

Summary
Duchenne muscular dystrophy (DMD) is a muscle degenerating disease caused by dystrophin deficiency, for which therapeutic options are limited. To facilitate drug development, it is desirable to develop in vitro disease models that enable the evaluation of DMD declines in contractile performance. Here, we show MYOD1-induced differentiation of hiPSCs into functional skeletal myotubes in vitro with collagen gel and electrical field stimulation (EFS). Long-term EFS training (0.5 Hz, 20 V, 2 ms, continuous for 2 weeks) mimicking muscle overuse recapitulates declines in contractile performance in dystrophic myotubes. A screening of clinically relevant drugs using this model detects three compounds that ameliorate this decline. Furthermore, we validate the feasibility of adapting the model to a 96-well culture system using optogenetic technology for large-scale screening. Our results support a disease model using patient-derived iPSCs that allows for the recapitulation of the contractile pathogenesis of DMD and a screening strategy for drug development.
Keywords: EFS training, muscle overuse, optogenetics, high-throughput, induced pluripotent stem cells, skeletal muscle cells, mature myotube differentiation model, Duchenne muscular dystrophy, contractile dysfunction
Graphical abstract
Highlights
Functional myotubes from iPSCs are generated on gels with electrical stimulations
Dystrophic myotubes show a muscle-fatigue-like decline in contractile performance
Small-scale screening detects compounds that ameliorate contractile performance
The feasibility of the screening platform using optogenetic technology is validated
Uchimura et al. establish an iPSC-based disease model using skeletal myotubes differentiated from Duchenne muscular dystrophy (DMD)-patients-derived iPSCs and demonstrate muscle-fatigue-like declines in contractile performance. They also detect compounds that ameliorate contractile declines and established a platform to screen potential therapeutics of DMD using optogenetic technology.
Introduction
Duchenne muscular dystrophy (DMD) is a progressive and fatal muscular degenerative disease caused by dystrophin deficiency, eventually leading to ambulatory and respiratory deficiency, whose symptoms typically begin in early childhood.1 Dystrophin serves as a shock absorber, stabilizing the sarcolemma and protecting the muscle from external stress.2 Although several decades have passed since dystrophin was identified,3 current treatment options remain markedly limited. A decline in muscle performance and fatigue accumulation are common symptoms across ages and disease stages of DMD patients.4 Despite their frequencies, they are still not well recapitulated and studied in vitro. In addition, the correlation between them and DMD onset remains controversial. Muscle fatigue refers to the decline of muscle performance over time. Patients with DMD under muscle fatigue conditions experience a gradual decline in muscle performance. Therefore, it could be hypothesized that patient muscles are under overuse conditions to produce a force, leading to increased stress on muscle fibers. As a result, the pathogenesis of DMD progresses with muscle contracture.5 However, the association between dystrophin deficiency, muscle fatigue, and pathogenesis progression remains unclear, and related treatment options targeting such mechanisms have not yet been developed.
Induced pluripotent stem cell (iPSC) technology has markedly advanced the field of regenerative medicine, including cell therapies, drug development, and basic science, and been utilized to understand the pathogenesis of intractable diseases.6 However, there are several aspects that need to be improved for iPSC-based disease modeling for DMD. In particular, a disease model that recapitulates the functional DMD phenotypes is urgently required for functional analysis. Thus, morphological and functional maturation of muscle cells is essential. Researchers have attempted to generate mature muscle cells using induced myogenic progenitor cells by transient overexpression of Pax7 from human-induced pluripotent stem cells (hiPSCs),7 neuromuscular junctions,8 electrical-field stimulation (EFS),9 and 3D matrices or molds.10 However, studies reporting the contractile phenotypes of DMD are limited.
EFS is traditionally used in skeletal muscle cell cultures to stimulate cells by inducing membrane depolarization and excitation-contraction coupling, as well as promoting maturation of cells.9,11, 12, 13, 14, 15, 16 EFS has also been widely used to mimic in vivo exercise in vitro for investigating the contractile performance, as well as the impact of contractile activities on muscle cells.17 Optogenetics is a combination of optical and genetic techniques using genetically encoded light-activated molecules and has been extensively employed to observe and control cellular functions in living cells.18,19 One of the optogenetic tools is channelrhodopsin-2 (ChR2),18 a light-driven cation channel from the green alga Chlamydomonas reinhardtii, which depolarizes the cell membrane in response to blue light (470 nm). This method has the advantages of fine spatial and temporal resolution, bi-directional control, parallel stimulations at multiple sites, and minimal invasiveness.
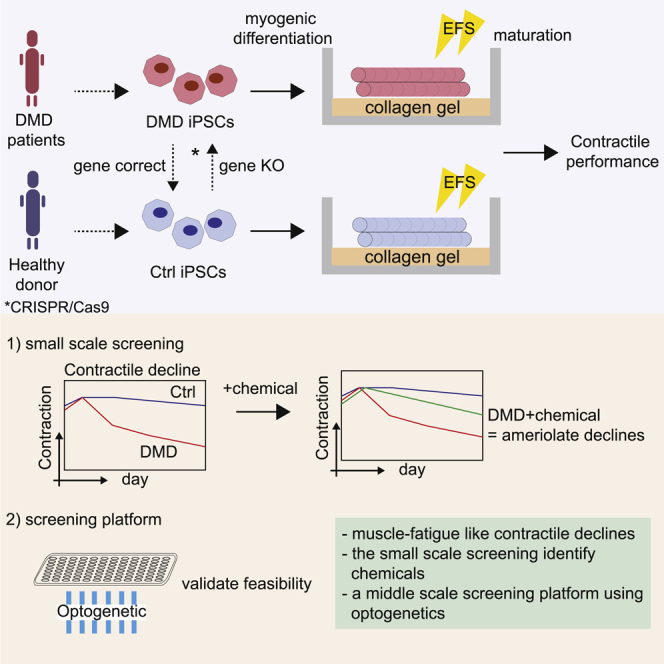
This study describes a modified replating method based on a previously established MYOD1 overexpression protocol, applying EFS with a collagen gel. The method was validated in three DMD models of iPSCs (exon 45 deletion, exon 44 deletion, and exon 46–47 deletion) and their isogenic controls generated by CRISPR-Cas9 genome editing. In this study, EFS was employed to induce functional maturation of myotubes differentiated from hiPSCs, and in vitro exercise models were designed to recapitulate functional dystrophic phenotypes. The EFS method improved myogenic differentiation and maturation of iPSC-myotubes, including cell fusion ability to form myotubes. Cells were further matured by continuous EFS and collagen gel culture, such that the cells showed sarcomere formation and excitation-contraction coupling responses. To recapitulate functional dystrophic phenotypes, two EFS-mediated training models, short-term and long-term, were developed (and patented), because many previous studies utilized EFS only short term (from several minutes to 24 h of stimulation).9 In the short-term training model (1 Hz, 20 V, 2 ms continuous for 24 h), the dystrophic myotubes’ contractile performance was significantly lower after 24 h of EFS. In the long-term training model (0.5 Hz, 20 V, 2 ms, continuous for 2 weeks), the dystrophic cell contractile performance gradually declined after 2 weeks, representing a contractile weakness phenotype of DMD seen under muscle fatigue conditions. Furthermore, a small-scale chemical screening in a 6-well format detected three compounds that ameliorate a decline in contractile performance. Finally, we established a 96-well format screening system using optogenetic technology and validated the culture system’s feasibility for large-scale screening. Our results propose a disease model using patient-derived iPSCs that enables the recapitulation of the functional pathogenesis of DMD and a screening platform to identify potential therapeutics for the disease.
Results
Modified replating method
Previously, we reported a competent myogenic differentiation screening method using hiPSCs.20 However, differentiated myotubes cannot be cultured for an extended period. As a result, cells are still quite limited in their maturation and functional abilities. Therefore, we decided to modify our previous replating method using the following hiPSCs: healthy donor-derived iPSCs 409B2;20 CRISPR-Cas9-mediated exon 45 deletion to create a model of DMD (409B2 ex45KO); and iPSCs from a DMD patient who lacks exon 44 (DMDΔ44)21 and corresponding CRISPR-Cas9-mediated exon 44 knockin to restore the DMD mutation (DMDΔ44-ctrl).22 Compared with our previous method, three changes improved the protocol in the modified method (Figure 1A). First, the dose of doxycycline during days 2–4 was reduced to adjust the premyogenic differentiation speed before replating. Second, replating was conducted on day 4 rather than day 3, which was the optimal time to replate cells for efficient myogenic differentiation after replating. Third, when replated, cells were treated with a low dose of Y-27632 to eliminate undifferentiated hiPSCs from the culture, increasing the myogenic differentiation efficiencies. By day 10, the cells had a spindle-shaped morphology, which is the typical shape of myotubes (Figure 1B), and restored dystrophin expression from DMDΔ44-ctrl was confirmed by immunocytochemistry (Figure 1C). To compare myogenic differentiation efficiencies, we performed a fusion index and gene expression analysis and observed slightly increased multinucleated myotubes and higher expression of myogenic differentiation and maturation-related markers than the previous method (Figures 1D, S1A, and S2).23 However, even with the modified replating differentiation protocol, we noted that the cells strongly expressed the embryonic marker, MYH3, and the neonatal marker, MYH8, as well as fast and slow muscle markers, MYH1, 2, and 7, but not MYH4 (Figure S1B).24 These data indicate successful improvement of the previous replating method; however, it was expected that myotubes differentiated by the modified protocol were still not sufficient to evaluate contractile performance due to insufficient maturation in the absence of an external stimulus that triggers an excitation-contraction coupling.
Figure 1.

The modified replating method improved the differentiation and maturation of hiPSC skeletal myotubes
(A) A schematic diagram of the modified replating method. iPSCs (409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44) were myogenically pre-differentiated in Primate ES Cell Medium (PECM) media in the presence of doxycycline. Cells were replated on day 4 in the presence of lower doses of Y-27632 and the absence of doxycycline in 2% horse serum media. The doxycycline-induced MyoD overexpression induced myogenic differentiation at day 6 for 4 days.
(B) Bright-field images of the time course of myogenic differentiation of 409B2 and DMDΔ44 lines. Scale bar represents 200 μm.
(C) Immunocytochemical analysis of pan-MHC and DYS1 in differentiated myotubes of 409B2 and DMDΔ44 lines on day 14. Scale bar represents 20 μm.
(D) Fusion index analysis of 409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44 myotubes differentiated by previous and modified replating methods. Data represent the mean ± SD and were analyzed by an unpaired t test from six biological replicates. ∗p < 0.05.
Cell maturation by EFS
Contraction is an essential endpoint for evaluating the contractile performance of muscles. As described previously, the modified method still has limitation in evaluating contractile performance due to insufficient maturation of the myotubes. Additionally, we noted that differentiated myotubes became shorter and thinner, atrophy, and die after reaching terminal differentiation in the current modified method. Thus, to overcome this issue, we hypothesized that EFS could be used to enhance the maturation of myotubes and improve cell survival, eventually leading to contraction. Because we observed that myotubes were easily dissociated upon EFS due to friction when cells were seeded directly onto the culture plate, we seeded cells onto a thick layer of collagen gel, which has the appropriate adherence properties and stiffness for smoother contractions. Moreover, we also designed an EFS protocol in which the EFS voltage was increased stepwise to allow cells to gradually adapt to the EFS culture. EFS stimulated the cells beginning on day 10 (D10), and the EFS voltage gradually increased until D17 (Figure 2A). To compare maturation of myotubes, we performed a gene expression analysis and observed much higher expression of myogenic differentiation and maturation-related markers than non-EFS-stimulated cells (Figure S2). Fusion index analysis revealed that EFS promoted maturation as myotubes containing more than 10 nuclei were observed in EFS-stimulated cells (Figures 1D and 2B). We immunostained cells with an α-actinin antibody and conducted a transmission electron microscopy analysis to confirm the sarcomere. In the modified method, approximately 70% of myotubes possessed a sarcomere-like pattern of α-actinin (Figure 2C), and sarcomere formation was confirmed (Figure 2D). Finally, cells were analyzed for contraction, and cells with the modified method showed higher motion speeds and total contracting area than those obtained by the previous method (Figure 2E; Videos S1, S2, S3, and S4). These data confirmed that myotubes cultured by the modified EFS and collagen gel method were morphologically and functionally matured.
Figure 2.

Electrical field stimulation (EFS) with a modified replating method induced the maturation, sarcomere formation, and contractile activities of hiPSC skeletal myotubes
(A) A schematic diagram of EFS with the modified replating method. EFS began at day 10 at 2 V, 2 ms, and 0.5 Hz, and the voltage was increased to 5 V and 10 V on days 12 and 14, respectively.
(B) Fusion index analysis of 409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44 myotubes was differentiated using modified replating methods with EFS and gel culture.
(C) Immunocytochemical analysis of α-actinin of matured 409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44 myotubes differentiated by the modified replating method with EFS. Scale bar represents 20 μM. The right panel is a quantitative analysis of the percentage of cells possessing a sarcomere-like pattern of α-actinin staining.
(D) Transmission electron microscopy analysis of mature 409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44 myotubes differentiated by the modified replating method with EFS. Rectangles denote magnified areas. Scale bar represents 3 μm.
(E) Heatmap analyses using a SI8000 motion imaging system of motion pixels of matured 409B2, 409B2 ex45KO, DMDΔ44-ctrl, and DMDΔ44 myotubes differentiated using the previous and modified replating methods with EFS. The right panel is a quantitative analysis of the contracting area. Scale bar represents 100 μm.
Data represent the mean ± SD and were analyzed by an unpaired t test from six biological replicates (B and C) and five biological replicates (E). ∗p < 0.05.
A representative bright-field video of DMDΔ44-ctrl after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of DMDΔ44 after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of 409B2 after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of 409B2 ex45KO after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
EFS training models
Next, we sought to develop an EFS-mediated in vitro muscle training program to recapitulate the phenotypes of DMD contractile performance. First, we performed CRISPR-Cas9-mediated knockin of exons 46 and 47 to generate the DMDΔ 46-47-ctrl clone as an isogenic control from the DMDΔ 46–47 iPSC21 to confirm the disease model’s reproducibility (Figure S3A). After we confirmed efficient myogenic differentiation and restoration of dystrophin protein using a modified method (Figures S3B–S3D), we then considered a previous report from our lab of a Ca2+ overload using DMDΔ44 and DMDΔ 46–47 lines combined with exon skipping,21 because it remains unclear whether similar Ca2+ overload occurs in dystrophic cells of three lines using the modified method. We conducted a Ca2+ mobilization assay to address this question and confirmed Ca2+ overload in dystrophic cells from three lines (Figure S3E). Therefore, we next proposed EFS-mediated short-term and long-term training models to recapitulate the phenotypes of DMD contractile performances.
Short-term EFS training model
In the short-term training model, cells were initially stimulated at 2 V and 5 V for 2 days and 10 V for 1 day and then the voltage was increased to 20 V for 1 day to evaluate the acute cell responses (Figure S4). Before the stimulation at day 15, there were no significant differences in contractile performances between dystrophic and control myotubes (Figures S4A–S4C). However, Ca2+ overload was observed in dystrophic cells, consistent with previous Ca2+ mobilization data (Figure S3E). Inflammatory response markers, interleukin-1β (IL-1β), tumor necrosis factor (TNF), IL-6 mRNA expression levels, and IL-6 protein levels were also not significantly different between the control and dystrophic cells (Figures S4D–S4G). These data indicate that dystrophin deficiency does not regulate the acquisition of basal contractile capability and inflammatory responses during physiological stimulation at 10 V.
Next, cells were stimulated for 24 h at 20 V to investigate the acute response of dystrophic cells. After 24 h stimulation beginning at D16, contractile performance and Ca2+ peaks were significantly lower in dystrophic cells (Figures 3A–3C). Cells were then analyzed for gene expression and apoptotic activities to characterize cellular phenotypes. TNF-α and IL-6 mRNA levels (but not IL-1β) were significantly induced in dystrophic cells (Figures 3D–3F). IL-6 protein levels were also significantly increased in dystrophic cells from 409B2 and DMDΔ44 lines (Figure 3G). Though not statistically significant, its level was also increased in dystrophic cells from the DMDΔ 46–47 line (Figure 3G). Dystrophic cells also had significantly increased active-caspase 8 and 9 activities compared to their control cells (Figure 3H), with no changes in mRNA expression (data not shown). Furthermore, we observed increased cyclic AMP (cAMP) levels in dystrophic cells (Figure 3I). These data indicate acute contractile phenotypes under dystrophic conditions from the three donor-derived iPSCs.
Figure 3.

Reduced muscle performance was observed in dystrophin-deficient myotubes at day 16 in the short-term training model using three donor-derived iPSCs
(A–C) Functional analyses and Ca2+ peak assay by the SI8000 system of differentiated and mature myotubes from three donor-derived iPSCs on day 16. (A) 409B2, (B) DMDΔ44, and (C) DMDΔ46-47 are shown.
(D–F) qRT-PCR analyses of inflammatory response-related genes on day 16. (D) IL-1β, (E) TNF-α, and (F) IL-6 are shown.
(G) ELISA of IL-6 protein levels in conditioned media harvested on day 16.
(H) An assay of active caspases 8 and 9 on day 16.
(I) cAMP level assay on day 16.
Data represent the mean ± SD and were analyzed by an unpaired t test from five biological replicates (A–C) and three biological replicates (D–I). ∗p < 0.05. N.D., non-detectable.
Long-term EFS training model
A previously described short-term training model revealed the acute response of dystrophic myotubes. However, we considered whether it is possible to recapitulate a muscle-fatigue-like decline in contractile performance. Thus, we designed another EFS program, a long-term training model, in which EFS gradually increased over the culture time, so that stimulated cells adapted to increasing EFS and became functionally mature. The cells were then further stimulated continuously at 20 V from day 16 to day 28 (Figure S4A). The model aimed to test the endurance and function of dystrophic myotubes over time and determine cellular and molecular responses to continuous stimulation. The contraction velocity was monitored every day up to approximately 2 weeks and gradually increased as EFS increased. A plateau was reached between days 17 and 20 in both dystrophic and control cells (Figure 4B). These data indicate that dystrophic cells can perform muscle functions at a level similar to that of control cells. However, after that, although control cells maintained the contraction velocity at the same level, dystrophic cells showed a gradual decline (Figure 4B). Similar trends were also observed for twitching distance, relaxation velocity, and acceleration (Figures S5A–S5C). Although the degree of reduction in contractile performance varied among iPSC clones, all clones showed a muscle-fatigue-like decline in contractile performance from dystrophic myotubes in the long-term training model. Because microscopic analyses did not identify distinct cellular damage or cell death, the observed phenotype was caused by a functional deterioration rather than a loss of contractile myotubes (Figure S5D).
Figure 4.

A gradual decline in DMD contractile performance was observed in dystrophin-deficient myotubes in the long-term training model using three donor-derived iPSCs
(A) A schematic diagram of the EFS-based long-term training model.
(B) Quantitative time-course analyses of the contraction velocity using the SI8000 motion imaging system.
(C–E) qRT-PCR and ELISA analyses of inflammatory response-related genes on day 28. Both cells and conditioned medium were harvested at day 28. (C) 409B2, (D) DMDΔ44, and (E) DMDΔ46–47 are shown.
(F) Ca2+ mobilization assay at day 28.
(G) The cAMP level assay at day 28.
Data represent the mean ± SD and were analyzed using an unpaired t test from at least three biological replicates. ∗p < 0.05.
At the end of culturing, cells were processed for gene expression and Ca2+ peak and cAMP level assays. Although mRNA expression levels of TNF-α, IL-1β, and IL-6 were not significantly induced, a protein level of TNF-α was induced in dystrophic cells of 409B2 and DMDΔ44, and IL-6 was induced from DMDΔ44 and DMDΔ 46–47 dystrophic cells (Figures 4C–4E). Consistent with the reduced contractile performance, the Ca2+ peaks were also decreased in dystrophic cells (Figure 4F). cAMP levels were also lower in dystrophic cells from the three iPSC lines (Figure 4G). Therefore, these data suggest a lack of, or limited, inflammatory responses and imply reduced secondary messenger signaling from Ca2+ and cAMP.
A small-scale screening in the long-term training model
The established long-term training model successfully recapitulated the decline in contractile performance seen in DMD patients. However, it remains unknown whether the observed phenotype represents a clinical symptom of DMD. To address this question, we performed small-scale screening to demonstrate the feasibility of the model for detecting chemicals that can ameliorate contractile phenotypes (Figure S6). Screened chemicals included compounds in current clinical use or under clinical development and commercially available Ca2+ modulators (Table 1). The first and second screening was conducted using a DMDΔ44 clone, which showed declines in contractile performance between day 17 and 28 and detected four hit compounds, dantrolene, creatine, 5 J 4, and ML9, that ameliorated this decline (Figure 5A). Conversely, chemicals such as an active form of deflazacort, an NHE1 inhibitor, BGP15, insulin-like growth factor I (IGF-I), and Rycal S107 failed to ameliorate contractile performance (Figure S6C). Therefore, compounds that failed to ameliorate contractile performance were terminated at this point. Next, a final screening was conducted using DMDΔ 46–47 clones and four positive compounds from the second screening. We found that dantrolene, ML9, and creatine also ameliorated the decline in contractile performance in DMDΔ 46–47 clones (Figure 5B). As a result, we detected three compounds that prevented declines in contractile performance in both DMDΔ44 and DMDΔ 46–47 clones in the long-term EFS training model.
Table 1.
A list of chemicals used for the screening
| Group | Chemical | Name | Dose used | Activity | Reference |
|---|---|---|---|---|---|
| I | clinically approved or under clinical development | steroid (deflazacort) | ∼0.1–1 μM | clinically used. anti-inflammation. | Griggs et al.25 |
| NHE1 inhibitor (rimeporide) | ∼1–10 μM | Na+/H+ exchanger inhibitor | Previtali et al.26 and Ghaleh et al.27 | ||
| BGP-15 | ∼1–10 μM | poly (ADP-ribose) polymerase inhibitor | Kennedy et al.28 | ||
| idebenone | ∼1–10 μM | antioxidant | Buyse et al.29 | ||
| dantrolene | 1 μM | muscle relaxant | Krause et al.30 | ||
| GsMTx4 (AT-300) | 1 μM | mechanosensitive channel blocker | Ward et al.31 | ||
| II | commercially available | 2-APB | 1 μM | Ca2+ modulator | Maruyama et al.32 |
| 5J4 | 10 μM | Ca2+ modulator | N/A | ||
| ML9 | 10 μM | Ca2+ modulator | Wu et al.33 | ||
| creatine | 100 μM | organic compound | Felber et al.34 and Tarnopolsky et al.35 | ||
| Rycal S107 | 10 μM | Ca2+ modulator | Capogrosso et al.36 | ||
| SKF 96365 | 10 μM | Ca2+ modulator | Esteve et al.37 | ||
| IGF-I | 100 nM | growth factor | Vandenburgh et al.38 | ||
| YM 58483 | 10 μM | Ca2+ modulator | Olah et al.39 | ||
| MRS1845 | 10 μM | Ca2+ modulator | N/A | ||
| Ochratoxin A | 10 μM | SERCA-ATP-Ca2+ pump stimulator | Fernandez-Tenorio et al.40 | ||
| istaroxime | 10 μM | Na+/K+ ATPase inhibitor | Huang41 |
Figure 5.

Amelioration of the declines in contractile performance upon the administration of compounds in the long-term training model
Quantitative time-course analyses of the contraction velocity upon the administration of compounds. (A) DMDΔ44 and (B) DMDΔ46-47 are shown. Data represent the mean ± SD and were analyzed using an unpaired t test from at least three biological replicates. ∗p < 0.05.
A 96-well system with optogenetic technology
Although we conducted small-scale screening using the long-term training model, there are limitations of using the system with EFS, including low throughput and the generation of toxic gases. In particular, low throughput needs to be improved for drug screening using a large-scale library. Thus, to address these limitations, we utilized optogenetic technology to scale up the system and minimize toxicity. ChR2,18 a light-driven cation channel from the green alga Chlamydomonas reinhardtii that depolarizes the cell membrane in response to blue light (470 nm), was used in this study, along with a ChR219 variant, CatCh+ (ChR2-L132C-T159C),42 which has improved membrane targeting, desensitization, and increased conductance of divalent cation compared with ChR2. The DMDΔ44 clone stably expressing both doxycycline-inducible MyoD and CatCh+ was established and cultured on a collagen gel (Figures S7A and S7B). Optogenetics stimulation (OS) training was applied to cells using a blue (470 nm) LED laser from the bottom of the plate, and myogenic differentiation efficiencies were determined at day 28 (Figures S7C–S7E). To validate the culture system’s feasibility for screening, we used and calculated a coefficient of validation (CV) value. In general, if the CV value is lower than 10%, it can be said that the culture system is suitable for screening with confidence.43 Because our model showed a CV value lower than 10% (Figure S7E), it meets the criterion as a screening platform. Next, to verify that OS can induce anatomical and functional maturation equivalent to the EFS model, we performed a fusion index analysis and immunohistochemical staining of α-actinin and analyzed the contractile performance on day 18 (Figures S7F–S7H). Fusion index analysis revealed that OS promoted maturation as myotubes containing more than 10 nuclei were observed in OS-stimulated cells, which was similar to findings from EFS-stimulated cells (Figures S7F and 2B). We found that approximately 70% of myotubes also possessed a sarcomere-like pattern of α-actinin, comparable to the EFS model (Figures S7G and 2C). In addition, myotubes stimulated with OS showed the same contractile performance at day 18 as cells stimulated with EFS (Figure S7H). These data suggest that myotubes stimulated with OS show maturation equivalent to that in cells stimulated with EFS.
Finally, we investigated whether a contractile performance phenotype observed from the long-term training model can be reproduced by OS in a 96-well culture system. First, we tried to reduce the assay variation by evaluating multiple adjacent wells together, because the variation between 96 wells tended to be larger, with a CV value higher than 10%. For this purpose, two different analysis methods, 2-well and 4-well combined assays, were used, and the CV value of the contractile performance at day 18 was determined (Figure 6A; Video S5). Both methods showed a CV value lower than 10% (Figure 6B), indicating that the analysis methods were validated. Next, OS-based long-term training was conducted, and contractile performance was analyzed using the 4-well combined assay method to determine whether OS-based long-term training can reproduce the decline in contractile performance in dystrophic myotubes as seen in EFS-based long-term training. We succeeded in reproducing the significantly reduced contractile performance at day 28 (Figures 6C and 6D), which is the same as that seen with the EFS-based long-term training model (Figure 4B). In addition, the results confirm that reduced contractile performance at day 28 can be a readout of the assay for the screening. Therefore, these results indicate that OS can be an alternative EFS method for inducing maturation and recapitulating a contractile phenotype from dystrophic myotubes and provides flexibility for adapting the model to a high-throughput culture system for large-scale screening.
Figure 6.

Establishment of the long-term training-based screening system in a 96-well platform using optogenetic technology
(A) A plate map indicating the analysis methods, 2- and 4-well combined.
(B) Heatmap analyses of the contraction velocity and statistics from 2-well and 4-well combined methods at day 28 in the long-term training model using the DMDΔ44 clone.
(C and D) Heatmap analyses of the reduced contraction velocity analyzed by the 4-well combined methods in the DMDΔ44 clone at day 28 in the (C) long-term training model and (D) its quantitative analyses.
Data represent the mean ± SD and were analyzed with an unpaired t test from twenty-four biological replicates. ∗p < 0.05.
A representative bright-field video of DMDΔ44 after 17 days of differentiation exhibiting continuous contractions under optogenetic stimulations. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
Discussion
In the first half of this study, we demonstrated contractile phenotypes from dystrophin-deficient myotubes using three patient-derived iPSCs with EFS-based training programs. In the short-term training model (1 Hz, 20 V, 2 ms, continuous for 24 h), dystrophic cells showed a significantly lower contractile performance after 24 h of EFS training accompanied by increased inflammatory responses and caspase activities. In the long-term training model (0.5 Hz, 20 V, 2 ms, continuous for 2 weeks), dystrophic cells’ contractile performance gradually declined over time under EFS training, representing a muscle-fatigue-like decline in contractile performance.
In this study, we aimed to engineer mature and functional myotubes with a combination culture using collagen gel and EFS, enabling us to evaluate a contractile performance to reveal the functional DMD phenotypes. A gel culture is beneficial as it can create suitable stiffness for muscle cells44 and cells contract more smoothly because of low friction. EFS is also employed in culture as it is known to stimulate muscle cell contraction and promote the maturation of cells.9,11, 12, 13, 14, 15, 16 Although it remains unknown whether EFS is also capable of promoting maturation and inducing the excitation-contraction of hiPSC-derived myotubes, our study demonstrated that, when hiPSC-muscle cells mature adequately, they show multinucleated myotubes, sarcomeres, and contract upon EFS exposure. Thus, our modified culture method with collagen gel and EFS can generate mature hiPSC-muscle cells in vitro for 2 weeks for further functional, cellular, and molecular analyses.
EFS is also traditionally used to mimic in vivo exercise in vitro. However, many studies using EFS training are limited because most are classified as short-term training models (from several minutes to 24 h of stimulation),9 patient-derived cells are rarely used,9 and these studies focused on signal transduction, myokines, or metabolic effects. The increased IL-6 expression levels and Ca2+ transients reported in previous studies are consistent with this study.9,11, 12, 13, 14,45,46 However, our study’s value lies in identifying functional phenotypes because such data are markedly limited.38,47,48 Nesmith et al.48 reported structural aberrations and contractile weakness using DMD myoblasts, although patient-to-patient variability cannot be excluded because of different healthy and DMD single donors and EFS was a short-term training model. Vandenburgh et al.38 evaluated the efficacy of chemicals on the tetanic force of constructed muscle tissues. Shimizu et al.49 evaluated dexamethasone’s effects on the contractile force of 3D cultured tissues. However, neither study demonstrated any dystrophic phenotypes. In our study, the short-term training model focusing on comparing cells’ responses between control and dystrophic cells within the 24-h EFS at 20 V revealed similar dystrophic phenotypes, consistent with previous reports.
Conversely, we also designed a long-term training model that focused on comparing cell contractile performance under long-term continuous EFS. This training revealed that dystrophin is dispensable for muscle fiber functional maturation because initially dystrophic myotubes were capable of performing similar contractile performance with respect to control myotubes. However, dystrophin is indispensable for cells to maintain contractile performance. This phenotype is markedly similar to the muscle fatigue conditions of DMD.50,51 Interestingly, there was no cellular damage when contractile performance declined. Thus, these data implicate functional deterioration rather than contractile cell loss.
We believe that our long-term training model is of considerable valuable for investigating further whether functional deterioration is an important factor triggering the onset of DMD, which is a degeneration of muscle fibers. Choi et al.52 discussed patient-to-patient variability by showing abnormal phenotypes using DMD-patient-derived iPSC myotubes. Our study used three different donor-derived iPSCs and their isogenic controls to identify concurrent phenotypes; thus, we believe that the phenotypes of contractile performance reported herein are convincing. Moreover, a decline in contractile performance resulting from the long-term training model is likely to recapitulate important common DMD symptoms, and targeting this decline may be a viable strategy for developing drugs for patients with various mutations.
Therefore, the second half of this study demonstrated the translational or clinical utility and the feasibility of scaling up our method as a drug discovery screening platform. Small-scale screening was conducted using clinically relevant compounds, and three chemicals that ameliorate dystrophic phenotypes were identified in the long-term training program. Furthermore, using optogenetic technology,19 we demonstrated the feasibility of scaling up an existing 6-well EFS system to a 96-well platform for future screening by showing the uniformity of myogenic differentiation and contractile performance as well as contractile reductions in the long-term training program with OS.
First, we conducted small-scale screening using clinically approved compound (steroids and dantrolene) drugs under clinical development (NHE1 inhibitor, idebenone) and commercially available chemicals (Ca2+ modulators and growth factors). Deflazacort failed to rescue the dystrophic phenotypes or exert negative effects.25 This finding can be explained because our model does not contain non-muscle cells, such as immune cells or inflammatory cells, because steroids are thought to exerts their effects through targeting immune cells and inflammation.53 Previously, IGF-I and deflazacort upregulated contractile ability in 3D-constructed tissues using immortalized mdx myoblasts;38 however, there were no additive effects observed in our screening. There were also no effects reported for BGP-1528 or rimeporide.26,27
Although most screened compounds failed due to no or adverse effects, at least three compounds ameliorated contractile performance decline. Creatine supplementation has previously been shown to possess positive activities in the muscles of DMD patients.34,35 In our model, adding creatine partially rescued the declines in contraction. However, because we did not observe cellular damage or leakage of creatine kinase in the conditioned medium from dystrophic cells (data not shown), creatine’s activities might be an additive effect rather than the supplementation of reduced internal creatine. ML9, an inhibitor of myosin light-chain kinase as well as store-operated calcium entry (SOCE), ameliorated the decline of contraction in both DMDΔ44 and DMDΔ 46–47 lines. Although ML9, from the myosin light-chain kinase inhibitor’s perspective, has been well studied,33 its activity as an SOCE inhibitor for DMD treatment remains elusive. Because dystrophic cells were under Ca2+ overload before the contraction declines started, ML9 may exert its effects by modulating Ca2+ mobilization. Finally, dantrolene was the most prominent among the three positive compounds in both DMDΔ44 and DMDΔ 46–47 lines. Dantrolene is a muscle relaxant that reduces excitation-contraction coupling.30 Consistent with ML9, dantrolene also modulates Ca2+ mobilization through different molecular mechanisms. Therefore, two positive compounds modulate Ca2+ mobilization.
Conversely, Rycal S107 was not effective in our model, although Rycal alleviated the pathologic Ca2+ leakage.36 One of the reasons why it did not work in our model is that our model captures a different DMD phase. For example, it is known that Rycal inhibits the S-nitrosylation of RYR1-induced SR Ca2+ leakage,54 which could be caused by pathological changes after the onset of DMD. Because our model mimicked the condition before the onset of DMD, in which the S-nitrosylation of RYR1 might not have progressed, it is likely that Rycal was not effective in our model. These data suggest that Ca2+ signaling could be one of the candidates leading to a decline in contractile performance, and using those negative and positive compounds, it might be possible to identify the target molecule that causes a decline in DMD contractile performance by regulating Ca2+ signaling. Because our model only contains differentiated myotubes and recapitulated a phenotype of the early phase of DMD, it is also possible to identify candidate chemicals with different mechanisms of action compared with current drugs. Dystrophin has previously been identified as a causative gene for DMD, and researchers have reported cellular and molecular dystrophic phenotypes in vitro.45,55, 56, 57, 58, 59, 60, 61, 62 However, the molecular mechanisms by which the loss of dystrophin leads to a decline in contractile performance or how these positive compounds ameliorate the phenotypes remain elusive. Nevertheless, we believe that our study is valuable in providing data to understand the molecular pathology of functional phenotypes further and facilitate the development of therapies targeting the phenotypes of early phases of DMD in skeletal muscle cells.
To facilitate the development of DMD therapies, it is also critical to adapt the long-term EFS training model to a high-throughput system for future screening. One of the limitations is that EFS systems in a 96-well format are rarely commercially available and concern is that these generate toxic gases.19 Thus, we adapted optogenetic technology as an alternative cell-stimulation method.19 Previously, few studies reported the screening of small molecules or established a screening platform using 3D tissue or iPSCs.38,63,64 For example, the Vanderburgh group conducted drug screening using 3D-constructed tissues from immortalized mdx myoblasts with EFS.38 In the last year, the Selvaraj group performed small-molecule screening using control iPSC myoblasts in a 2D model without EFS and identified four factors that promote maturation of iPSC myoblasts.63 Afshar and co-workers recently proposed a 96-well culture platform using 3D constructed microtissues composed of primary human myoblasts.64 Although our model shares some similarities with theirs, some differences make our model advanced. First, our high-throughput screening (HTS) assay utilizes optogenetics rather than EFS. As described previously, optogenetics is less toxic than EFS19 and quite flexible for HTS compared with electrodes. It is simple to apply stimulation to cultured cells for long periods, such as a month, using optogenetics. Second, our model was established using DMD-patient-derived iPSCs. The aim of establishing a screening platform using optogenetics is to detect compounds that ameliorate declines in contractile performance in dystrophic myotubes. We recapitulated a contractile decline in a 96-well culture system using OS and patient-derived iPSCs. Therefore, we believe that our HTS strategy for identifying compounds that ameliorate the contractile performance in a 96-well system with OS using a middle- to large-scale library of several hundred to thousand compounds is feasible. Because the feasibility of screening up to 20 compounds using a 6-well plate with EFS was confirmed in this study, a second screening will be conducted in the 6-well EFS system using the positive lead compounds identified in the first screening in a 96-well HTS assay. Finally, when designing a screening assay, we also utilized a coefficient of variation (CV) value to validate screening feasibility and reliability. A CV value is often used as a standard criterion for assessing the screening data variability and is critical for determining whether an assay can identify hit compounds with confidence.43,65 Our model achieved a CV value lower than 10% by evaluating multiple adjacent wells together, which is the most accepted standard for an HTS assay. Therefore, we succeeded in scaling up a 6-well EFS system to a 96-well HTS-compatible system and established an evaluation method for drug screening.
In conclusion, our data demonstrate the feasibility of culturing mature and functional myotubes from DMD patient iPSCs using a modified replating technique with a soft gel culture and EFS system in a fast, efficient, and versatile manner. We observed functional key phenotypes in the long-term training model, which may represent a muscle-fatigue-like decline in DMD contractile performance and present a DMD disease model for therapeutic purposes, such as drug screening or chemical and target evaluation. Furthermore, we demonstrated that the long-term training model could detect the agents that ameliorate declines in contractile performance in small-scale screening, implicating the model’s feasibility for detecting potential anti-DMD chemicals. We adapted an optogenetic technology for the long-term training model for future drug-screening purposes and established a 96-well screening platform. A middle-scale screening in a 96-well system or large-scale screening by further up-scaling to a 384-well system with an automated machine is expected to identify potential DMD therapeutics that will attenuate disease progression.
Limitations of the study
Many types of mutations on dystrophin gene have been reported from DMD patients.66 Because DMD patients do not express dystrophin protein, types of mutations may not cause different phenotypes except a degree or severity of them. Although three DMD models of iPSCs (exon 45 deletion, exon 44 deletion, and exon 46–47 deletion) and their isogenic controls were used in this study, it remains elusive whether the phenotype identified in this study represents concurrent phenotypes of all types of mutations in DMD patients.
One of the limitations of this study includes the utilization of doxycycline-induced MyoD overexpression system as well as maturation status of differentiated myotubes. Currently, several stepwise differentiation induction protocols utilizing small molecules and growth factors with cell surface markers are available.67,68 It is unclear whether the phenotypes identified by the MyoD overexpression system can be reproduced by the stepwise induction method. Our lab recently reported induction protocols69,70 by which differentiated myotubes could become more matured. Thus, this will require further detailed study to better understand the muscle fatigue phenotype using induction protocols.
Lastly, it is also controversial and unknown whether the muscle fatigue is a factor causing cellular damage and muscle fiber degeneration clinically. Although muscle fatigue is a common symptom in DMD patients, there is no evidence that modulating muscle fatigue prevents the onset or disease progression of DMD in patients. Currently, in vitro models that can evaluate cellular and muscle fiber damages are quite limited. Therefore, further studies are necessary to better understand the correlations between muscle fatigue and cellular and muscle fiber damages.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MHC antibody (MF20) | R&D | MAB4470; RRID: AB_1293549 |
| FASR/MYH1&2 antibody (MY-32) | Sigma | M4276; RRID: AB_477190 |
| MYH2 antibody | Millipore | MABT840 |
| MYH3 antibody | Atlas antibodies | A75757 |
| MYH7 antibody (A4.840) | Santa Cruz | sc-53089; RRID: AB_2147281 |
| MYH8 antibody | Novus Biologicals | NBP2-41309 |
| Sarcomeric α-actinin | Abcam | ab90776; RRID: AB_10670308 |
| Dystrophin antibody (rod-domain) | Leica | NCL-DYS1; RRID: AB_442080 |
| RYR1 antibody (D4E1) | Cell Signaling Technology | 8153; RRID: AB_2797637 |
| Myogenin antibody (F5D) | Santa Cruz | sc-12732; RRID: AB_627980 |
| TBP antibody (D5C9H) | Cell Signaling Technology | 44059; RRID: AB_2799258 |
| Chemicals, peptides, and recombinant proteins | ||
| StemFit AK02N | Reprocell | ECAK02N |
| Primate ES Cell Medium | Reprocell | RCHEMD001A |
| Knockout serum replacement | Thermo Fisher | 10828028 |
| αMEM | Nacalai | 21444-05 |
| Easy iMatrix-511 | Nacalai | 892011 |
| Matrigel growth factor reduced | BD | 356230 |
| Collagen solution | Nippi | 891500 |
| Blasticidin | Wako | 026-18711 |
| Puromycin | Sigma | P8833 |
| Y-27632 | Wako | 034-24024 |
| Cal-520 | AAT Bioquest | 21130 |
| Insulin | Wako | 093-06351 |
| Retinoic acid | Sigma | R2625 |
| Thyroid hormones | Sigma | T6397 |
| SB431542 | Wako | 198-16543 |
| Accutase | Nacalai | AT104 |
| Doxycycline | Nacalai | 19088-71 |
| Horse serum | Sigma | H1138 |
| 2-mercaptoethanol | Sigma | 21985023 |
| Glucose | Invitrogen | A24940-10 |
| Blocking one | Nacalai | 03953-95 |
| RIPA buffer | Pierce | 89900 |
| A complete mini protease inhibitor cocktail | Roche | 1183617001 |
| FluoroBrite DMEM | Invitrogen | A18967-01 |
| Critical commercial assays | ||
| RNeasy mini kit | QIAGEN | 74104 |
| PrimeScript RT reagent kit | Takara | RR037 |
| BAC protein assay kit | Pierce | 23228 |
| IL-6 Quantikine ELISA kit | R&D | D6050 |
| Caspase-Glo 8 | Promega | G8200 |
| Caspase-Glo 9 | Promega | G8210 |
| LANCE Ultra cAMP Detection kit | PerkinElmer | TRF0262 |
| Experimental models: Cell lines | ||
| DMDΔ44 hiPSC line | Shoji et al.21 | Shoji et al.21 |
| DMDΔ46-47 hiPSC line | Shoji et al.21 | Shoji et al.21 |
| 409B2 hiPSC line | Okita et al.71 | Okita et al. |
| Software and algorithms | ||
| GraphPad Prism 7.0e | GraphPad | N/A |
| Image Lab | Bio-Rad | N/A |
| Microsoft Excel | Microsoft | N/A |
| Microsoft Word | Microsoft | N/A |
| Adobe Photoshop | Adobe | N/A |
| Adobe Illustrator | Adobe | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hidetoshi Sakurai (hsakurai@cira.kyoto-u.ac.jp.)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact, Hidetoshi Sakurai, with a completed Material Transfer Agreement.
Data and code availability
This study did not generate/analyze [datasets/code].
Experimental model and subject details
Human iPSC lines
The 409B2 iPSC (healthy patient, female, 36 year old) line established from human fibroblasts which was obtained from Dr. Keisuke Okita of Kyoto University. The Δ44 DMD-iPSCs established from skin fibroblasts of a DMD patient (exon 44 deletion, male, 3 year old) and its isogenic control line, Δ44 DMD-ctrl-iPSCs were used21,22. The Δ46–47 DMD-iPSCs21 established from skin fibroblasts of a DMD patient (exon 46-47 deletion, male, 8yr old) were used for the experiments.
Maintenance of hiPSC lines
iPSCs were cultured on laminin-coated plates in StemFit AK02N (StemFit) (Reprocell, Beltsville, MD) containing 0.5 μg/mL puromycin (Sigma, St. Louis, MO, USA), depending on the antibiotic resistance of each line. Cells were passaged every seven days using Accutase72 (Nacalai, Kyoto, Japan) and seeded on laminin-coated plates in the presence of 10 μM Y-27632 (Nacalai, Kyoto, Japan) at a density of 1.5 × 104 cells/well in 6-well plates or 10 cm dishes for the first two days after plating. Forty-eight h after passaging, Y-27632 was removed and replaced with StemFit containing a suitable antibiotic. The media were changed at least every other day. All cell lines were tested for mycoplasma and were negative for mycoplasma contamination.
Ethical approval
This study was approved by the Ethics Committee of the Graduate School of Medicine, Kyoto University, Kyoto University Hospital (approval number #R0091 and #G259), and Takeda Pharmaceutical Company Ltd. (GEN-00000040-008), and was conducted according to the guidelines of the Declaration of Helsinki. All patient information was confidentially maintained, and written informed consent was obtained.
Method details
Plate preparation
For laminin-coated plates, 1.5 mL or 8 mL of Easy iMatrix-511 silk (Nippi) was added to each well of the 6-well plates or 10 cm dishes, after which the plates were incubated overnight at 4°C. Coated plates were stored at 4°C for up to 2 weeks until use. Laminin-coated plates were equilibrated at 24°C for at least 30 min before use in the experiments. For Matrigel-coated plates, all tubes, pipette tips, and reagents were pre-chilled in the refrigerator or on ice. Matrigel (BD Falcon, Franklin Lakes, NJ, USA) was prepared at a 1:100 dilution with pre-chilled media without any additives. An appropriate volume of diluted Matrigel was dispensed into each well of the 6-well or 96-well plates. Plates were incubated at 4°C overnight before use. Matrigel-coated plates were stored at 4°C for up to several weeks until use. For collagen gel plates, 1.2 mL or 10 μL of 0.5 mg/mL collagen solution (Nippi) was dispensed into a well of 6-well (BD Falcon) or a μPlate Angiogenesis 96 plate (Ibidi, Planegg, Germany), and a layer of 1-2 mm collagen gel was solidified in an incubator at 37°C overnight. Next day, collagen gel was further coated with Matrigel at 4°C overnight before the plating of the cells.
The generation of iPSC lines stably expressing Tet-inducible MyoD1
The generation of iPSC lines stably expressing Tet-inducible MyoD1 was performed as described previously20. iPSCs were dissociated into single cells with Accutase, and 1.0 × 106 cells were resuspended in Opti-MEM (Invitrogen, Carlsbad, CA). Doxycycline (Dox)-inducible MyoD1-expressing piggyBac vector, Tet-MyoD73, was co-electroporated with the piggyBac transposase vector PBaseII using NEPA 21 at 125 V and 5 ms (Nepagene, Chiba, Japan). After selecting the successfully differentiated clones from puromycin-resistant colonies, cells were established and used for experiments.
The generation of iPSC lines stably expressing CatCh+
Δ44 DMD-iPSCs, as described previously21,22, were used for the experiments. The generation of iPSC lines stably expressing Tet-inducible MyoD1 and channelrhodopsin-2 (ChR2)19 variant, CatCh+ (ChR2-L132C-T159C)42, which has an improved membrane targeting, desensitization and increased conductance of divalent cation compared with with-type ChR2, was performed as described previously20. iPSCs were dissociated into single cells with Accutase, and 1.0 × 106 cells were resuspended in Opti-MEM (Invitrogen). Tet-MyoD and CatCh+-expressing piggyBac vectors were co-electroporated with the piggyBac transposase vector PBaseII using NEPA 21 at 125 V and 5 ms (Nepagene). After selecting the successfully differentiated clones from puromycin and blasticidin-resistant colonies, cells were established and used for experiments.
CRISPR-Cas9-mediated exon 45 knockout
For the excision of exon 45 in healthy iPSCs 409B2, we utilized two sgRNAs that target the “5” and “3” ends of exon 45, as previously described22. Two sgRNAs (240 ng of each) and 1250 ng of Cas9 nuclease were transiently co-transfected into 409B2 iPSCs in wells of 24-well plates using Lipofectamine CRISPRMAX (Invitrogen). Several days after transfection, iPSC colonies were dissociated into single cells using Accutase and plated at a density of 100–1,000 cells in wells of 6-well plates to isolate single-cell clones. Each subclone was genotyped using genomic PCR to establish 409B2 ex45 KO iPSCs using the following primers: forward, 5-CAAGTTTAAAATAGCAGAAAACCACTAACTAGCCA-3 and reverse, 5-CTGACACATAAAAGGTGTCTTTCTGTCTTGTATCC-3′.
CRISPR-Cas9-mediated exon 46-47 knock-in
To insert exons 46 and 47 into DMD-iPSCs derived from a DMD patient who lacked exon 46 and 47, 5 μg of the donor vector was co-transfected with 5 μg Cas9 and 5 μg sgRNA expression plasmid vectors using NEPA21 as described previously22. Two days after electroporation, 0.5 μg/mL of puromycin selection was applied, and each puromycin-resistant colony was sub-cultured and genotyped by genomic PCR using the following sets of primers (Set 1: amplifying a fragment from upstream of the 5 arm to exon 45–46: forward, 5-GGTCTACTGTCAGCTTGCTATT −3 and reverse, 5-ATCTGCTTCCTCCAACCATAAA-3′. Set 2: amplify a fragment from exons to 45-46 to intron 45: forward, 5-AAGCCCAGAAGAGCAAGATAAA-3 and reverse, 5-AGAGAAGAATTTCAATCTGGGA −3′. Set 3: amplify a fragment from the EF1α promoter downstream of the 3 arm: forward, 5-CCAGCTTGGCACTTGATGTA −3 and reverse, 5-TGGATTAGATTGAGCCTAGTTCAG-3′. A puromycin cassette flanked by loxP sites was excised by transfection with 5 μg of the Cre expression vector, pCXW-Cre-puro, using NEPA21. Clone isolation was conducted, and excision of the puromycin-cassette was confirmed using genomic PCR with the sets of primers described above.
Skeletal muscle differentiation by the previous replating method
Replating was conducted as described previously20. iPSCs were treated with Accutase and plated on Matrigel-coated plates in StemFit+10μM Y-27632 at a density of 3.0 × 104 cells/cm2. After 24 and 48 h, the media were replaced with Primate ES Cell Medium (PECM) (Reprocell) and PECM+1 μg/mL Dox (Nacalai), respectively. The next day, pre-differentiated iPSCs were treated with Accutase and re-plated into Matrigel-coated microplates in 5% knockout serum replacement (KSR) (Thermo Fisher, Waltham, MA, USA) in αMEM (Nacalai)+Y-27632+1 μg/mL Dox. The medium was replaced every other day with fresh 5% KSR/αMEM+Y-27632+1 μg/mL Dox. To induce further myotube maturation, cells were cultured in 2% horse serum (Sigma) and αMEM. The media was replaced a few times per week.
Skeletal muscle differentiation using the modified replating method
iPSCs were treated with Accutase and plated on Matrigel-coated plates in StemFit+Y at a density of 3.0 × 104 cells/cm2. After 24 and 48 h, the media were replaced with PECM and PECM+0.3 μg/mL Dox, respectively. After an additional 48 h, pre-differentiated iPSCs were treated with Accutase and re-plated into Matrigel-coated microplates or hydrogels in 2% HS/αMEM supplemented with 200 μM 2-mercaptoethanol (Nacalai), 4.5 g/L glucose (Invitrogen), 10 μg/mL insulin (Wako, Richmond, VA), 5μM SB431542 (Wako), 20nM retinoic acid (Sigma), 20 ng/mL thyroid hormones (Sigma) +3μM Y-27632. After two days, the medium was replaced with 2% HS/αMEM+1 μg/mL Dox and changed every other day. To induce further maturation of myotubes, doxycycline was removed from the media, and the cells were cultured in 2% HS/αMEM for 3 weeks. The media were replaced every other day.
Immunocytochemistry
Cells were fixed with 2% paraformaldehyde (PFA)/PBS (Wako), methanol (Wako), blocked with Blocking One (Nacalai) for 45 min, and subsequently incubated with primary antibodies diluted in 5% Blocking One/ PBST (Wako) at 4°C overnight. Cells were washed three times in PBS and incubated with secondary antibodies diluted in 5% Blocking One/PBST for 1 h at room temperature. DAPI (Sigma) was used to counterstain the nuclei. Samples were visualized and photographed with BZ-710X (Keyence, Osaka, Japan) or the Opera Phenix System (PerkinElmer, Waltham, MA). The antibodies used for this study were as follows: mouse anti-myosin heavy chain (pan-MHC) monoclonal (MF20; 1:500, R&D, Minneapolis, MN), mouse anti-skeletal myosin (FAST/MYH1&2), monoclonal (MY-32; 1:100, Sigma), mouse anti-Myosin-2 (MYH2) monoclonal (MABT840; 1:100, Millipore, Burlington, MA), rabbit anti-MYH3 polyclonal (1:100, Atlas antibodies, Bromma, Sweden), mouse anti-MYH7 monoclonal (A4.840; 1:200, Santa Cruz, Dallas, TX), rabbit anti-MYH8 polyclonal (1:100, Novus Biologicals, Littleton, CO), rabbit anti-sarcomeric α actinin polyclonal (1:500, Abcam, Cambridge, UK), mouse anti-dystrophin (Rod domain) monoclonal (DYS1; 1:20, Leica, Buffalo Grove, IL), Alexa Fluor 488-conjugated anti-mouse/rabbit, and Alexa Fluor 647-conjugated anti-mouse/rabbit antibodies (1:500, Invitrogen).
Skeletal muscle-actinin positive fiber analysis
Cells were immunostained for α-actinin and counterstained with DAPI to visualize the nuclei. Cells were imaged with Opera Phenix, and the total numbers of α-actinin- positive fibers, which have a sarcomere-like staining pattern, were visualized. Next, the percentage of fibers with a sarcomere-like staining pattern per total number of fibers was calculated.
Fusion index analysis
Cells were immunostained for pan-MHC counterstained with DAPI to visualize the nuclei. Cells were imaged with either BZ-710X or Opera Phenix, and the total number of nuclei per MHC-positive myotube was counted. A fusion index was calculated depending on the number of nuclei in MHC-positive myotubes. The data were plotted as the percentage of myotubes with either single, double, triple, or more than four nuclei23.
RT-qPCR analysis
Total RNA was isolated using the RNeasy mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. Residual genomic DNA was digested and removed using DNase I (QIAGEN). First-strand cDNA was generated from 100 ng of total RNA using a PrimeScript RT reagent kit (Takara, Kyoto, Japan). Quantitative PCR was performed using PowerUp SYBR Green (Thermo Fisher) or TaqMan assay (Applied Biosystems, Foster City, CA, USA) and QuantStudio 7 Flex (Applied Biosystems). Ribosomal protein lateral stalk subunit P0 (RPLP) served as a reference gene for TaqMan assays, and TATA-binding protein (TBP) served as a reference gene for SYBR Green assays. The following TaqMan probes were used: MYH1 (Hs00428600), MYH2 (Hs00430042), MYH3 (Hs01074230), MYH4 (Hs00757977), MYH7 (Hs0110632), MYH8 (Hs00267293), IL-1β (Hs00174097), TNFα (Hs01113624), IL6 (Hs00985639), and RPLP0 (Hs99999902). The sequences for the primers used with SYBR Green were as follows: CKM, forward, 5-ACATGGCCAAGGTACTGACC-3, reverse, 5-TGATGGGGTCAAAGAGTTCC-3; Exo-MyoD, forward, 5-CCCCTTCACCATGGAGCTA-3, reverse, 5-AGTGCTCTTCGGGTTTCAGG-3; Endo-MyoD, forward, 5-CACTCCGGTCCCAAATGTAG-3, reverse, 5-TTCCCTGTAGCACCACACAC-3; MHC, forward, 5-GCAGATTGAGCTGGAAAAGG-3, reverse, 5-TCAGCTGCTCGATCTCTTCA-3; myogenin, forward, 5-TGGGCGTGTAAGGTGTGTAA-3′. Reverse, 5-CGATGTACTGGATGGCACTG-3; TBP, forward, 5-GCTGGCCCATAGTGATCTTT-3, reverse, 5-CATCTCCAGCACACTCTTCTC-3′.
Western blot analysis
Cells were lysed with a complete lysis-M-buffer (Roche, Basel, Switzerland) in the presence of a complete Mini protease inhibitor cocktail (Roche). Protein concentrations were determined using a BCA protein assay kit (Takara). For SDS-PAGE, 4x Laemmli sample buffer and KD mini-protean TGX precast gel with Mini-Protean Tetra Cell (Bio-Rad, Hercules, CA) were used to load samples except samples for blotting DYS1 and RYR1. The following were used for DYS1 and RYR1: 4x LDS sample buffer; 10x reducing agent and NuPAGE 3%–8%; and Tris-acetate protein precast gels with a Mini Gel Tank (Thermo Fisher). Western blot analysis was performed using the iBind Flex Western Device (Thermo Fisher), PVDF membranes (Bio-Rad), and the following primary antibodies: mouse anti-myosin heavy chain (pan-MHC) monoclonal antibody (MF20; 1:1,000, R&D), mouse anti-skeletal myosin (FAST/MYH1&2) monoclonal antibody (MY-32; 1:800, Sigma), mouse anti-Myosin-2 (MYH2) monoclonal antibody (MABT840; 1:1,000, Millipore), rabbit anti-MYH3 polyclonal antibody (1:1000, Atlas antibodies), mouse anti-MYH7 monoclonal antibody (A4.840; 1:500, Santa Cruz), rabbit anti-MYH8 polyclonal antibody (1:1,000, Novus Biologicals), rabbit anti-RYR1 monoclonal antibody (D4E1; 1:500, Cell Signaling Technology, Danvers, MA), mouse anti-myogenin monoclonal antibody (F5D; 1:200, Santa Cruz), mouse anti-dystrophin (Rod domain) monoclonal antibody (DYS1; 1:20, Leica), rabbit anti-TBP monoclonal antibody (D5C9H; 1:1000, Cell Signaling). A horseradish peroxidase-conjugated rabbit anti-mouse antibody (GE, Chicago, IL) was used as a secondary antibody, and ECL Select or Prime western blotting Detection Reagent (GE) was used for protein visualization.
ELISA
Conditioned media were harvested from each culture, and the samples were filtered using a Millex 0.22 μm, PVDF, and 33 mm filter unit (Millipore) to remove cellular debris before the assay. Filtered samples (2 mL) were concentrated using an Amicon ultra-2 centrifugal filter unit (2 mL, 10 kDa; Millipore). IL-6 and TNFα protein levels in the samples were measured using the IL-6 Quantikine ELISA kit (R&D) according to the manufacturer’s protocol. In all assays, the concentration of total proteins was used for normalization.
Caspase activity assay
Cells were collected and pelleted before the assay. Active-caspase 8 and 9 activity was determined using Caspase-Glo 8 and 9 (Promega, Madison, WI) according to the manufacturer’s protocol. Pellet samples were resuspended in 150 μL PBS; 15 μL of the samples and 15 μL of assay reagents were mixed and incubated for 20 min in 384-well solid white microplates, and the luminescence signal was measured using an Envision plate reader (PerkinElmer). Total DNA quantities were used for normalization.
cAMP assay
Cells were collected and pelleted before the assay. cAMP levels were determined using the LANCE Ultra cAMP Detection kit (PerkinElmer) according to the manufacturer’s protocol. Pellet samples were resuspended in 150 μL PBS; 5 μL of the samples were used for the assay in 384-well microplates, and the FRET signal was measured using an Envision plate reader (PerkinElmer). A standard curve was generated, and the cAMP level was calculated using GraphPad Prism.
DNA quantification
DNA content was measured based on a previously published protocol74. Cell pellets were lysed by freeze and thaw cycles in a −80°C freezer and resuspended in 150 μL PBS. DNA standards were prepared using calf thymus DNA (Sigma) and TNE buffer (TE buffer (Nacalai), 2 M NaCl). Next, 50 μL of samples and 50 μL of 200 ng/mL Hoechst 33258 were mixed and incubated in 96-well black clear-bottomed microplates. Fluorescence was measured at 360 nm excitation and 460 nm emission using an Envision plate reader (PerkinElmer). DNA concentrations were derived from a standard curve.
Calcium mobilization assay
The calcium mobilization assay was conducted as previously described21. Cells were differentiated on Matrigel-coated 96-well plates (PerkinElmer). Cells were loaded with Cal-520 AM, a fluorescent Ca2+ indicator (AAT Bioquest, Sunnyvale, CA), in FluoroBrite DMEM (Invitrogen), and incubated at 37°C for 1 h. Fluorescence was detected using an FDSS/μCELL system (Hamamatsu Photonics, Shizuoka, Japan). Electrical stimulation was applied at 15 V with a 50 ms interval and a 0.2Hz mono phase for 1 min after a 5 s resting phase. Fluorescence was measured at 480 nm excitation and 540 nm emission using an LED excitation light source and an electron-multiplying CCD (EMCCD) camera. Measurements for 96 wells were performed under uniform conditions at 37°C with simultaneous stimulation and detection.
EFS and motion imaging assays
Cells were differentiated on 6-well collagen gel plates prepared as described previously (refer “plate preparation”) and stimulated by EFS with C-Pace EP and 6-well C-Dish (IonOptics, Westwood, MA) for differentiated myotube maturation and muscle cell training under hypoxic conditions (5% O2, 5% CO2, 90% N2). The basic EFS stimulating protocol is described in Figure 2. A movie was recorded and analyzed using an SI8000 Motion Imaging System (SONY, Tokyo, Japan) equipped with an Eclipse Ti inverted fluorescent microscope (Nikon) and a Stage Top Incubator (Tokai Hit, Shizuoka, Japan) to maintain cells under humidified conditions at 37°C and 5% CO2. Six-well hydrogel or collagen gel plates with cells were cultured in an incubator equipped with a C-Dish connected to the C-Pace EP. An EFS was applied at between 2-20V with a 2 ms interval and a 0.5-1 Hz. The movie was recorded for 270 frames at a frame rate of 27 f/s (equal to 10 s) and analyzed using associated software.
Optogenetic stimulation and motion imaging assays
Cells were differentiated on μPlate Angiogenesis 96 collagen gel plates prepared as described previously (refer “plate preparation”) and stimulated with a train of pulses (470 nm; 7mw/mm2; duration, 50 ms; 0.5 Hz for continuous) using the Optogenetic LED array system (Bio Research Center, Nagoya, Japan) under hypoxic conditions (5% O2, 5% CO2, 90% N2). The optogenetic stimulating (OS) protocol is described in Figure S7C. A movie was recorded and analyzed using an SI8000 Motion Imaging System equipped with an Eclipse Ti inverted fluorescent microscope with an X-Cite XLED1 (Excelitas, Waltham, MA). A movie was recorded for a total of 270 frames at a frame rate of 27 f/s (equal to 10 s) and analyzed using associated software.
Microscopy
Cultured cells were visualized under a BZ-X710 fluorescence microscope (KeyENCE) and an Opera Phenix High-Content Screening System (PerkinElmer). Images were acquired and analyzed using associated software.
Transmission electron microscopy
Sample fixation was conducted according to the protocol provided by Tokai Electron Microscopy, Inc. Transmission electron microscopy analysis was performed by Tokai Electron Microscopy, Inc.
Quantification and statistical analysis
For all experiments, at least three independent experiments were performed in more than triplicate. Data are reported as the mean ± SD and were analyzed using an unpaired t test or one-way analysis of variance, followed post hoc by Dunnett’s test using GraphPad Prism. p < 0.05 is considered significant.
Acknowledgments
We thank Dr. Masataka Ifuku for technical assistance in establishing an ex45 knockout hiPSC line and Yukiyo Mikami, Taeko Yoshida, and Ryuichi Tozawa for performing experiments and research assistance. This work was funded by Takeda Pharmaceutical Company Limited and was partially supported by JSPS KAKENHI grant number JP18K14663 (to T.U.). A grant from The Acceleration Program for Intractable Diseases Research utilizing disease-specific iPSCs was provided by the Japan Agency for Medical Research and Development (nos. 19bm0804005h0003 and 20bm0804005h0004 to H.S. and A.H.).
Author contributions
T.U. and H.S. designed the research and wrote the manuscript. T.U. performed cell culture and cellular and molecular analyses. A.H. designed and constructed gRNAs and reagents for CRISPR-Cas9 knockin and knockout experiments. T.A., T.N., A.H., and H.S. provided advice throughout the project. All authors provided feedback on the manuscript.
Declaration of interests
The authors declare that the modified replating method with collagen gel and EFS culture protocol was patented in Japan (2018–204358). A culture protocol for the screening platform using OS has a patent pending in Japan (2020–212997). The authors also declare that T.U., A.H., and H.S. received research funding from Takeda Pharmaceutical Company Limited. The other authors declare no competing interests.
Published: June 4, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100298.
Contributor Information
Tomoya Uchimura, Email: tomoya.uchimura@cira.kyoto-u.ac.jp.
Hidetoshi Sakurai, Email: hsakurai@cira.kyoto-u.ac.jp.
Supplemental information
References
- 1.Fairclough R.J., Wood M.J., Davies K.E. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat. Rev. Genet. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 2.Le S., Yu M., Hovan L., Zhao Z., Ervasti J., Yan J. Dystrophin as a molecular shock absorber. ACS Nano. 2018;12:12140–12148. doi: 10.1021/acsnano.8b05721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 4.El-Aloul B., Speechley K.N., Wei Y., Wilk P., Campbell C. Fatigue in young people with Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2020;62:245–251. doi: 10.1111/dmcn.14248. [DOI] [PubMed] [Google Scholar]
- 5.Allen D.G., Whitehead N.P., Froehner S.C. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016;96:253–305. doi: 10.1152/physrev.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grskovic M., Javaherian A., Strulovici B., Daley G.Q. Induced pluripotent stem cells--opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011;10:915–929. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- 7.Rao L., Qian Y., Khodabukus A., Ribar T., Bursac N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 2018;9:126. doi: 10.1038/s41467-017-02636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osaki T., Uzel S.G.M., Kamm R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018;4:eaat5847. doi: 10.1126/sciadv.aat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikolić N., Görgens S.W., Thoresen G.H., Aas V., Eckel J., Eckardt K. Electrical pulse stimulation of cultured skeletal muscle cells as a model for in vitro exercise - possibilities and limitations. Acta Physiol. (Oxf.) 2017;220:310–331. doi: 10.1111/apha.12830. [DOI] [PubMed] [Google Scholar]
- 10.Maffioletti S.M., Sarcar S., Henderson A.B.H., Mannhardt I., Pinton L., Moyle L.A., Steele-Stallard H., Cappellari O., Wells K.E., Ferrari G. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 2018;23:899–908. doi: 10.1016/j.celrep.2018.03.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee I.H., Lee Y.J., Seo H., Kim Y.S., Nam J.O., Jeon B.D., Kwon T.D. Study of muscle contraction induced by electrical pulse stimulation and nitric oxide in C2C12 myotube cells. J. Exerc. Nutrition Biochem. 2018;22:22–28. doi: 10.20463/jenb.2018.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park H., Bhalla R., Saigal R., Radisic M., Watson N., Langer R., Vunjak-Novakovic G. Effects of electrical stimulation in C2C12 muscle constructs. J. Tissue Eng. Regen. Med. 2008;2:279–287. doi: 10.1002/term.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manabe Y., Miyatake S., Takagi M., Nakamura M., Okeda A., Nakano T., Hirshman M.F., Goodyear L.J., Fujii N.L. Characterization of an acute muscle contraction model using cultured C2C12 myotubes. PLoS ONE. 2012;7:e52592. doi: 10.1371/journal.pone.0052592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nedachi T., Fujita H., Kanzaki M. Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008;295:E1191–E1204. doi: 10.1152/ajpendo.90280.2008. [DOI] [PubMed] [Google Scholar]
- 15.Langelaan M.L., Boonen K.J., Rosaria-Chak K.Y., van der Schaft D.W., Post M.J., Baaijens F.P. Advanced maturation by electrical stimulation: differences in response between C2C12 and primary muscle progenitor cells. J. Tissue Eng. Regen. Med. 2011;5:529–539. doi: 10.1002/term.345. [DOI] [PubMed] [Google Scholar]
- 16.Ito A., Yamamoto Y., Sato M., Ikeda K., Yamamoto M., Fujita H., Nagamori E., Kawabe Y., Kamihira M. Induction of functional tissue-engineered skeletal muscle constructs by defined electrical stimulation. Sci. Rep. 2014;4:4781. doi: 10.1038/srep04781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W., Nyasha M.R., Koide M., Tsuchiya M., Suzuki N., Hagiwara Y., Aoki M., Kanzaki M. In vitro exercise model using contractile human and mouse hybrid myotubes. Sci. Rep. 2019;9:11914. doi: 10.1038/s41598-019-48316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagel G., Szellas T., Huhn W., Kateriya S., Adeishvili N., Berthold P., Ollig D., Hegemann P., Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asano T., Ishizuka T., Morishima K., Yawo H. Optogenetic induction of contractile ability in immature C2C12 myotubes. Sci. Rep. 2015;5:8317. doi: 10.1038/srep08317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchimura T., Otomo J., Sato M., Sakurai H. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. (Amst.) 2017;25:98–106. doi: 10.1016/j.scr.2017.10.023. [DOI] [PubMed] [Google Scholar]
- 21.Shoji E., Sakurai H., Nishino T., Nakahata T., Heike T., Awaya T., Fujii N., Manabe Y., Matsuo M., Sehara-Fujisawa A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015;5:12831. doi: 10.1038/srep12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T., Tanaka M., Amano N., Watanabe A., Sakurai H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millay D.P., O’Rourke J.R., Sutherland L.B., Bezprozvannaya S., Shelton J.M., Bassel-Duby R., Olson E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature. 2013;499:301–305. doi: 10.1038/nature12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiaffino S., Reggiani C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 25.Griggs R.C., Miller J.P., Greenberg C.R., Fehlings D.L., Pestronk A., Mendell J.R., Moxley R.T., 3rd, King W., Kissel J.T., Cwik V. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87:2123–2131. doi: 10.1212/WNL.0000000000003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Previtali S.C., Gidaro T., Díaz-Manera J., Zambon A., Carnesecchi S., Roux-Lombard P., Spitali P., Signorelli M., Szigyarto C.A., Johansson C. Rimeporide as a ðrst- in-class NHE-1 inhibitor: Results of a phase Ib trial in young patients with Duchenne muscular dystrophy. Pharmacol. Res. 2020;159:104999. doi: 10.1016/j.phrs.2020.104999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghaleh B., Barthélemy I., Wojcik J., Sambin L., Bizé A., Hittinger L., Tran T.D., Thomé F.P., Blot S., Su J.B. Protective effects of rimeporide on left ventricular function in golden retriever muscular dystrophy dogs. Int. J. Cardiol. 2020;312:89–95. doi: 10.1016/j.ijcard.2020.03.031. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy T.L., Swiderski K., Murphy K.T., Gehrig S.M., Curl C.L., Chandramouli C., Febbraio M.A., Delbridge L.M., Koopman R., Lynch G.S. BGP-15 improves aspects of the dystrophic pathology in mdx and dko mice with differing efficacies in heart and skeletal muscle. Am. J. Pathol. 2016;186:3246–3260. doi: 10.1016/j.ajpath.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Buyse G.M., Van der Mieren G., Erb M., D’hooge J., Herijgers P., Verbeken E., Jara A., Van Den Bergh A., Mertens L., Courdier-Fruh I. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur. Heart J. 2009;30:116–124. doi: 10.1093/eurheartj/ehn406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krause T., Gerbershagen M.U., Fiege M., Weisshorn R., Wappler F. Dantrolene--a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- 31.Ward C.W., Sachs F., Bush E.D., Suchyna T.M. GsMTx4-D provides protection to the D2.mdx mouse. Neuromuscul. Disord. 2018;28:868–877. doi: 10.1016/j.nmd.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maruyama T., Kanaji T., Nakade S., Kanno T., Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y., Erdodi F., Murányi A., Nullmeyer K.D., Lynch R.M., Hartshorne D.J. Myosin phosphatase and myosin phosphorylation in differentiating C2C12 cells. J. Muscle Res. Cell Motil. 2003;24:499–511. doi: 10.1023/b:jure.0000009810.36038.53. [DOI] [PubMed] [Google Scholar]
- 34.Felber S., Skladal D., Wyss M., Kremser C., Koller A., Sperl W. Oral creatine supplementation in Duchenne muscular dystrophy: a clinical and 31P magnetic resonance spectroscopy study. Neurol. Res. 2000;22:145–150. doi: 10.1080/01616412.2000.11741051. [DOI] [PubMed] [Google Scholar]
- 35.Tarnopolsky M.A., Mahoney D.J., Vajsar J., Rodriguez C., Doherty T.J., Roy B.D., Biggar D. Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology. 2004;62:1771–1777. doi: 10.1212/01.wnl.0000125178.18862.9d. [DOI] [PubMed] [Google Scholar]
- 36.Capogrosso R.F., Mantuano P., Uaesoontrachoon K., Cozzoli A., Giustino A., Dow T., Srinivassane S., Filipovic M., Bell C., Vandermeulen J. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: proof-of-concept study and independent validation of efficacy. FASEB J. 2018;32:1025–1043. doi: 10.1096/fj.201700182RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estève E., Eltit J.M., Bannister R.A., Liu K., Pessah I.N., Beam K.G., Allen P.D., López J.R. A malignant hyperthermia-inducing mutation in RYR1 (R163C): alterations in Ca2+ entry, release, and retrograde signaling to the DHPR. J. Gen. Physiol. 2010;135:619–628. doi: 10.1085/jgp.200910328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandenburgh H., Shansky J., Benesch-Lee F., Skelly K., Spinazzola J.M., Saponjian Y., Tseng B.S. Automated drug screening with contractile muscle tissue engineered from dystrophic myoblasts. FASEB J. 2009;23:3325–3334. doi: 10.1096/fj.09-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oláh T., Fodor J., Ruzsnavszky O., Vincze J., Berbey C., Allard B., Csernoch L. Overexpression of transient receptor potential canonical type 1 (TRPC1) alters both store operated calcium entry and depolarization-evoked calcium signals in C2C12 cells. Cell Calcium. 2011;49:415–425. doi: 10.1016/j.ceca.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Tenorio M., Niggli E. Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 2018;119:87–95. doi: 10.1016/j.yjmcc.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 41.Huang C.L. SERCA2a stimulation by istaroxime: a novel mechanism of action with translational implications. Br. J. Pharmacol. 2013;170:486–488. doi: 10.1111/bph.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prigge M., Schneider F., Tsunoda S.P., Shilyansky C., Wietek J., Deisseroth K., Hegemann P. Color-tuned channelrhodopsins for multiwavelength optogenetics. J. Biol. Chem. 2012;287:31804–31812. doi: 10.1074/jbc.M112.391185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed G.F., Lynn F., Meade B.D. Use of coefficient of variation in assessing variability of quantitative assays. Clin. Diagn. Lab. Immunol. 2002;9:1235–1239. doi: 10.1128/CDLI.9.6.1235-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engler A.J., Griffin M.A., Sen S., Bönnemann C.G., Sweeney H.L., Discher D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004;166:877–887. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henríquez-Olguín C., Altamirano F., Valladares D., López J.R., Allen P.D., Jaimovich E. Altered ROS production, NF-κB activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. Biochim. Biophys. Acta. 2015;1852:1410–1419. doi: 10.1016/j.bbadis.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furuichi Y., Manabe Y., Takagi M., Aoki M., Fujii N.L. Evidence for acute contraction-induced myokine secretion by C2C12 myotubes. PLoS ONE. 2018;13:e0206146. doi: 10.1371/journal.pone.0206146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacourpaille L., Gross R., Hug F., Guével A., Péréon Y., Magot A., Hogrel J.Y., Nordez A. Effects of Duchenne muscular dystrophy on muscle stiffness and response to electrically-induced muscle contraction: A 12-month follow-up. Neuromuscul. Disord. 2017;27:214–220. doi: 10.1016/j.nmd.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Nesmith A.P., Wagner M.A., Pasqualini F.S., O’Connor B.B., Pincus M.J., August P.R., Parker K.K. A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J. Cell Biol. 2016;215:47–56. doi: 10.1083/jcb.201603111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu K., Genma R., Gotou Y., Nagasaka S., Honda H. Three-dimensional culture model of skeletal muscle tissue with atrophy induced by dexamethasone. Bioengineering (Basel) 2017;4:E56. doi: 10.3390/bioengineering4020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mutlu A., Alkan H., Fırat T., Karaduman A.A., Yilmaz O.T. How do physical capacity, fatigue and performance differ in children with duchenne muscular dystrophy compared with their healthy peers? Neurosciences (Riyadh) 2018;23:39–45. doi: 10.17712/nsj.2018.1.20170242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellinger A.M., Mongillo M., Marks A.R. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J. Clin. Invest. 2008;118:445–453. doi: 10.1172/JCI34006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi I.Y., Lim H., Estrellas K., Mula J., Cohen T.V., Zhang Y., Donnelly C.J., Richard J.P., Kim Y.J., Kim H. Concordant but varied phenotypes among Duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 2016;15:2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 53.Rosenberg A.S., Puig M., Nagaraju K., Hoffman E.P., Villalta S.A., Rao V.A., Wakefield L.M., Woodcock J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015;7:299rv4. doi: 10.1126/scitranslmed.aaa7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bellinger A.M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., Marks A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Millay D.P., Goonasekera S.A., Sargent M.A., Maillet M., Aronow B.J., Molkentin J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA. 2009;106:19023–19028. doi: 10.1073/pnas.0906591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kameyama T., Ohuchi K., Funato M., Ando S., Inagaki S., Sato A., Seki J., Kawase C., Tsuruma K., Nishino I. Efficacy of prednisolone in generated myotubes derived from fibroblasts of Duchenne muscular dystrophy patients. Front. Pharmacol. 2018;9:1402. doi: 10.3389/fphar.2018.01402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans N.P., Misyak S.A., Robertson J.L., Bassaganya-Riera J., Grange R.W. Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R. 2009;1:755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Menazza S., Blaauw B., Tiepolo T., Toniolo L., Braghetta P., Spolaore B., Reggiani C., Di Lisa F., Bonaldo P., Canton M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010;19:4207–4215. doi: 10.1093/hmg/ddq339. [DOI] [PubMed] [Google Scholar]
- 59.Boldrin L., Zammit P.S., Morgan J.E. Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. (Amst.) 2015;14:20–29. doi: 10.1016/j.scr.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dumont N.A., Rudnicki M.A. Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. NPJ Regen. Med. 2016;1:16006. doi: 10.1038/npjregenmed.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaglianone R.B., Santos A.T., Bloise F.F., Ortiga-Carvalho T.M., Costa M.L., Quirico-Santos T., da Silva W.S., Mermelstein C. Reduced mitochondrial respiration and increased calcium deposits in the EDL muscle, but not in soleus, from 12-week-old dystrophic mdx mice. Sci. Rep. 2019;9:1986. doi: 10.1038/s41598-019-38609-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vila M.C., Rayavarapu S., Hogarth M.W., Van der Meulen J.H., Horn A., Defour A., Takeda S., Brown K.J., Hathout Y., Nagaraju K., Jaiswal J.K. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017;24:330–342. doi: 10.1038/cdd.2016.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selvaraj S., Mondragon-Gonzalez R., Xu B., Magli A., Kim H., Lainé J., Kiley J., Mckee H., Rinaldi F., Aho J. Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. eLife. 2019;8:e47970. doi: 10.7554/eLife.47970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afshar M.E., Abraha H.Y., Bakooshli M.A., Davoudi S., Thavandiran N., Tung K., Ahn H., Ginsberg H.J., Zandstra P.W., Gilbert P.M. A 96-well culture platform enables longitudinal analyses of engineered human skeletal muscle microtissue strength. Sci. Rep. 2020;10:6918. doi: 10.1038/s41598-020-62837-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 66.Flanigan K.M., Dunn D.M., von Niederhausern A., Soltanzadeh P., Gappmaier E., Howard M.T., Sampson J.B., Mendell J.R., Wall C., King W.M., United Dystrophinopathy Project Consortium Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009;30:1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hicks M.R., Hiserodt J., Paras K., Fujiwara W., Eskin A., Jan M., Xi H., Young C.S., Evseenko D., Nelson S.F. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol. 2018;20:46–57. doi: 10.1038/s41556-017-0010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shelton M., Metz J., Liu J., Carpenedo R.L., Demers S.P., Stanford W.L., Skerjanc I.S. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Reports. 2014;3:516–529. doi: 10.1016/j.stemcr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao M., Tazumi A., Takayama S., Takenaka-Ninagawa N., Nalbandian M., Nagai M., Nakamura Y., Nakasa M., Watanabe A., Ikeya M. Induced fetal human muscle stem cells with high therapeutic potential in a mouse muscular dystrophy model. Stem Cell Reports. 2020;15:80–94. doi: 10.1016/j.stemcr.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nalbandian M., Zhao M., Sasaki-Honda M., Jonouchi T., Lucena-Cacace A., Mizusawa T., Yasuda M., Yoshida Y., Hotta A., Sakurai H. Characterization of hiPSC-derived muscle progenitors reveals distinctive markers for myogenic cell purification toward cell therapy. Stem Cell Reports. 2021;16:883–898. doi: 10.1016/j.stemcr.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.. Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 72.Oceguera-Yanez F., Kim S.I., Matsumoto T., Tan G.W., Xiang L., Hatani T., Kondo T., Ikeya M., Yoshida Y., Inoue H., Woltjen K. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods. 2016;101:43–55. doi: 10.1016/j.ymeth.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka A., Woltjen K., Miyake K., Hotta A., Ikeya M., Yamamoto T., Nishino T., Shoji E., Sehara-Fujisawa A., Manabe Y. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi myopathy in vitro. PLoS ONE. 2013;8:e61540. doi: 10.1371/journal.pone.0061540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hasan M.S., Ahmed I., Parsons A., Walker G., Scotchford C. Cytocompatibility and mechanical properties of short phosphate glass fibre reinforced polylactic acid (PLA) composites: effect of coupling agent mediated interface. J. Funct. Biomater. 2012;3:706–725. doi: 10.3390/jfb3040706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A representative bright-field video of DMDΔ44-ctrl after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of DMDΔ44 after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of 409B2 after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of 409B2 ex45KO after 17 days of differentiation exhibiting continuous contractions under EFS. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
A representative bright-field video of DMDΔ44 after 17 days of differentiation exhibiting continuous contractions under optogenetic stimulations. The movie was recorded using an SI8000 motion imaging system equipped with an inverted microscope.
Data Availability Statement
This study did not generate/analyze [datasets/code].