

Epigenetic clocks reveal a rejuvenation event during embryogenesis followed by aging.
Abstract
The notion that the germ line does not age goes back to the 19th-century ideas of August Weismann. However, being metabolically active, the germ line accumulates damage and other changes over time, i.e., it ages. For new life to begin in the same young state, the germ line must be rejuvenated in the offspring. Here, we developed a multi-tissue epigenetic clock and applied it, together with other aging clocks, to track changes in biological age during mouse and human prenatal development. This analysis revealed a significant decrease in biological age, i.e., rejuvenation, during early stages of embryogenesis, followed by an increase in later stages. We further found that pluripotent stem cells do not age even after extensive passaging and that the examined epigenetic age dynamics is conserved across species. Overall, this study uncovers a natural rejuvenation event during embryogenesis and suggests that the minimal biological age (ground zero) marks the beginning of organismal aging.
INTRODUCTION
Aging is characterized by a progressive accumulation of damage, leading to the loss of physiological integrity, impaired function, and increased vulnerability to death (1). While the aging process affects the entire organism, it is often discussed that the germ line does not age, because this lineage is immortal in the sense that the germ line has reproduced indefinitely since the beginning of life (2–4). This notion dates to the 19th century when August Weismann proposed the separation of the ageless germ line and aging soma. However, being in the metabolically active state for two decades or more before its contribution to the offspring, the human germ line accumulates molecular damage, such as modified long-lived proteins, epimutations, metabolic by-products, and other age-related deleterious changes (5, 6). It was shown that sperm cells exhibit a distinct pattern of age-associated changes (7–9). Accordingly, it was recently proposed that germline cells may age and be rejuvenated in the offspring after conception (10, 11). If this is the case, there must be a point (or period) of the lowest biological age (here, referred to as the ground zero) during the initial phases of embryogenesis (Fig. 1A). Here, we carried out a quantitative, data-driven test of this idea.
Fig. 1. A rejuvenation event during early embryogenesis revealed by aging clocks.
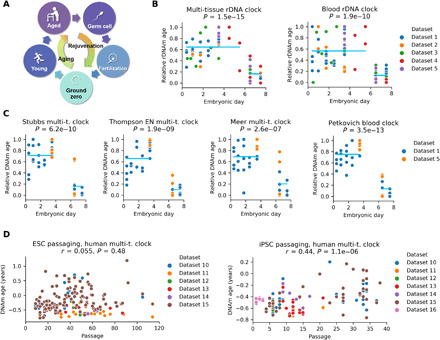
(A) Overview of the model, which posits that germline cells age during development and adulthood and are rejuvenated in the offspring after conception. The model also suggests that there is a time point corresponding to the lowest biological age (ground zero). (B) Multi-tissue and blood rDNA clocks applied to five datasets spanning the first 8 days of mouse embryogenesis (Table 1, datasets 1 to 5). We rescaled epigenetic age of each dataset to the interval [0,1] for comparison (“relative rDNA age”). 0 represents the lowest epigenetic age, and 1 represents the highest epigenetic age of each dataset. Blue lines indicate the mean of each group; P values of two-sided t test comparing the means of the two groups (before and after E6) are displayed. (C) Application of four genome-wide epigenetic aging clocks to two available mouse RRBS datasets. (D) Epigenetic age of human ESCs and iPSCs as a function of passage number. The Horvath human multi-tissue clock was applied.
Because of recent advances in technology, machine learning is flourishing and has led to breakthroughs in many areas of science by discovering multivariate relationships (12). Aging and developmental biology areas also exploited the potential of machine learning by developing algorithms (“aging clocks”) that can estimate chronological age or biological age (i.e., the age based on molecular markers) of an organism from a given data (13, 14). As epigenomic changes, which result in dysregulation of transcriptional and chromatin networks, are crucial components of aging (15), epigenetic clocks, based on methylation levels of certain CpG sites, emerged as a promising molecular estimator of biological age (16, 17). These clocks were shown to quantitatively measure numerous aspects of human aging (17–22). For example, epigenetic age acceleration was associated with age-related conditions, such as all-cause mortality (23, 24), cognitive performance (25), frailty (26), Parkinson’s disease (27), Down syndrome (28), and Werner syndrome (29). Epigenetic aging clocks were also developed for mice and could be used to evaluate longevity interventions, such as calorie restriction and growth hormone receptor knockout (KO) (30–36). The clocks developed on the basis of aging patterns of mostly adult tissues correctly predict much younger ages and report the effects of cell rejuvenation upon complete or partial reprogramming of adult fibroblasts into induced pluripotent stem cells (iPSCs) as demonstrated in both human and mouse (33, 35, 37–39). Although iPSCs correspond to an embryonic state and the transition to these cells involves major molecular changes, including changes in the epigenome, this rejuvenation event can be assessed by epigenetic clocks. Most recently, universal mammalian clocks have been developed on the basis of conserved cytosines, whose methylation levels change with age across mammalian species (40).
Considering that epigenetic clocks track the aging process, they may be applied to early development to characterize biological age dynamics during that period of life. Recent studies showed that clocks may be successfully applied to human fetal development using brain, retina, and cord blood samples (41–43). However, epigenetic age dynamics during the entire prenatal development for the entire organism remained unexplored. Here, we developed a new multi-tissue epigenetic clock using machine learning and applied it, together with other existing aging clocks, to assess prenatal development in mammals from the perspective of aging. This approach uncovered a rejuvenation period during early embryogenesis and the timing of the beginning of aging in mammals.
RESULTS
A rejuvenation event during early embryogenesis
To assess epigenetic age dynamics during embryogenesis, we collected available human and mouse DNA methylation (DNAm) datasets (Table 1) and subjected them to various epigenetic aging clocks (Table 2). We also developed a multi-tissue ribosomal DNAm (rDNAm) clock (figs. S1 and S2). The rDNA is characterized by a large number of age-associated CpG sites that exhibit high sequence coverage due to the multiplicity of rDNA in the genome (35, 44). The new clock is capable of predicting the epigenetic age of reduced representation bisulfite sequencing (RRBS), whole-genome bisulfite sequencing (WGBS), and even pseudo-bulk single-cell sequencing samples in various tissues. All the clocks we used showed high accuracy (r ≥ 0.8) in age prediction of test samples and were sensitive to age-related conditions and longevity interventions (Table 2).
Table 1. Embryonic DNA methylation datasets used in this study.
DKO, double knockout; E, gestational day; ESC, embryonic stem cell; ext ect, extraembryonic ectoderm; ext end, extraembryonic endoderm; GW, gestational weeks; iPSC, induced pluripotent stem cell; KO, knockout; P, passage; TKO, triple knockout; RRBS, reduced representation bisulfite sequencing; WGBS, whole-genome bisulfite sequencing; WT, wild type.
| Name | Reference | Accession | Platform | Species | Samples (number of biological replicates) |
| Dataset 1 | (63) | GSE34864 | RRBS | Mouse | Zygote (5), 2-cell (4), 4-cell (5), 8-cell (3), ICM (5), E6.5 (4), E7.5 (5) |
| Dataset 2 | (55) | GSE56697 | WGBS | Mouse | 2-cell (2), 4-cell (2), E3.5 (3), E6.5 (2), E7.5 (2), E13.5 (2) |
| Dataset 3 | (64) | GSE98151 | WGBS | Mouse | Zygote (1), early 2 cell (1), late 2 cell (1), 4 cell (1), 8 cell (1), morula (1), ICM (1), trophoblast (1), E6.5 epiblast (1), E6.5 ext ect (1), E7.5 epiblast (1), E7.5 ext ect (1) |
| Dataset 4 | (48) | GSE121690 | scNMT-seq | Mouse | 758 single cells from E4.5, E5.5, E6.5, E7.5 |
| Dataset 5 | (69) | GSE51239 | RRBS | Mouse | ICM (2), trophectoderm (2), E6.5 epiblast (2), E6.5 ext end (2), ESC P0–P5 (17) |
| Dataset 6 | (66) | ENCSR486XIX | WGBS | Mouse | Various tissues from E10.5 to birth (139) |
| Dataset 7 | (70) | GSE56515 | 450K array | Human | Various tissues from GW 9 to GW 22 (34) |
| Dataset 8 | (71) | GSE31848 | 450K array | Human | Various tissues from GW 14 to GW 20 (37) |
| Dataset 9 | (72) | GSE69502 | 450K array | Human | Various tissues from GW 14.5 to 23 (49) |
| Dataset 10 | (71) | GSE31848 | 450K array | Human | ESC P9–P105 (19), iPSC P5–P37 (29) |
| Dataset 11 | (73) | GSE34869 | 450K array | Human | ESC P32–P114 (19), iPSC P12–P21 (5) |
| Dataset 12 | (74) | GSE40909 | 450K array | Human | ESC P41–P49 (3), iPSC P6 (2) |
| Dataset 13 | (75) | GSE44424 | 27K array | Human | ESC P29–P87 (8), iPSC P9–P21 (21) |
| Dataset 14 | (76) | GSE51747 | 27K array | Human | ESC P52–P64 (3), iPSC P9–P17 (6) |
| Dataset 15 | (71) | GSE30653 | 27K array | Human | ESC P9–P114 (116), iPSC P4–P69 (46) |
| Dataset 16 | (77) | GSE54848 | 450K array | Human | iPSC P1–P3 (9) |
| Dataset 17 | (47) | GSE76261 | PBAT | Mouse | TET TKO (2) and WT E6.5 epiblast (2) |
| Dataset 18 | (48) | GSE133687 | scNMT-seq | Mouse | TET TKO (126) and WT ESCs (136) at days 2, 5, and 7 of differentiation |
| Dataset 19 | (51) | GSE130735 | RRBS | Mouse | KO of DNMT1 (3), DKO of DNMT3A/3B (6), WT (5), heterozygous control (6) |
| Dataset 20 | (46) | GSE60334 | RRBS | Mouse | E3.5-, E4.5 blastocyst (1-1), E5.5-, E6.5-, E7.5 epiblast (1-1-1), E8.5-, E10 whole embryos (3-2), E11.5 limbs (2) |
Table 2. Epigenetic aging clocks used in this study.
AD, Alzheimer’s disease; CR, calorie restriction; F, female; GHRKO, growth hormone receptor knockout; M, male; PD, Parkinson’s disease; WD, Werner syndrome.
| Clock | Reference | Species | Tissue | Method | Age range | Gender | Model |
No. clock sites |
Acc. on test |
Age-related condition sensitive for |
| Human multi-t. | (18) | Human | Multi | 27K array | Full lifespan | F & M | EN | 353 | r = 0.96 | E.g., mortality, cogn. perform., frailty, AD, PD, WD, centenarian status, iPSC |
| Petkovich blood | (30) | Mouse C57BL/6 |
Blood | RRBS | Full lifespan | M | EN | 90 | R2 = 0.9 | CR, GHRKO, SD, iPSC |
| Stubbs multi-t. | (31) | Mouse C57BL/6 |
Multi | RRBS | 1–41 weeks | M | EN | 329 | r = 0.84 | Low-fat diet |
| Meer multi-t. | (33) | Mouse C56BL/6 |
Multi | RRBS | Full lifespan | F & M | EN | 435 | R2 = 0.89 | GHRKO, iPSC |
| Thompson multi-t. EN |
(34) | Mouse mostly C57BL/6J, BALB/cBy |
Multi | RRBS | Full lifespan | F & M | EN | 582 | r = 0.89 | CR, Ames dwarf |
| Blood rDNA | (35) | Mouse C57BL/6 |
Blood | RRBS, only rDNA |
Full lifespan | M | EN | 72 | r = 0.92 | CR, GHRKO, iPSC |
| Multi-t. rDNA | This study | Mouse C57BL/6 |
Multi | RRBS, only rDNA |
1.7–21.3 months | F & M | EN | 355 | r = 0.94 | CR, GHRKO, iPSC |
First, we examined the behavior of rDNAm clocks when applied to five independent mouse early embryonic datasets. We found that the mean epigenetic age of embryonic day (E) 6.5/E7.5 embryos was consistently lower than in earlier stages of embryogenesis (Fig. 1B and figs. S3, A and B, and S4, A and B). We also applied four genome-wide RRBS-based epigenetic aging clocks to RRBS datasets (datasets 1 and 2). This again revealed that the epigenetic age of E6.5/E7.5 embryos is lower than during the period from zygote to blastocyst (Fig. 1C and figs. S3, C and D, and S4, C and D). Thus, epigenetic age decreases during early embryogenesis, and therefore, embryonic cells not only do not age during this period but also, at some point, get rejuvenated.
Previously, a near-zero epigenetic age of human embryonic stem cells (ESCs) and iPSCs was demonstrated by the Horvath multi-tissue clock even after extensive passaging (18). We analyzed iPSCs and ESCs on the basis of several currently available datasets to further assess whether these cells, which correspond to early embryogenesis, age (Fig. 1D). The epigenetic age of cells was very low (mostly below zero) even after more than 100 passages. Even under artificial culture conditions, at the level of oxygen above physiological, and with the number of passages well beyond physiological (which may lead to the accumulation of deleterious mutations), either no or very little increase in epigenetic age was observed. These findings support the notion that cells corresponding to the early stages of embryogenesis essentially do not age.
Organismal aging begins during mid-embryonic development in mouse and human
We quantified the epigenetic age by applying rDNA clocks to a mouse dataset that contains both early and late embryonic samples (Fig. 2A). The epigenetic age at E6.5 and E7.5 was significantly lower than at E13.5 (primordial germ cells that are the direct progenitors of sperm and oocytes).
Fig. 2. Organismal aging begins in mid-embryonic development in mice and humans.
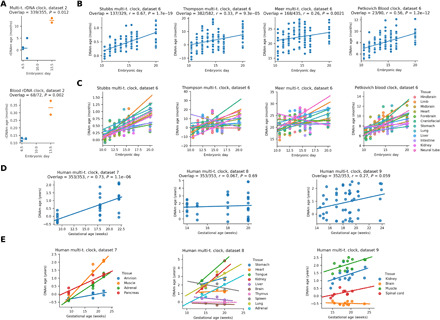
(A) Epigenetic age (multi-tissue and blood rDNA clocks) analysis of the dataset that contains both early and late mouse embryo samples (E13.5 samples are based on primordial germline cells). (B) Application of genome-wide epigenetic clocks to later stages of mouse embryogenesis (r, Pearson correlation coefficient; P, P value of the correlation). (C) The same data as above but separated by tissue. An increasing trend is observed for almost all tissues, with few nonsignificant exceptions. (D) Epigenetic age dynamics of four independent prenatal human 450K methylation array datasets based on the Horvath human multi-tissue clock. (E) The same data as above but separated by tissue (five significant increases, nine nonsignificant increases, four nonsignificant decreases, and zero significant decreases).
We also assessed the epigenetic age of mouse embryos across nine time points from E10.5 to birth (dataset 6) by genome-wide methylation clocks. A consistent increase in epigenetic age was observed during this period both when considering all data (Fig. 2B) and when separated by tissue (Fig. 2C). In addition, we analyzed human prenatal datasets by applying the Horvath multi-tissue DNAm clock to four independent human 450K methylation array datasets (datasets 7 to 9). An increase (or in some cases no change) in epigenetic age was observed both when considering all data (Fig. 2D) and tissue by tissue (Fig. 2E). Thus, at a certain point during embryonic development in mouse and human, the biological age begins to increase in most or all tissues. Considering that epigenetic clocks track the aging process, the data suggest that by then organisms already age.
Epigenetic age of mouse ESCs during early passaging
We assessed the epigenetic age of mouse ESCs after outgrowth (passage 0) and early passaging (passage 5) under three different culture conditions (Fig. 3, A and B). In the absence of two inhibitors [2i including PD0325901, which causes blockade of differentiation, and CHIR99021, which supports self-renewal (45)], we observed a lower epigenetic age after outgrowth compared to the condition when the two inhibitors were included (Fig. 1, A and B). We also found that the epigenetic age of all ESC samples is between the epigenetic age of E3.5 embryos and E6.5 epiblast embryos (fig. S3, A, B, and D). Principal components analysis (PCA) of the methylation profiles showed that ESC outgrowths cultured under 2i conditions clustered near E3.5 epiblast and E3.5 trophectoderm, while ESCs cultured without one or two inhibitors did not (Fig. 3C). Our analysis is consistent with the idea that ESCs under 2i conditions did not differentiate (or only slightly changed) and may serve as a model of E3.5 embryos, while ESCs without one or two inhibitors differentiated and may serve as a model of later-stage embryos. Overall, our data suggest that ESCs under incomplete self-renewal culture conditions may continue their development (without self-renewal) and rejuvenate, similar to what we observed in vivo.
Fig. 3. Epigenetic age of mouse ESCs during early passaging under different culture conditions.
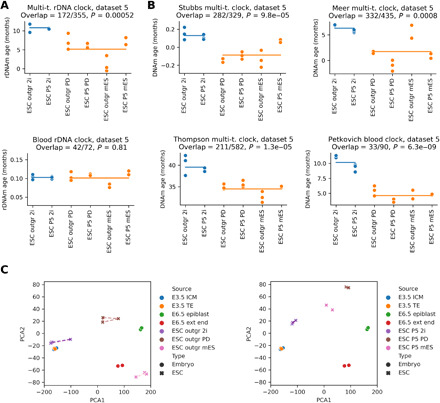
(A) Epigenetic age (by rDNA clocks) of mouse ESCs after outgrowth (passage 0) and passage 5 under three different culture conditions (2i, both self-renewal supporting inhibitors used; PD, only one inhibitor; mES, no inhibitor). (B) Application of genome-wide mouse epigenetic clocks to the same data. (C) Principal components analysis (PCA) of RRBS methylation profiles of embryo (circles) and ESC (crosses) sample groups after passage 0 (left) and passage 5 (right). Convex hull around the ESC samples in the same group (if n > 2) is displayed to help distinguish ESC samples from embryo samples.
Localization of the epigenetic age minimum (ground zero) during mouse embryonic development
We concatenated the results for early and later stages of embryogenesis by applying genome-wide mouse epigenetic clocks (Fig. 4A) and rDNA clocks (Fig. 4B). The variable number of overlapped clock sites across all stages caused a batch effect that resulted in a shift of the actual predicted age between early and late stages. However, the epigenetic age dynamics showed a clear U-shaped pattern in every case, with the minimum at E6.5/E7.5 in four cases and E10.5 in two cases. The results suggest that ground zero may lie in the range from E4.5 to E10.5. To refine the interval, we also analyzed a dataset containing mid-embryonic stages from E3.5 to E11.5 [dataset 20, (46)]. The epigenetic age minimum lied at E6.5/E7.5 in five cases and at E8.5 in one case (Fig. 4, C and D, and fig. S7). Together, our data suggest that ground zero may lie in the range from E4.5 to E10.5, most probably at E6.5/E7.5. Organismal aging begins after this rejuvenation event.
Fig. 4. Localization of the epigenetic age minimum (ground zero) during mouse embryonic development.
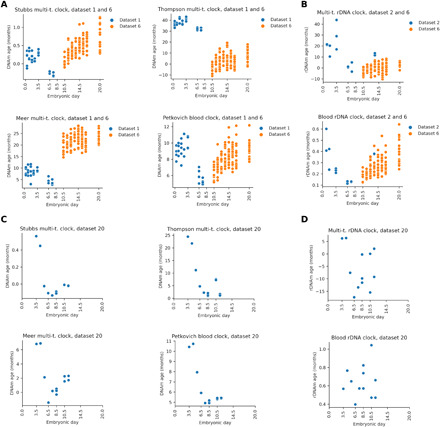
(A) We concatenated results for the entire period of embryogenesis by using the genome-wide mouse epigenetic clocks indicated. (B) We concatenated results for the entire period of embryogenesis by using the indicated mouse rDNA epigenetic clocks. (C) Application of genome-wide mouse epigenetic clocks to dataset 20 that contains mid-embryonic stages from E3.5 to E11.5. (D) Application of rDNA mouse epigenetic clocks to dataset 20.
Role of TET enzymes in the rejuvenation event
The observed epigenetic age minimum at E6.5/E7.5 corresponds to gastrulation, the period where the three germ layers are formed. Recent studies revealed that Ten-eleven translocation (TET) enzymes (Tet1, Tet2, and Tet3), which support demethylation by oxidizing 5-methylcytosines (5mCs), have a vital role in gastrulation (47, 48). Hence, we hypothesized that these enzymes may have a mechanistic role in the epigenetic age decrease. To investigate this possibility, we applied epigenetic clocks to available datasets of TET KO embryos and ESCs.
It was shown previously that until the onset of gastrulation (E6.5), TET triple KO (combined KO of Tet1, Tet2, and Tet3 enzymes) mutants were morphologically indistinguishable from control embryos and also that the global methylation level of the two groups was comparable (47). According to this finding, applying rDNAm clocks to the data (dataset 17), we found no epigenetic age difference between TET triple KO and control embryos at E6.5 (Fig. 5A).
Fig. 5. Role of TET enzymes in the rejuvenation event.
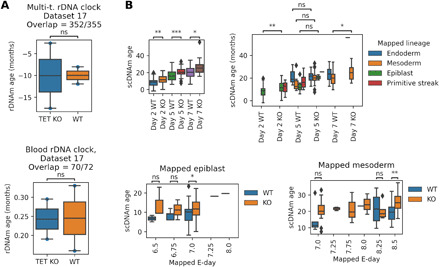
(A) Application of rDNAm clocks to TET triple KO (TET KO) and wild-type (WT) E6.5 epiblast samples (dataset 17). (B) Application of our recently developed single-cell clock [scAge; (49)] to TET KO and WT ESCs at days 2 and 5 of differentiation (dataset 18). Specific lineages and stages (Mapped lineage and Mapped E-day) were assigned by mapping the RNA expression profiles of the in vitro cells to an in vivo gastrulation atlas. Two-sided t tests were calculated (ns, P > 0.05; *, 1 × 10−2 < P ≤ 5 × 10−2; **, 1 × 10−3 < P ≤ 1 × 10−2; ***, 1 × 10−4 < P ≤ 1 × 10−3; ****P ≤ 1 × 10−4).
To investigate the role of TET enzymes in later stages of gastrulation, we analyzed a single-cell ESC dataset, wherein ESCs were allowed to differentiate to embryoid bodies until three time points (days 2, 5, and 7). Cells were assigned to specific lineages and stages (in E-days) by mapping their RNA expression profiles to an in vivo gastrulation atlas [dataset 18, (48)]. Application of our recently developed single-cell clock [scAge; (49)] showed an increased epigenetic age of the TET triple KO embryoid bodies assigned to E7 epiblast and E8 mesoderm compared to the corresponding controls (Fig. 5B). The epigenetic age increase of TET triple KO embryoid bodies is accompanied by a global average DNAm increase (fig. S6). Overall, our analysis of two TET KO methylation datasets revealed a possible mechanistic role of TET enzymes in the epigenetic age decrease during gastrulation.
Role of DNA methyltransferases in the rejuvenation event
DNA methyltransferases (DNMTs) have important roles in mouse development: DNMT1 is mainly responsible for DNAm maintenance after replication, while DNMT3A and DNMT3B perform de novo methylation of DNA during development (50, 51). We investigated the role of these enzymes in the rejuvenation event by applying epigenetic clocks to DNMT1 single KO, DNMT3A and DNMT3B double KO (DKO), and control embryos at E8.5 (dataset 19, Fig. 6). We observed a consistent increase in epigenetic age in the case of DNMT1−/− and DKO embryos (five of six epigenetic clocks showed similar dynamics). Our finding is in agreement with the observation that DNMT1−/− and DKO embryos show retardation at E8.5, accompanied by a global average methylation decrease compared to wild type and heterozygous KO controls (51). Overall, our findings suggest an important role of methylation maintenance and de novo methylation in the rejuvenation event.
Fig. 6. Role of DNMTs in the rejuvenation event.
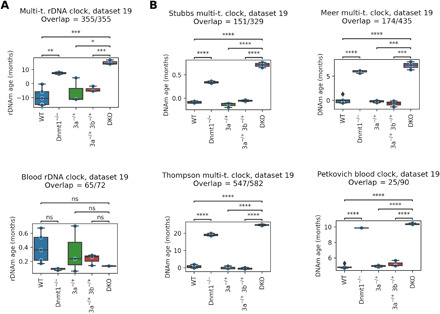
(A) We applied rDNA clocks to single KO of DNMT1 (Dnmt1−/−), double KO of DNMT3A and DNMT3B (DKO), wild type (WT), and heterozygous control (3a−/+ and 3a−/+ 3b−/+) embryos at E8.5 (dataset 19). (B) Application of four genome-wide epigenetic aging clocks to the same dataset. Two-sided t tests were calculated (ns, P > 0.05; *, 1 × 10−2 < P ≤ 5 × 10−2; **, 1 × 10−3 < P ≤ 1 × 10−2; ***, 1 × 10−4 < P ≤ 1 × 10−3; ****P ≤ 1 × 10−4).
Epigenetic ages of zygote, gametes, and somatic tissues
To investigate the epigenetic age of the germ line, we analyzed dataset 1 that includes zygote, sperm, oocyte, and adult tissue (heart, brain, and liver) samples. Most of the six applied epigenetic clocks showed no significant difference between zygote and somatic tissues (nonsignificant change in five of six cases), between zygote and oocytes (nonsignificant change in five of six cases), and between zygote and sperm (nonsignificant change in four of six cases) (fig. S4). These results suggest that the zygote is not yet fully rejuvenated; hence, it should undergo a further rejuvenation process, supporting our findings.
DISCUSSION
Back in the 19th century, August Weismann proposed the idea of the heritable non-aging germ line and the disposable aging soma. Yet, the germ line shows molecular changes characteristic of aging (7–9). Our study suggests that the germ line ages but is rejuvenated in the offspring at some point during early embryogenesis. This rejuvenation occurs during early post-implantation stages corresponding to gastrulation when the offspring reaches its minimal biological age. We propose that this minimum, the ground zero, marks the beginning of aging of an organism. The offspring proceeds naturally to ground zero from the zygote stage, but somatic cells may also be forced to this young stage, e.g., by reprogramming with Yamanaka factors (or by other methods), generating iPSCs. In vivo amelioration of age-associated hallmarks was also demonstrated by partial reprogramming (52). Most recently, partial reprogramming was found to restore vision in mice by resetting youthful epigenetic information (53). Thus, both soma and germ line may age and be rejuvenated.
Early embryogenesis, where we observed a rejuvenation period, is also accompanied by other molecular changes in preparation for organismal life, such as a gradual extension of telomeres (54), waves of global demethylation and methylation (55), transition from the use of maternal gene products to those of the embryo, inactivation of chromosome X, and development of monoallelic gene expression (56). Rejuvenation should also involve a decrease in molecular damage and other deleterious age-related changes that accumulate in the parental germ line (57, 58).
The data indicate that ground zero lies between E4.5 and E10.5 in mice, most probably at E6.5/E7.5. This period corresponds to the germ-layer specification (gastrulation) accompanied by the exit from pluripotency (48, 59). The global increase of methylation and the decrease in accessibility results in the repression of pluripotency markers such as Dppa4, Zfp42, Tex19.1, Pou3f1, Nodal, Otx2, and Dnmt3l (48, 59). TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signaling (47). At the onset of gastrulation (E6.5), TET triple KO embryos were morphologically indistinguishable from control, but by E7.5, they were much smaller than control (47). This observation suggests that, until E6.5, development may be independent of the function of TET enzymes, but they have a vital role in gastrulation (47, 48). Global average methylation rapidly increases (from approximately 25 to 75% in embryonic tissues) between E4.5 and E7.5 (46, 48), and the most marked change of de novo methylation occurs at E4.5 and E5.5 at the time of implantation (46). This period may be critical in rejuvenation, as our analysis of Dntm3a/b DKO embryos suggests an important role of de novo methylation in the rejuvenation event.
The mapped CpGs from traditional bisulfite sequencing methods are the sum of 5hmCs (5-hydroxymethylcytosines) and 5mCs (60); thus, the epigenetic clocks were trained on both types of methylation. However, the exact mechanisms behind epigenetic clocks are still unknown, including the role of 5hmCs in assessing the biological age (17). It would be interesting for the future to generate age-associated Tet-assisted bisulfite sequencing (TAB-seq) data and train an epigenetic clock that would distinguish between 5mC and 5hmC. It may result in more accurate predictions and help in the evaluation of epigenetic age dynamics during embryonic development.
The beginning of aging is subject to debate. It is often discussed that aging begins after completion of development, at the onset of reproduction, and at the time when strength of natural selection begins to decrease. However, our recent analysis of deleterious age-related changes revealed that aging begins early in life, even before birth (61). Our current work now pinpoints the beginning of aging to ground zero.
CpG sites associated with aging and lifespan may be both hypomethylated and hypermethylated upon aging (62). An attractive possibility is that rejuvenation may be supported by remethylation rather than by demethylation. Global remethylation was reported between E3.5 and E7.5 (48, 55, 63, 64), which is the same period where we observe rejuvenation. If global remethylation is associated with epigenetic age decrease, ground zero and global methylation maximum should correspond to the same developmental stage. This would make sense from the perspective that, to remove “epigenetic damage,” the genome should be first partially demethylated and then remethylated again.
We also found that cells corresponding to early embryogenesis, i.e., ESCs and iPSCs, do not age when cultured and passaged. However, early passaging seems to result in a partial epigenetic age reduction. Consistent with this age reduction, it was found that initial passaging induces telomere extension and that mice generated from these rejuvenated cells live longer and are better protected from age-related diseases than mice from the same cells that were not passaged (65). While the effect of early passaging and the rejuvenation associated with gastrulation may be different events, our study suggests an exciting possibility that the natural rejuvenation we uncover in this work may be targeted such that organisms may begin aging at a lower biological age and therefore may achieve a longer life span and extended health span.
Global cytosine methylation (average methylation level of CpG sites representing the whole genome) changes in waves during mammalian embryogenesis: An initial decrease from zygote to E3.5 is followed by an increase to E6.5/E7.5 (55) and unique tissue-specific dynamics during/after organogenesis (66). However, global cytosine methylation shows very little change after birth (67) and therefore is not highly predictive of biological age or its reduction. In contrast, the age predicted by epigenetic aging clocks (usually based on several hundred CpG sites) shows strong correlation with age (r ≥ 0.8), indicating that it can be used to predict biological aging and rejuvenation (Table 2).
Two epigenetic clocks (Petkovich et al. and rDNA clocks) were trained and validated on blood that is absent in early embryogenesis, yet we used them to analyze early embryonic samples. However, previous studies showed that these blood clocks were successfully applied to other tissues. The Petkovich clock accurately predicted the epigenetic age of a multi-tissue sample set excluding blood (33). Furthermore, both clocks were applied to kidney and lung fibroblasts and the corresponding iPSC, and reported the effect of cell rejuvenation during reprogramming (30, 35). In our analysis, blood clocks showed markedly similar dynamics in embryogenesis compared to multi-tissue clocks.
Overall, this work identifies a natural rejuvenation event during early life and suggests that organismal aging begins during embryogenesis, approximately at the time of gastrulation. These findings provide opportunities for understanding what this early rejuvenation process entails, whether it is similar to the Yamanaka reprogramming, and whether it may be induced in somatic cells to rejuvenate them.
MATERIALS AND METHODS
Acquisition and processing of sequencing datasets
Raw sequences were downloaded and extracted using the Sequence Read Archive (SRA) Toolkit. Reads were trimmed and quality-filtered by TrimGalore! v0.6.4 using the --rrbs option for RRBS. Methylation levels were extracted using Bismark v0.22.2 (68) in two separate processes: (i) For rDNA clocks, reads were mapped to the mouse rDNA sequence (BK000964.3), and (ii) for genomic clocks, reads were mapped to the complete mouse genome sequence (mm10/GRCm38.p6). Methylated and unmethylated cytosines of technical repeats were summed for each position. In the case of dataset 6, we directly used the available BED (browser extensible data) files [URLs were extracted from Gene Expression Omnibus (GEO) files]. For datasets 19 and 20, we used processed methylation files downloaded from GEO.
CpG site filtering and imputation of RRBS/WGBS datasets
Our filtering strategy was optimized for three competing goals: (i) highest possible read coverage, (ii) highest possible clock site overlap, and (iii) best (unbiased) sample comparison. We used only the CpG sites that are covered by at least 5 reads (or 50 in the two highest-covered cases when rDNA clocks were applied on datasets 2 and 3) in at least 90% of the samples of a given dataset. In the case of dataset 6, we just used the CpG sites that are covered by at least five reads without any other restriction (to avoid too many missing values). We omitted the two lowest-quality samples (both were oocytes) of dataset 1 for genomic analysis. Missing values of clock sites were imputed by the average methylation levels of the covered clock sites for all samples. Our imputation strategy resulted in a single number for each clock and dataset, and this allowed a less-biased comparison of the samples compared to a method that imputes different values for each sample. We used a clock-site-wise average instead of an all-site-wise average because we supposed that methylation levels of clock CpG sites are closer to each other than to other ones.
Application of mouse epigenetic clocks to RRBS/WGBS datasets
In the application of previously developed clocks, we followed the descriptions of authors. All available mouse clocks that we applied were based on linear regression (Table 2). We used the intercept and weights provided by the authors. Transformation was also applied in the case of Petkovich blood clock (30), Stubbs multi-tissue clock (31), and blood rDNA clock (35). In the case of the Stubbs multi-tissue clock, an initial normalization was applied by the published training set. Applying RRBS-based clocks with sites scattered across the genome is often challenging because of a varying number of covered clock sites, leading to missing values. Therefore, we required the clock site coverage to be at least 25%. RRBS-based clock sites had no sufficient coverage when we applied them to datasets 2, 3, and 4 (all of them are whole-genome datasets).
Single-cell clock workflow
Methylation levels were obtained by the rDNA mapping workflow described above. In the end, we obtained methylation data for each of the 758 cells separately. We used only the CpG sites that were covered by at least five reads and showed consistent methylation levels (higher than 0.8 or lower than 0.2). Then, we randomly assigned cells to two groups for each embryo stage (eight cell groups in total). We calculated the average methylation status for each CpG site of each of the eight groups (previously, methylation status was reassigned to 0 or 1). Last, we applied rDNA clocks to the eight groups as it would be eight samples from bulk sequencing.
Application of human DNAm clock to 450K methylation array datasets
Datasets were downloaded from the GEO database. We reindexed data to the 27K array and used the R script of the Horvath DNAm calculator as described in the tutorial (https://horvath.genetics.ucla.edu/html/dnamage/TUTORIAL1.pdf). In the case of pluripotent stem cell datasets, we only analyzed ESCs derived from normal fertilized blastocysts and iPSCs derived from normal cells (i.e., immortalized cells were excluded).
Development of a mouse multi-tissue rDNA clock
A multi-tissue mouse RRBS dataset (GSE120132) was used for training and testing the clock by cross-validation. An additional blood mouse RRBS dataset (GSE80672) was used for testing samples of an independent study. Raw sequences were downloaded from the SRA database. Reads were trimmed and quality-filtered by TrimGalore! v0.6.4, and methylation levels were extracted using Bismark v0.22.2. by mapping to the mouse rDNA sequence (BK000964.3). Fastq files of technical repeats were concatenated. We used only the CpG sites (and only positive-strand cytosines) that are covered in at least 50 reads in all of the samples of both datasets. We used only the C57BL/6J of the GSE120132 dataset and omitted muscle samples because we observed low age correlation of the CpG sites of muscle rDNA (fig. S5). After filtering, 166 samples remained for training and testing (fig. S1A). Then, fivefold cross-validations were performed on all of the 166 samples for different λ parameters of ElasticNet (Scikit-learn v0.23.2) to find the optimal λ parameter (fig. S1B). We retrained a final model (the actual clock) on the same dataset with the optimal parameter (λ = 0.0001) on the random 80% of all samples and tested on the remaining samples (fig. S1C and table S1). Our final model showed a similar performance to the average performance of the cross-validation. However, testing a model on the dataset of the cross-validation may lead to inflated performance. To resolve this issue, we also applied our model to the data of an independent study. That is, we tested the multi-tissue rDNA clock on 153 normal (control-fed, wild-type) C57BL/6J blood samples of the GSE80672 dataset (fig. S1C). We also applied it for age-related conditions of the same dataset (fig. S2).
Statistical analysis
We used the Python packages SciPy (v1.3.1) and Scikit-learn (v0.23.2) for statistical analysis. Two-sided t test was calculated for comparing two groups. If P values were indicated by asterisk, we used the notations as follows: ns, P > 0.05; *, 1 × 10−2 < P ≤ 5 × 10−2; **, 1 × 10−3 < P ≤ 1 × 10−2; ***, 1 × 10−4 < P ≤ 1 × 10−3; ****P ≤ 1 × 10−4. Correlations were evaluated by Pearson correlation coefficient (r) or Spearman rank correlation coefficient (Rho).
Acknowledgments
We thank S. H. Yim, A. Shindyapina, M. Meer, M. Mariotti, and M. Gerashchenko for discussion. Funding: This work was supported by NIH grants (AG064223, AG067782, AG065403) to V.N.G. Author contributions: V.N.G. conceived and supervised the study. C.K. acquired data, performed data analysis, and developed the multi-tissue rDNA clock. A.T. applied the single-cell clock. B.Z. created the schematic figure and helped with data analysis and discussions. All authors interpreted the results. V.N.G. and C.K. wrote the manuscript. B.Z., S.-G.L., and A.T. edited the manuscript. Competing interests: The authors declare th©at they have no competing interests. Data and materials availability: Methylation levels of rDNA CpG sites are available for all mouse datasets (GSE171332). Codes are available at https://github.com/kerepesi/MouseAgingClocks. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg6082/DC1
REFERENCES AND NOTES
- 1.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lepperdinger G., Open-ended question: Is immortality exclusively inherent to the germ line?—A mini-review. Gerontology 55, 114–117 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Sturm A., Ivics Z., Vellai T., The mechanism of ageing: Primary role of transposable elements in genome disintegration. Cell. Mol. Life Sci. 72, 1839–1847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rando T. A., Stem cells, ageing, and the quest for immortality. Nature 441, 1080–1086 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Gladyshev V. N., Aging: Progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15, 594–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rando T. A., Chang H. Y., Aging, rejuvenation, and epigenetic reprogramming: Resetting the aging clock. Cell 148, 46–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H. Y., Jung S. E., Oh Y. N., Choi A., Yang W. I., Shin K. J., Epigenetic age signatures in the forensically relevant body fluid of semen: A preliminary study. Forensic Sci. Int. Genet. 19, 28–34 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Jenkins T. G., Aston K. I., Pflueger C., Cairns B. R., Carrell D. T., Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLOS Genet. 10, e1004458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins T. G., Aston K. I., Cairns B., Smith A., Carrell D. T., Paternal germ line aging: DNA methylation age prediction from human sperm. BMC Genomics 19, 763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gladyshev V. N., The ground zero of organismal life and aging. Trends Mol. Med. 27, 11–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashapkin V. V., Kutueva L. I., Vanyushin B. F., Aging as an epigenetic phenomenon. Curr. Genomics 18, 385–407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan M. I., Mitchell T. M., Machine learning: Trends, perspectives, and prospects. Science 349, 255–260 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Fabris F., de Magalhães J. P., Freitas A. A., A review of supervised machine learning applied to ageing research. Biogerontology 18, 171–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galkin F., Mamoshina P., Aliper A., de Magalhães J. P., Gladyshev V. N., Zhavoronkov A., Biohorology and biomarkers of aging: Current state-of-the-art, challenges and opportunities. Ageing Res. Rev. 60, 101050 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Booth L. N., Brunet A., The aging epigenome. Mol. Cell 62, 728–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell C. G., Lowe R., Adams P. D., Baccarelli A. A., Beck S., Bell J. T., Christensen B. C., Gladyshev V. N., Heijmans B. T., Horvath S., Ideker T., Issa J. P. J., Kelsey K. T., Marioni R. E., Reik W., Relton C. L., Schalkwyk L. C., Teschendorff A. E., Wagner W., Zhang K., Rakyan V. K., DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 20, 249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath S., Raj K., DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Horvath S., DNA methylation age of human tissues and cell types. Genome Biol. 14, 3156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidner C. I., Lin Q., Koch C. M., Eisele L., Beier F., Ziegler P., Bauerschlag D. O., Jöckel K. H., Erbel R., Mühleisen T. W., Zenke M., Brümmendorf T. H., Wagner W., Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 15, R24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine M. E., Lu A. T., Quach A., Chen B. H., Assimes T. L., Bandinelli S., Hou L., Baccarelli A. A., Stewart J. D., Li Y., Whitsel E. A., Wilson J. G., Reinerl A. P., Avivl A., Lohman K., Liu Y., Ferrucci L., Horvath S., An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu A. T., Quach A., Wilson J. G., Reiner A. P., Aviv A., Raj K., Hou L., Baccarelli A. A., Li Y., Stewart J. D., Whitsel E. A., Assimes T. L., Ferrucci L., Horvath S., DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hannum G., Guinney J., Zhao L., Zhang L., Hughes G., Sadda S. V., Klotzle B., Bibikova M., Fan J. B., Gao Y., Deconde R., Chen M., Rajapakse I., Friend S., Ideker T., Zhang K., Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Q., Weidner C. I., Costa I. G., Marioni R. E., Ferreira M. R. P., Deary I. J., Wagner W., DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging 8, 394–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marioni R. E., Shah S., McRae A. F., Chen B. H., Colicino E., Harris S. E., Gibson J., Henders A. K., Redmond P., Cox S. R., Pattie A., Corley J., Murphy L., Martin N. G., Montgomery G. W., Feinberg A. P., Fallin M. D., Multhaup M. L., Jaffe A. E., Joehanes R., Schwartz J., Just A. C., Lunetta K. L., Murabito J. M., Starr J. M., Horvath S., Baccarelli A. A., Levy D., Visscher P. M., Wray N. R., Deary I. J., DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16, 25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marioni R. E., Shah S., McRae A. F., Ritchie S. J., Muniz-Terrera G., Harris S. E., Gibson J., Redmond P., Cox S. R., Pattie A., Corley J., Taylor A., Murphy L., Starr J. M., Horvath S., Visscher P. M., Wray N. R., Deary I. J., The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 44, 1388–1396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breitling L. P., Saum K.-U., Perna L., Schöttker B., Holleczek B., Brenner H., Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenetics 8, 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horvath S., Ritz B. R., Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 7, 1130–1142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath S., Garagnani P., Bacalini M. G., Pirazzini C., Salvioli S., Gentilini D., Di Blasio A. M., Giuliani C., Tung S., Vinters H. V., Franceschi C., Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maierhofer A., Flunkert J., Oshima J., Martin G. M., Haaf T., Horvath S., Accelerated epigenetic aging in Werner syndrome. Aging 9, 1143–1152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petkovich D. A., Podolskiy D. I., Lobanov A. V., Lee S.-G., Miller R. A., Gladyshev V. N., Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 25, 954–960.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stubbs T. M., Bonder M. J., Stark A.-K., Krueger F., von Meyenn F., Stegle O., Reik W., Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 18, 68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T., Tsui B., Kreisberg J. F., Robertson N. A., Gross A. M., Yu M. K., Carter H., Brown-Borg H. M., Adams P. D., Ideker T., Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction, and rapamycin treatment. Genome Biol. 18, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meer M. V., Podolskiy D. I., Tyshkovskiy A., Gladyshev V. N., A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson M. J., Chwiałkowska K., Rubbi L., Lusis A. J., Davis R. C., Srivastava A., Korstanje R., Churchill G. A., Horvath S., Pellegrini M., A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M., Lemos B., Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 29, 325–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Y., Eipel M., Franzen J., Sakk V., Dethmers-Ausema B., Yndriago L., Izeta A., de Haan G., Geiger H., Wagner W., Epigenetic age predictor for mice based on three CpG sites. eLife 7, e37462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olova N., Simpson D. J., Marioni R. E., Chandra T., Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 18, e12877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarkar T. J., Quarta M., Mukherjee S., Colville A., Paine P., Doan L., Tran C. M., Chu C. R., Horvath S., Qi L. S., Bhutani N., Rando T. A., Sebastiano V., Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 11, 1545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D. Gill, A. Parry, F. Santos, I. Hernando-Herraez, T. M. Stubbs, I. Milagre, W. Reik, Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. bioRxiv 2021.01.15.426786 [Preprint]. 17 January 2021. 10.1101/2021.01.15.426786. [DOI] [PMC free article] [PubMed]
- 40.MAMMALIAN METHYLATION CONSORTIUM, A. T. Lu, Z. Fei, A. Haghani, T. R. Robeck, J. A. Zoller, C. Z. Li, J. Zhang, J. Ablaeva, D. M. Adams, J. Almunia, R. Ardehali, A. Arneson, C. S. Baker, K. Belov, P. Black, D. T. Blumstein, E. K. Bors, C. E. Breeze, R. T. Brooke, J. L. Brown, A. Caulton, J. M. Cavin, I. Chatzistamou, H. Chen, P. Chiavellini, O.-W. Choi, S. Clarke, J. DeYoung, C. Dold, C. K. Emmons, S. Emmrich, C. G. Faulkes, S. H. Ferguson, C. J. Finno, J.-M. Gaillard, E. Garde, V. N. Gladyshev, V. Gorbunova, R. G. Goya, M. J. Grant, E. N. Hales, M. B. Hanson, M. Haulena, A. N. Hogan, C. J. Hogg, T. A. Hore, A. J. Jasinska, G. Jones, E. Jourdain, O. Kashpur, H. Katcher, E. Katsumata, V. Kaza, H. Kiaris, M. S. Kobor, P. Kordowitzki, W. R. Koski, B. Larison, S.-G. Lee, Y. C. Lee, M. Lehmann, J.-F. Lemaitre, A. J. Levine, C. Li, X. Li, D. T. Lin, N. Macoretta, D. Maddox, C. O. Matkin, J. A. Mattison, J. Mergl, J. J. Meudt, K. Mozhui, A. Naderi, M. Nagy, P. Narayan, P. W. Nathanielsz, N. B. Nguyen, C. Niehrs, A. G. Ophir, E. A. Ostrander, P. O. Ginn, K. M. Parsons, K. C. Paul, M. Pellegrini, G. M. Pinho, J. Plassais, N. A. Prado, B. Rey, B. R. Ritz, J. Robbins, M. Rodriguez, J. Russell, E. Rydkina, L. L. Sailer, A. B. Salmon, A. Sanghavi, K. M. Schachtschneider, D. Schmitt, T. Schmitt, L. Schomacher, L. B. Schook, K. E. Sears, A. Seluanov, D. Shanmuganayagam, A. Shindyapina, K. Singh, I. Sinha, R. G. Snell, E. Soltanmaohammadi, M. L. Spangler, L. Staggs, K. J. Steinman, V. J. Sugrue, B. Szladovits, M. Takasugi, E. C. Teeling, M. J. Thompson, B. Van Bonn, S. C. Vernes, D. Villar, H. V. Vinters, M. C. Wallingford, N. Wang, R. K. Wayne, G. S. Wilkinson, C. K. Williams, R. W. Williams, X. W. Yang, B. G. Young, B. Zhang, Z. Zhang, P. Zhao, Y. Zhao, J. Zimmermann, W. Zhou, J. Ernst, K. Raj, S. Horvath, Universal DNA methylation age across mammalian tissues. bioRxiv 2021.01.18.426733 [Preprint]. 19 January 2021. 10.1101/2021.01.18.426733. [DOI]
- 41.Hoshino A., Horvath S., Sridhar A., Chitsazan A., Reh T. A., Synchrony and asynchrony between an epigenetic clock and developmental timing. Sci. Rep. 9, 3770 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knight A. K., Craig J. M., Theda C., Bækvad-Hansen M., Bybjerg-Grauholm J., Hansen C. S., Hollegaard M. V., Hougaard D. M., Mortensen P. B., Weinsheimer S. M., Werge T. M., Brennan P. A., Cubells J. F., Newport D. J., Stowe Z. N., Cheong J. L. Y., Dalach P., Doyle L. W., Loke Y. J., Baccarelli A. A., Just A. C., Wright R. O., Téllez-Rojo M. M., Svensson K., Trevisi L., Kennedy E. M., Binder E. B., Iurato S., Czamara D., Räikkönen K., Lahti J. M. T., Pesonen A. K., Kajantie E., Villa P. M., Laivuori H., Hämäläinen E., Park H. J., Bailey L. B., Parets S. E., Kilaru V., Menon R., Horvath S., Bush N. R., LeWinn K. Z., Tylavsky F. A., Conneely K. N., Smith A. K., An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 17, 206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spiers H., Hannon E., Schalkwyk L. C., Smith R., Wong C. C. Y., O’Donovan M. C., Bray N. J., Mill J., Methylomic trajectories across human fetal brain development. Genome Res. 25, 338–352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McStay B., Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 30, 1598–1610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Der Jeught M., O’Leary T., Ghimire S., Lierman S., Duggal G., Versieren K., Deforce D., Chuva De Sousa Lopes S., Heindryckx B., De Sutter P., The combination of inhibitors of FGF/MEK/Erk and GSK3β signaling increases the number of OCT3/4-and NANOG-positive cells in the human inner cell mass, but does not improve stem cell derivation. Stem Cells Dev. 22, 296–306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auclair G., Guibert S., Bender A., Weber M., Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol. 15, 545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai H. Q., Wang B. A., Yang L., Chen J. J., Zhu G. C., Sun M. L., Ge H., Wang R., Chapman D. L., Tang F., Sun X., Xu G. L., TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 538, 528–532 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Argelaguet R., Clark S. J., Mohammed H., Stapel L. C., Krueger C., Kapourani C. A., Imaz-Rosshandler I., Lohoff T., Xiang Y., Hanna C. W., Smallwood S., Ibarra-Soria X., Buettner F., Sanguinetti G., Xie W., Krueger F., Göttgens B., Rugg-Gunn P. J., Kelsey G., Dean W., Nichols J., Stegle O., Marioni J. C., Reik W., Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.A. Trapp, C. Kerepesi, V. N. Gladyshev, Profiling epigenetic age in single cells. bioRxiv 2021.03.13.435247 [Preprint]. 15 March 2021. 10.1101/2021.03.13.435247. [DOI] [PMC free article] [PubMed]
- 50.Okano M., Bell D. W., Haber D. A., Li E., DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Dahlet T., Argüeso Lleida A., Al Adhami H., Dumas M., Bender A., Ngondo R. P., Tanguy M., Vallet J., Auclair G., Bardet A. F., Weber M., Genome-wide analysis in the mouse embryo reveals the importance of DNA methylation for transcription integrity. Nat. Commun. 11, 3153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E., Araoka T., Vazquez-Ferrer E., Donoso D., Roman J. L., Xu J., Rodriguez Esteban C., Nuñez G., Nuñez Delicado E., Campistol J. M., Guillen I., Guillen P., Izpisua Belmonte J. C., In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Y., Brommer B., Tian X., Krishnan A., Meer M., Wang C., Vera D. L., Zeng Q., Yu D., Bonkowski M. S., Yang J. H., Zhou S., Hoffmann E. M., Karg M. M., Schultz M. B., Kane A. E., Davidsohn N., Korobkina E., Chwalek K., Rajman L. A., Church G. M., Hochedlinger K., Gladyshev V. N., Horvath S., Levine M. E., Gregory-Ksander M. S., Ksander B. R., He Z., Sinclair D. A., Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L., Bailey S. M., Okuka M., Muñoz P., Li C., Zhou L., Wu C., Czerwiec E., Sandler L., Seyfang A., Blasco M. A., Keefe D. L., Telomere lengthening early in development. Nat. Cell Biol. 9, 1436–1441 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Wang L., Zhang J., Duan J., Gao X., Zhu W., Lu X., Yang L., Zhang J., Li G., Ci W., Li W., Zhou Q., Aluru N., Tang F., He C., Huang X., Liu J., Programming and inheritance of parental DNA methylomes in mammals. Cell 157, 979–991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng Q., Ramsköld D., Reinius B., Sandberg R., Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Zhang B., Gladyshev V. N., How can aging be reversed? Exploring rejuvenation from a damage-based perspective. Adv. Genet. 1, e10025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh K., Effect of parental ageing on offspring developmental and survival attributes in an aphidophagous ladybird, Cheilomenes sexmaculata. J. Appl. Entomol. 133, 500–504 (2009). [Google Scholar]
- 59.Kojima Y., Tam O. H., Tam P. P. L., Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol. 34, 65–75 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Yu M., Han D., Hon G. C., He C., Tet-assisted bisulfite sequencing (TAB-seq). Methods Mol. Biol. 1708, 645–663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kinzina E. D., Podolskiy D. I., Dmitriev S. E., Gladyshev V. N., Patterns of aging biomarkers, mortality, and damaging mutations illuminate the beginning of aging and causes of early-life mortality. Cell Rep. 29, 4276–4284.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y., Hapala J., Brenner H., Wagner W., Individual CpG sites that are associated with age and life expectancy become hypomethylated upon aging. Clin. Epigenetics 9, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith Z. D., Chan M. M., Mikkelsen T. S., Gu H., Gnirke A., Regev A., Meissner A., A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484, 339–344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang C., Liu X., Gao Y., Yang L., Li C., Liu W., Chen C., Kou X., Zhao Y., Chen J., Wang Y., Le R., Wang H., Duan T., Zhang Y., Gao S., Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 20, 620–631 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Muñoz-Lorente M. A., Cano-Martin A. C., Blasco M. A., Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 10, 4723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He Y., Hariharan M., Gorkin D. U., Dickel D. E., Luo C., Castanon R. G., Nery J. R., Lee A. Y., Zhao Y., Huang H., Williams B. A., Trout D., Amrhein H., Fang R., Chen H., Li B., Visel A., Pennacchio L. A., Ren B., Ecker J. R., Spatiotemporal DNA methylome dynamics of the developing mouse fetus. Nature 583, 752–759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sziráki A., Tyshkovskiy A., Gladyshev V. N., Global remodeling of the mouse DNA methylome during aging and in response to calorie restriction. Aging Cell 17, e12738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith Z. D., Chan M. M., Humm K. C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A., DNA methylation dynamics of the human preimplantation embryo. Nature 511, 611–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Slieker R. C., Roost M. S., van Iperen L., Suchiman H. E. D., Tobi E. W., Carlotti F., de Koning E. J. P., Slagboom P. E., Heijmans B. T., de Sousa Lopes S. M. C., DNA methylation landscapes of human fetal development. PLOS Genet. 11, e1005583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nazor K. L., Altun G., Lynch C., Tran H., Harness J. V., Slavin I., Garitaonandia I., Müller F. J., Wang Y.-C., Boscolo F. S., Fakunle E., Dumevska B., Lee S., Park H. S., Olee T., D’Lima D. D., Semechkin R., Parast M. M., Galat V., Laslett A. L., Schmidt U., Keirstead H. S., Loring J. F., Laurent L. C., Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 10, 620–634 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price E. M., Peñaherrera M. S., Portales-Casamar E., Pavlidis P., Van Allen M. I., McFadden D. E., Robinson W. P., Profiling placental and fetal DNA methylation in human neural tube defects. Epigenetics Chromatin 9, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mallon B. S., Chenoweth J. G., Johnson K. R., Hamilton R. S., Tesar P. J., Yavatkar A. S., Tyson L. J., Park K., Chen K. G., Fann Y. C., McKay R. D. G., StemCellDB: The human pluripotent stem cell database at the national institutes of health. Stem Cell Res. 10, 57–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurian L., Sancho-Martinez I., Nivet E., Aguirre A., Moon K., Pendaries C., Volle-Challier C., Bono F., Herbert J. M., Pulecio J., Xia Y., Li M., Montserrat N., Ruiz S., Dubova I., Rodriguez C., Denli A. M., Boscolo F. S., Thiagarajan R. D., Gage F. H., Loring J. F., Laurent L. C., Izpisua Belmonte J. C., Conversion of human fibroblasts to angioblast-like progenitor cells. Nat. Methods 10, 77–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zimmerlin L., Park T. S., Huo J. S., Verma K., Pather S. R., Talbot C. C., Agarwal J., Steppan D., Zhang Y. W., Considine M., Guo H., Zhong X., Gutierrez C., Cope L., Canto-Soler M. V., Friedman A. D., Baylin S. B., Zambidis E. T., Tankyrase inhibition promotes a stable human naïve pluripotent state with improved functionality. Development 143, 4368–4380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mallon B. S., Hamilton R. S., Kozhich O. A., Johnson K. R., Fann Y. C., Rao M. S., Robey P. G., Comparison of the molecular profiles of human embryonic and induced pluripotent stem cells of isogenic origin. Stem Cell Res. 12, 376–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ohnuki M., Tanabe K., Sutou K., Teramoto I., Sawamura Y., Narita M., Nakamura M., Tokunaga Y., Nakamura M., Watanabe A., Yamanaka S., Takahashi K., Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci. U.S.A. 111, 12426–12431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg6082/DC1