

Summary
Location of immune cells that form the germinal center reaction within secondary lymphoid tissues can be characterized using confocal microscopy. Here, we present an optimized immunofluorescence staining protocol to image germinal center structures in fixed/frozen spleen sections from ChAdOx1 nCoV-19 immunized mice. This protocol can be adapted to identify other cell types within secondary lymphoid tissues.
For complete information on the generation and use of this protocol to examine immune responses to the COVID vaccine ChAdOx1 nCoV-19, please refer to Silva-Cayetano et al. (2020).
Subject areas: Antibody, Cell Biology, Immunology, Microbiology, Microscopy, Model Organisms
Graphical abstract
Highlights
-
•
Immunostaining of tissues from ChAdOx1 nCoV-19 immunized mice
-
•
An optimized protocol for confocal imaging of splenic germinal centers
-
•
Protocol is also applicable for the identification of other cell types
-
•
Adaptable for processing other tissues
Location of immune cells that form the germinal center reaction within secondary lymphoid tissues can be characterized using confocal microscopy. Here, we present an optimized immunofluorescence staining protocol to image germinal center structures in fixed/frozen spleen sections from ChAdOx1 nCoV-19 immunized mice. This protocol can be adapted to identify other cell types within secondary lymphoid tissues.
Before you begin
Before culling mice and extracting spleens, prepare the necessary solutions according to the Materials and Equipment section. Here we describe the process of imaging germinal centers in the spleen after ChAdOx1 nCoV-19 immunization. The same protocol has also been used to process and acquire images of lymph nodes after vaccination or infection with multiple different immunogens (e.g., Vanderleyden et al., 2020).
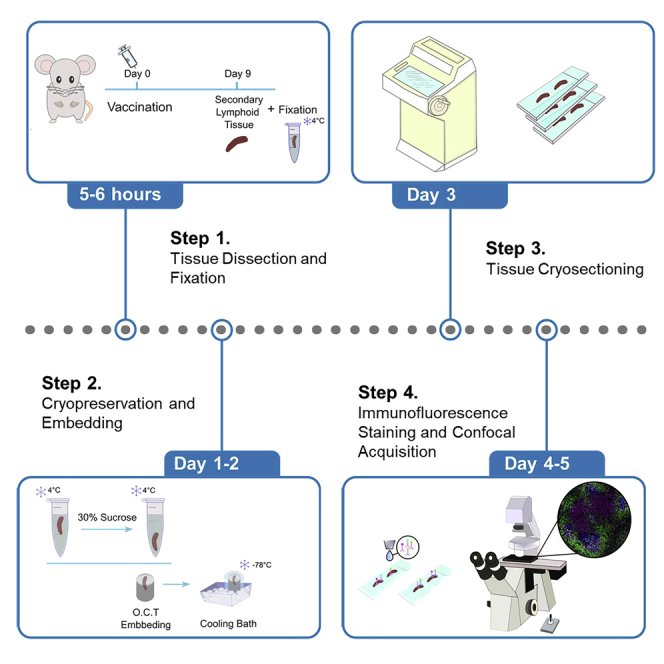
Tissue dissection, fixation and embedding
Timing: 0–3 days after culling mice
Here we describe the protocol from immunization, tissue dissection, until the spleen is ready for embedding in Optimal cutting temperature (OCT) compound (Figure 1).
Figure 1.

Schematic of the tissue dissection, fixation and embedding
The chemical fixation protocol includes a low concentration of paraformaldehyde which fixes and stabilizes the tissue by cross-linking proteins. The periodate included in Periodate–lysine–paraformaldehyde (PLP) solution oxidizes carbohydrates and forms aldehyde groups. The lysine, subsequently reacts with the aldehyde group thereby cross-linking the carbohydrate (Mclean and Nakane, 1974). Fixation steps and embedding of frozen tissue block described below help to maintain the tissue morphology and were adapted from a previous protocol (Vanderleyden and Linterman, 2017).
-
1.
Prepare the Fixative solution as outlined in Materials and Equipment section.
-
2.
Immunize adult mice (3-month-old and 22-month-old males) in the right quadriceps femoris muscle with 50μL of 1×108 infectious units of ChAdOx1 nCoV-19 vaccine in phosphate buffered saline (PBS).
-
3.
Cull mice 9 days after immunization and place them on a dissection board. Surface-sterilize the skin using 70% ethanol. Using sterile surgical instruments (forceps and scissors), cut through the skin just below the ribcage and visualize the spleen.
-
4.
Remove the spleen, trimming away any non-splenic tissue and place the spleen in PBS on ice.
Note: The inclusion of fat attached to the cryopreserved tissues makes the cryosectioning difficult, as it needs a different cutting temperature.
-
5.
Fix spleen in 1–2 mL of Fixative solution for 4–5 hours at 4°C.
CRITICAL: Respect fixation times as under or over-fixation can destroy tissue morphology, affecting the staining and imaging (Troubleshooting 1).
Note: The fixation protocol including type of fixative, duration and temperature must be optimized for every tissue type.
Alternatives: 4% Formaldehyde methanol-free solution can be used instead of 4% Paraformaldehyde.
-
6.
Remove Fixative solution and rinse tissue with 1× PBS three times for 5 min at 4°C.
-
7.
Incubate with 1–2 mL of 30% sucrose for 12–16 hours at 4°C until tissue sinks.
Alternatives: Tissue can be left immersed in 30% sucrose at 4°C for up to 5 days. Prevent evaporation and/or contamination by tightly wrapping the tissue container with Parafilm.
-
8.
Remove all sucrose solution from sample and dab-dry using a paper tissue.
-
9.
Transfer tissue to a cryomold containing OCT (the volume of OCT depends on the size of the mold but make sure the tissue is fully immersed and without bubbles surrounding it). (Figure 2A, Methods video S1).
Figure 2.

Steps for tissue preparation before cryosectioning
(A) Spleen embedded in OCT.
(B) 2-propanol cooling bath.
(C) Frozen Block in 2-propanol cooling bath.
(D) Frozen Block on dry ice before wrapped and stored at −80°C.
Alternatives: Cryomolds are commercially available but can also be made out of aluminum foil by wrapping the foil around an object of the desired mold size, for example the end of a marker pen.
Note: Placing the spleen in a transverse plane while embedding in OCT will make the cryosectioning and image acquisition easier.
Note: Avoid bubbles in the OCT, especially around the tissue itself as this will make the cryosectioning difficult.
-
10.
Prepare a 2-propanol/dry ice cooling bath by filling a box with dry ice and placing a glass beaker on top of the dry ice. Fill the bottom of the beaker with a small volume of 2-propanol until reach approximately −78°C (~30 min). (Figure 2B).
-
11.
Place cryomold into beaker containing 2-propanol/dry ice cooling bath (−78°C) until the OCT is solid. The rate of freezing must be fast, as slow freezing promotes ice crystal formation (Figure 2C, Methods video S1, Troubleshooting 2).
-
12.
Transfer frozen tissue block to dry ice (Figure 2D). Make sure it is tightly wrapped and then store at −80°C.
Note: Frozen embedded spleens can be stored at −80°C for long-term preservation.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor® 647 anti-mouse IgD (clone 11-26c.2a) | BioLegend | Cat# 405708; RRID:AB_893528 |
| Ki-67 Monoclonal Antibody, FITC, eBioscience™ (Clone SolA15) | Thermo Fisher Scientific | Cat# 11-5698-82; RRID:AB_11151330 |
| CD21/CD35 Rat anti-Mouse, Biotin, eBioscience™ (Clone: 8D9) | Thermo Fisher Scientific | Cat# 15239489 |
| Hamster Anti-CD3e Monoclonal Antibody, Unconjugated (Clone 500A2) | BD Biosciences | Cat# 553238; RRID:AB_394727 |
| Goat anti-Hamster IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-21112; RRID:AB_2535761 |
| SAv-Brilliant Violet 421™ | BioLegend | Cat# 405225 |
| Biological samples | ||
| Mouse C57BL/6Babr (spleen tissue) | Babraham Institute | C57BL/6Babr |
| Chemicals, peptides, and recombinant proteins | ||
| Triton™ X-100 | Sigma-Aldrich | Cat# T8787 |
| Tween® 20 | Sigma-Aldrich | Cat# P1379 |
| OCT Compound | Agar Scientific | AGR1180 |
| Hydromount mounting medium | National Diagnostics | Cat# HS-106 |
| Normal Goat Serum | Sigma-Aldrich | Cat# 566380 |
| Bovine Serum Albumin | Jackson ImmunoResearch | Cat# 001-000-161 |
| L-Lysine, Hydrochloride | Merck Life Sciences | 4400-100GM |
| Sodium phosphate monobasic | Sigma-Aldrich | Cat# S0751 |
| Sodium phosphate dibasic | Sigma-Aldrich | Cat# S3264 |
| Sodium periodate | Sigma-Aldrich | Cat# S1878 |
| Paraformaldehyde (PFA) | Sigma-Aldrich | Cat# P6148 |
| Streptavidin/Biotin Blocking Kit | Vector Laboratories | Cat# SP-2002 |
| Software and algorithms | ||
| ImageJ | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| ZEN black edition software | Zeiss | https://www.zeiss.com |
| Other | ||
| Superfrost Plus slides | VWR | Cat# 631-0108 |
| ImmEdge™ Hydrophobic Barrier PAP Pen | 2B Scientific Ltd | Cat# H-4000 |
| MX35 Premier Disposable Low-Profile Microtome Blades | Fisher Scientific UK Ltd | Cat# 3052835 |
| Cryostat, CM1850 | Leica | N/A |
| Zeiss LSM780 Confocal | Zeiss | N/A |
Materials and equipment
4% PFA
| Reagent | Final Concentration | Amount |
|---|---|---|
| PFA | 4% | 4 g |
| dH2O | n/a | Up to 100 mL |
| Total | n/a | 100 mL |
Note: Prepare 12.5 mL aliquots. Store at −20°C (stable for 1–2 months) and defrost at 4°C before adding into the fixative solution.
Fixative solution
Periodate–lysine–paraformaldehyde (PLP)
| Reagent | Final Concentration | Amount |
|---|---|---|
| L-lysine 0.2 M | 0.075 M | 18.75 mL |
| Sodium phosphate 0.1 M (pH 7.4) | 0.0375 M | 18.75 mL |
| NaIO4 | 0.01 M | 0.1069 g |
| 4% PFA | 1% | 12.5 mL |
| Total | n/a | 50 mL |
Note: 37.5 mL aliquots containing 0.075 M L-lysine, 0.0375 M Sodium phosphate and 0.01M NaIO4 can be stored at −20°C (stable for 3–4 months) and defrosted at 4°C before use.
Note: Prepare Fixative solution before use. Mix 12.5 mL 4% PFA aliquot with 37.5 mL aliquot containing 0.075 M L-lysine, 0.0375 M Sodium phosphate and 0.01M NaIO4.
30% Sucrose
| Reagent | Final Concentration | Amount |
|---|---|---|
| Sucrose | 30% | 30 g |
| 1× PBS | n/a | Up to 100 mL |
| Total | n/a | 100 mL |
Note: Store at 4°C well sealed (stable for 1–2 months).
Wash buffer solution
| Reagent | Final Concentration | Amount |
|---|---|---|
| Tween 20 | 0.5% (v/v) | 5 mL |
| 1× PBS | n/a | 995 mL |
| Total | n/a | 1000 mL |
Note: Prepare fresh the day of procedure and store at 4°C.
Blocking buffer
| Reagent | Final Concentration | Amount |
|---|---|---|
| Bovine Serum Albumin | 2% (w/v) | 0.4 g |
| Normal Goat Serum | 10% (v/v) | 2 mL |
| 1× PBS | n/a | Up to 20 mL |
| Total | n/a | 20 mL |
Alternatives: Aliquot the solution into 1000 μL per tube and store them at −20°C until further use (stable for 5–6 months). Defrost the amount needed at 4°C before use, avoiding freeze/thaw cycle.
Antibody diluents
For antibodies generated in goat use a solution made up 2% (v/v) Normal Goat Serum in Wash Buffer solution.
| Reagent | Final Concentration | Amount |
|---|---|---|
| Normal Goat Serum | 2% (v/v) | 0.4 mL |
| Wash Buffer solution | 0.5% (v/v) | 19.6 mL |
| Total | n/a | 20 mL |
For antibodies generated from other species use a solution made up 1% (w/v) Bovine Serum Albumin in Wash Buffer solution.
| Reagent | Final Concentration | Amount |
|---|---|---|
| Bovine Serum Albumin | 1% (v/v) | 0.2 g |
| Wash Buffer solution | 0.5% (v/v) | Up to 20 mL |
| Total | n/a | 20 mL |
Alternatives: Aliquot diluents into 1000 μL per tube and store them at −20°C until further use (stable for 5–6 months). Defrost the amount needed at 4°C before use, avoiding freeze/thaw cycle.
Permeabilization buffer
| Reagent | Final Concentration | Amount |
|---|---|---|
| Triton™ X-100 | 2% (v/v) | 0.2 mL |
| 1× PBS | n/a | 9.8 mL |
| Total | n/a | 10 mL |
Note: Freshly prepare the solution just before use.
Cryosectioning settings: Set the temperature of the cryostat chamber between −15°C and −20°C and the section thickness desired. This protocol works with thicknesses between 10 μm and 20 μm.
Note: Check cryostat manufacturer manual for set up and sectioning guidelines for the tissue in use. Adjust the temperature of the cutting chamber according to the cryostat model and tissue under study.
Note: Section thickness greater than 20 μm may be used but permeabilization times and antibodies concentration need to be validated.
Microscope and imaging: Images were acquired using Zeiss LSM780 Confocal harboring 405-, 488-, 561-, and 633-nm lasers. Image analysis was done using ImageJ software (Schneider et al., 2012). Details of image acquisition can be found on step-by-step method Section.
Step-by-step method details
Tissue cryosectioning
The following tissue processing steps are recommended for obtaining optimal frozen sections for confocal imaging.
-
1.
Set up the cryostat as described in Materials and Equipment section.
-
2.
Transport the OCT frozen tissue block in dry ice to a cryostat.
-
3.
Allow tissue block to reach cryostat chamber temperature for approximately 30 min.
-
4.
Use a razor blade to cut the OCT frozen tissue block to size (Methods video S2).
-
5.
Apply OCT to a specimen disc at 20°C–22°C and place the frozen tissue block on top. Transfer it immediately to the quick freeze shelf inside the cryostat until completely frozen (Methods video S2).
-
6.
Cut sections according to the desired thickness (Methods video S3).
Note: A brush can be useful to flatten the emerging section, in case the tissue sticks to the anti-roll plate when cutting (Troubleshooting 3).
-
7.
Transfer the tissue section to a slide at 20°C–22°C by gently touching the frozen tissue with the slide. The sections will immediately melt on the slide due to the temperature difference (Methods video S3, Troubleshooting 4).
Note: Try to collect each section in less than 30 seconds to avoid freeze-drying of the tissue.
Note: Minimum two sets of sections are collected, a first set for immunostaining control and a second set for germinal center staining.
Alternatives: Poly-L-Lysine coated slides improve adhesion of tissues to the slide.
-
8.
Leave the slide containing tissue sections air-dry for 20 min at 20–22°C. Insufficient drying may cause the section to detach from the slides.
-
9.
Sectioned tissues can be stored in an airtight slide holder inside a −80°C freezer until required for the immunofluorescence staining.
Note: Left-over tissue can be stored back into disposable cryomold with a layer of OCT on top to prevent freeze-drying during storage at −80°C.
Immunofluorescence staining
This segment describes how to stain spleen sections to identify germinal centers structures with the antibodies optimized for this protocol. If alternative primary and secondary antibodies are used they should be validated first.
-
10.
Remove slides from −80°C and air-dry them under airflow for 30 min at 20°C–22°C (e.g., hood, or fume cabinet).
-
11.
Draw an unbroken circle around tissue sections with a PAP pen to create a hydrophobic barrier, and leave for 15-30 min until the wax has completely dried (Figure 3A).
Note: Do not touch or get too close to the tissue with the PAP Pen.
-
12.
Rehydrate slides: Transfer slides to a staining jar or dish and wash them three times for 5 min with Wash Buffer at 20°C–22°C (Figure 3B).
Note: After rehydration it is important to keep slides hydrated at all times. This can be achieved by using a humidified chamber during the staining procedures (Figure 3C). High background levels can be observed during confocal images acquisition, if tissue is allowed to dry during the staining (Troubleshooting 5).
-
13.
Minimize non-specific binding of primary and secondary antibodies by adding blocking buffer for 60–120 min at 20°C–22°C. To prevent interactions with the tissue or with the primary/secondary antibodies, select the appropriated type of normal serum.
Note: We recommend using clones 8D9 or 8C12 when detecting Follicular Dendritic Cells (FDCs), as we observed other monoclonal antibodies produced non-specific binding, potentially due to the high levels of Fc receptors present in FDCs. To overcome this issue, we suggest the addition of Fc block into the blocking buffer.
-
14.
Wash slides with Wash Buffer three times for 5 min at 20°C–22°C.
-
15.Block endogenous biotin: to eliminate background caused by endogenous biotin we use a Streptavidin/Biotin Blocking Kit before adding the biotin-labeled probe (refer to Key resources table).
-
a.Incubate each tissue section with streptavidin solution for 15 min at 20°C–22°C.
-
b.Rinse quickly with Wash Buffer solution.
-
c.Incubate for 15 minutes with the biotin solution at 20°C–22°C.
-
d.Rinse quickly with Wash Buffer solution twice.
-
a.
Note: These steps should be performed prior to the addition of primary antibodies.
-
16.
Permeabilization: add Permeabilization buffer freshly prepared and incubate for 30 min at 20°C–22°C.
-
17.
Wash slides with Wash Buffer three times for 5 min at 20°C–22°C.
-
18.Primary antibody staining: Prepare primary antibody dilutions (refer to Key resources table) and pipette this mix onto the slide making sure each section is completely covered (depending on the size of the hydrophobic circle, 30–60 μL per section is usually enough).
-
a.The primary antibody cocktail includes: Alexa Fluor 647 rat anti-mouse IgD (1:200), FITC rat anti-mouse Ki-67 (1:100), biotin rat anti-mouse CD21/CD35 (1:400) and unconjugated hamster anti-CD3ε (1:200) in antibody diluent (1% BSA in Wash Buffer solution).
-
b.Incubate the covered slide in a dark humidified chamber for 60–120 min at 20°C–22°C (Figure 3D).
-
a.
Pause point: This step can be done for 12–16 hours at 4°C.
Note: Always include controls, e.g., minus primary antibody, secondary antibody alone and/or isotypes control.
-
19.
Wash slides with Wash Buffer three times for 5 min at 20°C–22°C to remove unbound antibodies.
-
20.Secondary antibody staining: Prepare secondary antibody dilutions (see Key resources table) and pipette this mix onto the slide making sure each section is completely covered.
-
a.The secondary antibody cocktail includes: Alexa Fluor 568 goat anti-hamster IgG (1:1000) and BV421-SAv (1:1000), in antibody diluent (2% Normal Goat serum in Wash Buffer solution).
-
b.Incubate the covered slide in a dark humidified chamber for 60–120 min at 20°C–22°C.
-
a.
Note: Fluorescently labeled secondary antibodies are stored in small aliquots at −20°C. Once thawed, they are kept at 4°C.
-
21.
Quickly rinse slides two times with Wash Buffer.
-
22.
Pour off the Wash Buffer and quickly rinse with 1×PBS.
-
23.
Wipe off excess 1×PBS without disrupting the tissue (Methods video S4).
-
24.Mount the tissue with mounting medium (Methods video S4).
-
a.Add a drop of mounting medium directly onto the tissue and gently place coverslip on top using forceps to get rid of any bubbles.
-
b.Dry slides at 20°C–22°C until mounting medium has set.
-
a.
Note: Be careful to avoid the formation of air bubbles, as this will make imaging more difficult.
-
25.
Store dried slides at 4°C for imaging.
Figure 3.

Immunofluorescence staining steps
(A) Hydrophobic barrier around spleen section.
(B) Slides rehydration in a staining jar.
(C) Example of humidified chamber containing slides.
(D) Dark humidified chamber kept in fridge for 12–16 hours.
Confocal image acquisition
In this section we describe in detail the microscope settings for experiments aimed at imaging of germinal center structures using the Zeiss LSM 780 system and Zen black edition software. Although each immunofluorescence staining will require some adjustment of these parameters, we outline settings that can be used as a starting point.
-
26.
Place the microscope slide onto the stage of the microscope ensuring the coverslip is facing the objective lens.
-
27.
Select a high Numerical Aperture (NA) plan apochromatic air objective, e.g., 20× 0.8 NA Plan Apo.
-
28.
Using the stage x/y controller (e.g., joystick) position the slide so that the tissue section is directly in front of the objective lens. If the tissue is very thin it can be helpful to mark the location of the section on the slide using a fine-tip pen.
-
29.
Using the “Locate” tab in Zen, select an appropriate filter set to observe the sample by eye e.g., “FITC”. Check that light is coming out of the objective lens (it will appear blue with the FITC filter set). Look down the eyepieces to focus the sample and position to a region of interest.
Note: Ki67+ cells-FITC labeled can be used to locate the region of interest easily.
-
30.
Using the “Acquisition” tab, set up the channel configuration (Figure 4A). The four-dye combination Brilliant Violet 421 (BV421), FITC, Alexa Fluor 568 (AF568) and Alexa Fluor 647 (AF647) can be imaged using a two-track configuration with minimal risk of bleed-through. Using the Imaging Setup and Channels windows, assign BV421+AF568 to Track 1 and FITC+AF647 to Track 2.
Note: For the fastest acquisition times the system needs to operate in “Line Switching” mode. This will only work if each dye is assigned to a separate detector and no filter/mirror changes are required between tracks (Figure 4A).
Note: BV421 may not appear as an option in the dye dropdown in the Imaging Setup window. Select DAPI as an alternative.
-
31.
Configure the scanning parameters in the Acquisition Mode window (Figure 4B, Methods video S5). With a 20× 0.8 NA lens, sufficient sampling will be achieved using a 1024×1024 scan at 1× zoom (pixel size 0.42 μm). Other parameters as follows: Scan Mode = Frame; Line Step = 1; Speed = 7–10 (a higher value indicates a faster speed, which will be possible if the signal is strong i.e., Gain <700); Averaging = 1, 2 or 4 (increasing averaging improves signal: noise, but increases the scan time. If the signal is strong i.e., gain <700, then use a value of 1); Mode = Line; Method = Mean; Bit Depth = 8 bit; Direction = <-> (this indicates bi-direction scanning which halves the time required to take the image but may result in “fringing” artifact. When scanning, look carefully at the image and use the Corr X & Corr Y sliders to remove any artifact).
Figure 4.

Confocal settings using of Zen black software for imaging germinal centers
(A) Channels configuration.
(B) Scanning parameters configuration.
(C) Detectors optimization.
-
32.
Optimize the detectors. As an approximate starting configuration, set each of the Channels to 2% laser power, Gain (Master) = 700, Digital Offset = 0, Digital Gain = 1. Deselect one Track and in the remaining active Track ensure only one detector is active. Start the scanning using the “Live” button. Use the fine focus on the microscope to ensure the focal plane is adjusted to give strongest signal. Apply the Range Indicator palette by ticking the Range Indicator checkbox (Figure 4C, Methods video S5). Adjust the laser power and or/Gain (Master) to give a bright image, but avoid saturation (red pixels). Ideally the laser power should be kept below ~5% to avoid bleaching and the Gain (Master) below ~750 to avoid high noise. Adjust the Digital Offset to maximize contrast in the image, but avoid zero value pixels (blue pixels). Repeat this process for all detectors in all tracks.
-
33.
Ensure all detectors and all tracks are turned on. Acquire images using the “Snap” button.
Expected outcomes
In Figures 5, 6, and 7, examples are given of possible confocal images obtained with this protocol. Cluster of Ki67+ cells showed in blue, allow a rapid identification of germinal center structures within the B cell follicle. A whole image overview of the spleen (Figure 5) can be acquired using a tiling mode and 10× objective, this will allow the identification of B cell follicles containing germinal centers structures.
Figure 5.

Confocal image of the spleen of ChAdOx1 nCoV-19 immunized 3-month-old mouse
A 10 μm splenic section was fixed and immunostained with anti-IgD (green), anti-CD3 (magenta), anti-Ki67 (blue) and anti-CD35 (white) antibodies. The image was tiled to reconstruct an image of the entire spleen. Boxed area focused on the splenic white pulp. The splenic B cell follicle (B) and T cells zone (TCZ) are indicated, germinal centers are indicated with white arrowheads. The scale bar represents 500 μm.
Figure 6.

Spleen of ChAdOx1 nCoV-19-immunized mice of the indicated ages
Confocal microscopy of spleen from ChAdOx1 nCoV-19 immunized mice where germinal center structures can be easily observed in adult mice (A), but mostly absent in the aged group (B). The scale bars represent 500 μm. IgD+ B cell follicle in green, CD3+ T cells in magenta, Ki67+ cells in blue and CD35+ follicular dendritic cells in white. Germinal centers are indicated with white arrowheads.
Figure 7.

Germinal Center structure after ChAdOx1 nCoV-19 immunization
Representative immunofluorescence of sections from spleens of 3-month-old (A) and 22-month-old (B) ChAdOx1 nCoV-19 immunized mice. Left side shows typical field of view of four channels and right side shows 2D composite image during a germinal center acquisition. The scale bars represent 50 μm. IgD+ B cell follicle in green, CD3+ T cells in magenta, Ki67+ cells in blue and CD35+ follicular dendritic cells in white.
The presence of germinal centers reactions in adult mice can be easily observed (Figure 6A), while it seems to be conspicuously absent in aged mice (Figure 6B).
Performing confocal imaging analysis helped us to define the localization of Follicular dendritic cells (CD35+) and T cells (CD4+) within the IgD+ B cell follicle in spleen sections (Figure 7). We observed how spleen from 3-month-old mice (Figure 7A) presented a higher number of germinal centers compare to aged mice (Figure 7B) upon vaccination.
Limitations
A major disadvantage of using frozen tissue sections compared to paraffin sections is the poor morphology of frozen tissues. Therefore, an appropriate fixation is crucial for the correct detection of the protein of interest. The fixation protocol described here improves tissue morphology. Nevertheless, we recommend to optimize the fixation step according to the tissue and cells of interest.
The confocal settings have been designed to acquire a 4-channel image in the shortest time possible (about 30 seconds per image) with minimal risk of bleed-through. Despite this, the Bi-directional scanning mode used to capture 20× images halves the capture timing, but may be prone to scan artefacts. Therefore, designing the staining panel needs to consider the confocal microscope configuration (North, 2006).
Another potential problem is the high triton X-100 concentration used during the permeabilization step, which may lead to loss or artifactual redistribution of the target (Jonkman et al., 2020). To prevent this, the duration of permeabilization step should be followed precisely, as this have been successfully validated in our laboratory.
Troubleshooting
Problem 1
Weak signal or lack of staining.
Potential solution
This problem could be due to different causes. Tissue over-fixation can mask epitopes and result in decreased immunoreactivity. To overcome this issue, try to reduce fixation times and/or optimize the fixation protocol.
Problem 2
Holes and distortion of tissue architecture.
Potential solution
Slow freezing can produce holes or a change in the tissue architecture due to ice crystal formation (Figure 8). Make sure the 2-propanol–dry ice cooling bath is cool enough and not in direct contact with the OCT containing the tissue sample.
Figure 8.

Representative images of tissues properly and improperly embedded
Confocal images of spleens section showing failed (A) and successful (B) rate of freezing while tissue embedding. Ice crystals formation produce the holes observed in (A). The scale bars represent 500 μm.
Problem 3
Tissue sticks or crumbles on the anti-roll plate (Figure 9).
Figure 9.

Representative sections of correctly and incorrectly cryosectioned tissues
Spleen tissue crumbles on anti-roll plate while cryosectioning (A) and a successful flat tissue section obtained (B).
Potential solution
This is a common problem that can be solved by repositioning and/or cooling down the anti-roll plate, removing rust and static electricity.
Another common issue that could cause tissues to crumble during cryosectioning is the presence of fat. To avoid this, make sure to trim the fat around the sample before embed the spleen in OCT.
Problem 4
The OCT frozen tissue block is difficult to section into flat sheets.
Potential solution
Tissue sections not properly flattened could be due to different causes, such as tissue block not cold enough, blunt blade, dirty or poorly positioned anti-roll plate. Under these circumstances, consider the following: lower cryostat temperature, align and clean anti-roll plate, use a different part of the blade cutting edge or replace it.
Problem 5
High background.
Potential solution
This problem can have different causes. It is important that sample remains covered in liquid during the staining procedure. Moreover, insufficient washing of tissue sections between steps can increase background levels.
Using the appropriated blocking agent and reducing antibodies concentration can also decrease background in tissue sections.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michelle Linterman (Michelle.Linterman@babraham.ac.uk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This project did not generate any new dataset or code.
Acknowledgments
This study was supported by funding from the Biotechnology and Biological Sciences Research Council (BBS/E/B/000C0427, BBS/E/B/000C0428, and the Campus Capability Core Grant to the Babraham Institute) and the Lister institute of Preventative Medicine through a Lister Institute Prize Fellowship to M.A.L. We acknowledge the contribution of the Babraham Institute Biological Support Unit staff, who performed in vivo treatments of our animals and took care of animal husbandry. We thank the staff of the Babraham Imaging Facility for technical assistance with imaging.
Author contributions
S.F.B. optimized and performed the experiments, created figures/videos, and wrote the manuscript. S.W. designed, wrote, and created the figure of confocal microscope settings. S.I. performed tissue dissections. M.L. supervised the project and revised and edited the manuscript. All authors read, edited, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100499.
Contributor Information
Sigrid Fra-Bido, Email: Sigrid.Fra-Bido@babraham.ac.uk.
Michelle A. Linterman, Email: Michelle.Linterman@babraham.ac.uk.
References
- Jonkman J., Brown C.M., Wright G.D., Anderson K.I., North A.J. Tutorial: guidance for quantitative confocal microscopy. Nat. Protoc. 2020;15:1585–1611. doi: 10.1038/s41596-020-0313-9. [DOI] [PubMed] [Google Scholar]
- Mclean I.W., Nakane P.K. Periodate-lysine-paraformaldehyde fixative a new fixative for immunoelectron microscopy. J. Histochem. Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- North A.J. Seeing is believing? A beginners' guide to practical pitfalls in image acquisition. J. Cell Biol. 2006;172:9–18. doi: 10.1083/jcb.200507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Cayetano A., Foster W.S., Innocentin S., Belij-Rammerstorfer S., Spencer A.J., Burton O.T., Fra-Bido S., Le Lee J., Thakur N., Conceicao C. A booster dose enhances immunogenicity of the COVID-19 vaccine candidate ChAdOx1 nCoV-19 in aged mice. Med (N Y) 2020 doi: 10.1016/j.medj.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderleyden I., Linterman M.A. Identifying follicular regulatory T cells by confocal microscopy. Methods Mol. Biol. 2017;1623:87–93. doi: 10.1007/978-1-4939-7095-7_7. [DOI] [PubMed] [Google Scholar]
- Vanderleyden I., Fra-Bido S., Innocentin S., Stebegg M., Okkenhaug H., Evans-Bailey N., Pierson W., Denton A.E., Linterman M.A. Follicular regulatory T cells can access the germinal center independently of CXCR5. Cell Rep. 2020;30:611–619. doi: 10.1016/j.celrep.2019.12.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This project did not generate any new dataset or code.