

Abstract
The transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) is widely recognized as a master regulator of the cellular stress response by facilitating the transcription of cytoprotective genes. As such, the Nrf2 pathway is critical in guarding the cell from the harmful effects of excessive reactive oxygen species/reactive nitrogen species (ROS/RNS) and in maintaining cellular redox balance. While excessive ROS/RNS are harmful to the cell, physiological levels of ROS/RNS play important roles in regulating numerous signaling pathways important for normal cellular function, including the synthesis of extracellular matrix (ECM). Recent advances have underscored the importance of ROS/RNS, and by extension, factors that influence redox-balance such as Nrf2, in regulating ECM production and deposition. In addition to reducing the oxidative burden in the cell, the discovery that Nrf2 can also directly target genes that regulate and form the ECM has cemented it as a multifaceted player in the regulation of ECM proteins, and provides new insight into its potential usefulness as a target for treating ECM-related pathologies.
Keywords: Nuclear factor-erythroid 2-related factor 2 (Nrf2), Matrisome, Extracellular matrix, Reactive oxygen species/reactive nitrogen species (ROS/RNS), Tissue repair, Senescence
Highlights
-
•
Reactive oxygen/nitrogen species regulate extracellular matrix.
-
•
Nrf2 can directly target extracellular matrix gene transcription.
-
•
Regulation of extracellular matrix by Nrf2 potentially impacts tissue repair/cancer.
Nuclear factor-erythroid 2-related factor 2 (Nrf2)
Since its initial discovery over 25 ago [1], nuclear factor-erythroid 2-related factor 2 (Nrf2) has become known as one of the most important factors in the cell that regulates cytoprotective responses to environmental stressors [2,3]. Nrf2 is a member of the cap'n'collar family of transcription factors whose major function is to facilitate the transcription of genes coding for antioxidant and detoxification enzymes [4]. In this way, the Nrf2 pathway serves as a principal mechanism through which the cell can respond to excessive accumulation of reactive oxygen species or reactive nitrogen species (ROS/RNS). Targeting the Nrf2 pathway is therefore seen as a promising strategy to combat a number of human pathologies where excessive ROS/RNS play a role, and several pharmaceuticals are being developed or already used in this regard [5].
The classical Nrf2 pathway has been extensively studied and its regulation and mechanisms of action have been reviewed previously in detail [2,3,6,7]. Nrf2 activity is primarily regulated at the protein level by its chief antagonist, Kelch-like ECH-associated protein 1 (Keap1) [8]. During homeostasis, Keap1 binds to Nrf2 in the cytoplasm, resulting in its ubiquitination and rapid proteasomal degradation. Electrophilic compounds that accumulate due to elevated environmental stress, or ROS/RNS themselves, react with cysteine residues on Keap1, rendering it ineffective and allowing newly synthetized Nrf2 to accumulate in the nucleus. Alternatively, other proteins such as p62, which accumulate when autophagic flux is reduced, may bind competitively to Keap1, and thereby promote nuclear accumulation of Nrf2 by weakening the Nrf2-Keap1 interaction [9,10]. Inside the nucleus, Nrf2 dimerizes with small MAF proteins and binds to specific regions of DNA called antioxidant response elements (AREs), resulting in transcription of its target genes [4,7]. In this way, the Nrf2 pathway can effectively combat accumulating toxic compounds as a result of excessive ROS/RNS and lessen the oxidative burden in the cell.
Restoring redox-balance by activating the Nrf2 pathway is central to many efforts at combating disease [5]. Numerous small molecules have been found to react with Keap1 similarly to endogenous electrophiles, and thereby activate Nrf2. Some of these compounds, such as dimethyl fumarate, are now approved for use in humans to treat multiple sclerosis and psoriasis, while several others are in clinical trials for the treatment of other chronic diseases or for cancer prevention [5]. Among the harmful effects of excessive ROS/RNS are their impact on ECM production and remodeling [11]. Dysregulation of normal ECM synthesis and/or remodeling are a hallmark feature of fibrotic diseases and can play a crucial role in cancer growth and metastasis [11,12].
It is worth noting that Nrf2, and ROS/RNS in their own right, affect inflammation by regulating gene expression or signaling in non-immune cells, or in immune cells themselves [13,14]. Inflammatory responses play crucial roles in chronic disease, including remodeling of the ECM via the action of proteases [15]. While Nrf2-mediated regulation of inflammation has been reviewed elsewhere [13,[16], [17], [18]], the remainder of this minireview will focus on mechanisms of Nrf2 in regulating the ECM in health and disease, either by its impact on ROS/RNS or by the recent discovery that Nrf2 can directly target and regulate ECM genes in certain cell types.
Regulation of ECM by ROS/RNS
ROS/RNS are often thought of as toxic and harmful molecules, as they cause tissue damage and contribute significantly to the pathogenesis of a number of chronic diseases [19]. While this is indeed the case for excessive ROS/RNS levels, lower levels of ROS/RNS play essential roles in physiological cellular processes and signaling [20]. Furthermore, transiently enhanced levels ROS/RNS help combat invading pathogens following injury [7,21]. As research on the role of ROS/RNS in health and disease continues to develop, the precise roles of ROS/RNS in regulating the ECM are becoming increasingly apparent. Modification of the ECM plays a fundamental role in the pathogenesis of various diseases, making ROS/RNS-mediated ECM modifications a subject of increasing relevance in the pursuit of improved therapies [22]. ROS/RNS can affect the ECM in multiple ways, including regulating fibroblast behavior and differentiation into myofibroblasts, ECM degradation and remodeling, facilitating post-translational modifications of ECM proteins and controlling the activity of ECM-regulating transcription factors (Fig. 1). Furthermore, the ECM can signal back to cells and regulate ROS/RNS production itself, allowing for a potential ECM-ROS/RNS feedback loop [23]. Redox-mediated regulation of various aspects of the ECM in health and disease has been explored and comprehensively reviewed in detail [11,12,24]. By regulating cellular redox-balance, Nrf2 may play an indirect role in ECM regulation by one or more of these mechanisms (briefly summarized below).
Fig. 1.

Actions of ROS/RNS on the ECM. ROS/RNS regulate the ECM in several ways. Degradation of glycosaminoglycans (GAGs) can be achieved by reacting directly with ROS/RNS or by the actions of ROS/RNS-regulated enzymes. ROS/RNS react with MMPs, thereby promoting their activation. Myofibroblast differentiation is regulated by ROS/RNS to facilitate intracellular TGF-β signaling, and can promote the release of sequestered TGF-β from the ECM (e.g. by reacting with LAP). Several transcription factors, including Nrf2, are regulated via the actions of ROS/RNS, and can promote the transcription of ECM genes. ROS/RNS also react with certain residues on ECM molecules to promote cross-linking.
Fibroblasts are the main producers of ECM in tissues, therefore factors regulating their behavior play major roles in the composition of ECM produced and deposited by these cells [25]. Upon activation, fibroblasts differentiate into myofibroblasts, which are responsible for the synthesis and deposition of ECM during healthy tissue repair and in a number of fibrotic diseases [26,27]. Several key factors are known to contribute to myofibroblast differentiation, including transforming growth factor-β (TGF-β), mechanical tension, and exposure to the extra domain A (EDA) form of fibronectin [26]. ROS, particularly those generated by NAD(P)H oxidases (Nox), are known to be crucial for TGF-β-mediated myofibroblast differentiation as shown, for example, in cardiac, pulmonary and dermal fibroblasts [[28], [29], [30]], although there may be exceptions to this in certain contexts [31]. Collectively however, these data and others [32] clearly demonstrate that ROS/RNS are crucial regulators of the myofibroblast phenotype.
Among the many functions of the ECM is its role as a reservoir for different growth factors and cytokines, which can either shield their activity or act synergistically to enhance their activity [33]. Targeted cleavage of ECM components can result in the release of sequestered growth factors, which are then free to signal to cells. For example, TGF-β is kept inactive by binding to latency-associated peptide (LAP) and stored within the ECM by linking to TGF-β binding protein. TGF-β remains in this inactive state until it is released by mechanical forces [[34], [35], [36]] or proteolytic cleavage [[37], [38], [39]]. ROS can also aid in this process by reacting directly with LAP, causing it to lose its ability to bind TGF-β, resulting in its eventual release and subsequent activation [[40], [41], [42], [43]]. Furthermore, ROS/RNS have been shown to indirectly promote TGF-β signaling by activating TGF-β liberating proteases [44,45], and by promoting its synthesis [[46], [47], [48], [49]].
Remodeling of the ECM is facilitated by proteases and is essential during embryonic development and tissue repair, but is pathological when the activity of certain proteases is excessive (e.g. in chronic wounds) or insufficient/dysregulated (e.g. in fibrosis) [50,51]. Matrix metalloproteinases (MMPs) are among the most prominent players when it comes to remodeling of the ECM, and their dysregulation is observed regularly in different chronic diseases featuring pathological ECM remodeling. ROS are capable of regulating the activation of MMPs, which are initially synthesized as inactive zymogens [52,53], by reacting with cystine residues bound to a zinc ion in the active site (referred to as the cystine switch) [45,54]. This reaction allows for the subsequent cleavage of the zymogen into the active enzyme, which can occur by autocatalytic cleavage or by a number of other proteases (including other MMPs) [53].
In addition to the regulation of MMP activity, ROS/RNS can react directly with ECM molecules, contributing to their degradation. One important example of this is ROS/RNS-mediated degradation of glycosaminoglycans (GAGs), polysaccharide chains that make up a large part of the ECM and exist independently (e.g. hyaluronan) or attached to proteins to form proteoglycans. GAGs play important roles in mediating cell signaling and are significant regulators of development and of multiple diseases, including cancer [[55], [56], [57], [58], [59], [60]]. Experiments performed more than three decades ago suggested that singlet oxygen can contribute to the degradation of hyaluronan [61]. ●NO2 and ●OH radicals resulting from the decomposition of ONOO− have also been shown to react with GAGs such as hyaluronan, which is degraded by ROS/RNS-mediated scission of the polymeric chains [[62], [63], [64]]. Numerous other reports have also confirmed the susceptibility of hyaluronan, as well as other GAGs such as chondroitin sulfate and heparan sulfate, to ROS/RNS-mediated degradation [[65], [66], [67], [68], [69], [70], [71], [72]]. In addition to GAGs, previous studies have demonstrated that superoxide can react with and fragment collagen, while other ROS/RNS-mediated modifications are capable of rendering collagen more susceptible to proteolysis [[73], [74], [75]].
Post-translational changes, such as ECM cross-linking, are known to require oxidative modifications. Cross-linking of collagen and elastin occurs enzymatically via lysyl oxidases, which form covalent bonds between lysyl and hydroxylysyl residues, but can also occur as a consequence of aging mediated by advanced glycation end-products [76]. The degree and type of cross-linking has important biological consequences such as alterations in the biomechanical strength of collagen in tissues and susceptibility to degradation by proteases [76]. ECM proteins, such as fibronectin, are also linked by ROS-mediated oxidation to form disulfide bridges, which contribute to the supramolecular assembly of different ECM components into large, insoluble structures. Multiple studies have demonstrated the importance of ROS, such as hydrogen peroxide (H2O2), and peroxidases in mediating ECM-cross-linking [31,[77], [78], [79], [80], [81], [82]]. This includes studies investigating ROS-mediated cross-linking in vivo, an effect that was suppressed with the administration of antioxidants or ROS-detoxifying enzymes such as catalase [83].
Finally, ROS/RNS are important regulators of transcription factor activity and can therefore influence the transcription of ECM genes and remodeling enzymes. In addition to TGF-β/SMAD signaling mentioned above, these include most other transcription factors such as, nuclear factor kappa B (NfkB), forkhead box, class O (Foxo), p53, activator protein-1 (AP-1), cAMP response-element binding protein (CREB), signal transducer and activator of transcription (STAT) and more [84,85]. H2O2, for example, regulates a number of cellular pathways either via increased transcription factor synthesis or modulation of transcription factor activity [84]. Nrf2 is one of the most well-known redox-regulated transcription factors which, in addition to being activated by electrophiles, can be activated directly by ROS/RNS through oxidation of Keap1 [86]. Several lines of evidence suggest that Nrf2 activation can impact the ECM, particularly in diseases like fibrosis [[87], [88], [89]]. Indeed, modulation of Nrf2 activity is thought to be a promising strategy for treatment of these ECM-regulated diseases due to its impact on intracellular ROS/RNS [5]. Furthermore, new advances suggest Nrf2 may, in addition, have a more direct role in regulating the ECM than once thought, extending its role in the body beyond the transcription of antioxidant genes.
Nrf2-mediated regulation of ECM
Regulation of intracellular redox-balance by Nrf2 has been reported to influence ECM deposition and remodeling in several cases. Experiments using Nrf2 knockout (KO) mice have shown that a deficiency in Nrf2 signaling results in greater inflammation and associated ECM degradation in mice exposed to UVB, likely due to the reduced expression of cytoprotective enzymes such as heme oxygenase 1 [90]. Other studies have used specific activators of Nrf2 to reduce the oxidative burden in the cell and reduce ECM production [91,92]. Multiple studies have observed that bleomycin-induced lung fibrosis is more severe in Nrf2 KO mice, and Nrf2 deficiency also promoted epithelial-mesenchymal transition (EMT) in vitro [87,[93], [94], [95]]. Interestingly, the antioxidant effects of Nrf2 have been suggested to specifically counteract the effects of Nox4 in mediating the myofibroblast phenotype and corresponding lung fibrosis [89]. Indeed, there is increasing evidence of a contribution of oxidative stress to tissue fibrosis, and links between Nrf2, TGF-β signaling and fibrosis have been the subject of several additional studies [88,96,97].
Regulation of ROS/RNS levels via the transcription of antioxidant genes is not the only mechanism by which Nrf2 can impact the ECM. Nrf2 is capable of directly regulating the expression of genes coding for ECM proteins, ECM-associated proteins and ECM remodeling enzymes, collectively referred to as the matrisome. This requires binding of Nrf2 to AREs in the promotor or enhancer regions of the respective target genes [4,7]. Efforts to uncover Nrf2 binding sites, and by extension, potential Nrf2 target genes, have typically involved chromatin immunoprecipitation followed by sequencing (ChIP-seq). To date, 21 ChIP-seq datasets can be found for human Nrf2 (NFE2L2) annotated on ChIP-Atlas (http://chip-atlas.org/ [98]) from four different cell types (lung epithelial adenocarcinoma cells [99], bronchial epithelial cells [100], aortic endothelial cells [101], lymphoblastoid cells [102,103]). Of all predicted Nrf2 binding sites within 1 kb of gene transcription start sites, 138 belong to genes encoding proteins that make up the human matrisome according to the list of matrisome genes generated by the Matrisome Project: http://matrisomeproject.mit.edu (32 core matrisome genes and 106 matrisome associated genes) [104]. Using the same methodology, Nrf2 binding is also observed near matrisome genes in mice according to 9 ChIP-seq datasets from 4 different cell types/tissues (myoblast cells [105], esophageal cells [106], hepatocytes [107], embryonic fibroblasts [108]). In these mouse experiments, Nrf2 binding can be observed in the regulatory region of 68 matrisome genes (11 core matrisome genes and 57 matrisome-affiliated genes). It is worth noting that Nrf2 binding sites detected in ChIP-seq experiments often vary considerably from cell type to cell type and may also depend on the nature of the Nrf2 activating agent used, supporting previous observations that certain genes are targets of Nrf2 only in specific cell types or under specific activation conditions [109]. Importantly, many of these Nrf2 binding sites remain to be validated by conventional ChIP followed by real-time PCR. Most importantly, the degree to which gene expression is changed as a result of Nrf2 binding, and the biological significance of this remains to be shown for most potential ECM targets identified in ChIP-seq experiments. Still, these data point to a potentially underappreciated role for matrisome gene transcription by Nrf2 in multiple cell types.
In our recent work, Nrf2-mediated control of the matrisome has been investigated in greater detail, and multiple direct matrisome gene targets have now been confirmed [110]. This has been done using transgenic mice that express a constitutively active form of Nrf2 (caNrf2) in mesenchymal cells, including skin fibroblasts [110]. The caNrf2 transgene codes for a mutant form of Nrf2 that lacks the Keap1 binding domain, resulting in its constitutive activation and transcription of target genes [111,112]. Importantly, the level of activation is comparable to the level achieved upon treatment of wild-type fibroblasts with Nrf2-activating compounds. Transcriptomic profiling of skin fibroblasts expressing caNrf2 showed substantial differences in the expression of matrisome genes when compared to control cells [110]. Nrf2 activation in these fibroblasts led to a significant reduction in the expression of genes coding for the major skin collagens (Col1a2, Col3a1) and an upregulation of genes coding mainly for glycoproteins and other ECM-affiliated proteins [110]. Mice with caNrf2 in fibroblasts also had less collagen in the skin in vivo, and reduced collagen deposition following wound healing, pointing to a potential role for Nrf2 signaling in the prevention of post-wound scarring [110].
To investigate the effect of caNrf2 on fibroblast-derived ECM at the protein level, proteomic analysis of ECM deposited by fibroblasts with caNrf2 was performed [110]. Similar to the differential gene expression in these cells, there was a marked difference in the type of ECM deposited. Importantly, the major ECM proteins in the skin, collagen I and collagen III were both reduced at the protein level as well [110]. Similar to the gene expression profile, among the upregulated ECM proteins were a number of glycoproteins and ECM-affiliated proteins. It is noteworthy that, despite numerous similarities between the RNA and protein data, a number of differentially regulated ECM genes did not show differences at the protein level (and vice versa), suggesting post-transcriptional and post-translational regulatory mechanisms are involved in their synthesis and ultimate incorporation into the ECM [110].
Bioinformatics analysis revealed that several of the matrisome genes regulated by caNrf2 in fibroblasts contain ARE sequences in their promoter or enhancer regions, and therefore, are potential direct Nrf2 targets [110]. ChIP followed by real-time PCR showed that Nrf2 indeed bound to AREs in the regulatory region of multiple genes that are overexpressed in caNrf2 fibroblasts [110]. These included the genes encoding the heparan sulfate proteoglycan glypican-1 (Gpc1), the glycoprotein angiopoietin-like protein 2 (Angptl2) and the ECM-affiliated protein plasminogen activator inhibitor-1 (PAI-1, Serpine1). Of these, PAI-1 was particularly interesting, since it has important functions that impact the ECM including during fibrosis, and is also a key regulator of fibroblast senescence [113,114]. Indeed, caNrf2 fibroblasts or wild-type fibroblasts treated with Nrf2 activating compounds, despite displaying reduced intracellular ROS and reduced evidence of DNA damage, undergo senescence much earlier than control fibroblasts in culture, or during wounding in vivo [110]. caNrf2 ECM, featuring increased PAI-1, was sufficient to accelerate the onset of senescence in wild-type fibroblasts compared to these same cells grown on ECM from control fibroblasts, an effect that was abolished using a PAI-1 inhibitor [110]. These data demonstrate the impact Nrf2 activation can have on the production and deposition of ECM, as well as the impact of the ECM in controlling important cell behaviors such as senescence. Interestingly, PAI-1 was previously shown to regulate ECM in the airways in a mouse model of asthma by controlling MMP-9 expression and collagen deposition, suggesting Nrf2-mediated regulation of PAI-1 may provide additional feedback signaling on ECM synthesis and/or remodeling by MMPs [115].
ECM-mediated control of senescence in fibroblasts was shown to have important consequences in vivo [110]. During wound healing, mice with caNrf2 in fibroblasts not only showed reduced fibroblast proliferation and elevated fibroblast senescence in the granulation tissue, but also increased keratinocyte proliferation, resulting in significantly faster wound re-epithelialization and closure [110]. This is likely a result of senescent fibroblasts producing a senescence-associated secretome, which was also capable of increasing keratinocyte proliferation in vitro [110,116]. The senescence-associated secretome, however, was also detrimental in the context of cancer [110]. Indeed, caNrf2 fibroblasts and their associated secretome resembled that of cancer associated fibroblasts (CAFs), and promoted tumor growth in a xenograft model of cutaneous squamous cell carcinoma [110]. As mentioned above, altered ECM regulation by Nrf2 also resulted in reduced collagen deposition and therefore reduced scarring in caNrf2 mouse wounds [110]. Taken together, the impact of Nrf2 on the ECM is clear and Nrf2-mediated regulation of ECM components may therefore have multiple pathophysiological consequences (Fig. 2).
Fig. 2.
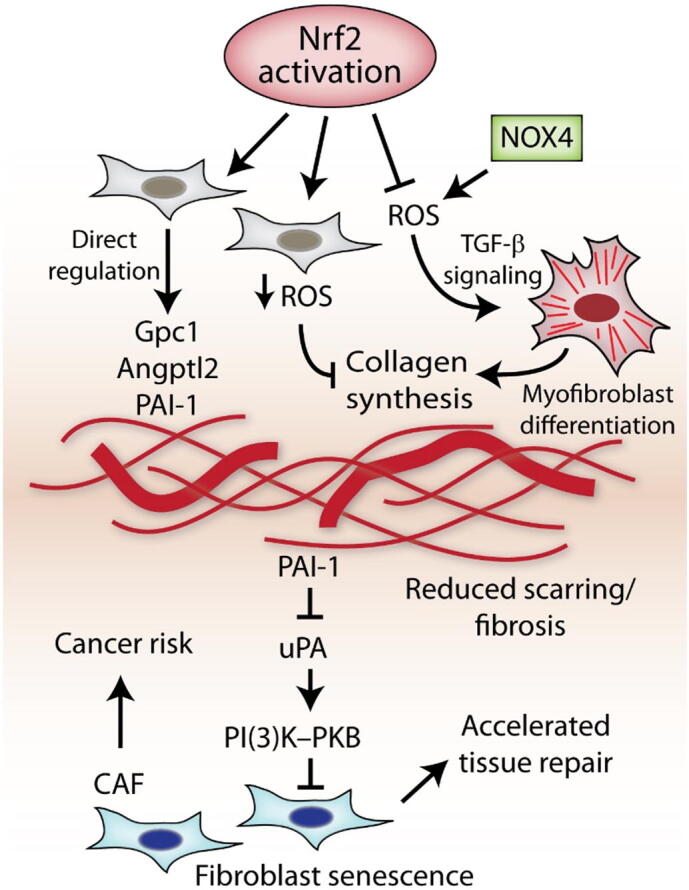
Potential consequences of Nrf2-mediated ECM alterations. Activation of Nrf2 in fibroblasts results in numerous changes in the production and deposition of ECM. The modified ECM typically features reduced collagen and increased levels of certain glycoproteins and other ECM-affiliated proteins such as PAI-1. Nrf2-mediated regulation of ECM can take place by directly targeting specific ECM genes, or indirectly by modulating ROS levels and ROS-dependent signaling pathways (e.g. TGF-b signaling) which can influence myofibroblast differentiation and ECM synthesis. This Nrf2-ECM can impact interacting cells and associated tissues, which may result in the development of a CAF phenotype and/or fibroblast senescence via PAI-1. Together, this may accelerate epithelial cell proliferation, aiding tissue repair and reducing scarring, but may also contribute to increased tumor growth. Abbreviations: PAI-1 (plasminogen activator inhibitor-1): Gcp1 (glypican-1); Angptl2 (angiopoietin-like protein 2).
It is intriguing to speculate that the effect of Nrf2 on the matrisome contributes to its cytoprotective activity. Indeed, ECM components can in some cases provide cytoprotective effects, including the induction of Nrf2 signaling [[117], [118], [119]]. Cellular senescence has long been thought of as a protective mechanism of sorts, eliminating the ability of a cell to further proliferate as a way of preventing malignant transformation and cancer progression [120]. By contrast, senescence of cells in the tumor stroma is frequently observed to have tumor promoting effects and is a common feature of CAFs [110,[121], [122], [123], [124]]. In the wound environment, however, limited induction of fibroblast senescence is beneficial, since this resulted in the production of growth factors that promote keratinocyte proliferation [110,125]. Some of the factors, which are released by caNrf2 fibroblasts (e.g. fibroblast growth factor 7) also protect keratinocytes from the harmful effects of ROS [126]. The influence of the ECM on senescence is significant, and has been documented in multiple reports. For example, in addition to senescence being regulated by the caNrf2 matrisome [110], ECM from old fibroblasts can promote senescence in young fibroblasts, while the reverse also holds true, with ECM from young cells preventing senescence in old fibroblasts [127]. The matricellular protein CCN1 was shown to contribute to reduced ECM deposition by inducing senescence in fibroblasts during wound healing [128]. Interestingly, the relationship between Nrf2 and cellular senescence has traditionally been an inverse one, where reduced Nrf2 activity, and the accompanied elevation in ROS, correlates with increased senescence [129,130]. Observations to the contrary [110] should lead to a rethinking of how Nrf2 contributes to senescence and cellular responses more broadly. It is possible that sustained, constitutive activation of Nrf2 has evolved as a signal of unresolved stress, triggering responses (such as deposition of PAI-1) as a way of inducing the senescence response. Undoubtedly, like much related to Nrf2 signaling, the effect of Nrf2 on senescence likely depends on a number of factors such as the level and/or duration of activation, cell type involved as well as co-factors and other contextual considerations, all of which will require additional studies to properly understand the potential role they may play.
Summary
Increasing evidence is pointing toward Nrf2 as having a multifaceted role in the body that can affect numerous cell and tissue processes beyond cytoprotection [6]. The use of Nrf2 activating compounds as therapeutic strategies to combat numerous chronic diseases or to prevent cancer development has been met with some success [5], however accumulating evidence of a potentially “dark side of Nrf2”, particularly with respect to cancer, should remain an important consideration [110,131,132]. As the ECM can contribute to the pathogenesis of almost all diseases [22], it is perhaps not surprising that recent studies uncovered important roles for Nrf2 in regulating the matrisome. While Nrf2-mediated control of redox status is surely a key mechanism through which Nrf2 can facilitate such regulation, a better understanding of what matrisome genes Nrf2 directly targets, and in what circumstances or cell types, will be of great interest. New discoveries into the roles of specific components of the matrisome regulated by Nrf2, and how they mediate these different cellular and tissue responses, have the potential to change our view on the role of the matrisome and its relationship to cytoprotection in both health and disease.
Abbreviations
- Nrf2
nuclear factor-erythroid 2-related factor 2
- Keap1
kelch-like ECH-associated protein 1
- ARE
antioxidant response element
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- ECM
extracellular matrix
- TGF-β
transforming growth factor beta
- EDA
extra domain A
- Nox
NAD(P)H oxidases
- LAP
latency-associated peptide
- MMP
matrix metalloproteinase
- GAG
glycosaminoglycan
- KO
knockout
- ChIP
chromatin immunoprecipitation
- PAI-1
plasminogen activator inhibitor-1
Declaration of competing interest
The author declares no conflicts of interest.
Acknowledgments
Acknowledgements
Research on NRF2 in our laboratory was/is supported by grants from the Swiss National Science Foundation (grant 31003A_169204 and 31003B-189364 to Sabine Werner), and a Banting postdoctoral fellowship (to Paul Hiebert) and an ETH Career Seed Grant (No. SEED-03 19-1 to Paul Hiebert).
References
- 1.Moi P., Chan K., Asunis I., Cao A., Kan Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U. S. A. 1994;91(21):9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taguchi K., Motohashi H., Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16(2):123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 3.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sykiotis G.P., Bohmann D. Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal. 2010;3(112) doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuadrado A., Rojo A.I., Wells G., Hayes J.D., Cousin S.P., Rumsey W.L., Attucks O.C., Franklin S., Levonen A.L., Kensler T.W., Dinkova-Kostova A.T. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019;18(4):295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 6.Hiebert P., Werner S. Regulation of wound healing by the NRF2 transcription factor-more than cytoprotection. Int. J. Mol. Sci. 2019;20(16) doi: 10.3390/ijms20163856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T., Yamamoto M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017;292(41):16817–16824. doi: 10.1074/jbc.R117.800169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 10.Lau A., Wang X.J., Zhao F., Villeneuve N.F., Wu T., Jiang T., Sun Z., White E., Zhang D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010;30(13):3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosche J., Meissner J., Eble J.A. More than a syllable in fib-ROS-is: the role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol. Asp. Med. 2018;63:30–46. doi: 10.1016/j.mam.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Nikitovic D., Corsini E., Kouretas D., Tsatsakis A., Tzanakakis G. ROS-major mediators of extracellular matrix remodeling during tumor progression. Food Chem. Toxicol. 2013;61:178–186. doi: 10.1016/j.fct.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed S.M., Luo L., Namani A., Wang X.J., Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. basis Dis. 2017;1863(2):585–597. doi: 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Blaser H., Dostert C., Mak T.W., Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26(4):249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Parks W.C., Wilson C.L., Lopez-Boado Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 16.Mohan S., Gupta D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018;108:1866–1878. doi: 10.1016/j.biopha.2018.10.019. [DOI] [PubMed] [Google Scholar]
- 17.Ruiz S., Pergola P.E., Zager R.A., Vaziri N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83(6):1029–1041. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hennig P., Garstkiewicz M., Grossi S., Di Filippo M., French L.E., Beer H.D. The crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018;19(2) doi: 10.3390/ijms19020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z., Ren Z., Zhang J., Chuang C.C., Kandaswamy E., Zhou T., Zuo L. Role of ROS and nutritional antioxidants in human diseases. Front. Physiol. 2018;9:477. doi: 10.3389/fphys.2018.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawagishi H., Finkel T. Unraveling the truth about antioxidants: ROS and disease: finding the right balance. Nat. Med. 2014;20(7):711–713. doi: 10.1038/nm.3625. [DOI] [PubMed] [Google Scholar]
- 21.Schafer M., Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008;58(2):165–171. doi: 10.1016/j.phrs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Iozzo R.V., Gubbiotti M.A. Extracellular matrix: the driving force of mammalian diseases. Matrix Biol. 2018;71-72:1–9. doi: 10.1016/j.matbio.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H., Du W., Brekken R.A. Extracellular matrix induction of intracellular reactive oxygen species. Antioxid. Redox Signal. 2017;27(12):774–784. doi: 10.1089/ars.2017.7305. [DOI] [PubMed] [Google Scholar]
- 24.Eble J.A., de Rezende F.F. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid. Redox Signal. 2014;20(13):1977–1993. doi: 10.1089/ars.2013.5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLeon-Pennell K.Y., Barker T.H., Lindsey M.L. Fibroblasts: the arbiters of extracellular matrix remodeling. Matrix Biol. 2020;91-92:1–7. doi: 10.1016/j.matbio.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz B., Phan S.H., Thannickal V.J., Galli A., Bochaton-Piallat M.L., Gabbiani G. The myofibroblast: one function, multiple origins. Am. J. Pathol. 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 28.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S., Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 29.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C., Pennathur S., Martinez F.J., Thannickal V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15(9):1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alili L., Sack M., Puschmann K., Brenneisen P. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci. Rep. 2014;34(1) doi: 10.1042/BSR20130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levigne D., Modarressi A., Krause K.H., Pittet-Cuenod B. NADPH oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic. Biol. Med. 2016;96:374–384. doi: 10.1016/j.freeradbiomed.2016.04.194. [DOI] [PubMed] [Google Scholar]
- 32.Sampson N., Berger P., Zenzmaier C. Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease. Biomed. Res. Int. 2014;2014:131737. doi: 10.1155/2014/131737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz G.S., Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17(2):153–162. doi: 10.1111/j.1524-475X.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 34.Maeda T., Sakabe T., Sunaga A., Sakai K., Rivera A.L., Keene D.R., Sasaki T., Stavnezer E., Iannotti J., Schweitzer R., Ilic D., Baskaran H., Sakai T. Conversion of mechanical force into TGF-beta-mediated biochemical signals. Curr. Biol. 2011;21(11):933–941. doi: 10.1016/j.cub.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wipff P.J., Rifkin D.B., Meister J.J., Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007;179(6):1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buscemi L., Ramonet D., Klingberg F., Formey A., Smith-Clerc J., Meister J.J., Hinz B. The single-molecule mechanics of the latent TGF-beta1 complex. Curr. Biol. 2011;21(24):2046–2054. doi: 10.1016/j.cub.2011.11.037. [DOI] [PubMed] [Google Scholar]
- 37.Maeda S., Dean D.D., Gay I., Schwartz Z., Boyan B.D. Activation of latent transforming growth factor beta1 by stromelysin 1 in extracts of growth plate chondrocyte-derived matrix vesicles. J. Bone Miner. Res. 2001;16(7):1281–1290. doi: 10.1359/jbmr.2001.16.7.1281. [DOI] [PubMed] [Google Scholar]
- 38.Lyons R.M., Keski-Oja J., Moses H.L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 1988;106(5):1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abe M., Oda N., Sato Y. Cell-associated activation of latent transforming growth factor-beta by calpain. J. Cell. Physiol. 1998;174(2):186–193. doi: 10.1002/(SICI)1097-4652(199802)174:2<186::AID-JCP6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Jobling M.F., Mott J.D., Finnegan M.T., Jurukovski V., Erickson A.C., Walian P.J., Taylor S.E., Ledbetter S., Lawrence C.M., Rifkin D.B., Barcellos-Hoff M.H. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006;166(6):839–848. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 41.Pociask D.A., Sime P.J., Brody A.R. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab. Investig. 2004;84(8):1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 42.Barcellos-Hoff M.H., Dix T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996;10(9):1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 43.Vodovotz Y., Chesler L., Chong H., Kim S.J., Simpson J.T., DeGraff W., Cox G.W., Roberts A.B., Wink D.A., Barcellos-Hoff M.H. Regulation of transforming growth factor beta1 by nitric oxide. Cancer Res. 1999;59(9):2142–2149. [PubMed] [Google Scholar]
- 44.Yu Q., Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–176. [PMC free article] [PubMed] [Google Scholar]
- 45.Van Wart H.E., Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U. S. A. 1990;87(14):5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaffer O.A., Carter A.B., Sanders P.N., Dibbern M.E., Winters C.J., Murthy S., Ryan A.J., Rokita A.G., Prasad A.M., Zabner J., Kline J.N., Grumbach I.M., Anderson M.E. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 2015;52(1):106–115. doi: 10.1165/rcmb.2013-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shvedova A.A., Kisin E.R., Murray A.R., Kommineni C., Castranova V., Fadeel B., Kagan V.E. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol. Appl. Pharmacol. 2008;231(2):235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 48.Montorfano I., Becerra A., Cerro R., Echeverria C., Saez E., Morales M.G., Fernandez R., Cabello-Verrugio C., Simon F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Lab. Investig. 2014;94(10):1068–1082. doi: 10.1038/labinvest.2014.100. [DOI] [PubMed] [Google Scholar]
- 49.Bellocq A., Azoulay E., Marullo S., Flahault A., Fouqueray B., Philippe C., Cadranel J., Baud L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am. J. Respir. Cell Mol. Biol. 1999;21(1):128–136. doi: 10.1165/ajrcmb.21.1.3379. [DOI] [PubMed] [Google Scholar]
- 50.McCarty S.M., Percival S.L. Proteases and delayed wound healing. Adv Wound Care (New Rochelle) 2013;2(8):438–447. doi: 10.1089/wound.2012.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Menou A., Duitman J., Crestani B. The impaired proteases and anti-proteases balance in idiopathic pulmonary fibrosis. Matrix Biol. 2018;68-69:382–403. doi: 10.1016/j.matbio.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Hibbs M.S., Hasty K.A., Seyer J.M., Kang A.H., Mainardi C.L. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J. Biol. Chem. 1985;260(4):2493–2500. [PubMed] [Google Scholar]
- 53.Ra H.J., Parks W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26(8):587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Springman E.B., Angleton E.L., Birkedal-Hansen H., Van Wart H.E. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc. Natl. Acad. Sci. U. S. A. 1990;87(1):364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wight T.N. A role for proteoglycans in vascular disease. Matrix Biol. 2018;71-72:396–420. doi: 10.1016/j.matbio.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caon I., Bartolini B., Parnigoni A., Carava E., Moretto P., Viola M., Karousou E., Vigetti D., Passi A. Revisiting the hallmarks of cancer: the role of hyaluronan. Semin. Cancer Biol. 2020;62:9–19. doi: 10.1016/j.semcancer.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Pan J., Qian Y., Zhou X., Lu H., Ramacciotti E., Zhang L. Chemically oversulfated glycosaminoglycans are potent modulators of contact system activation and different cell signaling pathways. J. Biol. Chem. 2010;285(30):22966–22975. doi: 10.1074/jbc.M109.063735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin X., Perrimon N. Role of heparan sulfate proteoglycans in cell-cell signaling in Drosophila. Matrix Biol. 2000;19(4):303–307. doi: 10.1016/s0945-053x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 59.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131(24):6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 60.Garantziotis S., Savani R.C. Hyaluronan biology: a complex balancing act of structure, function, location and context. Matrix Biol. 2019;78-79:1–10. doi: 10.1016/j.matbio.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andley U.P., Chakrabarti B. Role of singlet oxygen in the degradation of hyaluronic acid. Biochem. Biophys. Res. Commun. 1983;115(3):894–901. doi: 10.1016/s0006-291x(83)80019-9. [DOI] [PubMed] [Google Scholar]
- 62.Hassan M.S., Mileva M.M., Dweck H.S., Rosenfeld L. Nitric oxide products degrade chondroitin sulfates. Nitric Oxide. 1998;2(5):360–365. doi: 10.1006/niox.1998.0198. [DOI] [PubMed] [Google Scholar]
- 63.Li M., Rosenfeld L., Vilar R.E., Cowman M.K. Degradation of hyaluronan by peroxynitrite. Arch. Biochem. Biophys. 1997;341(2):245–250. doi: 10.1006/abbi.1997.9970. [DOI] [PubMed] [Google Scholar]
- 64.Vilar R.E., Ghael D., Li M., Bhagat D.D., Arrigo L.M., Cowman M.K., Dweck H.S., Rosenfeld L. Nitric oxide degradation of heparin and heparan sulphate. Biochem. J. 1997;324(Pt 2):473–479. doi: 10.1042/bj3240473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghael D., Mileva M., Dweck H.S., Rosenfeld L. The nitric oxide donor S-nitroso-N-acetyl-D, L-penicillamine degrades heparan sulfate and heparin. Biochem Mol Biol Int. 1997;43(1):183–188. doi: 10.1080/15216549700203951. [DOI] [PubMed] [Google Scholar]
- 66.Agren U.M., Tammi R.H., Tammi M.I. Reactive oxygen species contribute to epidermal hyaluronan catabolism in human skin organ culture. Free Radic. Biol. Med. 1997;23(7):996–1001. doi: 10.1016/s0891-5849(97)00098-1. [DOI] [PubMed] [Google Scholar]
- 67.Rees M.D., Hawkins C.L., Davies M.J. Hypochlorite and superoxide radicals can act synergistically to induce fragmentation of hyaluronan and chondroitin sulphates. Biochem. J. 2004;381(Pt 1):175–184. doi: 10.1042/BJ20040148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rees M.D., Pattison D.I., Davies M.J. Oxidation of heparan sulphate by hypochlorite: role of N-chloro derivatives and dichloramine-dependent fragmentation. Biochem. J. 2005;391(Pt 1):125–134. doi: 10.1042/BJ20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawkins C.L., Davies M.J. Degradation of hyaluronic acid, poly- and monosaccharides, and model compounds by hypochlorite: evidence for radical intermediates and fragmentation. Free Radic. Biol. Med. 1998;24(9):1396–1410. doi: 10.1016/s0891-5849(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 70.Al-Assaf S., Navaratnam S., Parsons B.J., Phillips G.O. Chain scission of hyaluronan by peroxynitrite. Arch. Biochem. Biophys. 2003;411(1):73–82. doi: 10.1016/s0003-9861(02)00724-5. [DOI] [PubMed] [Google Scholar]
- 71.Moseley R., Waddington R., Evans P., Halliwell B., Embery G. The chemical modification of glycosaminoglycan structure by oxygen-derived species in vitro. Biochim. Biophys. Acta. 1995;1244(2–3):245–252. doi: 10.1016/0304-4165(95)00010-9. [DOI] [PubMed] [Google Scholar]
- 72.Fuchs B., Schiller J. Glycosaminoglycan degradation by selected reactive oxygen species. Antioxid. Redox Signal. 2014;21(7):1044–1062. doi: 10.1089/ars.2013.5634. [DOI] [PubMed] [Google Scholar]
- 73.Henrotin Y., Deberg M., Mathy-Hartert M., Deby-Dupont G. Biochemical biomarkers of oxidative collagen damage. Adv. Clin. Chem. 2009;49:31–55. doi: 10.1016/s0065-2423(09)49002-4. [DOI] [PubMed] [Google Scholar]
- 74.Monboisse J.C., Braquet P., Randoux A., Borel J.P. Non-enzymatic degradation of acid-soluble calf skin collagen by superoxide ion: protective effect of flavonoids. Biochem. Pharmacol. 1983;32(1):53–58. doi: 10.1016/0006-2952(83)90651-2. [DOI] [PubMed] [Google Scholar]
- 75.Monboisse J.C., Gardes-Albert M., Randoux A., Borel J.P., Ferradini C. Collagen degradation by superoxide anion in pulse and gamma radiolysis. Biochim. Biophys. Acta. 1988;965(1):29–35. doi: 10.1016/0304-4165(88)90147-x. [DOI] [PubMed] [Google Scholar]
- 76.Robins S.P. Biochemistry and functional significance of collagen cross-linking. Biochem. Soc. Trans. 2007;35(Pt 5):849–852. doi: 10.1042/BST0350849. [DOI] [PubMed] [Google Scholar]
- 77.Edens W.A., Sharling L., Cheng G., Shapira R., Kinkade J.M., Lee T., Edens H.A., Tang X., Sullards C., Flaherty D.B., Benian G.M., Lambeth J.D. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 2001;154(4):879–891. doi: 10.1083/jcb.200103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Larios J.M., Budhiraja R., Fanburg B.L., Thannickal V.J. Oxidative protein cross-linking reactions involving L-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J. Biol. Chem. 2001;276(20):17437–17441. doi: 10.1074/jbc.M100426200. [DOI] [PubMed] [Google Scholar]
- 79.Lardinois O.M., Medzihradszky K.F., Ortiz de Montellano P.R. Spin trapping and protein cross-linking of the lactoperoxidase protein radical. J. Biol. Chem. 1999;274(50):35441–35448. doi: 10.1074/jbc.274.50.35441. [DOI] [PubMed] [Google Scholar]
- 80.Lazar E., Peterfi Z., Sirokmany G., Kovacs H.A., Klement E., Medzihradszky K.F., Geiszt M. Structure-function analysis of peroxidasin provides insight into the mechanism of collagen IV crosslinking. Free Radic. Biol. Med. 2015;83:273–282. doi: 10.1016/j.freeradbiomed.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 81.Cheng G., Li H., Cao Z., Qiu X., McCormick S., Thannickal V.J., Nauseef W.M. Vascular peroxidase-1 is rapidly secreted, circulates in plasma, and supports dityrosine cross-linking reactions. Free Radic. Biol. Med. 2011;51(7):1445–1453. doi: 10.1016/j.freeradbiomed.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clark R.A., Szot S., Williams M.A., Kagan H.M. Oxidation of lysine side-chains of elastin by the myeloperoxidase system and by stimulated human neutrophils. Biochem. Biophys. Res. Commun. 1986;135(2):451–457. doi: 10.1016/0006-291x(86)90015-x. [DOI] [PubMed] [Google Scholar]
- 83.Elgawish A., Glomb M., Friedlander M., Monnier V.M. Involvement of hydrogen peroxide in collagen cross-linking by high glucose in vitro and in vivo. J. Biol. Chem. 1996;271(22):12964–12971. doi: 10.1074/jbc.271.22.12964. [DOI] [PubMed] [Google Scholar]
- 84.Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kohlgruber S., Upadhye A., Dyballa-Rukes N., McNamara C.A., Altschmied J. Regulation of transcription factors by reactive oxygen species and nitric oxide in vascular physiology and pathology. Antioxid. Redox Signal. 2017;26(13):679–699. doi: 10.1089/ars.2016.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kobayashi M., Li L., Iwamoto N., Nakajima-Takagi Y., Kaneko H., Nakayama Y., Eguchi M., Wada Y., Kumagai Y., Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009;29(2):493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cho H.Y., Reddy S.P., Yamamoto M., Kleeberger S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18(11):1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 88.Walters D.M., Cho H.Y., Kleeberger S.R. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid. Redox Signal. 2008;10(2):321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 89.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y.Y., Thannickal V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6(231) doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saw C.L., Yang A.Y., Huang M.T., Liu Y., Lee J.H., Khor T.O., Su Z.Y., Shu L., Lu Y., Conney A.H., Kong A.N. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci. 2014;4:39. doi: 10.1186/2045-3701-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiao S., Liu R., Lv C., Miao Y., Yue M., Tao Y., Wei Z., Xia Y., Dai Y. Bergenin impedes the generation of extracellular matrix in glomerular mesangial cells and ameliorates diabetic nephropathy in mice by inhibiting oxidative stress via the mTOR/beta-TrcP/Nrf2 pathway. Free Radic. Biol. Med. 2019;145:118–135. doi: 10.1016/j.freeradbiomed.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 92.Yao H., Zhang N., Zhang W., Li J., Hua H., Li Y. Discovery of polypodiside as a Keap1-dependent Nrf2 activator attenuating oxidative stress and accumulation of extracellular matrix in glomerular mesangial cells under high glucose. Bioorg. Med. Chem. 2020;28(24):115833. doi: 10.1016/j.bmc.2020.115833. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Z., Qu J., Zheng C., Zhang P., Zhou W., Cui W., Mo X., Li L., Xu L., Gao J. Nrf2 antioxidant pathway suppresses numb-mediated epithelial-mesenchymal transition during pulmonary fibrosis. Cell Death Dis. 2018;9(2):83. doi: 10.1038/s41419-017-0198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kikuchi N., Ishii Y., Morishima Y., Yageta Y., Haraguchi N., Itoh K., Yamamoto M., Hizawa N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010;11:31. doi: 10.1186/1465-9921-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou W., Mo X., Cui W., Zhang Z., Li D., Li L., Xu L., Yao H., Gao J. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci. Rep. 2016;6:38646. doi: 10.1038/srep38646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Latella G. Redox imbalance in intestinal fibrosis: beware of the TGFbeta-1, ROS, and Nrf2 connection. Dig. Dis. Sci. 2018;63(2):312–320. doi: 10.1007/s10620-017-4887-1. [DOI] [PubMed] [Google Scholar]
- 97.Gong Y., Yang Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-beta1/Smad3 pathway: a promising therapeutic strategy for hepatic fibrosis - a review. Life Sci. 2020;256:117909. doi: 10.1016/j.lfs.2020.117909. [DOI] [PubMed] [Google Scholar]
- 98.Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C. ChIP-atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19(12) doi: 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Namani A., Liu K., Wang S., Zhou X., Liao Y., Wang H., Wang X.J., Tang X. Genome-wide global identification of NRF2 binding sites in A549 non-small cell lung cancer cells by ChIP-Seq reveals NRF2 regulation of genes involved in focal adhesion pathways. Aging (Albany NY) 2019;11(24):12600–12623. doi: 10.18632/aging.102590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X., Campbell M.R., Lacher S.E., Cho H.Y., Wan M., Crowl C.L., Chorley B.N., Bond G.L., Kleeberger S.R., Slattery M., Bell D.A. A polymorphic antioxidant response element links NRF2/sMAF binding to enhanced MAPT expression and reduced risk of Parkinsonian disorders. Cell Rep. 2016;15(4):830–842. doi: 10.1016/j.celrep.2016.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hogan N.T., Whalen M.B., Stolze L.K., Hadeli N.K., Lam M.T., Springstead J.R., Glass C.K., Romanoski C.E. Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. Elife. 2017;6 doi: 10.7554/eLife.22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chorley B.N., Campbell M.R., Wang X., Karaca M., Sambandan D., Bangura F., Xue P., Pi J., Kleeberger S.R., Bell D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40(15):7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levings D.C., Wang X., Kohlhase D., Bell D.A., Slattery M. A distinct class of antioxidant response elements is consistently activated in tumors with NRF2 mutations. Redox Biol. 2018;19:235–249. doi: 10.1016/j.redox.2018.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A., Hynes R.O. The extracellular matrix: tools and insights for the “omics” era. Matrix Biol. 2016;49:10–24. doi: 10.1016/j.matbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Uruno A., Yagishita Y., Katsuoka F., Kitajima Y., Nunomiya A., Nagatomi R., Pi J., Biswal S.S., Yamamoto M. Nrf2-mediated regulation of skeletal muscle glycogen metabolism. Mol. Cell. Biol. 2016;36(11):1655–1672. doi: 10.1128/MCB.01095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fu J., Xiong Z., Huang C., Li J., Yang W., Han Y., Paiboonrungruan C., Major M.B., Chen K.N., Kang X., Chen X. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019;294(1):327–340. doi: 10.1074/jbc.RA118.005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nault R., Doskey C.M., Fader K.A., Rockwell C.E., Zacharewski T. Comparison of hepatic NRF2 and aryl hydrocarbon receptor binding in 2,3,7,8-Tetrachlorodibenzo-p-dioxin-treated mice demonstrates NRF2-independent PKM2 induction. Mol. Pharmacol. 2018;94(2):876–884. doi: 10.1124/mol.118.112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Malhotra D., Portales-Casamar E., Singh A., Srivastava S., Arenillas D., Happel C., Shyr C., Wakabayashi N., Kensler T.W., Wasserman W.W., Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38(17):5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018;29(17):1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hiebert P., Wietecha M.S., Cangkrama M., Haertel E., Mavrogonatou E., Stumpe M., Steenbock H., Grossi S., Beer H.D., Angel P., Brinckmann J., Kletsas D., Dengjel J., Werner S. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev Cell. 2018;46(2):145–161. doi: 10.1016/j.devcel.2018.06.012. (e10) [DOI] [PubMed] [Google Scholar]
- 111.Schafer M., Farwanah H., Willrodt A.H., Huebner A.J., Sandhoff K., Roop D., Hohl D., Bloch W., Werner S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol Med. 2012;4(5):364–379. doi: 10.1002/emmm.201200219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schafer M., Dutsch S., Keller U. auf dem, Navid F., Schwarz A., Johnson D.A., Johnson J.A., Werner S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010;24(10):1045–1058. doi: 10.1101/gad.568810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kortlever R.M., Higgins P.J., Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006;8(8):877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghosh A.K., Vaughan D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012;227(2):493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oh C.K., Ariue B., Alban R.F., Shaw B., Cho S.H. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem. Biophys. Res. Commun. 2002;294(5):1155–1160. doi: 10.1016/S0006-291X(02)00577-6. [DOI] [PubMed] [Google Scholar]
- 116.Coppe J.P., Desprez P.Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu J., Ravikumar P., Nguyen K.T., Hsia C.C., Hong Y. Lung protection by inhalation of exogenous solubilized extracellular matrix. PLoS One. 2017;12(2) doi: 10.1371/journal.pone.0171165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kappler B., Anic P., Becker M., Bader A., Klose K., Klein O., Oberwallner B., Choi Y.H., Falk V., Stamm C. The cytoprotective capacity of processed human cardiac extracellular matrix. J Mater Sci Mater Med. 2016;27(7):120. doi: 10.1007/s10856-016-5730-5. [DOI] [PubMed] [Google Scholar]
- 119.Gallorini M., Berardi A.C., Gissi C., Cataldi A., Osti L. Nrf2-mediated cytoprotective effect of four different hyaluronic acids by molecular weight in human tenocytes. J. Drug Target. 2020;28(2):212–224. doi: 10.1080/1061186X.2019.1648476. [DOI] [PubMed] [Google Scholar]
- 120.Campisi J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Procopio M.G., Laszlo C., Al Labban D., Kim D.E., Bordignon P., Jo S.H., Goruppi S., Menietti E., Ostano P., Ala U., Provero P., Hoetzenecker W., Neel V., Kilarski W.W., Swartz M.A., Brisken C., Lefort K., Dotto G.P. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 2015;17(9):1193–1204. doi: 10.1038/ncb3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Papadopoulou A., Kletsas D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int. J. Oncol. 2011;39(4):989–999. doi: 10.3892/ijo.2011.1132. [DOI] [PubMed] [Google Scholar]
- 123.Krtolica A., Parrinello S., Lockett S., Desprez P.Y., Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. U. S. A. 2001;98(21):12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu D., Hornsby P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67(7):3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 125.Demaria M., Ohtani N., Youssef S.A., Rodier F., Toussaint W., Mitchell J.R., Laberge R.M., Vijg J., Van Steeg H., Dolle M.E., Hoeijmakers J.H., de Bruin A., Hara E., Campisi J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell. 2014;31(6):722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Braun S., Krampert M., Bodo E., Kumin A., Born-Berclaz C., Paus R., Werner S. Keratinocyte growth factor protects epidermis and hair follicles from cell death induced by UV irradiation, chemotherapeutic or cytotoxic agents. J. Cell Sci. 2006;119(Pt 23):4841–4849. doi: 10.1242/jcs.03259. [DOI] [PubMed] [Google Scholar]
- 127.Choi H.R., Cho K.A., Kang H.T., Lee J.B., Kaeberlein M., Suh Y., Chung I.K., Park S.C. Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell. 2011;10(1):148–157. doi: 10.1111/j.1474-9726.2010.00654.x. [DOI] [PubMed] [Google Scholar]
- 128.Jun J.I., Lau L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010;12(7):676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fulop G.A., Kiss T., Tarantini S., Balasubramanian P., Yabluchanskiy A., Farkas E., Bari F., Ungvari Z., Csiszar A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40(5–6):513–521. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yuan H., Xu Y., Luo Y., Wang N.X., Xiao J.H. Role of Nrf2 in cell senescence regulation. Mol. Cell. Biochem. 2020;476(1):247–259. doi: 10.1007/s11010-020-03901-9. [DOI] [PubMed] [Google Scholar]
- 131.Wang X.J., Sun Z., Villeneuve N.F., Zhang S., Zhao F., Li Y., Chen W., Yi X., Zheng W., Wondrak G.T., Wong P.K., Zhang D.D. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rolfs F., Huber M., Kuehne A., Kramer S., Haertel E., Muzumdar S., Wagner J., Tanner Y., Bohm F., Smola S., Zamboni N., Levesque M.P., Dummer R., Beer H.D., Hohl D., Werner S., Schafer M. Nrf2 activation promotes keratinocyte survival during early skin carcinogenesis via metabolic alterations. Cancer Res. 2015;75(22):4817–4829. doi: 10.1158/0008-5472.CAN-15-0614. [DOI] [PubMed] [Google Scholar]