

Summary
Necrotizing enterocolitis (NEC) is a deadly intestinal inflammatory disorder that primarily affects premature infants and lacks adequate therapeutics. Interleukin (IL)-22 plays a critical role in gut barrier maintenance, promoting epithelial regeneration, and controlling intestinal inflammation in adult animal models. However, the importance of IL-22 signaling in neonates during NEC remains unknown. We investigated the role of IL-22 in the neonatal intestine under homeostatic and inflammatory conditions by using a mouse model of NEC. Our data reveal that Il22 expression in neonatal murine intestine is negligible until weaning, and both human and murine neonates lack IL-22 production during NEC. Mice deficient in IL-22 or lacking the IL-22 receptor in the intestine display a similar susceptibility to NEC, consistent with the lack of endogenous IL-22 during development. Strikingly, treatment with recombinant IL-22 during NEC substantially reduces inflammation and enhances epithelial regeneration. These findings may provide a new therapeutic strategy to attenuate NEC.
Keywords: interleukin-22, necrotizing enterocolitis, epithelial cells, microbiome, regeneration, intestinal immunity, neonatal
Graphical abstract
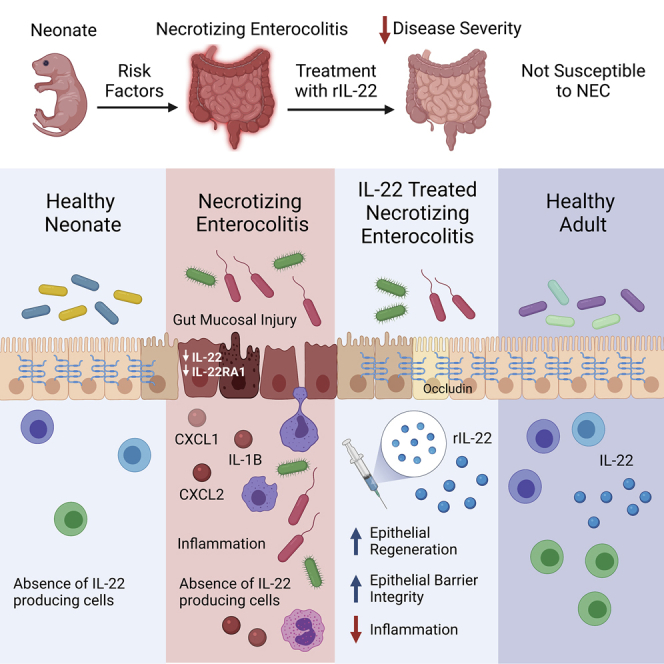
Highlights
Intestinal prematurity is characterized by the lack of efficient IL-22 production
rIL-22 enhances the regeneration and integrity of intestinal epithelium during NEC
rIL-22-driven antimicrobial response does not affect neonatal microbiome composition
Necrotizing enterocolitis (NEC) is a deadly intestinal disease with limited treatment options. Interleukin (IL)-22 plays a role in controlling intestinal inflammation in adult animal models of gastrointestinal disease. Mihi et al. demonstrate that IL-22 can attenuate intestinal inflammation by promoting epithelial regeneration in a neonatal mouse model of NEC.
Introduction
The length of gestation in mammals has evolved to allow appropriate fetal growth suitable for postnatal life. Shortened in utero development, such as in the context of prematurity, exposes the newborns to a plethora of pathologies inherent to the immaturity of their organs.1,2 One such devastating disease is necrotizing enterocolitis (NEC), one of the most prevalent and lethal neonatal emergencies associated with prematurity.3 NEC is an intestinal inflammatory disorder characterized by the impairment of the epithelial barrier and leading, in the most extreme cases, to intestinal perforation and tissue necrosis, which require surgical intervention.3 Despite decades of intensive research, the pathogenesis of NEC remains obscure, thus hampering the development of effective therapeutic strategies and leaving the clinicians in the neonatal intensive care unit (NICU) unarmed against this deadly disease. It is widely accepted that NEC is triggered by dysbiosis of colonizing microorganisms, which invade the immature intestine of premature neonates.4,5 In the setting of prematurity, intestinal epithelial cells express high levels of Toll-like receptor 4 (TLR4), rendering them prone to the induction of an exaggerated inflammatory response upon colonization with microorganisms.6 Several studies have demonstrated that the onset of NEC is preceded by a significant increase of Gammaproteobacteria.7, 8, 9 More specifically, a recent metagenomic study conducted by Olm et al. revealed that an abundance of Klebsiella and Fimbriae-encoding bacteria are present in the stools of infants prior to the clinical symptoms of NEC.9 However, the impact of these changes on intestinal homeostasis and the integrity of the intestinal mucosa remains to be explored.
Interleukin-22 (IL-22), a cytokine belonging to the IL-10 family, is an essential modulator of intestinal microbial communities and can control intestinal homeostasis by preventing harmful inflammatory responses in adults.10 IL-22 is mainly produced by CD4+ T cells and group 3 innate lymphoid cells (ILC3s) and promotes the expression of several antimicrobial factors, including regenerating islet-derived proteins (Reg) REG3B and REG3G, β-defensins, and S100 proteins.11, 12, 13, 14 These antimicrobial proteins shape the intestinal microbiome to prevent the development of chronic inflammatory disorders and contribute to the clearance of harmful pathogens.12, 13, 14 Similarly, by enhancing the fucosylation of epithelial cells,15, 16, 17 IL-22 obviates the expansion of the opportunistic pathogen Enterococcus faecalis and dampens the expression of bacterial virulence factors during murine experimental colitis.16,17
In addition to the prevention of intestinal inflammation through the modulation of the microbiome, IL-22 can stimulate epithelial cell regeneration following intestinal insults via the proliferation of epithelial cells and the activation of anti-apoptotic pathways in adult animals.18, 19, 20, 21, 22 While the importance of IL-22 in protecting against adult intestinal inflammation has been extensively documented, the role of IL-22 in neonates has not been widely investigated. Investigators have shown that an influx of IL-22-producing ILC3s can provide resistance against neonatal pneumonia.23 Moreover, IL-22 reduced pancreatic enzyme expression, leading to the lethality and growth retardation of newborn IL-23 transgenic mice.24 Interestingly, Shindo et al. found that mice carrying a CFLAR transgene encoding for the caspase-8/FADD-like IL-1β-converting enzyme (FLICE)-inhibitory proteins that regulate cell-death-receptor-mediated apoptosis displayed severe neonatal ileitis.25 They further discovered that activated ILC3s produced IL-22 and were responsible for apoptosis, but not necroptosis, of intestinal epithelial cells.25 Taken together, these reports highlight the versatile function of IL-22 in different neonatal pathological settings; however, the role of IL-22 in the context of NEC has not been studied. In the current study, we aimed to investigate the role of IL-22 signaling during neonatal NEC.
Results
The neonatal period is characterized by a lack of IL-22 production in the intestine
To unravel the role of IL-22 in the pathogenesis of NEC, we first determined the expression of Il22 in the small intestine of mice throughout the first weeks of life. We demonstrated that the pre-and postnatal expression of Il22 in the terminal ileum is relatively low and starts to increase at the time of weaning (Figure 1A). In addition, the expression of the IL-22 receptor subunit alpha 1 (IL22ra1) slightly increased during development (Figure 1B). These data were further confirmed by assessing the percentage of IL-22-producing cells in the small intestine lamina propria (LP) of neonatal pups and adult mice. The percentages of CD45+ cells that express IL-22 in LP of newborn mice were significantly lower (0.03% ± 0.01%) than those found in the LP of adult mice (1.17% ± 0.94%) (Figures 1C, 1D, and S1). Interestingly, we found that the damaged terminal ileum of neonatal mice subjected to an experimental NEC protocol through formula feeding and hypoxia treatment (NEC) did not exhibit increased expression levels of Il22 when compared to the dam-fed (DF) pups (Figures 2A and 2B). We also found that the induction of NEC in mouse pups was associated with a significant reduction of the IL-22 receptor expression (Figure 2B). On the other hand, the analysis of human NEC intestinal samples did not reveal any difference in IL-22RA1 expression between NEC and non-NEC (control) groups (Figure 2D). These data suggest that intestinal prematurity is associated with the inability to efficiently produce IL-22 under both homeostatic and inflammatory conditions.
Figure 1.

Neonatal intestinal immune cells display low levels of IL-22 production
(A and B) Il22 (A) and Il22ra1 (B) gene expression was quantified in the terminal ileum of embryonic day 15 (e15) and e17, as well as postnatal days 1, 4, 7, 14, 21, 28, and 49, mice (n = 3 embryonic mice, n = 10 postnatal mice as biological replicates per group).
(C) IL-22 intracellular staining of LP cells isolated from the small intestine of 1- and 8-week-old mice. Data are representative of two independent experiments.
(D) Percentage of CD45+IL22+ cells in the small intestine.
Data are displayed as mean ± SEM. Results were analyzed using a Mann-Whitney test. ∗∗p < 0.01.
Figure 2.

IL-22 signaling deficiency does not increase NEC susceptibility
(A) Representative histology of the small intestine of dam-fed (DF) pups (left panel) or pups that have been subjected to a NEC protocol (NEC, right panel). Scale bar, 200 μm.
(B) Il22 and Il22ra1 expression in the terminal ileum of DF mice and pups subjected to experimental NEC.
(C) Representative histology of non-NEC control and NEC human infant small intestine. Scale bar, 200 μm.
(D) Expression levels of IL22 (left panel) or IL22RA1 (right panel) in the ileum of infants with NEC and non-NEC controls.
(E) Effect of intestinal epithelial-specific IL22ra1 deficiency on Il1b expression in the intestine of DF pups and mice subjected to experimental NEC. Data were combined from independent experiments.
(F) Representative H&E staining of terminal ileum sections from each genotype. Scale bar, 200 μm.
(G) Histological score of the intestinal lesions in pups subjected to NEC (n = 11–15 per group). Histological scores are displayed as mean ± SEM as the percentage of lesion extent from the total length of the entire tissue section.
(H) Effect of IL-22 deficiency on Il1b expression in the intestine of DF pups and neonatal mice subjected to NEC. Data were combined from at least three independent experiments.
(I) Representative H&E staining of terminal ileum sections from each genotype. Scale bar, 200 μm.
(J) Histological score of the intestinal lesions in the experimental NEC pups (n = 7–8 per group).
All data are presented as mean ± SEM percentage of lesion extent from the total length of the tissue section. All gene expression data are displayed as mean ± SEM and were analyzed using a Mann-Whitney test. ∗∗p < 0.01, ∗∗∗p < 0.001.
Deficiency in intestinal epithelial IL-22 signaling does not affect the susceptibility to NEC
Previous studies have shown that IL-22 deficiency leads to increased susceptibility of adult mice to intestinal inflammation.12, 13, 14, 15, 16, 17 Given that experimental NEC was not associated with an upregulation of Il22, we reasoned that a deficiency in IL-22 signaling might not increase the severity of experimental NEC. To test this hypothesis, we subjected newborn pups lacking the expression of the Il22 receptor in intestinal epithelial cells (IL-22ra1fl/fl; Vil cre+) and their wild-type (WT) littermates (IL-22ra1fl/fl; Vil cre−) to experimental NEC. Induction of experimental NEC resulted in a significant upregulation of Il1b in WT mice compared to DF mice (Figure 2E). In addition, mice subjected to experimental NEC that carried a conditional deletion of Il22ra1, specifically in intestinal epithelial cells (IL-22ra1fl/fl; Vil cre+), had similar expression levels of Il1b to their WT littermates (IL-22ra1fl/fl; Vil cre−; Figure 2E). Histological assessment of NEC-like small intestinal injury showed that the disease severity was similar in IL-22ra1fl/fl Vil cre+ and IL-22ra1fl/fl Vil cre− mice (Figures 2F and 2G). We next sought to investigate the effect of IL-22 germline deletion on the development of NEC. In accordance with our previous observations, IL-22 deficiency did not alter the susceptibility of neonatal mice to NEC, as IL-22KO mice exhibited similar small intestine expression of Il1b (Figure 2H) and mucosal injury as compared to mice in the WT group (Figures 2I and 2J). Interestingly, WT and IL-22 KO pups did not display a significant difference in epithelial cell proliferation during NEC (Figure S2). Taken together, these data demonstrate that IL-22 deficiency does not affect the outcome of NEC in newborn mice, likely because of the inefficiency of the neonatal immune cell compartment to mount an appropriate IL-22-mediated anti-inflammatory response with IL-22 to counteract the detrimental inflammation.
rIL-22 administration reduces NEC severity and enhances epithelial barrier regeneration
Given the inability of neonates to mount an efficient IL-22 response during NEC, we asked whether treatment with IL-22 could protect against the development of the disease. In mice subjected to NEC, those treated with recombinant murine IL-22 (rIL-22) displayed significantly lower mRNA levels of the pro-inflammatory markers Il1b and C-X-C motif chemokine ligand 2 (Cxcl2) compared to littermates injected with phosphate-buffered saline (PBS) (Figure 3A). Moreover, we found that IL-22 treatment was associated with a significant reduction of IL-1β production in ileal samples when compared to the PBS control group (Figure 3A). On the other hand, the expression levels of Cxcl1 in the intestines of IL-22-treated pups were similar to those in the vehicle control group (Figure 3A). In addition to the reduction of inflammatory factors, IL-22 treatment resulted in reduced disease severity, as shown by the decreased percentage of mucosal damage in comparison to PBS-treated pups (Figures 3B and 3C). To determine whether the protective role of rIL-22 is mediated through the activation of IL-22 signaling in epithelial cells, we experimentally induced NEC in IL-22ra1fl/fl; Vil cre+ and IL-22ra1fl/fl; Vil cre− pups along with IL-22 or PBS treatments (Figure S3). In line with our hypothesis, we found that IL-22 treatment in WT pups (IL-22ra1fl/fl; Vil cre−) resulted in a significant downregulation of Il1b and Cxcl2 (Figures S3A and S3B) and improved histologic appearance (Figure S3C) compared to the PBS control group. In contrast, the deletion of the Il22ra1 gene in IL-22ra1fl/fl; Vil cre+ pups resulted in the suppression of IL-22-mediated protection, as suggested by the similar expression levels of Il1b and Cxcl2 (Figures S3A and S3B) and the similar appearance of epithelial destruction seen in the rIL-22- and PBS-treated pups (Figure S3C).
Figure 3.

IL-22 treatment attenuates experimental NEC by enhancing epithelial cell regeneration
(A) Four-day-old pups were either subjected to the NEC protocol or kept with the dams as controls for 3 days. The pups received a daily i.p. injection of recombinant IL-22 (rIL-22, 100 μg/kg) or an equivalent volume of PBS. Expression levels of Il1b, Cxcl1, and Cxcl2 were measured in the terminal ileum of the pups after 3 days of PBS or IL-22 treatment. Data were combined from at least three independent experiments.
(B) Representative H&E staining of terminal ileum sections from each experimental group. Scale bar, 200 μm.
(C) Histological score of the intestinal lesions in pups subjected to NEC and either PBS or rIL-22 injections (n = 6–9 per group). Data are presented as mean ± SEM percentage of lesion extent from the total length of the tissue section.
(D) Representative confocal images of ileal samples of pups with NEC and indicated treatments stained with antibodies against proliferating cell nuclear antigen (PCNA, green) and Olfactomedin 4 (OLFM4, red). The nuclei were stained with Hoechst (blue). Scale bar, 50 μm.
(E) Quantification of epithelial cell proliferation in the terminal ilea of pups with NEC treated with rIL-22 or PBS.
All gene expression data are displayed as mean ± SEM and were analyzed using Mann-Whitney test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
In light of published reports that demonstrate that IL-22 protects the intestine of adult mice through the enhancement of epithelial cell regeneration,20 we sought to assess the impact of IL-22 treatment on the regeneration of the neonatal epithelial barrier. Immunostaining of the ileum revealed that IL-22 treatment promotes a significant expansion of proliferating cell nuclear antigen (PCNA)+ cells (Figures 3D and 3E), whereas the number of Olfactomedin 4 (OLFM4+) stem cells was unchanged (Figure 3D). These data suggest that treatment with IL-22 attenuates experimental NEC by promoting the proliferation of epithelial cells and improving epithelial regeneration. To further understand the impact of IL-22 treatment on the intestinal epithelial barrier, we have also analyzed the integrity of the tight junctions (TJs) in the intestinal epithelium through immunofluorescent staining of the intestinal specimens with anti-occludin antibody (Figure S4). The cross-sectional images of the epithelial cells in DF pups showed a clear membrane localization of occludin. A similar staining pattern was observed in intestinal epithelial cells of pups subjected to NEC and treated with IL-22. On the other hand, the occludin staining of epithelial cells in the PBS-treated NEC group displayed a more diffused cytoplasmic localization, suggesting impairment of the TJs. Taken together, our data demonstrate that in addition to the enhancement of epithelial cell regeneration, IL-22 treatment protects the integrity of the intestinal epithelial barrier during NEC.
IL-22 treatment triggers an antimicrobial transcriptional program in the neonatal small intestine
To better understand the cellular modifications initiated by IL-22, we investigated the differential gene expressions induced by IL-22 treatment in intestinal enteroids and compared them to those observed in the small intestine of adult mice lacking IL-22ra1 in the epithelial cell compartment (IL-22ra1fl/fl; Vil cre+ versus IL-22ra1fl/fl; Vil cre−). RNA sequencing transcriptional analysis showed that treatment of enteroids with rIL-22 triggers an antimicrobial response in vitro and in vivo, as shown by the upregulation of several genes such as Retnlb, Reg3g, Fut2, and S100 genes, which are all known to be involved in the regulation of the microbiome (Figure 4A). To validate these findings, we treated enteroids cultured from the small intestine of neonatal mice with rIL-22 and found that treatment upregulated Retnlb, Reg3g, and Fut2 (Figure 4B). We also found that IL-22 treatment of neonatal mice leads to a significant upregulation of antimicrobial genes including Retnlb, Reg3g, and Fut2 in both DF pups and pups subjected to experimental NEC (Figure 4C). Similarly, IL-22 treatment of human enteroids cultured from premature infant ileum led to an upregulation of REG3G, whereas it slightly reduced the expression of RETNLB (Figure 4D). These data indicate that IL-22 treatment can trigger an antimicrobial response in the small intestine of neonatal mice and premature infants.
Figure 4.

IL-22 treatment induces an antimicrobial transcriptional program in the small intestine of neonates
(A) Heatmap analysis showing the differential gene expression of adult mouse small intestinal enteroids cultured in the presence of rIL-22 (100 ng/mL) or an equivalent volume of PBS for 24 h (left column) as well as the small intestine of adult of IL22ra1fl/fl Vil cre+ and IL22ra1fl/fl Vil cre− mice (right column).
(B) qPCR quantification of Retnlb, Reg3g, and Fut2 expression in neonatal mouse intestinal enteroids cultured in the presence of rIL-22 (2 ng/mL) or an equivalent volume of PBS for 24 h. Data are representative of three independent experiments.
(C) Impact of IL-22 treatment on ileal expression of Retnlb, Reg3g, and Fut2 in DF pups and pups subjected to NEC. All gene expression data are displayed as mean ± SEM and were analyzed using Mann-Whitney test. Data were combined from at least three independent experiments.
(D) Gene expression of RETNLB and REG3G in human neonatal intestinal enteroids treated for 24 h with PBS, 1, 10, or 100 ng/mL of human rIL-22. Unpaired Student’s t test was used for this analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
IL-22 treatment is not associated with a modification of intestinal microbial communities during experimental NEC
Our data showed that IL-22 treatment upregulates the antimicrobial peptide Reg3g in the intestinal epithelium. Reg3g has been shown to play an essential role in protecting the intestinal mucosa of adult mice via the modulation of different bacterial species.12,13 Therefore, we next assessed the impact of Reg3g treatment on the severity of experimental NEC. We observed no difference in small intestine Il1b expression in the pups subjected to NEC that received PBS injections when compared to pups treated with recombinant Reg3g (rReg3g) (Figure 5A). Moreover, the histological analysis revealed no impact of Reg3g treatment on intestinal injury, suggesting that Reg3g antimicrobial activity is not required for the IL-22-mediated protection against NEC (Figures 5B and 5C). In a complementary approach, pups were given a Reg3g neutralizing antibody or an isotype control via oral gavage along with an intraperitoneal (i.p.) injection of IL-22 or PBS (Figure S5). In line with our previous findings, our data showed that the neutralization of Reg3g did not abrogate the protection mediated by IL-22, as shown by the similar expression levels of Il1b in the pups with experimental NEC treated with rIL-22 or PBS. Taken together, these data suggest that Reg3g antimicrobial activity is not required for IL-22-mediated protection against NEC. We next performed a genomic analysis of microbial composition and diversity to gain further insight into the impact of the IL-22-driven antimicrobial response on the composition of the microbiome. As expected, the most frequent species detected in the experimental NEC groups belong to the Enterobacter genus, with Enterobacter cloacae being the most abundant species (Figure 5D). This is consistent with the fact that Enterobacteriaceae is the predominant species in the intestinal content isolated from the infant with severe NEC that has been used to induce a NEC-like disease in our neonatal mouse model.26 The comparison of the microbial species between the IL-22-treated group and the PBS control pups that were subjected to NEC did not show any significant differences (Figure 5D). These data were further confirmed by the calculation of the Shannon, Chao1, and Simpson indices, which reflect the diversity and the richness of microbial species (Figures 5E and 5F). These parameters were not significantly altered by IL-22 treatment when compared to the experimental NEC group treated with PBS. Our data revealed no difference in the relative abundance of Enterobacter, Escherichia, or Klebsiella species between rIL-22- and PBS-treated groups during experimental NEC (Figure S6). Taken together, these data demonstrate that IL-22-mediated protection against NEC is not associated with an alteration of intestinal microbial composition in an experimental model of NEC and suggest that it is independent of the IL-22-driven antimicrobial response.
Figure 5.

Antimicrobial response initiated by IL-22 in the small intestine does not impact the composition of the intestinal microbiome
(A) DF pups and pups subjected to NEC received a daily i.p. injection of recombinant Reg3g (rReg3g, 2 ng/pup) or an equivalent volume of PBS. Expression levels are shown of Il1b in the terminal ileum of either DF or pups subjected to NEC plus PBS or rReg3g.
(B) Representative H&E staining of terminal ileal sections from each experimental group. Scale bar, 200 μm.
(C) Histological score of the intestinal lesions in pups subjected to NEC and treated with PBS or IL-22 (n = 8–11 per group).
(D) Microbial community composition of colon contents collected from pups with NEC injected with PBS or rIL-22 (NEC + rIL-22).
(E) Shannon, Chao1, and Simpson indices demonstrating overall bacterial diversity in the microbiome of a subset of pups with NEC injected with either PBS or rIL-22 (NEC + rIL-22).
(F) Boxplots of alpha diversity indices (Shannon, Chao1, and Simpson, respectively) reflecting the abundance and the diversity of the operational taxonomic units (OTUs) in the colon contents of pups subjected to NEC and injected with either PBS or rIL-22. Data are displayed as mean ± SEM and were analyzed using Mann-Whitney test. ∗p < 0.05, ∗∗p < 0.01.
Discussion
In the absence of an effective therapeutic, NEC remains a major challenge in the NICU with a high mortality rate. With the growing literature supporting the important role of IL-22 in protecting the adult intestinal barrier against inflammatory insults, it seems intuitive to hypothesize a similar protective role of IL-22 in premature infants affected by NEC. Therefore, a better understanding of the role of IL-22 in preserving the intestinal barrier of neonates may lay the ground for a future therapeutic strategy against this devastating disease. Hence, in the current study, we aimed to explore the role of IL-22 in neonatal intestinal epithelial cells and to determine the impact of IL-22 on the outcome of NEC in a murine model of the disease. Over the last decades, several rodent models have been developed to decipher the pathobiology of NEC.27 Different methods have been adopted to induce a NEC-like disease consisting of chemical ablation of Paneth cells, the use of dextran sulfate sodium (DSS) and 2,4,6-trinitrobenzene sulfonic acid (TNBS) or a combination of asphyxia and formula feeding along with oral administration of enteric bacteria and/or lipopolysaccharide. The mouse model used in the current study, which uses a human enteric dysbiotic bacteria mixture cultured from a human NEC patient, offers the advantage of studying the interaction between the human microbiota and the immature intestine in neonatal mice. Moreover, a transcriptional analysis carried out in our laboratory revealed that our mouse NEC model (unpublished data) shares several pathological signaling pathways with the human disease.28
Our developmental time course study revealed that IL-22 expression levels in the intestine were very low in embryonic mouse intestine and increased slightly after birth, but significantly increased at weaning, which coincides with a dramatic change of the enteric microbial composition. Indeed, studies with germ-free mice have shown that the induction of IL-22 depends on intestinal colonization with commensal microbes.29,30 It is well accepted that early life is characterized by a low microbial diversity consisting mainly of facultative anaerobic species, especially in preterm infants.31 As a consequence, it is possible that neonates lack the microbial species responsible for the development of IL-22-producing cells. These data are in line with a previous study that utilized an IL-22 reporter mouse to show that IL-22 expression was low in the intestinal T cells, NKp46+ ILC3s, and lymphoid-tissue inducer (LTi) cells of newborn pups when compared to those found in the intestine of adult mice.32 In addition, the authors have shown that a subset of intestinal macrophages—characterized by their high expression levels of Il23a, Il6, and Tnfa upon activation with the TLR9 agonist CpG oligonucleotide—harbored the most potential of inducing IL-22 in neonatal mice prior to weaning. Interestingly, Gribar et al. have shown that CpG-mediated activation results in reduced NEC severity in a murine model.33 Moreover, we have previously shown that oral treatment with the probiotic Lactobacillus rhamnosus prevents the development of NEC in neonatal mice and premature piglets through the activation of TLR9 by unmethylated CpG dinucleotide of L. rhamnosus DNA.34 However, whether this TLR9-mediated protection conferred by L. rhamnosus against NEC is achieved through the enhancement of IL-22 production remains unknown.
The inability of neonates to mount an effective response to produce IL-22 is also likely inherent to postnatal immunosuppression. An analysis of the LP cells in newborn mice shows that the vast majority of CD4+ T cells display a naive phenotype prior to weaning.35 This effect was attributed to different cell types such as T regulatory cells, CD71+ erythroid cells, and myeloid-derived suppressor cells, which are particularly enriched in newborns and likely to play a key role in preventing the development of detrimental inflammation upon colonization with enteric microbes.35, 36, 37 The role of IL-22 in preventing intestinal injury in murine models of adult inflammatory bowel disease has been well documented over the last decade.10 As a result of intestinal inflammation, the induction of IL-22 promotes the maintenance of mucosal integrity and prevents the damage caused by colitogenic bacteria.12 Hence, the impairment of IL-22 signaling in adult mice lacking the expression of IL-22 or IL-22 receptor (IL-22ra1) leads to increased intestinal damage upon Citrobacter rodentium infection.12,17 Our data demonstrated that unlike adult mice, neonates do not possess the ability to trigger the production of IL-22 during NEC-mediated inflammation. Consequently, neonatal pups deficient in either Il-22 or the receptor IL-22Ra1 do not exhibit enhanced susceptibility to the disease. Therefore, this inability to mount an appropriate IL-22 response during intestinal inflammation might increase the susceptibility of the premature infant to intestinal damage and rapid onset of necrosis in the setting of NEC. This assumption is supported by the amelioration of NEC-mediated inflammation following treatment with exogenous IL-22, as defined by the downregulation of pro-inflammatory marker expression, improvement in histologic architecture, and reduction in histological scores. These findings are supported by previous reports showing the benefit of IL-22 treatment in reducing the severity of adult intestinal inflammatory disorders.12, 13, 14, 15, 16, 17
IL-22 signaling has been shown to trigger a regeneration program leading to the repair of an impaired epithelial barrier.18, 19, 20 Early investigations suggested that this is achieved by the expansion of the intestinal stem cell compartment,18, 19, 20 while recent studies challenged these findings and identified that epithelial barrier regeneration is the result of enhanced proliferation of the transit amplifying (TA) cells, which are defined by the expression of a proliferation marker such as PCNA.21,22 Our data were in accordance with the second line of evidence, as the protective role of IL-22 against NEC was dependent on IL-22Ra1 signaling in the intestinal epithelium and was associated with increased numbers of proliferating cells rather than an expansion of the intestinal stem cells. In parallel to the implication of IL-22 involvement in mucosal wound healing, we demonstrate that IL-22 treatment also induces the expression of several genes involved in antimicrobial activities and glycosylation in the gut of neonates such Reg3g and Fut2. In adults, these IL-22-mediated changes contribute largely to the protection of the gut from the harm of colitogenic bacteria.12, 13, 14, 15, 16, 17,38 One of the most studied IL-22 target genes is Reg3g, which has been shown to protect the intestinal mucosa via the maintenance of microbial-host segregation by modulating the composition of the microbiota.12,13 On the other hand, Pham et al. have demonstrated that Reg3g is dispensable in preventing Citrobacter rodentium infection.17 Our data similarly showed that treatment with rReg3g did not influence the outcome of experimental NEC. The injection of pups with Reg3g as well as the neutralization of this antimicrobial peptide during experimental NEC did not reduce the expression of inflammatory factors or attenuate epithelial cell damage.
To understand the influence of IL-22 treatment on the intestinal microbiome, we analyzed the diversity of the microbial communities with 16S rRNA sequencing. The analysis did not show any substantial impact of IL-22 treatment on the diversity and the community composition of intestinal microbiota. These findings were rather surprising, as previous studies conducted in adult mice have shown the role of IL-22 in shaping the microbiome.17,38,39 This may be due to the profound differences in the composition of the neonatal and the adult microbiomes. It is well accepted that the first postnatal waves of colonizing microbes consist of facultative anaerobes, which are present in relatively high abundance in the intestine of neonates and prepare the intestine for the colonization of more diverse, strict anaerobic species.31 Therefore, it is reasonable to speculate that the IL-22-driven antimicrobial response in neonates does not affect the facultative anaerobes that are present in the first days of life.
In conclusion, the current study demonstrates that the neonatal period is associated with a reduced ability to produce IL-22 in the intestine, which could render the premature infant susceptible to the extensive epithelial cell damage seen during NEC. In this study, we demonstrate that the exogenous administration of IL-22 results in the protection of the mucosal barrier during experimental NEC by promoting intestinal epithelial cell regeneration. Surprisingly, despite the transcriptional increase of antimicrobial genes, the administration of IL-22 neonates does not affect the composition of the intestinal microbiome. Altogether, our pre-clinical data demonstrate that IL-22 treatment has the potential to be used as a therapeutic to attenuate NEC in infants. This study demonstrated that similar to rodents, the human premature intestine is characterized by the absence of an IL-22 response during NEC, which might explain the susceptibility of premature infants to the disease. Given the protective role of IL-22 in the experimental murine model, it seems intuitive that the administration of IL-22 to infants displaying NEC symptoms may prevent intestinal injury through the enhancement of epithelial cell regeneration. This idea is supported by our in vitro experiments, which suggest that the epithelial cell response to IL-22 is at least conserved in premature human intestinal organoids. Therefore, the exploration of the protective role of IL-22 in clinical settings may set the ground for a new therapeutic strategy to prevent NEC.
Limitations of study
The current study utilizes a neonatal mouse model of NEC to study the effects of IL-22 treatment. A limitation of this study is that these experiments do not establish the maximal tolerated dose of rIL-22 cytokine in the neonatal mouse NEC model, which may provide additional insights for clinical translatability. Given that clinical NEC represents a spectrum of disease, additional studies are necessary to investigate whether treatment with rIL-22 can rescue experimental NEC and at which time points. Another limitation of the study is that we did not monitor the long-term effects of rIL-22 administration to neonatal mice subjected to experimental NEC, which may require further consideration.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-OLFM4 | Cell Signaling Technologies | Cat# 39141S; RRID: AB_2650511 |
| Mouse Anti-PCNA | Santa Cruz | Cat# sc-56; RRID: AB_628110 |
| Mouse Anti-Occludin | Invitrogen | Cat# 33-1500; RRID: AB_2533101 |
| Hoechst 33342 | Invitrogen | Cat# H1399 |
| CD16/CD32 antibodies | Miltenyi Biotec | Cat# 130-102-429; RRID: AB_2660222 |
| Zombie NIR Fixable Viability Dye | BioLegend | Cat# 423105 |
| Anti-mouse CD45 | BD Bioscience | Cat# 561487; RRID: AB_10697046 |
| Anti-mouse IL22 | BioLegend | Cat# 516409; RRID: AB_2563355 |
| Anti-mouse Reg3g | Abcepta, Inc. | Cat# AP5606C; RRID: AB_10817069 |
| Rabbit Immunoglobulin G | Sigma Aldrich | Cat# I5006; RRID: AB_1163659 |
| Donkey anti-mouse 647 | Invitrogen | Cat# A31571; RRID: AB_162542 |
| Donkey anti-mouse 488 | Invitrogen | Cat# A21202; RRID: AB_141607 |
| Donkey anti-rabbit 488 | Invitrogen | Cat# A21206; RRID: AB_2535792 |
| Donkey anti-rabbit 594 | Invitrogen | Cat# A21207; RRID: AB_141637 |
| Biological samples | ||
| Human: healthy small intestine tissue | St. Louis Children’s Hospital | IRB Protocol 201804040 |
| Mouse: healthy small intestine tissue | Washington University School of Medicine | Protocol 20190190 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse IL-22 | BioLegend | Cat# 576204 |
| Recombinant Mouse Reg3g | R&D Systems | Cat# 8189-RG-050 |
| Recombinant Human IL-22 | BioLegend | Cat# 571302 |
| Critical commercial assays | ||
| V-PLEX Mouse IL-1b Kit | Meso Scale Discovery | Cat# K152QPD-1 |
| Deposited data | ||
| Raw and analyzed data | This paper | N/A |
| RNA sequencing data | Deposited in Gene expression omnibus Database: https://www.ncbi.nlm.nih.gov/geo/ | GEO Accession: GSE155172 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | The Jackson Laboratory | JAX Stock No. 000664 |
| Villin cre mice | The Jackson Laboratory | JAX Stock No. 021504 |
| IL22ra1fl/fl mice | Zheng et al., 2016 | Gift from Dr. Jay Kolls |
| IL22−/− mice | Trevejo-Nunez et al., 2016 | Gift from Dr. Jay Kolls |
| Oligonucleotides | ||
| Primers for RT-PCR analysis, see Table 1 | This paper | N/A |
| Primer sequences for 16 s rRNA analysis, see Table 2 | This paper | N/A |
| Software and algorithms | ||
| Volocity v6.3.5 | Quorum Technologies | https://quorumtechnologies.com |
| ZEN 2.3 pro, blue edition | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij |
| FlowJo | TreeStar | https://www.flowjo.com/ |
| GraphPad Prism Software, Version 8.0 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Similac® Advance® OptigroTM with Iron infant formula | Abbott Nutrition | N/A |
| Esbilac Puppy Milk Replacer | PetAg | N/A |
| LWRN 50%CM | VanDussen, et al., 2019 | N/A |
| Y-27632 | R&D Systems | Cat# 1254/10 |
| SB-431542 | R&D Systems | Cat# 1614/10 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the lead contact, Misty Good (mistygood@wustl.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The datasets generated during this study are available at Gene Expression Omnibus (GEO): accession number GSE155172; Database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155172.
Experimental model and subject details
Mice
C57BL/6 and villin cre mice were purchased from the Jackson Laboratory. IL22ra1fl/fl and IL22−/− mice were obtained as a kind gift from Dr. Jay Kolls and have been previously described.11,40,41 Mice were bred and maintained in a specific pathogen-free animal facility at Washington University School of Medicine in St. Louis. All animal experiments were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) (protocol #20190190).
Human specimens
Human small intestinal samples were obtained from infants undergoing intestinal resection for NEC or non-NEC conditions (controls) at St. Louis Children’s Hospital. All the tissue specimens were obtained in a de-identified manner and processed as discarded tissue via a waiver of consent with approval of the Washington University in St. Louis School of Medicine Institutional Review Board and in accordance with the Washington University in St. Louis anatomical tissue procurement guidelines.
Induction of neonatal NEC
Experimental NEC induction was performed as previously described with slight modifications.34 Briefly, four-day-old neonatal mice were separated from the dams, randomly assigned into experimental NEC groups and maintained in an infant incubator at 37°C. Age-matched dam-fed control pups remained with their mothers. Experimental NEC was induced by oral gavage of a formula mixture containing Similac® Advance® Optigro™ with Iron infant formula (Abbott Nutrition) and Esbilac canine milk replacer (ratio 2:1). The formula was supplemented with 5 μg/g of body weight of lipopolysaccharide (Sigma Aldrich) and enteric bacteria isolated from a human infant with NEC totalis, the most severe form of NEC.34 Pups subjected to this NEC protocol were fed six times daily at regular intervals with the formula described above using 1.9 French Single-Lumen Silicon Peripherally Inserted Central Catheter (PICC). The pups were subjected to 10 minutes of hypoxic stress twice a day in a hypoxia chamber (Billups-Rothenberg) containing 95% nitrogen and 5% oxygen. After 72 hours, pups were euthanized in accordance with IACUC policy. The terminal ileal samples were collected during necropsy for analysis.
Animal treatments
Where indicated, dam-fed or experimental pups received either a daily intraperitoneal injection of PBS (vehicle control), recombinant mouse IL-22 (100 μg/kg; Biolegend) or recombinant Reg3g (2 ng/pup; R&D Systems). Additionally, where indicated, dam-fed or experimental pups received daily enteral gavage of anti-Reg3g neutralizing antibody (2 μg/gram body weight; Abcepta, Inc.) or rabbit immunoglobulin G (IgG) (2 μg/g body weight; Sigma Aldrich).
Histological assessment of the disease severity
Segments of terminal ilea were fixed in formalin, paraffin-embedded and stained with hematoxylin and eosin by the Digestive Disease Research Core Center at the Washington University School of Medicine in St. Louis. Histological slides were scanned at the Washington University Center for Cellular Imaging using a Zeiss AxioScan Z1. Using ImageJ software, disease severity was assessed based on the extent of histological lesions by an investigator blinded to the study conditions. Normal tissue sections were characterized by intact villi. Mild lesions were characterized by areas with mild villi destruction reflected by a substantial reduction of the villi length. Severe lesions were defined as areas with complete villus necrosis and significant disruption to the architecture of the villi (Figure S7). The severity score reflects the extent of the lesion as a percentage of the total length of the histological specimen.
Method details
Immunofluorescence
Tissue sections (5 μm) were deparaffinized in xylenes, rehydrated in isopropanol, and boiled in antigen unmasking solution (Vector Laboratories). After washing with water, the tissues were blocked in Tris Buffered Saline (Boston BioProducts) supplemented with 1% bovine serum albumin (BSA, Sigma), 10% normal donkey serum (NDS, Sigma) and 0.1% Tween-20 (Sigma) for an hour at room temperature. Tissue specimens were stained overnight with a rabbit anti-OLMF4 (Cell Signaling Technologies), mouse anti-PCNA (Santa Cruz) or mouse anti-Occludin (Invitrogen) diluted 100X in the blocking buffer at 4°C. Following washing in Tris Buffered Saline supplemented with 1% bovine serum albumin and 0.1% Tween-20, the tissue sections were incubated for an hour at room temperature with secondary antibodies (donkey anti-mouse 647, donkey anti-mouse 488, donkey anti-rabbit 488 or donkey anti-rabbit 594, Invitrogen) diluted 200X in blocking buffer. Nuclear staining was performed using Hoechst 33342 (Invitrogen) for 5 minutes, and slides were mounted with Prolong Gold (Thermo Fisher). Images were acquired using either a Leica SP8 tandem scanning confocal and processed using Volocity (v6.3.5, Quorum) or a Zeiss Axio Observer.Z1 microscope and processed using ZEN 2.3 pro, blue edition software (Zeiss). Image analysis was performed with NIH ImageJ software. The number of PCNA stained nuclei were counted in 15 to 20 intestinal crypts in each sample. The data were displayed as the average of PCNA+ cells per crypt.
Lamina propria cell isolation and flow cytometry
Lamina propria cells were isolated from the last 5 cm of the distal small intestine. Briefly, intestinal specimens were opened longitudinally, washed in ice cold phosphate-buffered saline (PBS) and incubated in 2 mM EDTA/PBS for 30 minutes at 4°C in a rotating device. After transferring the tissue samples to ice cold PBS, the epithelial cells were removed by pipetting 3 times up and down. The remaining intestinal tissues were digested in RPMI supplemented with 2.5% fetal bovine serum (FBS), 1 mg/ml collagenase type 4 (Worthington-Biochem) and 0.05 mg/ml DNase (Sigma) at 4°C for 40 minutes in an orbital shaker. The digestion was quenched with RPMI supplemented with 10% FBS and passed successively through 70 μm and 40 μm strainers to obtain a single cell suspension. Cells were stimulated with ionomycin phorbol-12-myristate 13-acetate activation cocktail (Biolegend) in the presence of BD GolgiStop (BD Biosciences) for 4 hours in RPMI supplemented with 10% FBS. After washing the cells in RPMI supplemented with 10% FBS, they were resuspended in PBS and stained with Zombie NIR Fixable Viability dye (BioLegend) following the manufacturer’s instructions. After washing in FACS buffer (PBS supplemented with 3% FBS), the cells were sequentially blocked on ice with CD16/CD32 antibodies (Miltenyi Biotec) for 10 minutes and stained with of 200X diluted anti-mouse CD45 antibody (BD Bioscience) for 20 minutes. Intracellular staining of IL-22 was carried out with a 100X diluted anti-mouse IL-22 antibody (Biolegend) using BD Biosciences Cytofix/Cytoperm and Perm/Wash solutions following the manufacturer’s protocol. Cell acquisition was carried out using MACSQuant Analyzer 10 (Miltenyi Biotec) and the data were analyzed FlowJo software (TreeStar).
Human and murine intestinal enteroid culture
Human intestinal tissue biopsies were obtained from premature infants with approval from the Washington University in St. Louis Institutional Review Board (no. 201804040). Intestinal tissue samples were digested Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and 1 mg/ml of collagenase I (Invitrogen, Waltham, MA) for 10 minutes at 37°C. The enzymatic digestion was followed by a mechanical dissociation. The resulting crypts were filtered through a 70-μm cell strainer, washed in DMEM supplemented with 10% FBS and resuspended in growth factor-reduced Matrigel (Becton Dickinson, Franklin Lakes, NJ). The resuspended crypts were seeded into culture plates and cultured in 50% L-WRN conditioned medium supplemented with 10 μM Y-27632 and SB 202190 (R&D Systems). After 7 days, the resulting enteroids were treated with recombinant human IL-22 (Biolegend) at 1, 10 and 100 ng/ml for 24 hours.
Murine enteroids were generated from isolated crypts from the small intestines of adult (4 weeks) or neonatal mice (7 days) as previously described.42 The crypts were resuspended in Matrigel and cultured for 24 hours in 50% L-WRN conditioned medium43 supplemented with 10 μM Y-27632 and SB 202190. The following day, the enteroids were differentiated in Advanced DMEM/F12 medium (GIBCO) supplemented with 2 mM GlutaMax (Invitrogen), 10 mM HEPES (Thermo Fisher Scientific). For differentiation, the enteroids were cultured in differentiation media consisting of basal culture medium with (1X) N2 supplement (Invitrogen), (1X) B27 supplement (Invitrogen), and 1 mM N-acetylcysteine (Sigma Aldrich), 50 ng/mL murine recombinant EGF (Invitrogen), 10 ng/mL murine recombinant Noggin (PeproTech), 10 ng/mL human recombinant R-spondin 1 (Miltenyi Biotec). After 4 days in the differentiation medium, the enteroids were treated with 2 ng/ml of recombinant mouse IL-22 (Biolegend) for 24 hours before being further processed.
Quantitative PCR
Intestinal enteroids were directly lysed in Trizol (Thermo Fisher Scientific) by pipetting up and down, while intestinal tissue samples were homogenized in Trizol using Tissue Lyzer II (QIAGEN). Total RNA was extracted according to the manufacturer’s instructions and was quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific). RNA was converted into cDNA using QuantiTech Reverse Transcription Kit (QIAGEN). Quantitative real-time PCR (qRT-PCR) was performed on the CFX Connect™ Real-Time PCR Detection System (Bio-Rad) using IQ SYBR Green Supermix and the primers listed in Table 1. The reaction protocol includes an initial preincubation at 95°C for 3 minutes to denature the DNA, with amplification performed for 40 cycles (10 s at 95°C, 10 s at 55°C and 30 s at 72°C). Gene expression levels were expressed as normalized values relative to the housekeeping gene Rplo.
Table 1.
Primers used in the present study for RT-PCR analysis of gene expression
| Gene | Species | Forward primer | Tm forward,°C | Reverse primer | Tm reverse,°C | Amplicon size, bp |
|---|---|---|---|---|---|---|
| Rpl0 | mouse | GGCGACCTGGAAGTCCAACT | 59.7 | CCATCAGCACCACAGCCTTC | 58.7 | 143 |
| Il1b | mouse | AGTGTGGATCCCAAGCAATACCCA | 60.4 | TGTCCTGACCACTGTTGTTTCCCA | 60.5 | 175 |
| Reg3g | mouse | GTACCCTGTCAAGAGCCTCA | 56.3 | TGTGGGGAGAATGTTCCCTT | 56.1 | 182 |
| Il22 | mouse | CGACCAGAACATCCAGAAGAA | 54.5 | GAGACATAAACAGCAGGTCCA | 54.5 | 110 |
| Il22ra1 | mouse | CAGCGGATCACCCAGAAGTT | 57.4 | CGTGGAGCTCTAAGCGGTAG | 57.2 | 298 |
| Cxcl1 | mouse | TGGCTGGGATTCACCTCAAG | 57.2 | CCGTTACTTGGGGACACCTT | 57.1 | 179 |
| Cxcl2 | mouse | CCAGACAGAAGTCATAGCCACT | 56.3 | GGCACATCAGGTACGATCCA | 56.9 | 217 |
| Fut2 | mouse | CAGAAGAGCCATGGCGAGTG | 58.4 | CGTTGCTGGAGGTGGATGAT | 57.4 | 102 |
| Retnlb | mouse | CCATTTCCTGAGCTTTCTGG | 53.6 | AGCACATCCAGTGACAACCA | 56.8 | 320 |
| RETNLB | human | AGCTCTCGTGTGCTAGTGTC | 56.6 | TGAACATCCCACGAACCACA | 56.7 | 109 |
| REG3G | human | TATCTGTGTGTCCTCCCGCT | 57.8 | AGGAAAGCAGCATCCAGGAC | 57.5 | 118 |
| IL22 | human | AACCCCCTTTCCCTGCTAGA | 58.1 | ACGCAGGGGTTCATTTGGAA | 57.4 | 119 |
| IL22RA1 | human | ACGTACGGAGAGAGGGACTG | 57.9 | GAGGGTAGTGTGCTGCAGAG | 57.6 | 189 |
Tm, melting temperature
V-plex assay
A mouse IL-1β V-plex assay (Meso Scale Diagnostics) was performed by the Immunomonitoring Laboratory (IML) at Washington University.
RNA sequencing
Total RNAs were isolated from the terminal ileum of 4 week old IL22ra1fl/fl; Vil cre+ and IL22ra1fl/fl; Vil cre- mice. In addition, total RNAs were also isolated from intestinal enteroids cultured from the small intestine of WT mice which have treated for 24 hours with PBS or 100 ng/ml of mouse IL-22. mRNA was purified with oligo-dT beads, and used for cDNA synthesis with random primers after a fragmentation step with magnesium followed by heat-catalyzed hydrolysis. The cDNA 3′ ends were adenylated, followed by adaptor ligation and a 15-cycle PCR to enrich DNA fragments. cDNA libraries were generated using a v2 Guide Quantification of cDNA libraries was performed by using Kapa Biosystems primer premix kit with Illumina-compatible DNA primers. The cDNA libraries were pooled at a final concentration 1.8 pM. Single-read sequencing was performed on Illumina Genome Analyzer IIx and NextSeq 500. The raw reads were processed following the methods described by Kumar et al.44 to generate Reads per Mapped Million per Kilobase (RPKM) values. RNA sequencing data is deposited in Gene Expression Omnibus (Geo); accession number GSE155172.
16S rRNA microbial community analysis
Mouse fecal samples were collected from the distal portion of the colon during necropsy. DNA was isolated using QIAamp PowerFecal Pro stool DNA extraction kit (QIAGEN) and the 16S rRNA sequencing and analysis were performed by Genome Technology Access Center at Washington University in St. Louis. Seven PCR amplicons representative of all nine 16S variable regions using the primers indicated in Table 2 were generated using the Fluidigm Access Array System. Reaction mixture components (all from Fluidigm) included 10X FastStart High Fidelity buffer without MgCl, 25mM MgCl, dimethyl sulfoxide, 10 mM PCR Grade Nucleotide Mix, 0.05 U/μL of 5U/μL FastStart High Fidelity Enzyme Blend, 20X Access Array Loading Reagent, 1 μL DNA, and molecular grade water. The BioMark HD system from Fluidigm was employed for PCR amplification. Reaction products were indexed with unique 10 base pair sequences via 7 rounds of PCR in order to combine each index sequence. 48 sample libraries were constructed via sample pooling and bead purification (Fluidigm) used for cleaning as in Schriefer et al.45 Illumina MiSeq sequencer (2 × 150 base pair kit) was used for library sequencing. Demultiplexed reads from the 7 amplicons were analyzed using the MVRSION pipeline described by Schriefer et al.,45 to generate a list of microbial species with their corresponding number of reads for each sample. Default parameters were employed for the MVRSION analysis in conjunction with the Silva 16S database. Diversity analysis was run for the following sample groupings: Experimental NEC + PBS control (NEC + PBS) versus NEC + rIL-22.
Table 2.
Primers sequences associated with the seven PCR amplicons covering nine variable regions in the bacterial 16S rRNA gene
| Name | Sequence |
|---|---|
| V1-V2_F | TCGTCGGCAGCGTCAGAGTTTGATCCTGGCTCAG |
| V2_F | TCGTCGGCAGCGTCAGYGGCGIACGGGTGAGTAA |
| V3_2_F | TCGTCGGCAGCGTCCCTACGGGAGGCAGCAG |
| V4_F | TCGTCGGCAGCGTCGTGCCAGCMGCCGCGGTAA |
| V5-V6_F | TCGTCGGCAGCGTCAGGATTAGATACCCTGGTA |
| V6_1_F | TCGTCGGCAGCGTCAAACTCAAAKGAATTGACGG |
| V7-V8_F | TCGTCGGCAGCGTCGYAACGAGCGCAACCC |
| V1-V2_R | GTCTCGTGGGCTCGGTGCTGCCTCCCGTAGGAGT |
| V2_R | GTCTCGTGGGCTCGGCYIACTGCTGCCTCCCGTAG |
| V3_2_R | GTCTCGTGGGCTCGGGTATTACCGCGGCTGCTGG |
| V4_R | GTCTCGTGGGCTCGGGGACTACHVGGGT WTCTAAT |
| V5-V6_R | GTCTCGTGGGCTCGGCRRCACGAGCTGACGAC |
| V6_1_R | GTCTCGTGGGCTCGGACGAGCTGAC GACARCCATG |
| V7-V8_R | GTCTCGTGGGCTCGGGACGGGCGGTGWGTRC |
Quantification and statistical analysis
Statistical analyses with Mann Whitney U-tests, analysis of variance, and two-tailed Student’s t tests were performed where indicated using GraphPad Prism software version 8.0. Statistical significance was set at a P value of < 0.05.
Acknowledgments
This manuscript was supported by R01DK118568 (M. Good), R03DK111473 (M. Good), K08DK101608 (M. Good), and 5T32HD043010 (L.S.N.) from the National Institutes of Health; March of Dimes Foundation grant 5-FY17-79 (M. Good); American Academy of Pediatrics Marshall Klaus Award (L.S.N.); the St. Louis Children’s Hospital Foundation (M. Good); The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (M. Good); and the Department of Pediatrics at Washington University School of Medicine, St. Louis.
We would like to thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with 16S analysis. The Center is partially supported by NCI Cancer Center Support grant P30 CA91842 to the Siteman Cancer Center and by the ICTS/CTSA grant UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. We would also like to thank the Washington University Digestive Diseases Research Center Core for histology services supported by NIH grant P30 DK052574. Histological imaging was performed in part through the use of Washington University Center for Cellular Imaging (WUCCI) supported by The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2019-813). This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR, NIH, or any of the funders.
Author contributions
Conceptualization, B.M., J.K.K., and M. Good; methodology, all authors; investigation, all authors; writing – original draft, B.M. and M. Good; writing – review & editing, all authors; funding acquisition, L.S.N. and M. Good; resources, J.K.K. and M. Good; supervision, B.M. and M. Good.
Declaration of interests
Drs. J.K.K. and M. Good have a patent pending on the use of IL-22 to prevent or treat NEC. Dr. M. Good has received sponsored research agreement funding from Astarte Medical, Evive Biotech, and Takeda Pharmaceuticals. Dr. M. Good also serves on the Scientific Advisory Council of the NEC Society and the Clinical and Technology Advisory Board of Astarte Medical. She also participated in a neonatal microbiome advisory board for Abbott Laboratories in 2019. None of these sources had any role in this study.
Published: June 15, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100320.
Supplemental information
References
- 1.Widdowson E.M. Effects of Prematurity and Dysmaturity in Animals. In: Jonxis J.H.P., Visser H.K.A., Troelstra J.A., editors. Aspects of Praematurity and Dysmaturity: Groningen 10–12 May 1967. Nutricia Symposium. Springer Netherlands; 1968. pp. 127–137. [Google Scholar]
- 2.Institute of M. Preterm Birth . Causes, Consequences, and Prevention. In: Richard E.B., Adrienne Stith B., editors. The National Academies Press; 2007. [Google Scholar]
- 3.Neu J., Walker W.A. Necrotizing enterocolitis. N. Engl. J. Med. 2011;364:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel R.M., Denning P.W. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr. Res. 2015;78:232–238. doi: 10.1038/pr.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rusconi B., Good M., Warner B.B. The microbiome and biomarkers for necrotizing enterocolitis: Are we any closer to prediction? J. Pediatr. 2017;189:40–47.e2. doi: 10.1016/j.jpeds.2017.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mihi B., Good M. Impact of Toll-like receptor 4 signaling in necrotizing enterocolitis: The state of the science. Clin. Perinatol. 2019;46:145–157. doi: 10.1016/j.clp.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warner B.B., Deych E., Zhou Y., Hall-Moore C., Weinstock G.M., Sodergren E., Shaikh N., Hoffmann J.A., Linneman L.A., Hamvas A. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: a prospective case-control study. Lancet. 2016;387:1928–1936. doi: 10.1016/S0140-6736(16)00081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pammi M., Cope J., Tarr P.I., Warner B.B., Morrow A.L., Mai V., Gregory K.E., Kroll J.S., McMurtry V., Ferris M.J. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome. 2017;5:31. doi: 10.1186/s40168-017-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olm M.R., Bhattacharya N., Crits-Christoph A., Firek B.A., Baker R., Song Y.S., Morowitz M.J., Banfield J.F. Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci. Adv. 2019;5:eaax5727. doi: 10.1126/sciadv.aax5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parks O.B., Pociask D.A., Hodzic Z., Kolls J.K., Good M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front. Cell Dev. Biol. 2016;3:85. doi: 10.3389/fcell.2015.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aujla S.J., Chan Y.R., Zheng M., Fei M., Askew D.J., Pociask D.A., Reinhart T.A., McAllister F., Edeal J., Gaus K. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y., Valdez P.A., Danilenko D.M., Hu Y., Sa S.M., Gong Q., Abbas A.R., Modrusan Z., Ghilardi N., de Sauvage F.J., Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 13.Vaishnava S., Yamamoto M., Severson K.M., Ruhn K.A., Yu X., Koren O., Ley R., Wakeland E.K., Hooper L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonnenberg G.F., Monticelli L.A., Alenghat T., Fung T.C., Hutnick N.A., Kunisawa J., Shibata N., Grunberg S., Sinha R., Zahm A.M. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goto Y., Obata T., Kunisawa J., Sato S., Ivanov I.I., Lamichhane A., Takeyama N., Kamioka M., Sakamoto M., Matsuki T. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. 2014;345:1254009. doi: 10.1126/science.1254009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickard J.M., Maurice C.F., Kinnebrew M.A., Abt M.C., Schenten D., Golovkina T.V., Bogatyrev S.R., Ismagilov R.F., Pamer E.G., Turnbaugh P.J., Chervonsky A.V. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514:638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham T.A., Clare S., Goulding D., Arasteh J.M., Stares M.D., Browne H.P., Keane J.A., Page A.J., Kumasaka N., Kane L., Sanger Mouse Genetics Project Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 2014;16:504–516. doi: 10.1016/j.chom.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanash A.M., Dudakov J.A., Hua G., O’Connor M.H., Young L.F., Singer N.V., West M.L., Jenq R.R., Holland A.M., Kappel L.W. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. 2012;37:339–350. doi: 10.1016/j.immuni.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aparicio-Domingo P., Romera-Hernandez M., Karrich J.J., Cornelissen F., Papazian N., Lindenbergh-Kortleve D.J., Butler J.A., Boon L., Coles M.C., Samsom J.N., Cupedo T. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015;212:1783–1791. doi: 10.1084/jem.20150318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindemans C.A., Calafiore M., Mertelsmann A.M., O’Connor M.H., Dudakov J.A., Jenq R.R., Velardi E., Young L.F., Smith O.M., Lawrence G. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528:560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwarycz B., Gracz A.D., Rivera K.R., Williamson I.A., Samsa L.A., Starmer J., Daniele M.A., Salter-Cid L., Zhao Q., Magness S.T. IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cell. Mol. Gastroenterol. Hepatol. 2018;7:1–17. doi: 10.1016/j.jcmgh.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zha J.-M., Li H.-S., Lin Q., Kuo W.-T., Jiang Z.-H., Tsai P.-Y., Ding N., Wu J., Xu S.-F., Wang Y.-T. Interleukin 22 Expands Transit-Amplifying Cells While Depleting Lgr5+ Stem Cells via Inhibition of Wnt and Notch Signaling. Cell. Mol. Gastroenterol. Hepatol. 2019;7:255–274. doi: 10.1016/j.jcmgh.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray J., Oehrle K., Worthen G., Alenghat T., Whitsett J., Deshmukh H. Intestinal commensal bacteria mediate lung mucosal immunity and promote resistance of newborn mice to infection. Sci. Transl. Med. 2017;9:eaaf9412. doi: 10.1126/scitranslmed.aaf9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L., Strohmeier V., He Z., Deshpande M., Catalan-Dibene J., Durum S.K., Moran T.M., Kraus T., Xiong H., Faith J.J. Interleukin 22 disrupts pancreatic function in newborn mice expressing IL-23. Nat. Commun. 2019;10:4517. doi: 10.1038/s41467-019-12540-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shindo R., Ohmuraya M., Komazawa-Sakon S., Miyake S., Deguchi Y., Yamazaki S., Nishina T., Yoshimoto T., Kakuta S., Koike M. Necroptosis of Intestinal Epithelial Cells Induces Type 3 Innate Lymphoid Cell-Dependent Lethal Ileitis. iScience. 2019;15:536–551. doi: 10.1016/j.isci.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Good M., Sodhi C.P., Yamaguchi Y., Jia H., Lu P., Fulton W.B., Martin L.Y., Prindle T., Nino D.F., Zhou Q. The human milk oligosaccharide 2′-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br. J. Nutr. 2016;116:1175–1187. doi: 10.1017/S0007114516002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ares G.J., McElroy S.J., Hunter C.J. The science and necessity of using animal models in the study of necrotizing enterocolitis. Semin. Pediatr. Surg. 2018;27:29–33. doi: 10.1053/j.sempedsurg.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Good M., Chu T., Shaw P., McClain L., Chamberlain A., Castro C., Rimer J.M., Mihi B., Gong Q., Nolan L.S. Global hypermethylation of intestinal epithelial cells is a hallmark feature of neonatal surgical necrotizing enterocolitis. Clin. Epigenetics. 2020;12:190. doi: 10.1186/s13148-020-00983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satoh-Takayama N., Vosshenrich C.A., Lesjean-Pottier S., Sawa S., Lochner M., Rattis F., Mention J.J., Thiam K., Cerf-Bensussan N., Mandelboim O. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Sanos S.L., Bui V.L., Mortha A., Oberle K., Heners C., Johner C., Diefenbach A. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderickx J.G.E., Zwittink R.D., van Lingen R.A., Knol J., Belzer C. The Preterm Gut Microbiota: An Inconspicuous Challenge in Nutritional Neonatal Care. Front. Cell. Infect. Microbiol. 2019;9:85. doi: 10.3389/fcimb.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savage A.K., Liang H.-E., Locksley R.M. The development of steady-state activation hubs between adult LTi ILC3s and primed macrophages in small intestine. J. Immunol. 2017;199:1912–1922. doi: 10.4049/jimmunol.1700155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gribar S.C., Sodhi C.P., Richardson W.M., Anand R.J., Gittes G.K., Branca M.F., Jakub A., Shi X.H., Shah S., Ozolek J.A., Hackam D.J. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J. Immunol. 2009;182:636–646. doi: 10.4049/jimmunol.182.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Good M., Sodhi C.P., Ozolek J.A., Buck R.H., Goehring K.C., Thomas D.L., Vikram A., Bibby K., Morowitz M.J., Firek B. Lactobacillus rhamnosus HN001 decreases the severity of necrotizing enterocolitis in neonatal mice and preterm piglets: evidence in mice for a role of TLR9. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;306:G1021–G1032. doi: 10.1152/ajpgi.00452.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torow N., Yu K., Hassani K., Freitag J., Schulz O., Basic M., Brennecke A., Sparwasser T., Wagner N., Bleich A. Active suppression of intestinal CD4(+)TCRαβ(+) T-lymphocyte maturation during the postnatal period. Nat. Commun. 2015;6:7725. doi: 10.1038/ncomms8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elahi S., Ertelt J.M., Kinder J.M., Jiang T.T., Zhang X., Xin L., Chaturvedi V., Strong B.S., Qualls J.E., Steinbrecher K.A. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. 2013;504:158–162. doi: 10.1038/nature12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Y.-M., Li X., Perego M., Nefedova Y., Kossenkov A.V., Jensen E.A., Kagan V., Liu Y.-F., Fu S.-Y., Ye Q.-J. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat. Med. 2018;24:224–231. doi: 10.1038/nm.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zenewicz L.A., Yin X., Wang G., Elinav E., Hao L., Zhao L., Flavell R.A. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 2013;190:5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo B.C., Shin S.B., Canals Hernaez D., Refaeli I., Yu H.B., Goebeler V., Cait A., Mohn W.W., Vallance B.A., McNagny K.M. IL-22 Preserves Gut Epithelial Integrity and Promotes Disease Remission during Chronic Salmonella Infection. J. Immunol. 2019;202:956–965. doi: 10.4049/jimmunol.1801308. [DOI] [PubMed] [Google Scholar]
- 40.Zheng M., Horne W., McAleer J.P., Pociask D., Eddens T., Good M., Gao B., Kolls J.K. Therapeutic Role of Interleukin 22 in Experimental Intra-abdominal Klebsiella pneumoniae Infection in Mice. Infect. Immun. 2016;84:782–789. doi: 10.1128/IAI.01268-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trevejo-Nunez G., Elsegeiny W., Aggor F.E.Y., Tweedle J.L., Kaplan Z., Gandhi P., Castillo P., Ferguson A., Alcorn J.F., Chen K. Interleukin-22 (IL-22) Binding Protein Constrains IL-22 Activity, Host Defense, and Oxidative Phosphorylation Genes during Pneumococcal Pneumonia. Infect. Immun. 2019;87 doi: 10.1128/IAI.00550-19. e00550-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato T., Clevers H. Primary mouse small intestinal epithelial cell cultures. Methods Mol. Biol. 2013;945:319–328. doi: 10.1007/978-1-62703-125-7_19. [DOI] [PubMed] [Google Scholar]
- 43.VanDussen K.L., Sonnek N.M., Stappenbeck T.S. L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res. (Amst.) 2019;37:101430. doi: 10.1016/j.scr.2019.101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar P., Monin L., Castillo P., Elsegeiny W., Horne W., Eddens T., Vikram A., Good M., Schoenborn A.A., Bibby K. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity. 2016;44:659–671. doi: 10.1016/j.immuni.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schriefer A.E., Cliften P.F., Hibberd M.C., Sawyer C., Brown-Kennerly V., Burcea L., Klotz E., Crosby S.D., Gordon J.I., Head R.D. A multi-amplicon 16S rRNA sequencing and analysis method for improved taxonomic profiling of bacterial communities. J. Microbiol. Methods. 2018;154:6–13. doi: 10.1016/j.mimet.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus (GEO): accession number GSE155172; Database: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155172.