang J, Nie J, Ma X, et al. : Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol Cancer 18:26, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanker AB, Kaklamani V, Arteaga CL: Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov 9:482-491, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Bono JS, De Giorgi U, Rodrigues DN, et al. : Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res 25:928-936, 2019 [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Bracarda S, Sternberg CN, et al. : LBA4: IPATential150: Phase III study of ipatasertib (ipat) plus abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). ESMO Virtual Congress; 2020
- 10.Liu W, Laitinen S, Khan S, et al. : Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med 15:559-565, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grasso CS, Wu YM, Robinson DR, et al. : The mutational landscape of lethal castration-resistant prostate cancer. Nature 487:239-243, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran H, Yelensky R, Frampton GM, et al. : Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 63:920-926, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson D, Van Allen EM, Wu YM, et al. : Integrative clinical genomics of advanced prostate cancer. Cell 161:1215-1228, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A, Coleman I, Morrissey C, et al. : Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 22:369-378, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abida W, Armenia J, Gopalan A, et al. : Prospective genomic profiling of prostate cancer across disease States reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 2017:1-3, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armenia J, Wankowicz SAM, Liu D, et al. : The long tail of oncogenic drivers in prostate cancer. Nat Genet 50:645-651, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quigley DA, Dang HX, Zhao SG, et al. : Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174:758-769.e9, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abida W, Cyrta J, Heller G, et al. : Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 116:11428-11436, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung JH, Dewal N, Sokol E, et al. : Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol 3:PO.18.00283, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilson C, Ingleby F, Gilbert DC, et al. : Genomic profiles of de novo high- and low-volume metastatic prostate cancer: Results from a 2-stage feasibility and prevalence study in the STAMPEDE trial. JCO Precision Oncol 882-897, 2020 [DOI] [PubMed] [Google Scholar]
- 21.Sumanasuriya S, Omlin A, Armstrong A, et al. : Consensus statement on circulating biomarkers for advanced prostate cancer. Eur Urol Oncol 1:151-159, 2018 [DOI] [PubMed] [Google Scholar]
- 22.Husain H, Velculescu VE: Cancer DNA in the circulation: The liquid biopsy. JAMA 318:1272-1274, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang CC, Du M, Wang L: Bioinformatics analysis for circulating cell-free DNA in cancer. Cancers (Basel) 11:805, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyatt AW, Azad AA, Volik SV, et al. : Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol 2:1598-1606, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wyatt AW, Annala M, Aggarwal R, et al. : Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst 109:djx118, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Annala M, Struss WJ, Warner EW, et al. : Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur Urol 72:34-42, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Annala M, Vandekerkhove G, Khalaf D, et al. : Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov 8:444-457, 2018 [DOI] [PubMed] [Google Scholar]
- 28.Torquato S, Pallavajjala A, Goldstein A, et al. : Genetic alterations detected in cell-free DNA are associated with enzalutamide and abiraterone resistance in castration-resistant prostate cancer. JCO Precision Oncol 3:1-14, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohli M, Tan W, Zheng T, et al. : Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine 54:102728, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herberts C, Murtha AJ, Fu S, et al. : Activating AKT1 and PIK3CA mutations in metastatic castration-resistant prostate cancer. Eur Urol 78:834-844, 2020 [DOI] [PubMed] [Google Scholar]
- 31.Carreira S, Romanel A, Goodall J, et al. : Tumor clone dynamics in lethal prostate cancer. Sci Transl Med 6:254ra125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azad AA, Volik SV, Wyatt AW, et al. : Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res 21:2315-2324, 2015 [DOI] [PubMed] [Google Scholar]
- 33.Romanel A, Gasi Tandefelt D, Conteduca V, et al. : Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med 7:312re310, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conteduca V, Wetterskog D, Sharabiani MTA, et al. : Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann Oncol 28:1508-1516, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jayaram A, Wingate A, Wetterskog D, et al. : Plasma androgen receptor copy number status at emergence of metastatic castration-resistant prostate cancer: A pooled multicohort analysis. JCO Precis Oncol 3:1-13, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia S, Kohli M, Du M, et al. : Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget 6:16411-16421, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fettke H, Kwan EM, Docanto MM, et al. : Combined cell-free DNA and RNA profiling of the androgen receptor: Clinical utility of a novel multi-analyte liquid biopsy assay for metastatic prostate cancer. Eur Urol 78:173-180, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adalsteinsson VA, Ha G, Freeman SS, et al. : Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 8:1324, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carver BS, Chapinski C, Wongvipat J, et al. : Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19:575-586, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulholland DJ, Tran LM, Li Y, et al. : Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19:792-804, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crumbaker M, Khoja L, Joshua AM: AR signaling and the PI3K pathway in prostate cancer. Cancers (Basel) 9:34, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferraldeschi R, Nava Rodrigues D, Riisnaes R, et al. : PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur Urol 67:795-802, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chae YK, Davis AA, Jain S, et al. : Concordance of genomic alterations by next-generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol Cancer Ther 16:1412-1420, 2017 [DOI] [PubMed] [Google Scholar]
- 44.Taavitsainen S, Annala M, Ledet E, et al. : Evaluation of commercial circulating tumor DNA test in metastatic prostate cancer. JCO Precision Oncol 3:1-9, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crabb SJ, Griffiths G, Marwood E, et al. : Pan-AKT inhibitor capivasertib with docetaxel and prednisolone in metastatic castration-resistant prostate cancer: A randomized, placebo-controlled phase II trial (ProCAID). J Clin Oncol 39:190-201, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia S, Liu Z, Zhang S, et al. : Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454:776-779, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wee S, Wiederschain D, Maira SM, et al. : PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 105:13057-13062, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni J, Liu Q, Xie S, et al. : Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov 2:425-433, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia S, Gao X, Lee SH, et al. : Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov 3:44-51, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berenjeno IM, Guillermet-Guibert J, Pearce W, et al. : Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J 442:151-159, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmit F, Utermark T, Zhang S, et al. : PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A 111:6395-6400, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearson HB, Li J, Meniel VS, et al. : Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with pten loss to accelerate progression and castration-resistant growth. Cancer Discov 8:764-779, 2018 [DOI] [PubMed] [Google Scholar]
- 53.Carpten JD, Faber AL, Horn C, et al. : A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448:439-444, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Cancer Genome Atlas Research Network : The molecular taxonomy of primary prostate cancer. Cell 163:1011-1025, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gundem G, Van Loo P, Kremeyer B, et al. : The evolutionary history of lethal metastatic prostate cancer. Nature 520:353-357, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Etten JL, Dehm SM: Clonal origin and spread of metastatic prostate cancer. Endocr Relat Cancer 23:R207-R217, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanayama M, Luo J: Predictive biomarkers in prostate cancer: Is it time to go “all in” on liquid biopsies? Eur Urol 78:181-183, 2020 [DOI] [PubMed] [Google Scholar]
- 58.Miyamoto DT, Lee RJ: PIK carefully, AKT accordingly: Towards precision medicine in prostate cancer. Eur Urol 78:845-846, 2020 [DOI] [PubMed] [Google Scholar]
- 59.De Laere B, van Dam PJ, Whitington T, et al. : Comprehensive profiling of the androgen receptor in liquid biopsies from castration-resistant prostate cancer reveals novel intra-AR structural variation and splice variant expression patterns. Eur Urol 72:192-200, 2017 [DOI] [PubMed] [Google Scholar]
- 60.Kohli M, Li J, Du M, et al. : Prognostic association of plasma cell-free DNA-based androgen receptor amplification and circulating tumor cells in pre-chemotherapy metastatic castration-resistant prostate cancer patients. Prostate Cancer Prostatic Dis 21:411-418, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Laere B, Oeyen S, Mayrhofer M, et al. : TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration-resistant prostate cancer. Clin Cancer Res 25:1766-1773, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Laere B, Rajan P, Gronberg H, et al. : Androgen receptor burden and poor response to abiraterone or enzalutamide in TP53 wild-type metastatic castration-resistant prostate cancer. JAMA Oncol 5:1060-1062, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hodara E, Morrison G, Cunha A, et al. : Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight 4:e125529, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fettke H, Kwan EM, Docanto MM, et al. : Combined cell-free DNA and RNA profiling of the androgen receptor: Clinical utility of a novel multianalyte liquid biopsy assay for metastatic prostate cancer. Eur Urol 78:173-180, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Markowski MC, Silberstein JL, Eshleman JR, et al. : Clinical utility of CLIA-grade AR-V7 testing in patients with metastatic castration-resistant prostate cancer. JCO Precis Oncol 10.1200/PO.17.00127, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]