

Lysine methylation camouflages a bacterial pathogen from the host ubiquitin attack.
Abstract
Many intracellular pathogens avoid detection by their host cells. However, it remains unknown how they avoid being tagged by ubiquitin, an initial step leading to antimicrobial autophagy. Here, we show that the intracellular bacterial pathogen Rickettsia parkeri uses two protein-lysine methyltransferases (PKMTs) to modify outer membrane proteins (OMPs) and prevent their ubiquitylation. Mutants deficient in the PKMTs were avirulent in mice and failed to grow in macrophages because of ubiquitylation and autophagic targeting. Lysine methylation protected the abundant surface protein OmpB from ubiquitin-dependent depletion from the bacterial surface. Analysis of the lysine-methylome revealed that PKMTs modify a subset of OMPs, including OmpB, by methylation at the same sites that are modified by host ubiquitin. These findings show that lysine methylation is an essential determinant of rickettsial pathogenesis that shields bacterial proteins from ubiquitylation to evade autophagic targeting.
INTRODUCTION
Intracellular pathogens generally evade the host immune surveillance machinery. This includes avoidance of surface targeting by the host ubiquitylation machinery and subsequent formation of a polyubiquitin coat, a first step in cell-autonomous immunity (1–5). The ubiquitin coat recruits autophagy receptors that engage with the autophagy machinery to target cytosol-exposed microbes for destruction (2, 3, 5–8). Although bacterial outer membrane proteins (OMPs) are targets for the host ubiquitylation machinery (9–11), the detailed mechanisms that pathogens use to block lysine ubiquitylation of surface proteins, including OMPs, are unknown.
Rickettsia species are vector-borne obligate intracellular bacterial pathogens of humans that have evolved strategies to evade host detection and block ubiquitylation of their surface proteins (12). For example, Rickettsia parkeri uses the conserved and abundant surface protein OmpB to protect other OMPs, including OmpA, from host ubiquitin detection. This OmpB-mediated mechanism is critical for R. parkeri to avoid autophagic targeting in both cultured macrophages in vitro and in immune cells in vivo (11). Nevertheless, whether and how OmpB itself avoids ubiquitylation is unknown, and it is also unclear how OmpB prevents ubiquitylation of other bacterial surface proteins at the molecular level.
Beyond OmpB, additional surface structures or modifications could provide protection from ubiquitylation at the molecular level. One such modification is lysine methylation, which is widespread in all domains of life. Lysine methylation involves the transfer of one, two, or three methyl groups to the amino group of lysine, the same group that is modified by ubiquitin (13). Rickettsia are known to use two protein-lysine methyltransferase (PKMT) enzymes, PKMT1 and PKMT2, to methylate lysines in OmpB (14–16). PKMT1 primarily monomethylates, and PKMT2 primarily trimethylates lysines in OmpB, but both can mono- and trimethylate OmpB (16, 17). Although it has been implied that the PKMTs promote Rickettsia virulence (16, 18–20), whether lysine methylation of bacterial surfaces prevents host detection and promotes intracellular survival has not been explored.
Here, we describe a forward genetic screen to identify R. parkeri genes involved in avoiding ubiquitylation and autophagy. We found that PKMT-mediated lysine methylation is critical to protect R. parkeri from ubiquitin and autophagic targeting and to cause disease in mice models. This virulence mechanism involves shielding OmpB’s lysines from host ubiquitin, thereby revealing an undescribed role for lysine methylation in avoiding cell-autonomous immunity and in bacterial pathogenesis.
RESULTS AND DISCUSSION
R. parkeri surface modifications block ubiquitylation
To identify bacteria-derived surface modifications that protect against ubiquitin coating, we screened a library of R. parkeri transposon mutants (table S1) (21) for increased polyubiquitylation (pUb) relative to wild type (WT) in Vero cells (an epithelial cell line commonly used to propagate and study intracellular pathogens) by immunofluorescence microscopy (Fig. 1A). We subsequently analyzed individual mutants and identified four that were ubiquitylated, similar to ompB mutant bacteria (Fig. 1, B and D) (11). Two of the strains had insertions in the PKMT genes pkmt1 and pkmt2, which are located in two distinct chromosomal regions. The remaining two strains had insertions in the wecA and rmlD genes, which are required for the biosynthesis of O-antigen (fig. S1), a common surface molecule in Gram-negative bacteria. The O-antigen also protects Francisella tularensis from ubiquitylation (3), suggesting that it performs a conserved function in this process. These four mutant strains expressed OmpB (pkmt1::tn, pkmt2::tn, and wecA::tn bacteria displayed reduced OmpB levels; fig. S2). As a control, we analyzed a strain with a mutation in the mrdA gene, which is required for peptidoglycan biosynthesis and cell shape in other bacteria (22). This mutant strain had altered shape but was not polyubiquitylated (Fig. 1, B and D), suggesting that not all bacterial cell envelope structures are required to avoid ubiquitylation. Collectively, these data indicate that OmpB, PKMTs, and the O-antigen protect R. parkeri from ubiquitylation.
Fig. 1. The O-antigen and lysine methylation protect R. parkeri from ubiquitylation.
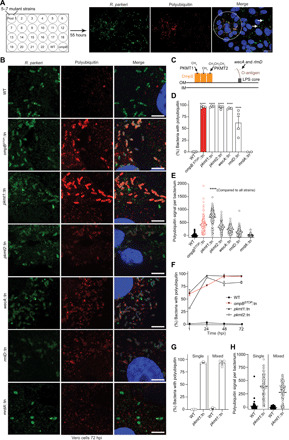
(A) Pools of R. parkeri (anti–Rickettsia-I7205 antibody; green) mutants screened for increased pUb (FK1 antibody; red) via immunofluorescence microscopy. DNA (Hoechst; blue). Circle, pUb-positive bacteria; arrows, pUb-negative bacteria. Scale bar, 20 μm. (B) Infected cells at 72 hpi stained as in (A) (n = 4). Scale bars, 5 μm. (C) The biological function of genes identified. IM, inner membrane; LPS, lipopolysaccharide. (D) Percentage of bacteria colocalized with pUb at 72 hpi (n = 4 for WT and mutants; mrdA::tn, n = 2). Statistical comparisons between WT and mutants were performed using a one-way analysis of variance (ANOVA) with Dunnett’s post hoc test. (E) Polyubiquitin signal per bacterium from (B) (≥150 bacteria counted). Statistical comparisons between strains were performed using a Kruskal-Wallis test with Dunn’s post hoc test. (F) Percentage polyubiquitylated bacteria at times between 1 and 72 hpi (n = 3). (G) Percentage bacteria polyubiquitylated in single and mixed infections (n = 3). (H) Quantifications of ≥80 bacteria in (G). Ubiquitylation of pkmt1::tn was not statistically significant (P = 0.28) between single and mixed infections, as determined by a Kruskal-Wallis test with Dunn’s post hoc test. ****P < 0.0001. All data are means ± SEM.
To assess the relative contribution of the identified genes in protecting R. parkeri from ubiquitylation, we next quantified pUb levels on individual bacteria. We observed the highest levels in the pkmt1::tn mutant, followed by the ompBSTOP::tn and pkmt2::tn mutants (Fig. 1E). Kinetic experiments with the WT and mutant strains revealed that >75% of the mutant bacteria were ubiquitylated at 24, 48, and 72 hours post-infection (hpi), with pkmt1::tn having high ubiquitylation frequencies at all time points analyzed (Fig. 1F). The previously described biochemical properties of the PKMTs suggest that they act in the bacterial cytoplasm, before or during protein secretion (16, 17, 19). To ascertain whether the PKMTs promote ubiquitylation avoidance in a cell-intrinsic manner, we assessed polyubiquitin frequencies and levels in coinfection experiments with WT and pkmt1::tn bacteria. We observed that the frequencies and levels of ubiquitylation on pkmt1::tn bacteria were similar between single and mixed infections and that WT bacteria remained ubiquitin negative after coinfection with pkmt1::tn (Fig. 1, G and H). These data support the notion that the PKMTs act in the bacterial cytoplasm to promote ubiquitylation avoidance in a cell-intrinsic manner.
PKMT1 and PKMT2 are R. parkeri virulence factors
OmpB was previously found to be required for R. parkeri to cause lethal disease in Ifnar−/−Ifngr−/− mice lacking the type I interferon receptor (IFNAR) and IFN-γ receptor (23). We therefore examined whether PKMT1 or PKMT2 are important for causing disease in vivo. We observed that Ifnar−/−Ifngr−/− mice succumbed to infection with WT but not to pkmt1::tn or pkmt2::tn bacteria after intravenous delivery (Fig. 2A). Mice infected with the pkmt1::tn mutant showed no signs of disease, whereas mice infected with pkmt2::tn showed a transient loss in body weight (Fig. 2B). We further analyzed the roles of PKMT1 and PKMT2 by intradermal inoculations in Ifnar−/−Ifngr−/− mice, which result in skin lesion formation after infection with WT R. parkeri (24), similar to what is observed in humans after a tick bite (25). Mice infected with the pkmt1::tn displayed no or reduced skin lesion formation (Fig. 2, C and D), as well as no lethality (Fig. 2E), and they did not lose as much weight as mice infected with WT bacteria (Fig. 2F). In contrast, mice infected with pkmt2::tn mutant bacteria developed skin lesions, albeit smaller and significantly slower than those elicited by WT bacteria (Fig. 2C). Together, these data indicate that PKMT1 and PKMT2 are determinants of R. parkeri pathogenesis.
Fig. 2. Lysine methylation is critical for R. parkeri pathogenesis.
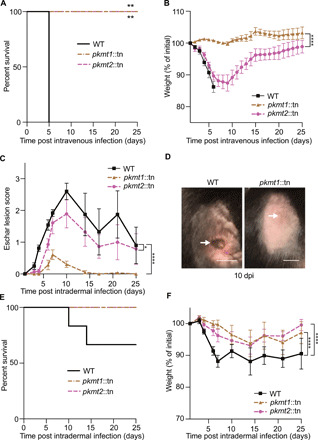
(A) Survival of Ifnar−/−Ifngr−/− mice intravenously infected with 5 × 106 WT bacteria (n = 5 mice, WT; n = 6, pkmt1::tn and pkmt2::tn). Statistical comparison between WT and mutants was performed using a log-rank (Mantel-Cox) test. Mice were humanely euthanized if body temperature dropped below 32.2 °C. (B) Weight changes of mice infected in (A). A two-way ANOVA from 0 to 25 days post-infection (dpi) with Sidak’s post hoc test was used to compare the pkmt1::tn and pkmt2::tn mutants. (C) Eschar lesion score for Ifnar−/−Ifngr−/− mice intradermally infected with 1 × 106 bacteria (n = 6, WT; n = 7, pkmt1::tn and pkmt2::tn) (24). (D) Images of mice infected intradermally in (C), at 10 dpi. Arrows indicate eschar (WT, left) or injection site (pkmt1::tn, right). Scale bars, 1 cm. Photo credit: Patrik Engström, UC Berkeley. (E) Survival of the mice infected in (C). (F) Weight changes of the mice infected in (C). A two-way ANOVA from 0 to 25 dpi with Tukey’s post hoc test was used to compare strains statistically in (C) and (F). *P < 0.05, **P < 0.01, and ****P < 0.0001. All data are means ± SEM.
Lysine methylation protects OmpB and OmpA from ubiquitylation
Because PKMT1 or PKMT2 had previously been shown to methylate OmpB in vitro (15–17, 19), we next examined whether methylation protects OmpB, or another abundant OMP, OmpA (26), from ubiquitylation. First, Vero cells overexpressing 6xHis-tagged ubiquitin were infected with WT, ompBSTOP::tn, pkmt1::tn, and pkmt2::tn strains. 6xHis-tagged ubiquitin was recruited to the surface of all the mutants but not WT bacteria, as observed by immunofluorescence microscopy (Fig. 3A). Then, 6xHis-ubiquitylated proteins were affinity-purified from infected cells, and OmpA and OmpB were detected by Western blotting. OmpA was shifted toward higher molecular weights in cells infected with mutant but not WT bacteria, indicating that OmpA is ubiquitylated (Fig. 3B). Similarly, in comparison with WT bacteria, OmpB was also shifted toward higher and lower molecular weights in cells infected with the pkmt1::tn and pkmt2::tn mutants, suggesting that OmpB is ubiquitylated. The observed lower molecular weight products of OmpB, but not OmpA, implied that methylation protects OmpB from ubiquitin-dependent proteolysis (Fig. 3B). To confirm that methylation protects OmpB and OmpA from ubiquitylation on the bacterial surface, we performed pUb enrichments of surface fractions from purified bacteria followed by Western blotting. This revealed that both OmpB and OmpA shifted toward higher molecular weights in the methyltransferase mutants but not in WT bacteria (Fig. 3C). Thus, methylation is critical to protect OMPs from ubiquitylation on the bacterial surface.
Fig. 3. Lysine methylation protects OMPs from ubiquitylation.
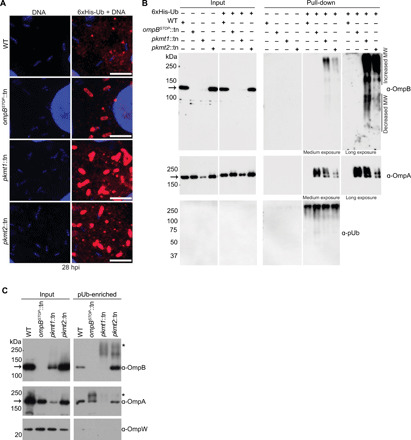
(A) Micrographs of infected Vero cells expressing 6xHis-ubiquitin stained with anti-His antibody (red) and Hoechst (bacterial and host DNA; blue) at 28 hpi (n = 2). Scale bars, 5 μm. (B) Western blot of His-Ub input and pull-down samples from infected control and 6xHis-ubiquitin expressing cells probed for OmpB (affinity-purified anti-OmpB antibody), OmpA (monoclonal anti-OmpA antibody, 13-3), and pUb (FK1, Enzo) (n = 3). (C) pUb-enriched (TUBE-1, pan-specific) samples from purified bacteria probed for OmpB, OmpA as above, and OmpW [anti-OmpW serum; OmpB and OmpA of endogenous molecular weight (MW) represent nonspecific binding to TUBE-1 beads; n = 3]. Asterisks indicate OmpB and OmpA that exhibit increased molecular weight, indicating ubiquitylation. Arrows indicate OmpB and OmpA of endogenous molecular weight.
Lysine methylation blocks ubiquitin-mediated depletion of OmpB
On the basis of our observations implying that methylation protects OmpB from ubiquitin-dependent proteolysis (Fig. 3B) and that OmpB levels are reduced on pkmt1::tn bacteria (fig. S2), we assessed whether the levels of ubiquitylation correlated with the amount of suface-associated OmpB. We infected Vero and human microvascular endothelial cells [HMECs; included because bacterial ubiquitylation is reduced in this cell type compared to Vero cells (11)] with WT and pkmt1::tn bacteria. Ubiquitin and OmpB levels were then quantified on individual bacteria by immunofluorescence microscopy. In HMECs, OmpB levels on pkmt1::tn bacteria were similar to WT bacteria (Fig. 4, A to C), correlating with low levels of ubiquitin (Fig. 4D). In Vero cells, however, OmpB levels on pkmt1::tn bacteria were lower than WT (Fig. 4, A to C), correlating with increased ubiquitylation (Fig. 4D). When plotted together, OmpB and ubiquitin levels on individual pkmt1::tn bacteria had an inverse relationship (Fig. 4E). These data indicate that PKMT1 function is linked to OmpB levels in some but not all host cell lines.
Fig. 4. Lysine methylation blocks host-mediated reduction of OmpB from the bacterial surface.
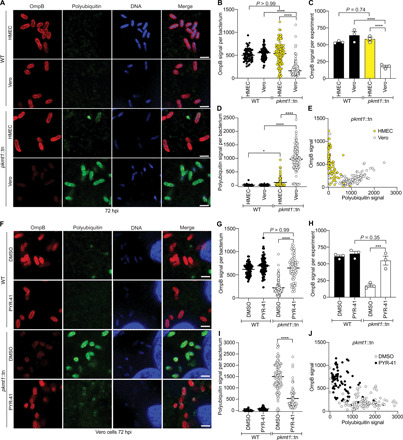
(A) HMEC and Vero cells infected WT and pkmt1::tn bacteria at 72 hpi, fixed with 4% paraformaldehyde (PFA), postfixed with methanol and stained with an anti-OmpB antibody (red) and sequentially with the FK1 antibody (green) and Hoechst (DNA; blue) (n = 3). Scale bars, 2 μm. (B) Quantification of OmpB signal per bacterium from (A) (≥63 bacteria counted). (C) Average OmpB signal per bacterium from n = 3. (D) Quantification of polyubiquitin signal on individual bacteria from (A) (≥63 bacteria). (E) OmpB and polyubiquitin signal plotted together on individual bacteria. (F) Micrographs of infected Vero cells, treated with 100 μM ubiquitin inhibitor PYR-41 between 66 and 72 hpi, stained as in (A) (n = 3). Scale bars, 2 μm. (G to J) Quantifications of (F) as in (B) to (E) (≥72 bacteria). Statistical comparisons between cell types and conditions in (B), (D), (G), and (I) were performed using a Kruskal-Wallis test with Dunn’s post hoc test and in (C) and (H) with a one-way ANOVA with Tukey’s post hoc test. ****P < 0.0001, ***P < 0.001, and *P < 0.05. All data are means ± SEM.
To further assess whether PKMTs influence OmpB levels via a ubiquitin-dependent or ubiquitin-independent mechanism, we treated infected Vero cells with PYR-41, an inhibitor of the host ubiquitin-activating enzyme E1 that is required for the ubiquitylation cascade. Treatment with PYR-41 restored OmpB levels on pkmt1::tn to that of WT bacteria (Fig. 4, F to H) and was also accompanied by reduced ubiquitin levels (Fig. 4I). Inhibition of the host ubiquitin machinery also shifted the relationship between ubiquitin and OmpB levels on individual pkmt1::tn mutant bacteria (Fig. 4J) to what was observed in HMECs (Fig. 4E). These data indicate that lysine methylation blocks a ubiquitin-dependent mechanism that depletes OmpB from the bacterial surface.
Lysine methylation of OMPs is common in R. parkeri
On the basis of our observation that methylation protects both OmpA and OmpB from ubiquitylation, we set out to determine how frequently, and to what extent, lysines of R. parkeri OMPs are methylated. Peptides with methylated lysines from whole WT bacteria were quantified using label-free liquid chromatography–mass spectrometry (LC-MS). We then analyzed lysine methylation frequency in abundant OMPs and other abundant R. parkeri proteins (table S2). This analysis revealed that R. parkeri OmpB, OmpA, and surface cell antigen 2 (Sca2) proteins had the highest abundance of methylated peptides. Lysine methylation was also detected in b-barrel assembly machinery (BAM) outer membrane protein BamA and in a predicted OMP porin (WP_014410329.1; from here on referred to as OMP-porin) (Fig. 5A, fig. S3, and tables S2 and S3). We next mapped both methylated and unmethylated lysines on the abovementioned OMPs and found that more than 50% of lysines detected from OmpB and OmpA were methylated, and a significant fraction of lysines were also methylated in Sca2 (31%), OMP-porin (30%), and BamA (27%) (Fig. 5B and fig. S3). We further mapped methylated lysines onto the predicted structures of the transmembrane domains of the above OMPs and a fragment of Sca2’s surface-exposed passenger domain, for which the structure of Rickettsia conorii protein has been determined (no structural information is available for the passenger domains of OmpB or OmpA) (27). This analysis suggested that most modified lysines are located at surface-exposed regions (fig. S4), similar to methylated residues in Salmonella Typhimurium flagellin (28). This is consistent with previous reports that lysines often are located at protein surfaces and are susceptible to posttranslational modification (29). Collectively, these observations indicate that lysine methylation of R. parkeri OMPs is common and likely to occur on residues positioned at protein surfaces.
Fig. 5. Methylation camouflages lysines in OmpB from ubiquitylation.
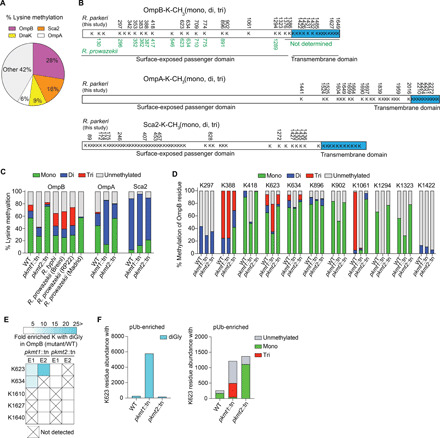
(A) Percentage lysine methylation of the total lysine methylation abundance from all the analyzed proteins in WT R. parkeri as determined by LC-MS (data are combined from n = 2). (B) Methylated lysines (K) are indicated with numbers [black, R. parkeri from this work; green, Rickettsia prowazekii OmpB from previous work (16)] (data are combined from n = 5). (C) Percentage lysine methylation of the total abundance (only from lysines with methylation abundance) in (B) that is unmethylated, monomethylated, dimethylated, or trimethylated (data are means of n = 2). Rickettsia typhi and R. prowazekii data were from published work (19). (D) Percentage of OmpB residue that is unmethylated, monomethylated, dimethylated, or trimethylated. Only lysines repeatedly detected were analyzed (data are means of n = 2). (E) Heatmap representation of OmpB residues from pkmt1::tn or pkmt2::tn that have similar levels of K-diGly (diglycine) peptides (white boxes) compared to WT or with ≥5-fold increase (cyan) after pUb-enrichments (n = 2). (F) Abundance of ubiquitylated (diGly), unmethylated, monomethylated, dimethylated, or trimethylated OmpB K623 (data are means of n = 2). Each experiment (n) was performed in technical triplicate.
PKMT1-mediated methylation camouflages lysines in OmpB from ubiquitylation
To identify OMPs that are methylated by PKMT1 and PKMT2 during infection, we compared lysine methylation frequencies in WT with those of pkmt1::tn or pkmt2::tn mutant bacteria using LC-MS. We found that monomethylation of OmpB, OmpA, the predicted OMP-porin, and another surface cell antigen protein Sca1 were reduced in pkmt1::tn compared to WT bacteria (Fig. 5C and fig. S5). Dimethylation of rickettsial surface proteins was not reduced in the mutants (fig. S5). Although trimethylation was rare and therefore difficult to analyze at the individual protein level (fig. S6), OmpB had reduced trimethylation levels in both methyltransferase mutants (Fig. 5C). Notably, the frequency of unmethylated lysines in OmpB was specifically increased in pkmt1::tn bacteria, comparable to what was observed for the avirulent Rickettsia prowazekii Madrid E strain (Fig. 5C and fig. S5) (19). Lysine methylation of five other outer membrane-associated proteins (Sca1, Sca2, BamA, LomR, and Pal-lipoprotein), and 21 of 23 abundant proteins with different predicted subcellular distributions, was not affected by mutations in the pkmt1 or pkmt2 genes (fig. S5). Whether proteins unaffected by mutations in pkmt1 and pkmt2 are methylated by other bacterial or host enzymes remains to be determined. Together, these data indicate that the PKMTs are required for methylation of a subset of OMPs including OmpB.
To determine which OmpB residues are modified by PKMT1 and PKMT2 during R. parkeri infection, we analyzed the methylation frequency of individual lysines in the mutants compared to WT using LC-MS. We observed reduced monomethylation frequencies of OmpB K418, K623, K902, K1061, K1294, and K1323 in pkmt1::tn bacteria compared with WT and reduced trimethylation frequencies on K1061 and K388 in the pkmt2::tn strain (Fig. 5D). These data indicate that several lysines in R. parkeri OmpB are methylated by PKMT1 and PKMT2 during infection. Although these results were consistent with previous biochemical results indicating that PKMT1 monomethylates and PKMT2 trimethylates OmpB’s lysines (16, 19), we found that total OmpB methylation is unaffected in the pkmt2::tn mutant. This suggests that PKMT1 is a primary methyltransferase for OmpB and that it can compensate, at least partly, for a deficiency in PKMT2.
To test the hypothesis that methylation of specific lysines in OmpB shields the same residues from ubiquitylation, we performed pUb enrichments of bacterial surface fractions followed by LC-MS to quantify lysines with diglycine (diGly) remnants, a signature for ubiquitin after trypsin digestion. A prediction of this hypothesis is that individual lysines that are heavily methylated in OmpB of Rickettsia species (16), including WT R. parkeri (Fig. 5D), are targets for ubiquitylation in pkmt1::tn bacteria. In support of this hypothesis, OmpB K634 and K623 in pkmt1::tn exhibited 7- to 10,000-fold increased ubiquitylation compared with WT bacteria (Fig. 5, E and F; fig. S7; and table S4). Furthermore, we observed a 13-fold increase in ubiquitylation of the OMP-porin in pkmt1::tn bacteria (table S4), indicating that methylation also protects additional OMPs. However, in the pkmt2::tn mutant, differential OMP-ubiquitylation was below detection limits (Fig. 5, E and F, and table S4). Together, these data indicate that methylation by PKMT1 camouflages lysines in OMPs from ubiquitylation.
Lysine methylation protects R. parkeri from autophagic targeting
Because pUb promotes recruitment of the autophagy receptors p62/SQSTM1 (Sequestosome-1) and NDP52 (nuclear dot protein 52) (6, 7), we hypothesized that lysine methylation shields OMPs from ubiquitylation to block recruitment of these proteins. Consistent with this hypothesis, we observed that the majority of pkmt1::tn, pkmt2::tn, and ompB mutant bacteria colocalized with p62 and NDP52 by immunofluorescence microscopy (Fig. 6, A to D). These data demonstrate that PKMTs protect R. parkeri from autophagy recognition.
Fig. 6. Lysine methylation blocks the recruitment of autophagy receptors to R. parkeri.
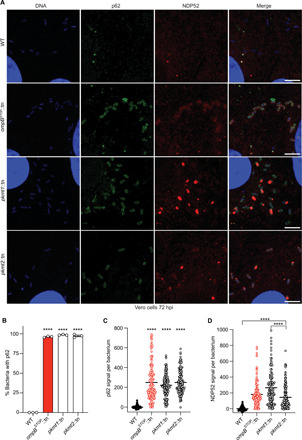
(A) Micrographs of Vero cells infected with the indicated strains at 72 hpi. Fixed with 4% PFA and stained for bacterial and host DNA (Hoechst; blue), p62 (anti-p62 antibody; green), and NDP52 (anti-NDP52 antibody; red) (representative of n = 4 for p62; n = 2 for NDP52). Scale bars, 5 μm. Image adjustments for each marker were applied equally for all the bacterial strains [except DNA (blue), which was adjusted slightly differently for WT bacteria]. (B) Percentage of bacteria that show rim-like surface localization with p62 at 72 hpi (n = 3). Statistical comparisons between all strains were performed using a one-way ANOVA with Tukey’s post hoc test. (C) Quantification of p62 signal per bacterium from a representative experiment (≥142 bacteria counted). Statistical comparisons were performed using a Kruskal-Wallis test with Dunn’s post hoc test. (D) Quantification and statistical comparisons of NDP52 signal per bacterium as in (C). ****P < 0.0001. Data are means ± SEM.
Many pathogenic bacteria including R. parkeri grow in immune cells such as macrophages (11, 30, 31), although microbial detection in such cells triggers antibacterial pathways. We therefore investigated whether PKMT1 or PKMT2 was required for evading autophagy targeting and bacterial growth in cultured bone marrow–derived macrophages (BMDMs), as was observed for OmpB (11). BMDMs were generated from control mice and mice lacking the gene encoding for autophagy-related 5 (ATG5), a protein required for optimal membrane envelopment around pathogens targeted by autophagy and for their subsequent destruction (8). We observed that pkmt1::tn mutant bacteria were unable to grow in control BMDMs (Atg5flox/flox) and that growth was rescued in Atg5-deficient BMDMs (Atg5−/−). Furthermore, >91% of pkmt1::tn bacteria were labeled with both pUb and p62 in Atg5-deficient BMDMs (Fig. 7, A to D), suggesting that ubiquitin-tagged bacteria were not restricted when the autophagy cascade was prevented. In contrast, the pkmt2::tn mutant did not have a major growth defect compared with WT bacteria, and 50% of the bacteria were labeled with p62, irrespective of host genotype (Fig. 7, A to D), consistent with less pronounced ubiquitylation phenotypes compared to pkmt1::tn bacteria. Collectively, these data indicate that methylation is required for R. parkeri growth in macrophages by avoiding autophagic targeting.
Fig. 7. Methylation prevents ATG5-dependent R. parkeri killing in macrophages.
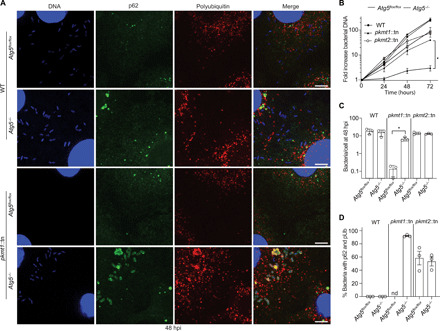
(A) Micrographs of infected control (Atg5flox/flox) and Atg5-deficient (Atg5−/−) BMDMs at 48 hpi. Fixed with 4% PFA and stained for bacterial and host DNA (Hoechst; blue), polyubiquitin (FK1, Enzo; red), and p62 (anti-p62 antibody; green) (n = 3). Scale bars, 5 μm. (B) Bacterial growth curves in control and Atg5−/− BMDMs, as measured by genomic equivalents using quantitative polymerase chain reaction (PCR; n = 3). (C) Quantification of the mean number of bacteria per cell (n = 3; bacteria from five fields of vision per strain, host genotype, and experiment). (D) Quantification of the percentage of bacteria that colocalize with p62 and pUb (n = 3). nd, not determined. Statistical comparisons for each bacterial strain between the different host genotypes in (B) and (C) were performed using a Brown-Forsyth and Welch ANOVA with Dunnett’s post hoc test. *P < 0.05. All data are means ± SEM.
Our work reveals a detailed molecular mechanism that camouflages bacterial surface proteins from host detection. In particular, we found that lysine methylation was essential for blocking ubiquitylation, a first step in cell-autonomous immunity (1, 2, 4, 5). This highlights an intricate evolutionary arms race between pathogens and hosts and reveals a strategy that pathogens can adapt to counteract host responses. The lysine methyltransferases PKMT1 and PKMT2 are conserved between rickettsial species (figs. S8 and S9) and contain a core Rossmann fold found in the broader superfamily of class I methyltransferases that exist in diverse organisms (13, 17). Thus, we propose that lysine methylation and potentially other lysine modifications could be used by pathogens and symbionts and perhaps even in eukaryotic organelles to prevent undesirable surface ubiquitylation and downstream consequences, including elimination by autophagy. Further study of microbial surface modifications will continue to enhance our understanding of the pathogen-host interface and could ultimately lead to new therapeutic targets for treating human diseases including those caused by infectious agents.
MATERIALS AND METHODS
Cell lines and primary mouse macrophages
Vero and HMEC-1 cells were purchased from the UC Berkeley Cell Culture Facility. The identity of Vero cells was repeatedly confirmed by MS analysis, and the identity of HMEC-1 cells was authenticated by short-tandem repeat analysis at the UC Berkeley Cell Culture Facility. Vero cells were grown at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium (Gibco, catalog no. 11965) with high glucose (4.5 g liter−1) and 2% heat-inactivated (Hi) (20 min, 65°C, in a water bath) fetal bovine serum (FBS; Gemcell). Low-passage HMEC cells were grown at 37°C and 5% CO2 in MCDB 131 media containing 10 mM l-glutamine (Sigma-Aldrich, M8537), supplemented with epidermal growth factor (10 ng ml−1; Fisher Scientific, CB40001; Corning, 354001), hydrocortisone (1 μg ml−1; Spectrum Chemical, CO137), and 10% Hi-FBS (30 min, 56°C, in a water bath; HyClone). Vero and HMEC cells were confirmed to be mycoplasma negative by 4′,6-diamidino-2-phenylindole staining and fluorescence microscopy screening at the UC Berkeley Cell Culture Facility.
BMDMs generated from the femurs of mutant Atg5flox/flox and matched Atg5−/− C57BL/6 mice were a gift from the laboratory of J. S. Cox (UC Berkeley), and they were prepared as previously described (11) although in the absence of antibiotics. Genotypes were confirmed by polymerase chain reaction (PCR) and Sanger sequencing at the UC Berkeley DNA Sequencing Facility, as previously described (11).
R. parkeri strain generation and validation
WT R. parkeri strain Portsmouth [National Center for Biotechnology Information (NCBI) accession no. NC_017044.1; originally a gift from C. Paddock, Center for Disease Control and Prevention] and bacterial stocks of ompBSTOP::tn [the genome sequences of these bacterial strains are available at the Sequence Read Archive as accession no. SRP154218 (WT, SRX4401164; ompBSTOP::tn, SRX4401167)], pkmt1::tn, pkmt2::tn, wecA::tn, and rlmD::tn mutants were propagated and purified every ~6 to 10 months as described below. Side-by-side experimental comparisons were made between stocks prepared at similar times.
R. parkeri pkmt1::tn, pkmt2::tn, wecA::tn, and 114 other transposon insertion mutant strains, screened for pUb, were previously isolated in a screen for small-plaque (Sp) mutants (21, 32). The ompBSTOP::tn was previously isolated in a suppressor screen and lacked expression of OmpB (11). The rmlD::tn and 132 other transposon insertion mutant strains, screened for pUb, were isolated in an independent screen in which mutants were isolated without regard for plaque size (table S1). The genomic locations of transposon insertion sites for all mutants were determined by semirandom nested PCR. To verify the insertions and clonality, we used PCRs that amplified the transposon insertion site using primers for flanking chromosomal regions: 5′-GCTCACTAGATAGCACTCG-′3 and 5′-GCTCGATTTATCTCACTTTATG-′3 for rlmD::tn, 5′-CGTTTAATAGTCCAGTTAATTTGT-′3 and 5′-CCGTCTATACCGTCCATAAAAT-′3 for wecA::tn, 5′-GCATCGAAATAACCCTGAG-′3 and 5′-GCAAACTTCTCAAAGAAATTAACG-′3 for pkmt1::tn, 5′-GCTAAGAAATCTTCTAATTTGATATTTTAC-′3 and 5′-CGAAAATTTACCTGAGCCTT-′3 for pkmt2::tn, and 5′-CGACACATAATAGCACAAACTAC-′3 and 5′-GCGGAGGCGGTAGTAAAG-′3 for mrdA::tn (fig. S10).
Screening for pUb-positive strains
To prepare the mutant library for screening, passage 1 (P1) transposon insertion mutants were amplified one time in Vero cells using 24-well cell culture plates. At 5 to 12 days post-infection, when 50 to 70% of the infected cells appeared to be rounded up (as a sign of infection) by visual inspection using a light microscope, cell culture media were completely removed, and cells were subsequently lysed in 500 μl of cold sterile water for 2 to 3 min. Next, 500 μl of cold 2× sterile brain-heart infusion (BHI) broth (BD Difco, 237500) was added to the lysed cells and resuspended, and P2 bacteria were transferred to cryogenic storage vials and frozen at −80°C.
To screen for pUb-positive strains, 10 to 40 μl of each of five to seven P2 mutant bacterial strains was diluted in 1 ml of room temperature (RT) cell culture media supplemented with 2% FBS. Subsequently, the pooled bacterial suspension was centrifuged at 250g, 4 min at RT onto confluent Vero cells grown on coverslips in 24-well plates. Cells were then incubated at 33°C and fixed at 50 to 55 hpi with prewarmed (37°C) 4% paraformaldehyde (PFA; Ted Pella Inc., 18505) for 10 min at RT. If cells were overinfected (i.e., individual infection foci had grown together) as determined by immunofluorescence microscopy, then infections of that specific pool were repeated using reduced volumes of P2 bacteria. Next, fixed cells were permeabilized with 0.2% Triton X-100 for 5 min and then stained with the anti-Rickettsia I7205 antibody (1:500 dilution; a gift from T. Hackstadt) and anti-polyubiquitin FK1 antibody (1:250 dilution; Enzo Life Sciences, BML-PW8805-0500), followed by Alexa Flour 488 anti-rabbit antibody (1:500 dilution; Invitrogen, A11008) or goat anti-mouse Alexa Flour 568 (Invitrogen, A11004). Whole coverslips were manually inspected on a Nikon Ti Eclipse microscope with 60× (1.4 numerical aperture) Plan Apo objective. The initial screen revealed that 5 of 39 mutant pools contained pUb-positive areas.
In a secondary screen, individual strains from the pUb-positive pools were used to infect Vero cells, as stated above. Infected cells were also fixed and stained as above except that a postfixation step using 100% methanol (RT) for 5 min was included and an OmpB antibody (11) and Hoechst (1:2500 dilution; Sigma-Aldrich, B2261) were used instead of the anti-Rickettsia I7205 antibody to detect bacteria. Samples were inspected as above, and strains were scored as follows: (i) pUb negative (49 strains); (ii) a few infection foci were pUb positive (two strains: Sp mutant 24, insertion at base pair position 753916; Sp mutant 94, insertion at base pair position 774831); (iii) bacteria in the center of foci were pUb positive but not on the edges (two strains: Sp mutant 43, insertion at base pair positions 651602 to 651604; Sp mutant 45, insertion at base pair position 751156); and (iv) almost all bacteria in all foci were pUb positive [four strains: pkmt1::tn, insertion at base pair position 1161553 (gene MC1_RS06185); pkmt2::tn, insertion at base pair position 34100 (MC1_RS00180); wecA::tn, insertion at base pair position 1223170 (MC1_RS06510); and rmlD::tn, insertion at base pair position 455753 (MC1_RS02345)].
Rickettsia purification
R. parkeri strains were propagated as described previously (11). “Purified bacteria” were from five T175 flasks of Vero cells that after 5 to 8 days of infection (normally ~75% infected as observed by light microscopy) were harvested in the media using a cell scraper. Bacteria were then centrifuged at 12,000g for 15 min at 4°C in prechilled tubes. Pellets were resuspended in cold K-36 buffer [0.05 M KH2PO4, 0.05 M K2HPO4 (pH 7), 100 mM KCl, and 15 mM NaCl], and a prechilled Dounce homogenizer (tight fit) was used for 60 strokes to release bacteria from host cells. The homogenate was then centrifuged at 200g for 5 min at 4°C to remove cellular debris. The supernatant was overlaid onto cold 30% v/v MD-76R (Mallinckrodt Inc., 1317-07) diluted in K-36 and centrifuged at 58,300g for 20 min at 4°C in an SW-28 swinging-bucket rotor. The bacterial pellet was resuspended in cold 1× BHI broth (0.5 ml of BHI per infected T175 flask). After letting DNA precipitates sediment to the bottom of the tubes, bacterial suspensions were collected, aliquoted, and frozen at −80°C.
“Gradient-purified bacteria” were from 10 T175 flasks of Vero cells, purified as above with the addition of a 40/44/54% v/v MD-76R (diluted in K-36 buffer) gradient step centrifuged at 58,300g for 25 min at 4°C using the SW-28 swinging-bucket rotor. The bacteria were then collected from the 44 to 54% interface, diluted in K-36 buffer, and pelleted by centrifugation at 12,000g for 15 min at 4°C. The pellet was resuspended in cold 1× BHI broth and subsequently aliquoted and frozen at −80°C.
OmpW and elongation factor Tu antibody production
The sequence encoding amino acids 22 to 224 of OmpW (WP_014411122.1; a protein that lacks the signal peptide), or full-length elongation factor Tu (EF-Tu; WP_004997779.1), were amplified by PCR from R. parkeri genomic DNA and subsequently cloned into plasmid pETM1, which encodes N-terminal 6xHis and maltose-binding protein (MBP) tags. From the resulting plasmids, fusion proteins were expressed in Escherichia coli strain BL21 codon plus RIL-Camr (DE3) (QB3 MacroLab, UC Berkeley) by induction with 1 mM isopropyl-β-d-thiogalactoside for 2 to 2.5 hours at 37°C. Bacterial pellets were resuspended in lysis buffer [50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol (DTT)] and stored at −80°C. For protein purification, bacteria were thawed, lysozyme was added to 1 mg/ml (Sigma-Aldrich, L4919), and lysis was carried out by sonication. Lysates containing 6xHis-MBP-OmpW and 6xHis–MBP–EF-Tu were incubated on amylose resin (New England Biolabs, E8021L), and bound proteins were eluted in lysis buffer lacking EDTA and DTT but containing 10 mM maltose. Fractions were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and those with the highest concentrations of fusion proteins were pooled to generate rabbit antibodies against OmpW and EF-Tu. Purified 6xHis-MBP-OmpW (1.2 mg) and 6xHis–MBP–EF-Tu proteins (1.2 mg) were sent to Pocono Rabbit Farm and Laboratory (Canadensis, PA), and immunization was carried out according to their 91-day protocol.
Western blotting
To determine the levels of bacterial and host proteins in purified bacterial samples, 30% purified bacterial samples were boiled in 1× SDS loading buffer [150 mM tris (pH 6.8), 6% SDS, 0.3% bromophenol blue, 30% glycerol, and 15% β-mercaptoethanol] for 10 min; then, 5 × 106 plaque-forming units (PFU) were resolved on an 8 to 12% SDS-PAGE gel and transferred to an Immobilon-FL polyvinylidene difluoride membrane (Millipore, IPEL00010). Membranes were probed for 30 min at RT or 4°C overnight with antibodies as follows: affinity-purified rabbit anti-OmpB antibody (11) diluted 1:200 to 30,000 in TBS-T [20 mM tris, 150 mM NaCl (pH 8.0), and 0.05% Tween 20 (Sigma-Aldrich, P9416)] plus 5% dry milk (Apex, 20-241), mouse monoclonal anti-OmpA 13-3 antibody diluted 1:10,000 to 50,000 in TBS-T plus 5% dry milk, rabbit anti-OmpW serum diluted 1:8000 in TBS-T plus 5% dry milk, mouse monoclonal FK1 anti-polyubiquitin antibody diluted 1:2500 in TBS-T plus 2% bovine serum albumin (BSA), rabbit anti–EF-Tu serum diluted 1:15,000 in TBS-T plus 5% dry milk, or rabbit anti–O-antigen serum diluted 1:5000 in TBS-T plus 5% dry milk. Secondary antibodies were mouse anti-rabbit horseradish peroxidase (HRP; Santa Cruz Biotechnology, sc-2357) or goat anti-mouse HRP (Santa Cruz Biotechnology, sc-2005), all diluted 1:1000 to 2500 in TBS-T plus 5% dry milk. Secondary antibodies were detected with ECL Western Blotting Detection Reagents (GE Healthcare, RPN2106) for 1 min at RT and developed using BioMax Light Film (Carestream, 178-8207).
Immunofluorescence microscopy
R. parkeri infections were carried out in 24-well plates with sterile circle 12-mm coverslips (Thermo Fisher Scientific, 12-545-80). To initiate infection, 30% purified bacteria were diluted in cell culture media at RT to reach a multiplicity of infection (MOI) of 0.01 for Vero and HMEC cells [for 1 hpi in Fig. 1F, an MOI of 2 was used; for other time points (24, 48, and 72 hpi) in Fig. 1F, an MOI of 0.01 was used] and an MOI of 0.1 for BMDMs. Bacteria were centrifuged onto cells at 300g for 5 min at RT and subsequently incubated at 33°C. For experiments with the ubiquitin inhibitor PYR-41 (Millipore, 662105), the cell culture media were exchanged at 66 hpi to media containing 100 μM PYR-41 [dimethyl sulfoxide (DMSO) dissolved; PYR-41 was diluted in prewarmed media (supplemented with 10% FBS), swirled, and incubated at 37°C for 5 min] or the corresponding amount of DMSO and fixed at 72 hpi. We found that supplementing the Vero cell culture media with 10% FBS, instead of 2%, is required for the inhibitory activity of PYR-41. Next, infected cells were fixed for 10 min at RT in prewarmed (37°C) 4% PFA [diluted in PBS (pH 7.4)] and then washed three times with PBS. Cells were permeabilized with 0.5% Triton X-100 for 5 min before staining. Primary antibodies were as follows: for staining with the guinea pig polyclonal anti-p62 antibody (1:500 dilution; Fitzgerald, 20R-PP001), mouse polyclonal anti-NDP52 antibody (1:100 dilution; Novus Biologicals, H00010241-B01P), a rabbit anti-Rickettsia I7205 antibody (1:500 dilution; a gift from T. Hackstadt), or anti-polyubiquitin FK1 antibody (both at 1:250 dilutions; Enzo Life Sciences, BML-PW8805-0500, or Millipore, 04-262). For staining with mouse monoclonal anti-OmpA 13-3 antibody (1:5000 dilution), affinity-purified rabbit anti-OmpB antibody (1:1000 dilution) (11) or rabbit anti–O-antigen serum (1:500 dilution) (33) infected cells were postfixed in methanol for 5 min at RT (no Triton X-100). Cells were then washed three times with PBS and incubated with the primary antibodies for 30 min at RT. To detect the primary antibodies, secondary goat anti-rabbit Alexa Flour 568 (Invitrogen, A11036), goat anti-mouse Alexa Flour 568 (Invitrogen, A11004) or goat anti–guinea pig Alexa Flour 568 (Invitrogen, A11075), Alexa Flour 488 anti-rabbit antibody (Invitrogen, A11008), and Alexa Flour 488 anti-mouse antibody (Invitrogen, A11001) antibodies were incubated at RT for 30 min (all 1:500 in PBS with 2% BSA). Images were captured as 15 to 30 z stacks (0.1- or 0.2-μm step sizes) on a Nikon Ti Eclipse microscope with a Yokogawa CSU-XI spinning disc confocal with 100× (1.4 numerical aperture) Plan Apo objectives and a Clara Interline CCD Camera (Andor Technology) using MetaMorph software (Molecular Devices). Images were processed using ImageJ using z stack average maximum intensity projections and assembled in Adobe Photoshop. The pkmt1::tn mutant expressed low green fluorescent protein levels, which was used to distinguish pkmt1::tn from WT bacteria in the mixed infection samples in Fig. 1 (G and H), similar to previously described (32). For quantification of the percentage of bacteria with pUb and p62, only bacteria colocalized with rim-like patterns of the respective marker were scored as positive for staining. Image adjustments for each marker [pUb, p62, NDP52, OmpB, and OmpA (red or green)] were applied equally for the bacterial strains, host cell lines, and experimental conditions [except DNA (blue), which, in rare occasions, was adjusted slightly differently between conditions or bacterial strains]. To quantify pUb, p62, NDP52, OmpB, and OmpA signal per bacterium, z stacks were projected as stated above, and the edges of individual bacteria were marked by the freehand region of interest (ROI) function in ImageJ. Subsequently, the average pixel intensity within that ROI was measured. pUb/p62/NDP52 signal intensities were calculated by subtracting the average pUb/p62/NDP52 signal of WT bacteria from the pUb/p62/NDP52 value of each bacterium. OmpB signal intensity was calculated by subtracting the average OmpB signal of ompBSTOP::tn bacteria, or the average background signal (areas with no bacteria) when ompBSTOP::tn was not included, from the OmpB value of each bacterium. OmpA signal intensity was calculated by subtracting the average background signal (areas with no bacteria) from the OmpA value of each bacterium.
Sample preparation for MS to determine the lysine methylome
A total of 5 × 107 gradient-purified WT (P6), pkmt1::tn (P4), and pkmt2::tn (P4) bacteria were centrifuged at 11,000g for 3 min. Each pellet was resuspended in 50 μl of tris (10 mM)–EDTA (10 mM) (pH 7.6) and incubated for 45 min in a 45°C water bath. Bacterial surface fractions were recovered from the supernatant after centrifugation at 11,000g for 3 min. Pellet was resuspended as above and incubated for an additional 45 min at 45°C. Both pellet and surface fractions were boiled at 95°C for 10 min. Samples were cooled to RT before the addition of 20 μl of 50 mM NH4HCO3 (pH 7.5) and 50 μl of a 0.2% solution of RapiGest (diluted in NH4HCO3; Waters, 186001861). Next, samples were heated at 80°C for 15 min and cooled to RT before the addition of 1 μg of trypsin (Promega, V511A). Samples were digested at 37°C overnight. To hydrolyze the RapiGest, 20 μl of 5% trifluoroacetic acid (TFA) was added to samples, which were incubated at 37°C for 90 min before centrifugation at 15,000g for 25 min at 4°C. Samples were desalted using C18 OMIX tips (Agilent Technologies, A57003100) according to the manufacturer’s instructions, and sample volume was decreased to 20 μl using a SpeedVac vacuum concentrator. Samples were stored at 4°C before analysis.
TUBE (tandem ubiquitin binding entity) assay and sample preparation for MS
To enrich for polyubiquitylated proteins, 3 × 108 PFU of “purified” WT (P6), ompBSTOP::tn (P6), pkmt1::tn (P4), and pkmt2::tn (P3) bacteria were centrifuged at 14,000g for 3 min at RT. Next, to release the surface protein fraction, the bacterial pellets were resuspended in lysis buffer [50 mM tris-HCI (pH 7.5), 150 mM NaCl, 1 mM EDTA and 10% glycerol]; supplemented with 0.0031% v/v Lysonase (Millipore, 71230), the deubiquitylase inhibitor PR619 (Life Sensor, SI9619) at a final concentration of 20 μM, and 0.8% w/v octyl β-d-glucopyranoside (Sigma-Aldrich, O8001); and incubated on ice for 10 min with occasional pipetting of samples to break pellet into smaller pieces. Subsequently, the lysate was cleared by centrifugation at 14,000g at 4°C for 5 min and incubated with equilibrated agarose TUBE-1 (Life Sensor, UM401) for 3 hours at 4°C. After binding of polyubiquitylated proteins to TUBE-1, agarose beads were washed once with TBS supplemented with 0.05% Tween 20 and 5 mM EDTA and, subsequently, three times with TBS only (no Tween or EDTA) and centrifuged at 5000g for 5 min. To prepare samples for MS analysis, enriched proteins were digested at 37°C overnight on agarose beads in RapiGest SF solution (Waters, 186001861) supplemented with 0.75 μg of trypsin (Promega, V511A). The reaction was stopped using 1% TFA (Sigma-Aldrich, T6508). Octyl β-d-glucopyranoside was extracted using water-saturated ethyl acetate (Sigma-Aldrich, 34858). Before the submission of samples for MS analysis, samples were desalted using C18 OMIX tips (Agilent Technologies, A57003100) according to the manufacturer’s instructions.
Liquid chromatography–mass spectrometry
Samples of proteolytically digested proteins were analyzed using a Synapt G2-Si ion mobility mass spectrometer that was equipped with a nanoelectrospray ionization source (Waters). The mass spectrometer was connected in line with an ACQUITY M-class ultraperformance LC system that was equipped with trapping (Symmetry C18; inner diameter, 180 μm; length, 20 mm; and particle size, 5 μm) and analytical (HSS T3; inner diameter, 75 μm; length, 250 mm; and particle size, 1.8 μm) columns (Waters). Data-independent, ion mobility–enabled, high-definition mass spectra and tandem mass spectra were acquired in the positive ion mode (34–36). Raw data acquisition was controlled using MassLynx software (version 4.1), and tryptic peptide identification and relative quantification using a label-free approach (37, 38) were performed using Progenesis QI for Proteomics software (version 4.0; Waters). Raw data were searched against R. parkeri and Chlorocebus sabaeus protein databases (NCBI) to identify tryptic peptides, allowing for up to three missed proteolytic cleavages, with diGly-modified lysine (i.e., ubiquitylation remnant) and methylated lysine as variable posttranslational modifications. Calculation of the percentage of lysine methylation (mono-, di-, tri-, or unmethylated), for each bacterial strain, was performed by dividing the abundance of a residue/protein bearing a modification by the total abundance and multiplying by 100. Data-dependent analysis was performed using an UltiMate 3000 RSLCnano LC system that was connected in line with an LTQ Orbitrap XL mass spectrometer equipped with a nanoelectrospray ionization source and Xcalibur (version 2.0.7) and Proteome Discoverer (version 1.3; Thermo Fisher Scientific, Waltham, MA) software, as described elsewhere (39).
Localization of tagged ubiquitin and ubiquitin pull-down
To assess the localization of 6xHis-ubiquitin during infection, confluent Vero cells grown in 24-well plates with coverslips were transfected with 2 μg of pCS2-6xHis-ubiquitin plasmid DNA using Lipofectamine 2000 (Invitrogen, 11668-019) for 6 hours in Opti-MEM (Gibco, 31985-070). Subsequently, media were exchanged to media without transfection reagent, and cells were incubated overnight at 37°C and 5% CO2. The following day (~16 hours after transfection), transfected cells were infected with purified WT or mutant bacteria at an MOI of 1. At 28 hpi, infected cells were fixed with 4% PFA diluted in PBS (pH 7.4) for 10 min and then washed three times with PBS. Primary anti-6xHis monoclonal mouse antibody (diluted 1:1000; Clontech, 631212) was used to detect 6xHis-ubiquitin in samples permeabilized with 0.5% Triton X-100 and a goat anti-mouse Alexa Flour 568 (Invitrogen, A11004) to detect the primary 6xHis antibody. Hoechst (diluted 1:2500; Thermo Fisher Scientific, 62249) was used to detect host and bacterial DNA. Samples were imaged as already described.
For ubiquitin pull-downs, confluent Vero cells grown in six-well plates were transfected and infected as described above. At 28 hpi, cells were washed once with 1× PBS (pH 7.4), and subsequently lysed in urea lysis buffer [8 M urea, 50 mM tris-HCI (pH 8.0), 300 mM NaCl, 50 mM Na2HPO4, and 0.5% Igepal CA-630 (Sigma-Aldrich, I8896)] for 20 min at RT. Subsequently, samples were sonicated, and the lysate was cleared by centrifugation at 15,000g for 15 min at RT. Before incubation with Ni-NTA (nitrilotriacetic acid) resin (QIAGEN, 1018244), an aliquot was saved for the input sample. 6xHis-ubiquitin conjugates were purified by incubation and rotation with Ni-NTA resin for 3 hours at RT in the presence of 10 mM imidazole. Beads were washed three times with urea lysis buffer and once with urea lysis buffer lacking Igepal CA-630. Ubiquitin conjugates were eluted at 65°C for 15 min in 2× Laemmli buffer containing 200 mM imidazole and 5% 2-mercaptoetanol (Sigma-Aldrich, M6250), vortexed for 90 s, and centrifuged at 5000g for 5 min at RT. Eluted and input proteins were detected by SDS-PAGE followed by Western blotting, as described above.
Animal experiments
Animal research using mice was conducted under a protocol approved by the UC Berkeley Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act. The UC Berkeley IACUC is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and adheres to the principles of the Guide for the Care and Use of Laboratory Animals. Infections were performed in a biosafety level 2 facility. Mice were age-matched between 8 and 18 weeks old. Mice were selected for experiments on the basis of their availability, regardless of sex. All mice were healthy at the time of infection and were housed in microisolator cages and provided chow and water. Littermates of the same sex were randomly assigned to experimental groups. For intravenous infections, R. parkeri was prepared by diluting 30% prep bacteria into cold sterile 1× PBS to 5 × 106 PFU per 200 μl. Bacterial suspensions were kept on ice during injections. Mice were exposed to a heat lamp while in their cages for approximately 5 min, and then, each mouse was moved to a mouse restrainer (Braintree Scientific, TB-150 STD). The tail was sterilized with 70% ethanol, and 200 μl of bacterial suspension was injected using 30.5-gauge needles into the lateral tail vein. For intradermal infections, R. parkeri was prepared by diluting purified bacteria into cold sterile 1× PBS to 1 × 106 PFU per 50 μl. Mice were shaved and wiped with 70% ethanol, and 50 μl of bacterial suspension was injected intradermally using a 30.5-gauge needle. Body weights and temperatures were monitored. The skin lesions were scored as follows: 0, no visible lesion; 1, moderate redness; 2, extensive redness and 1- to 4-mm-diameter lesion; 3, 4- to 10-mm lesion; and 4, lesion >10 mm. The temperatures were measured using a rodent rectal thermometer (Braintree Scientific, RET-3). Mice were also monitored daily for clinical signs of disease, such as hunched posture, lethargy, or scruffed fur. If a mouse displayed severe signs of infection, as defined by a reduction in body temperature below 32.2 °C or an inability to move around the cage normally, then the animal was immediately and humanely euthanized using CO2 followed by cervical dislocation according to IACUC-approved procedures (23, 24).
Structure prediction
Primary amino acid sequences of lysine-methylated proteins were submitted to the Protein Homology/analogY Recognition Engine version 2.0 (Phyre2) to detect homolog proteins with known structure (40). This generated predicted structural models as obtained in PDB files. Template structure is indicated in the figure legend of fig. S4. Predicted homology structure with the highest protein identity and with a confidence value of 100% was visualized in EzMol (41), using the obtained PDF files. Methylated Lys residues were highlighted in red; protein backbone was highlighted in purple, and protein surface was highlighted in white.
Statistical analysis, experimental variability, and reproducibility
Statistical parameters and significance are reported in the legends. Data were considered to be statistically significant when P < 0.05, as determined by a one-way analysis of variance (ANOVA) with Dunnett’s or Tukey’s post hoc tests, a Kruskal-Wallis test with Dunn’s post hoc test, a Brown-Forsyth and Welch ANOVA with Dunnett’s post hoc test, or a two-way ANOVA (all two sided). Statistical analysis was performed using Prism 6 software (GraphPad Software). If not otherwise described, n indicates the number of independent biological experiments executed at different times.
Acknowledgments
We thank D. Portnoy (UC Berkeley) and L. Radoshevich (University of Iowa) for providing comments on this manuscript and N. Fischer (UC Berkeley) for critically reading the manuscript. We also thank H. Kim (Stony Brook University) for providing the O-antigen antibody and for fruitful discussions. We are also grateful for the Atg5flox/flox and Atg5−/− BMDMs provided by G. Golovkine (UC Berkeley) and J. S. Cox and M. Rape (both UC Berkeley) for fruitful discussions. Funding: P.E. was supported by a postdoctoral fellowship from the Sweden-America Foundation. M.D.W. was supported by NIH/NIAID grants R01 AI109044 and R21 AI138550. A mass spectrometer used in this study was purchased with support from NIH grant 1S10 OD020062-01. Author contributions: P.E. conceived the study with the assistance of M.D.W. P.E. performed laboratory work and analysis, except for MS, which was conducted together with A.T.I, and animal experiments, which were conducted together with T.P.B. and C.J.T. P.E. drafted the initial and revised manuscript, and A.T.I., T.P.B., C.J.T., and M.W.D. provided editorial feedback. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The Himar1 pMW1650 transposon plasmid can be provided by the University of South Alabama pending scientific review and a completed material transfer agreement. Requests for the pMW1650 transposon plasmid should be submitted to the University of South Alabama Tech transfer office. The O-antigen serum can be provided by the University of Chicago pending scientific review and a completed material transfer agreement. Requests for the O-antigen serum should be submitted to H. Kim, Stony Brook University. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg2517/DC1
REFERENCES AND NOTES
- 1.Perrin A. J., Jiang X., Birmingham C. L., So N. S. Y., Brumell J. H., Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr. Biol. 14, 806–811 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Randow F., Youle R. J., Self and nonself: How autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Case E. D., Chong A., Wehrly T. D., Hansen B., Child R., Hwang S., Virgin H. W., Celli J., The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell. Microbiol. 16, 862–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Wijk S. J. L., Fricke F., Herhaus L., Gupta J., Hötte K., Pampaloni F., Grumati P., Kaulich M., Sou Y.-s., Komatsu M., Greten F. R., Fulda S., Heilemann M., Dikic I., Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat. Microbiol. 2, 17066 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Noad J., von der Malsburg A., Pathe C., Michel M. A., Komander D., Randow F., LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2, 17063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng Y. T., Shahnazari S., Brech A., Lamark T., Johansen T., Brumell J. H., The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Thurston T. L. M., Ryzhakov G., Bloor S., von Muhlinen N., Randow F., The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Huang J., Brumell J. H., Bacteria-autophagy interplay: A battle for survival. Nat. Rev. Microbiol. 12, 101–114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiskin E., Bionda T., Dikic I., Behrends C., Global analysis of host and bacterial ubiquitinome in response to Salmonella Typhimurium infection. Mol. Cell 62, 967–981 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Polajnar M., Dietz M. S., Heilemann M., Behrends C., Expanding the host cell ubiquitylation machinery targeting cytosolic Salmonella. EMBO Rep. 18, 1572–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engström P., Burke T. P., Mitchell G., Ingabire N., Mark K. G., Golovkine G., Iavarone A. T., Rape M., Cox J. S., Welch M. D., Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nat. Microbiol. 4, 2538–2551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salje J., Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nat. Rev. Microbiol. 19, 375–390 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Lanouette S., Mongeon V., Figeys D., Couture J.-F., The functional diversity of protein lysine methylation. Mol. Syst. Biol. 10, 724 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodionov A. V., Eremeeva M. E., Balayeva N. M., Isolation and partial characterization of the M(r) 100 kD protein from Rickettsia prowazekii strains of different virulence. Acta Virol. 35, 557–565 (1991). [PubMed] [Google Scholar]
- 15.Abeykoon A. H., Chao C.-C., Wang G., Gucek M., Yang D. C., Ching W.-M., Two protein lysine methyltransferases methylate outer membrane protein B from Rickettsia. J. Bacteriol. 194, 6410–6418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang D. C. H., Abeykoon A. H., Choi B.-E., Ching W.-M., Chock P. B., Outer membrane protein OmpB methylation may mediate bacterial virulence. Trends Biochem. Sci. 42, 936–945 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Abeykoon A. H., Noinaj N., Choi B.-E., Wise L., He Y., Chao C.-C., Wang G., Gucek M., Ching W.-M., Chock P. B., Buchanan S. K., Yang D. C. H., Structural insights into substrate recognition and catalysis in outer membrane protein B (OmpB) by protein-lysine methyltransferases from Rickettsia. J. Biol. Chem. 291, 19962–19974 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J.-Z., Hao J.-F., Walker D. H., Yu X.-J., A mutation inactivating the methyltransferase gene in avirulent Madrid E strain of Rickettsia prowazekii reverted to wild type in the virulent revertant strain Evir. Vaccine 24, 2317–2323 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Abeykoon A., Wang G., Chao C.-C., Chock P. B., Gucek M., Ching W.-M., Yang D. C. H., Multimethylation of Rickettsia OmpB catalyzed by lysine methyltransferases. J. Biol. Chem. 289, 7691–7701 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y., Wu B., Weinstock G., Walker D. H., Yu X. J., Inactivation of SAM-methyltransferase is the mechanism of attenuation of a historic louse borne typhus vaccine strain. PLOS ONE 9, e113285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamason R. L., Kafai N. M., Welch M. D., A streamlined method for transposon mutagenesis of Rickettsia parkeri yields numerous mutations that impact infection. PLOS ONE 13, e0197012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spratt B. G., Pardee A. B., Penicillin-binding proteins and cell shape in E. coli. Nature 254, 516–517 (1975). [DOI] [PubMed] [Google Scholar]
- 23.Burke T. P., Engström P., Chavez R. A., Fonbuena J. A., Vance R. E., Welch M. D., Inflammasome-mediated antagonism of type I interferon enhances Rickettsia pathogenesis. Nat. Microbiol. 5, 688–696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.T. P. Burke, P. Engström, C. J. Tran, D. R. Glasner, D. A. Espinosa, E. Harris, M. D. Welch, Interferon receptor-deficient mice are susceptible to eschar-associated rickettsiosis. bioRxiv 2020.09.23.310409 [Preprint]. 2020. 10.1101/2020.09.23.310409. [DOI] [PMC free article] [PubMed]
- 25.Paddock C. D., Finley R. W., Wright C. S., Robinson H. N., Schrodt B. J., Lane C. C., Ekenna O., Blass M. A., Tamminga C. L., Ohl C. A., McLellan S. L., Goddard J., Holman R. C., Openshaw J. J., Sumner J. W., Zaki S. R., Eremeeva M. E., Rickettsia parkeri rickettsiosis and its clinical distinction from Rocky Mountain spotted fever. Clin. Infect. Dis. 47, 1188–1196 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Noriea N. F., Clark T. R., Hackstadt T., Targeted knockout of the Rickettsia rickettsii OmpA surface antigen does not diminish virulence in a mammalian model system. MBio 6, e00323-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madasu Y., Suarez C., Kast D. J., Kovar D. R., Dominguez R., Rickettsia Sca2 has evolved formin-like activity through a different molecular mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, E2677–E2686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horstmann J. A., Lunelli M., Cazzola H., Heidemann J., Kühne C., Steffen P., Szefs S., Rossi C., Lokareddy R. K., Wang C., Lemaire L., Hughes K. T., Uetrecht C., Schlüter H., Grassl G. A., Stradal T. E. B., Rossez Y., Kolbe M., Erhardt M., Methylation of Salmonella Typhimurium flagella promotes bacterial adhesion and host cell invasion. Nat. Commun. 11, 2013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo M., Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 118, 6656–6705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curto P., Simões I., Riley S. P., Martinez J. J., Differences in intracellular fate of two spotted fever group Rickettsia in macrophage-like cells. Front. Cell. Infect. Microbiol. 6, 80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristof M. N., Allen P. E., Yutzy L. D., Thibodaux B., Paddock C. D., Martinez J. J., Significant growth by Rickettsia species within human macrophage-like cells is a phenotype correlated with the ability to cause disease in mammals. Pathogens 10, 228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamason R. L., Bastounis E., Kafai N. M., Serrano R., Del Alamo J. C., Theriot J. A., Welch M. D., Rickettsia Sca4 reduces vinculin-mediated intercellular tension to promote spread. Cell 167, 670–683 e610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H. K., Premaratna R., Missiakas D. M., Schneewind O., Rickettsia conorii O antigen is the target of bactericidal Weil–Felix antibodies. Proc. Natl. Acad. Sci. U.S.A. 116, 19659–19664 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plumb R. S., Johnson K. A., Rainville P., Smith B. W., Wilson I. D., Castro-Perez J. M., Nicholson J. K., UPLC/MSE; A new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Spectrom. 20, 1989–1994 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Shliaha P. V., Bond N. J., Gatto L., Lilley K. S., Effects of traveling wave ion mobility separation on data independent acquisition in proteomics studies. J. Proteome Res. 12, 2323–2339 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Distler U., Kuharev J., Navarro P., Levin Y., Schild H., Tenzer S., Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 11, 167–170 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., Haynes P. A., Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Nahnsen S., Bielow C., Reinert K., Kohlbacher O., Tools for label-free peptide quantification. Mol. Cell. Proteomics 12, 549–556 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlström C. I., Loutey D., Bauer S., Clark I. C., Rohde R. A., Iavarone A. T., Lucas L., Coates J. D., (Per)chlorate-reducing bacteria can utilize aerobic and anaerobic pathways of aromatic degradation with (per)chlorate as an electron acceptor. MBio 6, e02287-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynolds C. R., Islam S. A., Sternberg M. J. E., EzMol: A web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J. Mol. Biol. 430, 2244–2248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg2517/DC1