

Summary:
Adipose tissue invariant natural killer T (iNKT) cells are phenotypically different from other iNKT cells because they produce IL-10 and control metabolic homeostasis. Why that is the case is unclear. Here, using single-cell RNA-sequencing we found several adipose iNKT clusters, which we grouped into two functional populations based on NK1.1 expression. NK1.1NEG cells almost exclusively produced IL-10 and other regulatory cytokines, while NK1.1POS iNKT cells predominantly produced IFNγ. Mechanistically, biochemical fractionation revealed that free fatty acids drive IL-10 production primarily in NK1.1NEG iNKT cells via the IRE1α-XBP1s arm of the unfolded protein response. Correspondingly, adoptive transfer of adipose tissue NK1.1NEG iNKT cells selectively restored metabolic function in obese mice. Further, we found an unexpected role for NK1.1POS iNKT cells in lean adipose tissue, as IFNγ licenses natural killer cell-mediated macrophage killing to limit pathological macrophage expansion. Together, these two iNKT cell populations utilize non-redundant pathways to preserve metabolic integrity.
Introduction
Restraining local inflammation is essential for proper adipose tissue function, and an increase in adipose inflammation during overfeeding drives obesity-related metabolic disorders like type 2 diabetes and cardiovascular disease (Gregor and Hotamisligil, 2011). To ensure homeostasis, the adipose tissue of lean animals is replete with anti-inflammatory immune cells such as alternatively activated (M2-like) macrophages, regulatory T cells (Tregs) and eosinophils that preserve metabolic tenor (Kohlgruber and Lynch, 2015). These immune cells cross-regulate each other and interact with the adipose microenvironment, thus maintaining a dynamic immunometabolic dialogue within the tissue. (DiSpirito and Mathis, 2015).
Invariant natural killer T (iNKT) cells are highly enriched in the visceral adipose tissue of lean mice and humans, constituting between 10–30% of all adipose tissue-resident T cells (Lynch et al., 2015; Lynch et al., 2009; Schipper et al., 2012). iNKT cells are innate-like αβ T cells that utilize a semi-invariant T cell receptor (TCR) to recognize lipid antigens in the context of the antigen-presenting molecule CD1d (Godfrey et al., 2010). While these cells are typically associated with proinflammatory immune responses during microbial infection, we found that adipose tissue iNKT cells are highly anti-inflammatory and essential for metabolic homeostasis (Lynch et al., 2012). Mice that lack iNKT cells gain weight more rapidly when placed on a high-fat diet (HFD) and have poorer metabolic function compared to wild-type (WT) counterparts (Ji et al., 2012; Lynch et al., 2012). Furthermore, activation of adipose iNKT cells with their prototypical lipid antigen α-galactosylceramide (αGalCer) drives weight loss and reverses metabolic disorder in obesity (Lynch et al., 2016; Lynch et al., 2012). Mechanistically, iNKT cells protect against metabolic disease by limiting adipose tissue inflammation through regulatory cytokine production. Several reports have shown that adipose tissue iNKT cells produce IL-10, IL-4, IL-13, and IL-2, which serve to expand and enhance the functions of alternatively activated M2-like macrophages, Tregs and adipocytes (Huh et al., 2017; Ji et al., 2012; Lynch et al., 2016; Lynch et al., 2015; Lynch et al., 2012; Sag et al., 2014; Schipper et al., 2012). Thus, iNKT cells play a central and unique role in adipose tissue, by controlling the phenotype and functions of several important adipose tissue-resident cells (LaMarche et al., 2019). Interestingly, several studies have observed the opposite of these results; namely, that adipose tissue iNKT cells are proinflammatory and drive metabolic dysfunction (Satoh et al., 2016; Wu et al., 2012). The mechanisms behind these divergent findings are currently unknown.
We previously observed that adipose tissue iNKT cells are phenotypically distinct from iNKT cells in other organs. For example, these cells lack expression of the Promyelocytic Leukemia Zinc Finger (PLZF) transcription factor, which is highly expressed in iNKT cells in all other organs and thought to be required for iNKT cell development (Lynch et al., 2015). Additionally, they produce IL-10, a characteristic not seen in other iNKT cells from other tissues, which is critical for iNKT cell-mediated control of adipose tissue inflammation (Lynch et al., 2012). IL-10 expression is driven by the E4BP4 transcription factor (encoded by Nfil3), which is not expressed by other iNKT cells at steady state (Lynch et al., 2015; Motomura et al., 2011). However, the mechanism by which adipose tissue iNKT cells express E4BP4 and upregulate IL-10 production is not known. Nor is it known whether there is functional heterogeneity within the adipose tissue iNKT compartment.
Here, we address these questions by using single-cell transcriptomics, biochemical fractionation of adipose tissue, and in vivo functional studies. We found that the first iNKT cells to arrive in the adipose tissue in perinatal mice expressed PLZF, but then rapidly downregulated this transcription factor as mice mature. Single-cell RNA-sequencing (ScRNA-seq) of adipose iNKT cells revealed surprising heterogeneity, with distinct clusters characteristically distinguished by expression of NK1.1. Unexpectedly, we found that the anti-inflammatory cytokine IL-10, as well as the majority of regulatory cytokines previously attributed to the bulk adipose tissue iNKT compartment, were produced by NK1.1NEG iNKT cells, which represent 50% of the total adipose iNKT cell population. NK1.1POS iNKT cells, in contrast, produce large amounts of TNF and IFNγ, two cytokines reported to drive adipose inflammation and tissue dysfunction during obesity. Importantly, we found that iNKT cell exposure to adipose tissue-derived free fatty acids (FFAs) drove E4BP4 expression and IL-10 production downstream of the inositol-requiring enzyme 1 α (IRE1α)-XBP1s arm of the unfolded protein response (UPR). This pathway was predominantly active in NK1.1NEG iNKT cells in situ, accounting for their preferential production of IL-10. Correspondingly, only adoptive transfer of NK1.1NEG adipose iNKT cells into obese adipose tissue restored metabolic control and reduced local inflammation.
We also uncovered an independent role for IFNγ-producing NK1.1POS iNKT cells in promoting adipose tissue homeostasis in lean animals. We found that iNKT cell-derived IFNγ transactivates adipose natural killer (NK) cells and facilitates their cytotoxicity against adipose tissue macrophages (ATMs). Mice lacking either iNKT cells or IFNγ exhibited defective NK cell cytotoxic capacity resulting in ATM accumulation at steady state, which was associated with greater adipose tissue inflammation and metabolic dysfunction. Specific activation of adipose tissue iNKT cells drove NK cell-mediated killing of ATMs in an IFNγ-dependent manner. Collectively, these findings provide new insights into the signals that govern adipose tissue iNKT cell function and define the mechanisms by which these cells exert control over adipose tissue-resident immune cells.
Results
Adipose tissue iNKT cells downregulate PLZF as mice age
Three thymically imprinted iNKT cell lineages have been described in mice, each defined by graded expression of PLZF: Th1-like NKT1 cells, which are PLZFLOTbetPOSRORγTNEG; Th17-like NKT17 cells, which are PLZFINTTbetNEGRORγTPOS; and Th2-like NKT2 cells, which are PLZFHITbetNEGRORγTNEG (Lee et al., 2013). The discovery of PLZF-negative adipose tissue iNKT cells raised the possibility that this novel population was similarly imprinted in the thymus. However, scRNA-seq of thymic iNKT cells has so far failed to identify a PLZF-negative precursor of the adipose tissue iNKT cell lineage (Engel et al., 2016). Therefore, we hypothesized that iNKT cells might home to adipose tissue in a PLZF-positive state and subsequently downregulate PLZF over time.
Previously, we used fate-mapping mice to determine if adipose tissue iNKT cells derived from a PLZF-positive precursor; however, the majority of hematopoietic cells transiently express low levels of PLZF during development, so it was not possible to answer this question (Lynch et al., 2015). To determine if iNKT cells home to adipose tissue in a PLZF-negative state, or if they downregulate PLZF after arrival, we analyzed the first iNKT cells to arrive in the adipose tissue during perinatal development. The earliest time point at which we could identify iNKT cells in white adipose tissue was nine days after birth (data not shown). Strikingly, at this age adipose tissue iNKT cells uniformly expressed high levels of PLZF. Importantly, PLZF levels then dropped to barely detectable levels by adulthood (Figure 1a). In contrast, PLZF expression did not change significantly in splenic iNKT cells over the same time period (Figure 1b). When we analyzed biaxial co-expression of PLZF against Tbet and RORγT, the data were consistent with all three populations of TbetHI, PLZFHI and RORγTPOS cells being present in the adipose tissue of neonatal mice. By adulthood, however, the PLZFHI population had dropped to near zero numbers, while the relative frequencies of the TbetHI and RORγTPOS populations remained unchanged (Figure 1c). To further examine this phenomenon, we adoptively transferred splenic iNKT cells, which express high levels of PLZF, into congenically marked mice and recovered the transferred cells from recipient spleen and adipose tissue two weeks later (Figure 1d). Transferred cells recovered from spleen maintained high PLZF expression, while those recovered from adipose tissue had uniformly downregulated PLZF (Figure 1e), suggesting that exposure to the adipose microenvironment drives PLZF downregulation. Collectively, these data indicate that adipose iNKT cells do not derive from PLZF-negative precursors, but rather downregulate PLZF expression in situ from tissue cues.
Figure 1. Adipose tissue iNKT cells lose PLZF expression with age.
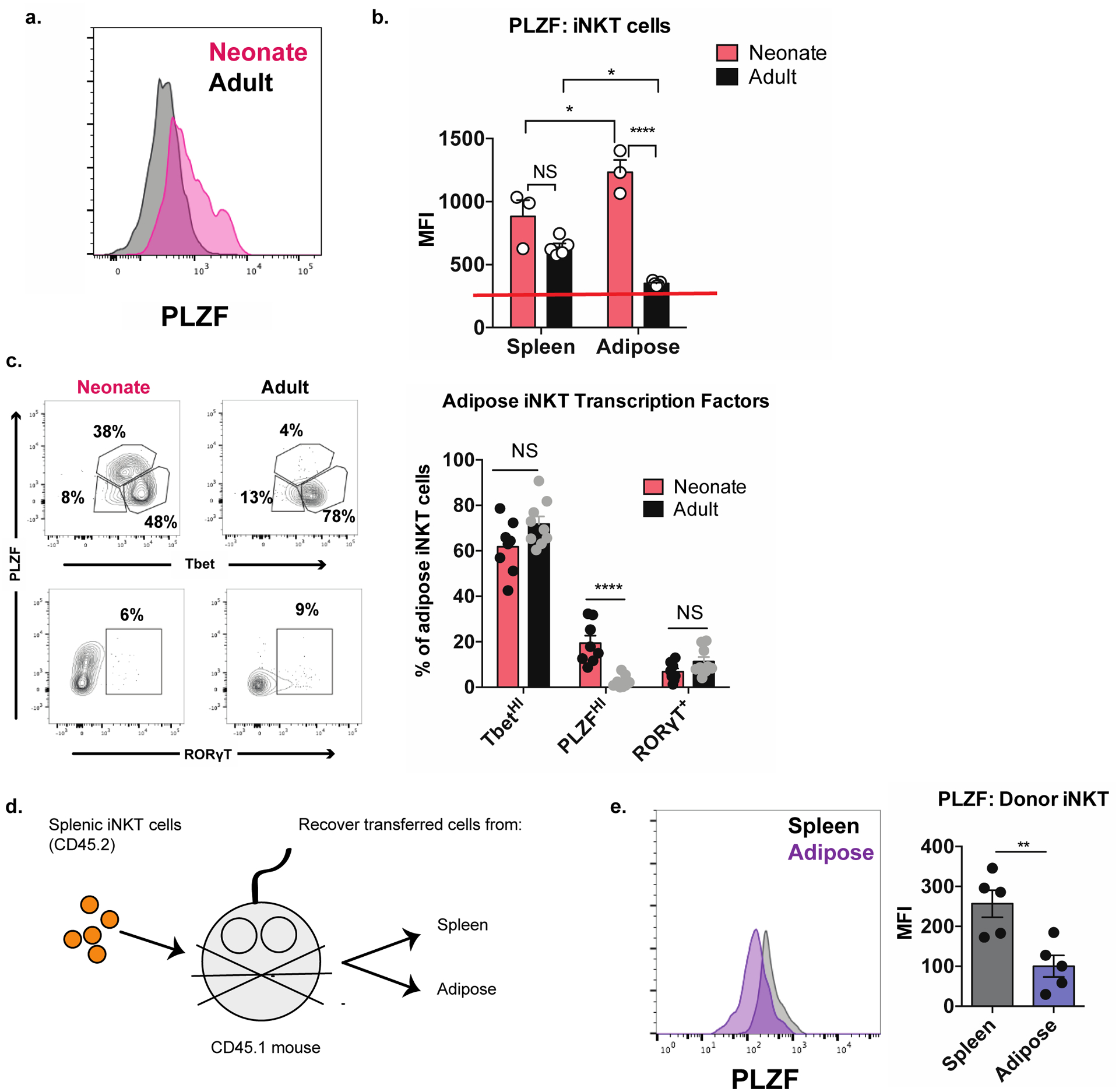
(a) Representative histogram of PLZF expression in adipose iNKT cells from neonatal and adult mice (n = 3–5 histograms analyzed per group).
(b) Quantification of PLZF expression in splenic and adipose iNKT cells from neonatal and adult mice (n = 3–5 mice or pooled mice per group). Red line indicates the baseline PLZF mean fluorescence intensity of PLZFNEG T cells.
(c) Adipose tissue iNKT cells in neonatal and adult mice were analyzed by biaxial gating of PLZF against either Tbet or RORγT (n = 8 mice).
(d) Schematic diagram outlining adoptive transfer experiment.
(e) Expression of PLZF in transferred splenic iNKT cells recovered from recipient spleen and adipose tissue (n = 5 mice).
NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Two-tailed Student’s t-test. Error bars indicate mean (± S.E.M.). Data are representative of three or more independent experiments (a-c) or two experiments (e).
ScRNA-seq identifies transcriptionally distinct adipose tissue iNKT cell populations
Past studies of adipose tissue iNKT cells assumed that they were a homogeneous PLZF-negative lineage. Given our new finding that all iNKT cells downregulate PLZF upon adipose tissue ingress, we next asked whether there was unappreciated heterogeneity within this cellular compartment. To address this question, we performed droplet-capture based scRNA-seq of iNKT cells from the adipose tissue of 8-week-old male C57BL/6 mice, compared with splenic iNKT cells. iNKT cells grouped largely by tissue of origin using Uniform Manifold Approximation and Projection (UMAP) (Figure S1a), consistent with our previous microarray analysis that identified adipose tissue iNKT cells as a transcriptionally distinct from other tissue populations (Lynch et al., 2015). Importantly, several defining transcriptional characteristics of the bulk adipose tissue iNKT cell population (Lynch et al., 2015) were present among all adipose cells that we analyzed, including uniformly high expression of the activation-associated genes Cd69 and Nr4a1 (Figure S1b). However, UMAP and Seurat V3 analysis also revealed unexpected heterogeneity within adipose tissue iNKT cells and highlighted four transcriptionally distinct cell clusters (Figure 2a). Similar to our flow cytometric studies, we observed a large population of Tbx21 (Tbet)-expressing NKT1 cells, and a smaller cluster of Rorc-expressing NKT17 cells (Figure 2b). We recently identified a role for IL-17A producing γδ T cells in adipose Treg expansion and thermogenesis (Kohlgruber et al., 2018), and these NKT17 cells may serve a similar function. Another cluster (“Cycling”) was dominated by genes encoding cell cycle-related proteins, which is consistent with our observation that adipose tissue iNKT cells are highly proliferative (Figure S1c). Unexpectedly, the NKT1 cells segregated into two clusters, which we labeled “NKT1-A” and “NKT1-B”. Half of the NKT1-A cells expressed Klrg1 while the NKT1-B cells expressed Klrb1c (encoding NK1.1) (Figure 2c). When we performed differentially-expressed gene (DEG) analysis, we found that each cluster had an easily identifiable transcriptional signature defined by genes encoding cytokines (eg Ccl5, Il17a), surface markers (eg Klrk1, Il18rap), and transcription factors (eg Maf, Satb1) (Figure 2d and Table S1).
Figure 2. Transcriptional and functional diversity among adipose tissue iNKT cells.
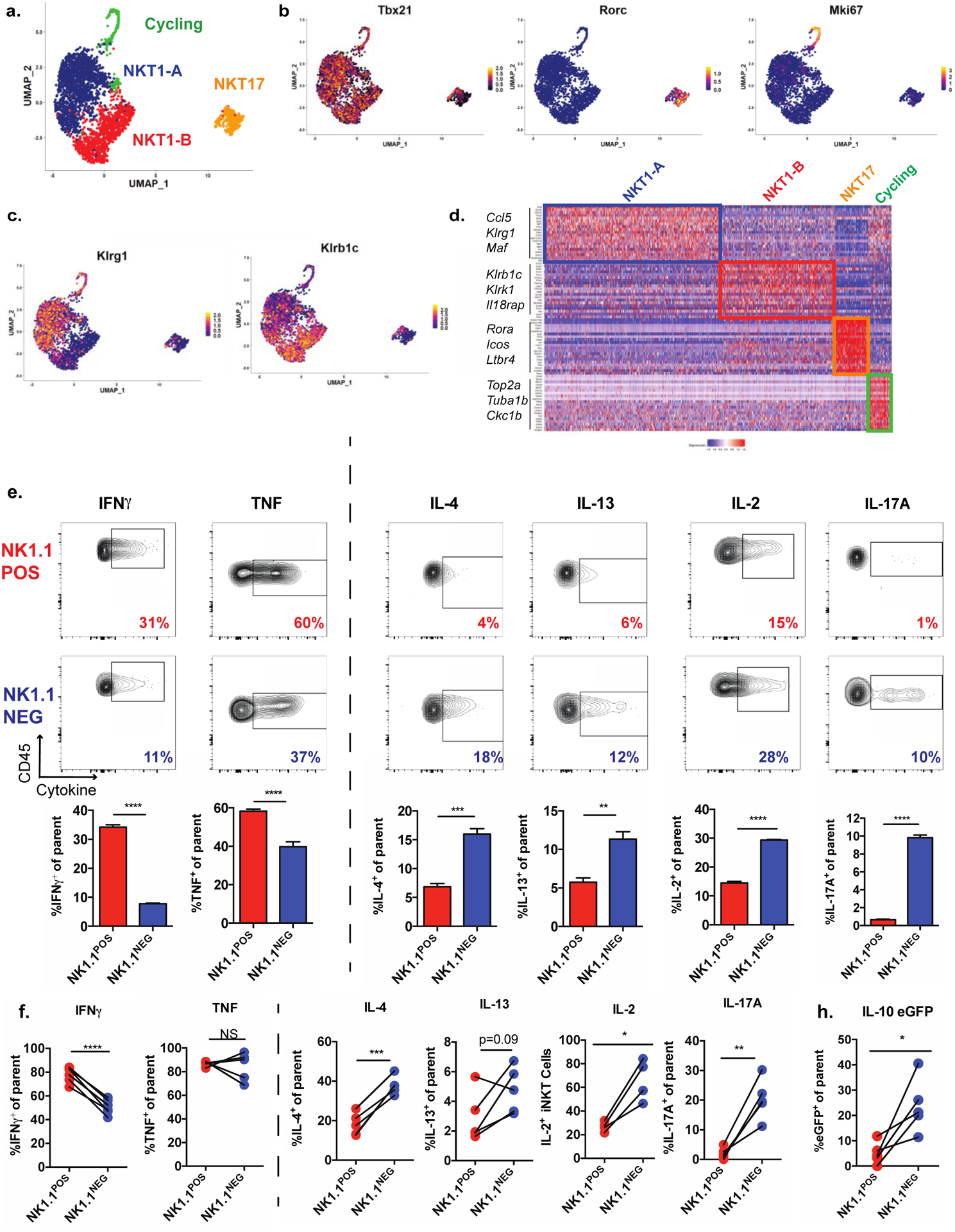
(a) UMAP of scRNA-seq data from adipose tissue iNKT cells, showing cell clusters.
(b-c) Normalized expression of cluster-defining transcription factors (b) and surface markers distinguishing the NKT1-A and NKT1-B clusters (c).
(d) Heatmap representing top 20 overexpressed genes per cell cluster relative to all other cells.
(e) Cytokine levels, as evaluated by intracellular cytokine staining of adipose tissue iNKT cells from Vα14 TN mice pre-sorted based on expression of NK1.1 and then stimulated for 4 hours with PMA/I. CD45 is a pan leukocyte marker used on the Y axes (n = 3 biological replicates plated in triplicate).
(f) Cytokine levels by NK1.1POS and NK1.1NEG iNKT cells, as evaluated by ICS, from the stromal vascular fraction (SVF) from WT mice stimulated with PMA/I for two hours. Each pair of connected dots represents paired cell populations in a single SVF sample (n = 4–6 mice/group).
(g) Percentage of eGFP+ cells among NK1.1POS and NK1.1NEG iNKT cells from the SVF from IL-10 eGFP (TIGER) mice stimulated as in (g) (n = 5 mice).
NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.. Two-tailed Student’s t-test in e-g; Error bars indicate mean (± S.E.M.). Data are representative of one experiment (a-d) or two or more independent experiments (g-d).
NK1.1 expression defines functionally distinct populations of adipose tissue iNKT cells
During thymic development, iNKT cells sequentially upregulate CD44 and NK1.1, correlating with enhanced proinflammatory cytokine production capacity (Pellicci et al., 2002). Several studies have shown that NK1.1POS and NK1.1NEG iNKT cells in peripheral organs are distinct populations that do not interconvert (McNab et al., 2007). As NK1.1 (Klrb1c) expression discriminated the two large NKT1 sub-clusters (Figure 2c), we hypothesized that this marker might define functionally distinct populations of adipose tissue iNKT cells.
While the majority of peripheral iNKT cells in C57BL/6 mice express NK1.1, we found that adipose tissue was enriched for NK1.1NEG iNKT cells (33.5 ± 2.9% in spleen versus 48.02 ± 2.7% in adipose tissue) (Figure S2a), which was stable with age (Fig. S2b). Earlier work has shown that iNKT cells rapidly but temporarily downregulate NK1.1 in response to strong antigen-driven stimulation by αGalCer (Wilson et al., 2003). Given that iNKT cells may receive chronic, low-grade TCR stimulation from adipocytes at steady state (Huh et al., 2013; Satoh et al., 2016), we analyzed NK1.1 expression in adipose tissue iNKT cells from adipocyte-specific CD1d knockout mice (CD1d1ΔAD). In these mice, adipose tissue iNKT cells expressed significantly lower levels of the NFAT-driven transcription factor Nur77, but their expression of NK1.1 was unchanged compared to controls (Figure S2c). Additionally, the expression of Nur77 or CD69 did not differ between NK1.1POS and NK1.1NEG adipose tissue iNKT cells (Figure S2d), suggesting that these populations do not represent two different activation states. We furthermore found that the frequencies of these populations were dynamically regulated by physiological signals in adipose tissue. Namely, fasting, which drives adipose lipolysis downstream of β-adrenergic signaling, drove an expansion of NK1.1NEG adipose tissue iNKT cells (Figure S2e), while, in contrast, the frequencies of these populations were not affected by overnutrition in the form of short-term HFD (Figure S2f).
Next, we queried whether NK1.1POS and NK1.1NEG adipose tissue iNKT cells differed in their ability to produce key cytokines that regulate adipose tissue functions. Sorting and activating these populations from Vα14 transnuclear (TN) mice, which contain enriched numbers of iNKT cells, revealed distinct cytokine profiles. NK1.1POS iNKT cells dominantly produced IFNγ and TNF, which have been described to drive inflammation and adipose tissue dysfunction during obesity (Gregor and Hotamisligil, 2011; O’Sullivan et al., 2016), while NK1.1NEG iNKT cells produced the metabolically protective cytokines IL-4, IL-13, IL-2, and IL-17 (Brestoff et al., 2015; Ji et al., 2012) (Kohlgruber et al., 2018; Lynch et al., 2015) (Figure 2e and Figure S2g). We verified these results in WT mice by flow cytometry after iNKT cell activation (Figure 2f). Furthermore, we analyzed stromovascular fraction (SVF) cells from IL-10 eGFP reporter mice and found that this regulatory cytokine was dominantly produced by the NK1.1NEG population (Figure 2g). NK1.1 expression did not change during this short time frame (Figure S2h). Collectively, these results demonstrate that NK1.1 expression discriminates adipose tissue iNKT cell populations that produce different classes of cytokines.
Exposure to adipose tissue lipids drives E4BP4 expression and IL-10 production in iNKT cells
We were intrigued by our observation that IL-10 is differentially produced among adipose tissue iNKT cells. Single-cell analysis revealed that Nfil3, encoding the IL-10-inducing transcription factor E4BP4, was highly expressed in NK1.1NEG iNKT cells, but much less so in the NK1.1POS population (Figure 3a). In fact, only half of the NKT1-B cluster (containing the majority of NK1.1POS cells) highly expressed this transcript (Figure 3b). Given that non-adipose iNKT express no E4BP4, we first sought to determine what factors drove E4BP4 expression in the bulk adipose tissue iNKT compartment. We reasoned that understanding this mechanism would give us insight into why this protein was more highly expressed in the NK1.1NEG population.
Figure 3. Exposure to adipose tissue lipids induces regulatory iNKT cells.
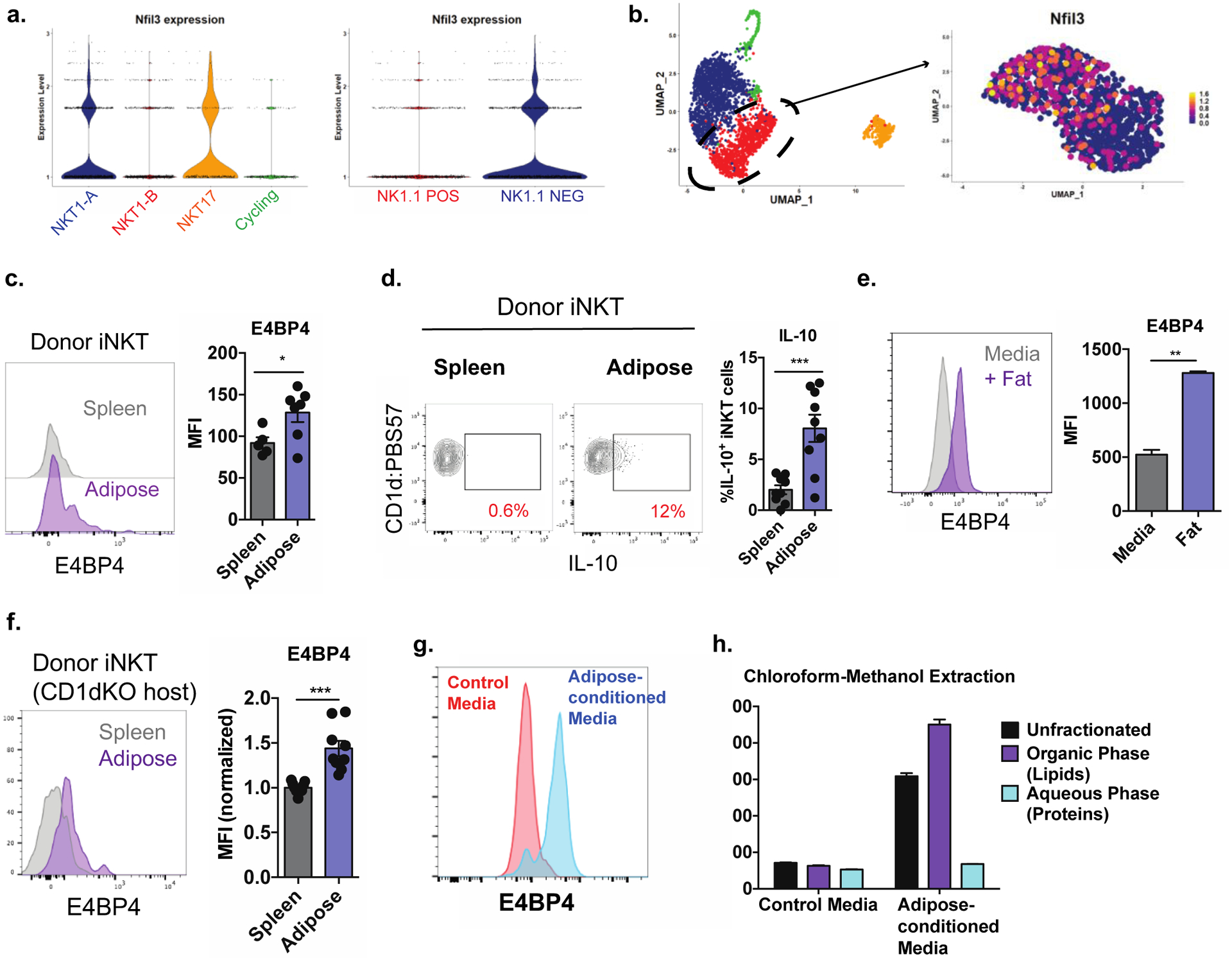
(a) Expression of Nfil3 in scRNA-seq clusters (left) or in cells from scRNA-seq experiment segregated based on Klrb1c (NK1.1) expression (right).
(b) Expression of Nfil3 within fine-clustered NKT1-B cells.
(c) E4BP4 expression in splenic iNKT cells adoptively transferred into congenically marked mice and recovered from spleen and adipose tissue one week later (n = 5–6 organs per group).
(d) Percentage of IL-10+ donor iNKT cells recovered from spleen and adipose tissue after αGalCer stimulation (n = 9 organs per group).
(e) Expression of E4BP4 in splenic iNKT cells cultured in control media or in the presence of adipose tissue. (n = 2 technical replicates per group).
(f) Expression of E4BP4 in donor splenic iNKT cells recovered from recipient spleen and adipose tissue (n = 9 organs per group).
(g) Expression of E4BP4 in iNKT cells cultured in control media or adipose-conditioned media.
(h) E4BP4 expression in iNKT cells exposed to adipose-conditioned media left unfractionated or fractionated into aqueous and organic phases (n = 3 technical replicates per group).
Error bars indicate mean (± S.E.M.). NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001. Two-tailed student’s t-test (c, d, e, f). Data representative of (c) or combined from (d, f) two independent experiments or representative of three or more experiments (e, g, h).
We hypothesized that E4BP4 and IL-10 could be induced by exposure to the adipose microenvironment. To test this, we adoptively transferred Vα14 TN splenic iNKT cells, which lack E4BP4 and do not produce IL-10, into congenically marked mice and recovered the transferred cells from the spleen and adipose tissue one week later. Surprisingly, iNKT cells recovered from adipose tissue robustly upregulated E4BP4 expression compared to those recovered from spleen (Figure 3c). Correspondingly, these cells produced IL-10 upon αGalCer stimulation (Figure 3d). To rule out the possibility that these results were due to homing of a specialized progenitor population to the fat, we directly cultured splenic iNKT cell lines with congenically marked adipose tissue overnight and found that virtually all iNKT cells cultured with fat robustly upregulated E4BP4 (Figure 3e) with no signs of increased proliferation or death of a singular population (data not shown). Collectively, these data indicate that exposure to the adipose microenvironment drives E4BP4 expression in adipose iNKT cells.
Next, we sought to determine what factors in the adipose microenvironment induced E4BP4 in iNKT cells and endowed them with regulatory capacity. We found that this was unrelated to TCR stimulation, as splenic iNKT cells transferred into CD1d-deficient mice still upregulated E4BP4 in adipose tissue (Figure 3f). Supporting this, adipose tissue iNKT cells from Cd1d1ΔAD mice expressed similar levels of E4BP4 (Figure S3a) and produced similar levels of IL-10 (Figure S3b) as WT controls. Additionally culture of iNKT cells with CD1d KO adipose tissue sufficiently drove E4BP4 upregulation (Figure S3c). Thus, it is unlikely that cognate CD1d-TCR interaction drives E4BP4 expression in adipose iNKT cells.
To determine if exposure to fat induced E4BP4 via a cell-cell contact mechanism or through a soluble factor, we cultured iNKT cells in the presence of control media or cell-free adipose tissue-conditioned media and found that conditioned media induced robust upregulation of E4BP4 in all iNKT cells (Figure 3g). We then separated the adipose-conditioned media into aqueous (protein) and organic (lipid) fractions and found that all E4BP4-inducing activity was in the lipid phase (Figure 3h). Together, these results implicate nonantigenic lipids in adipose tissue in driving E4BP4 expression in iNKT cells.
FFAs induce regulatory iNKT cells via the IRE1α-XBP1s arm of the Unfolded Protein Response
We hypothesized that E4BP4, and IL-10 are induced in adipose iNKT cells as a consequence of their residence in a lipid-rich microenvironment. Indeed, adipose tissue contains the highest concentration of extracellular lipids in the body, 85% of which are the FFAs palmitate, oleate, and linoleate (Tan et al., 2015). Consistently, we noted that iNKT cells in adipose tissue contained over twice as many intracellular lipids as splenic iNKT cells, as measured by LipidTox staining (Figure 4a). To model this lipid-rich microenvironment, we cultured splenic iNKT cells with 200 μM palmitate, which reflects physiological concentrations in adipose tissue and was sufficient to increase intracellular lipid content by roughly two-fold (Figure S4a). Strikingly, we found that exposure to palmitate drove a rapid upregulation of Nfil3 (E4BP4) and Il10 expression in splenic iNKT cells (Figure 4b). We confirmed this upregulation by flow cytometry by culturing whole splenocytes from IL-10 eGFP reporter mice with palmitate and then stimulating them with the protein kinase C / NFAT activating cocktail PMA/I and analyzing eGFP fluorescence in iNKT cells (Figure 4c). This was specific to iNKT cells, as other T cells did not upregulate IL-10 production in response to palmitate (Figure 4c). Interestingly, we also found that exposure to palmitate induced splenic iNKT cells to downregulate PLZF (Figure S4b and S4c).
Figure 4. Adipose tissue FFAs drive E4BP4 expression and IL-10 production in iNKT cells via the IRE1α-XBP1s arm of the UPR.
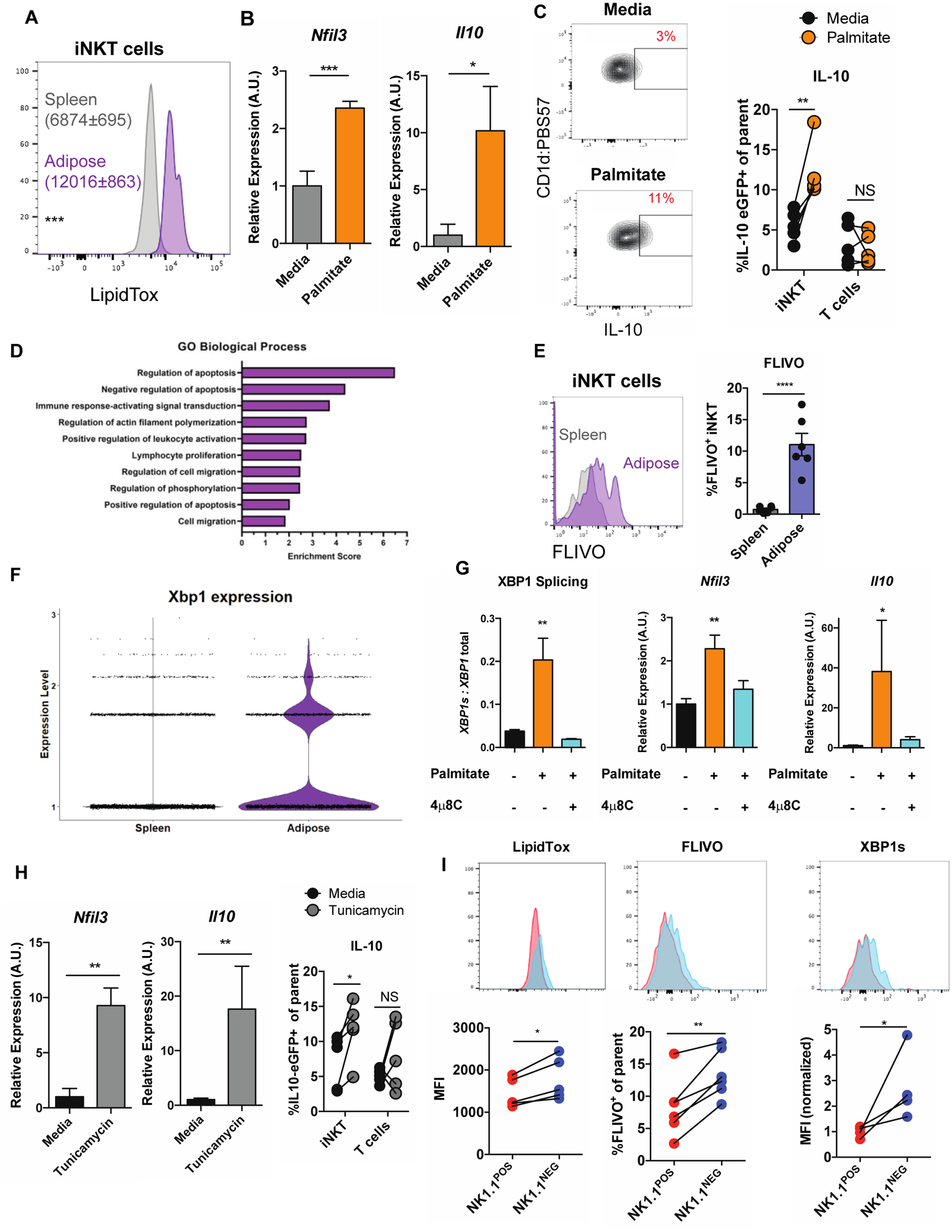
(a) LipidTox expression in splenic versus adipose iNKT cells (n = 5 mice).
(b) Relative expression of Nfil3 and Il10 in iNKT cells cultured in control media or in the presence of palmitate (n = 6 technical replicates/group).
(c) Percentage of IL-10 eGFP+ adipose iNKT cells, based on NK1.1 expression, after PMA/I stimulation (n = 5 paired biological replicates).
(d) Top enriched GO terms in adipose iNKT cells over splenic iNKT cells
(e) Percentage of FLIVO+ iNKT cells in spleen and adipose tissue (n = 6 mice).
(f) Expression of XBP1 in splenic and adipose iNKT cells from scRNA-seq.
(g) Relative expression of indicated genes in iNKT cells cultured in the presence of control media, palmitate, or palmitate and 4μ8c (n = 6 technical replicates / group).
(h) Relative expression of Nfil3 and Il10 in iNKT cells cultured in control media or tunicamycin (n = 4–9 technical replicates/group, representative of 3 biological replicates).
(i) Flow cytometry staining of LipidTox, FLIVO and XBP1s in NK1.1POS versus NK1.1NEG adipose iNKT cells (n =4–5 mice per group).
Error bars indicate mean (± S.E.M.). NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001. Two-tailed unpaired Student’s t-test (a, b, e, g). Two-tailed paired Student’s t-test (c, h). MAST test (f). ANOVA with post hoc Tukey’s test (g). Data representative of three (a, b, g) or two (i), or one (f) or combined from two (c) or three (h) independent experiments.
To understand why exposure to FFAs induces regulatory iNKT cells in fat, we analyzed transcriptional differences between splenic and adipose tissue iNKT cell populations from our scRNA-seq dataset (Table S2). Interestingly, after Gene Ontogeny (GO) analysis we found that adipose tissue iNKT cells were highly enriched with transcripts associated with apoptosis and cellular stress (Figure 4d). In vivo analysis with the FAM-FLIVO reagent, which labels cells with active caspases, confirmed this; at any point in time an average of 10% of adipose tissue iNKT cells were undergoing apoptosis, compared to 1% in the spleen (Figure 4e).
In addition to its role in the immune system, E4BP4 has been extensively characterized as an apoptosis-associated factor induced in several different cell types subjected to physiological stressors (Beach et al., 2011; Yu et al., 2002; Zhao et al., 2013). A potential connection between the role of E4BP4 in stress and in adipose tissues was found when it was reported that exposing iNKT cells to palmitate activated the IRE1α-XBP1s axis of the UPR, one of three homeostatic pathways engaged by cells undergoing apoptotic or endoplasmic reticulum (ER) stress. (Ko et al., 2017). Here, ER stress drives activation of the RNAse/kinase IRE1α, which then splices a single mRNA XBP1 into its active translatable form XBP1s, encoding a potent transcription factor. While one study recently probed the role of the kinase domain of IRE1α in iNKT cells (Govindarajan et al., 2018), the role of the RNAse domain and downstream XBP1 signaling has remained largely unexplored. We found that the XBP1 gene was highly upregulated in adipose tissue iNKT cells over splenic iNKT cells (P adj = 4.73E-212, MAST test) (Figure 4f).
Given that the Nfil3 gene was recently described to be an XBP1s target gene in T cells (Pramanik et al., 2018), we hypothesized that ER stress driven by intracellular lipid accumulation may drive E4BP4 expression and subsequently IL-10 production in iNKT cells. Thus, to interrogate the role of the IRE1α-XBP1s axis, we cultured iNKT cells with palmitate in the presence or absence of the chemical compound 4μ8c, which specifically inhibits the RNAse domain of IRE1α and thus XBP1 splicing. Exposure to physiological levels of palmitate drove XBP1 splicing in iNKT cells, which was blocked by 4μ8c treatment (Figure 4g, left). Strikingly, 4μ8C treatment also completely blocked Nfil3 and Il10 upregulation in response to palmitate, demonstrating that this pathway was completely driven by IRE1α-XBP1s (Figure 4g, middle, right). This effect was mirrored when we exposed palmitate-treated cells to the chemical chaperone 4-phenylbutyrate (4-PBA), which nonspecifically attenuates ER stress (Figure S4d). Conversely, when we pharmacologically induced ER stress with tunicamycin, both Nfil3 and Il10 were rapidly upregulated in iNKT cells (Figure 4h). Thus, exposure to adipose tissue FFAs drives E4BP4 and IL-10 expression downstream of IRE1α-XBP1s, thereby endowing adipose iNKT cells with regulatory functions.
We next sought to determine why, despite existing in the same environment, NK1.1POS and NK1.1NEG iNKT cells differed in their expression of E4BP4 and production of IL-10. We hypothesized that these two populations differed in their engagement of the IRE1α-XBP1s signaling and started by analyzing the initiating signal of this pathway; namely, intracellular lipid accumulation. Strikingly, LipidTox staining revealed that NK1.1POS iNKT cells contained significantly lower levels of intracellular lipids than did NK1.1NEG cells (Figure 4i, left). From a developmental standpoint, NK1.1POS iNKT cells have been shown to be highly resistant to apoptotic stress-inducing stimuli compared to NK1.1NEG iNKT cells (Engel et al., 2016). Consistent with this, in the adipose tissue NK1.1POS iNKT cells underwent apoptosis at half the rate of NK1.1NEG counterparts (Figure 4i, middle). Correspondingly, when we analyzed IRE1α signaling by flow cytometric staining for XBP1s, we found that this pathway was less engaged in NK1.1POS iNKT cells (MFI ~ 25% of NK1.1NEG) (Figure 4i, right). Collectively, these results demonstrate that adipose FFAs drive iNKT production of IL-10 via the IRE1α-XBP1s arm of the UPR, and that this pathway is reduced in NK1.1POS iNKT cells, likely due to both an lower intracellular lipid content and a developmental propensity to resist stressful stimuli.
NK1.1NEG adipose tissue iNKT cells selectively restore glycemic control and reduce inflammation in obesity
The majority of reports have shown that adipose tissue iNKT cells protect against metabolic disease during obesity by producing the regulatory cytokines IL-10, IL-4, IL-13, and IL-2 (Ji et al., 2012; Lynch et al., 2015; Schipper et al., 2012). As NK1.1NEG iNKT cells predominantly produced these cytokines, we hypothesized that the NK1.1NEG iNKT cell population was responsible for the protective metabolic effects. To test this, we placed Jα18-knockout (Jα18 KO) mice, which lack iNKT cells, on a HFD for 8 weeks, and then adoptively transferred either NK1.1POS or NK1.1NEG adipose iNKT cells obtained from lean Vα14 TN mice, or a vehicle control, into obese mice, and two weeks later, performed metabolic testing (Figure 5a). Transfer of either cell type did not significantly alter whole body mass or fat pad mass (data not shown). However, we found that transfer of NK1.1NEG iNKT cells restored glycemic control in obese mice, as measured by fasting blood glucose (Figure 5b) and glucose tolerance test (Figure 5c), but that transfer of NK1.1POS iNKT cells had no effect. Furthermore, transfer of NK1.1NEG iNKT cells drove the expansion of CD301+ M2 macrophages and the contraction of CD11c+ M1 macrophages in obese adipose tissue (Figure 5d). Mice receiving NK1.1NEG iNKT cells also had a smaller average adipocyte size compared to those receiving NK1.1POS iNKT cells or vehicle control (Figure 5e). Thus, in the context of obesity NK1.1NEG iNKT cells selectively reverse metabolic dysfunction by controlling adipose tissue inflammation. The fact that we found only half of all adipose iNKT cells to be protective against obesity may explain why some studies have characterized the bulk population as being proinflammatory and metabolically pathogenic.
Figure 5. NK1.1NEG iNKT cells selectively restore glycemic control during obesity.
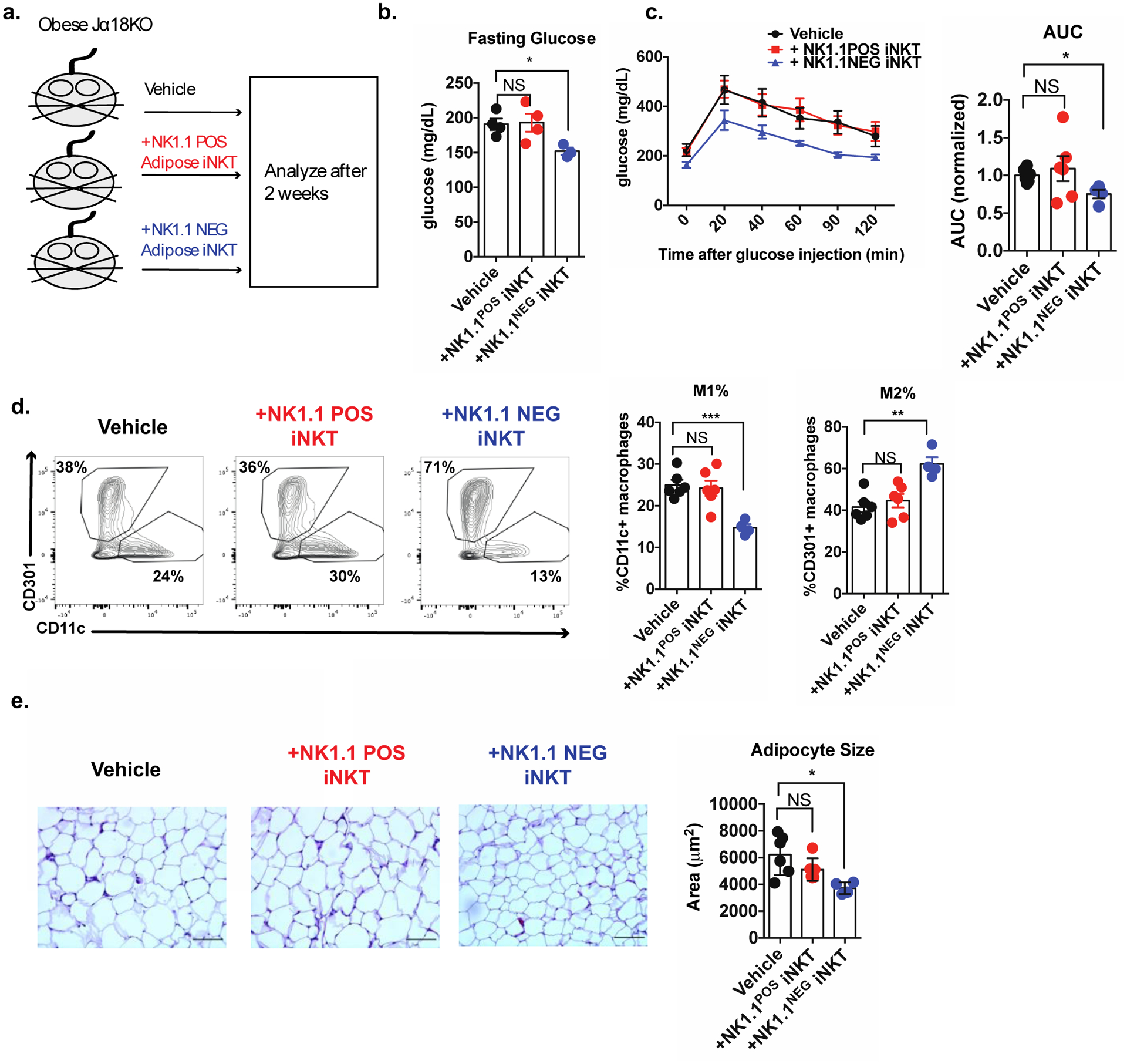
(a) Schematic diagram outlining the adoptive transfer experiment.
(b-c) Fasting glucose (b) and glucose tolerance test (c) of mice receiving treatments indicated in (a).
(d) Percentage of ATMs (CD45+CD11b+F4/80+) expressing canonical M1 (CD11c) and M2 (CD301) markers.
(e) Average adipocyte area of mice receiving indicated treatments. Scale bars = 100 μm. (representative images of 4–6 mice per group).
Error bars indicate mean (± S.E.M.). NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001. Two tailed Student’s t-test with Bonferroni correction (b, d) or ANOVA with post-hoc Dunnet test for multiple comparisons (c). n = 4–6 mice per group, representative of (b) or pooled from (c, d, e) two independent experiments.
iNKT cell-derived IFNγ is critical for ATM homeostasis and metabolic health in lean mice
Our data so far suggest that the positive effect of regulatory adipose tissue iNKT cells on metabolism is mediated exclusively by the NK1.1NEG population. However, NK1.1POS iNKT cells constitute 50% of all adipose iNKT cells (and up to 15% of all adipose T cells) at steady state, and therefore likely play a significant role in adipose tissue physiology. We found that NK1.1POS iNKT cells dominantly produced TNF and IFNγ, (Fig. 3e) cytokines typically associated with inflammation in obesity. However, these cytokines also may have beneficial effects. For example, low levels of TNF production have been described to be beneficial in lean adipose tissue, as it facilitates healthy expansion of the tissue in response to food intake (Wernstedt Asterholm et al., 2014). To our knowledge, a role for IFNγ in lean adipose tissue has not been described. Because all other cytokines that are dominantly produced by adipose iNKT cells have an important role in lean adipose, we tested if iNKT cell-derived IFNγ was harmful or beneficial in adipose tissue physiology at steady state.
iNKT cells are a major source of IFNγ in adipose tissue, comprising up to 20% of all IFNγ-competent cells in the tissue (Figure S5a). We found that adipose tissue iNKT cells produced three times the amount of IFNγ as tetramer-negative T cells (Figure S5b) and could produce this cytokine in response to both TCR- and cytokine-driven activation signals (Fig. 6a). To determine if IFNγ signaling regulates the immunological tenor in adipose tissue at steady state, we quantified adipose immune cell populations in WT and IFNγ KO mice. The majority of adipose immunocytes were unaltered in IFNγ KO mice, with the exception of Tregs and eosinophils, which were slightly higher in number (Figure S5c). However, the expression of Il10, Il4, and Il13, the key cytokines these cells produce to control adipose tissue inflammation (Feuerer et al., 2009; Wu et al., 2011) was unchanged (Figure S5d), suggesting that the marginal difference in cell number was inconsequential.
Figure 6. Adipose tissue iNKT cells produce IFNγ, which is required for proper metabolic function in lean fat.
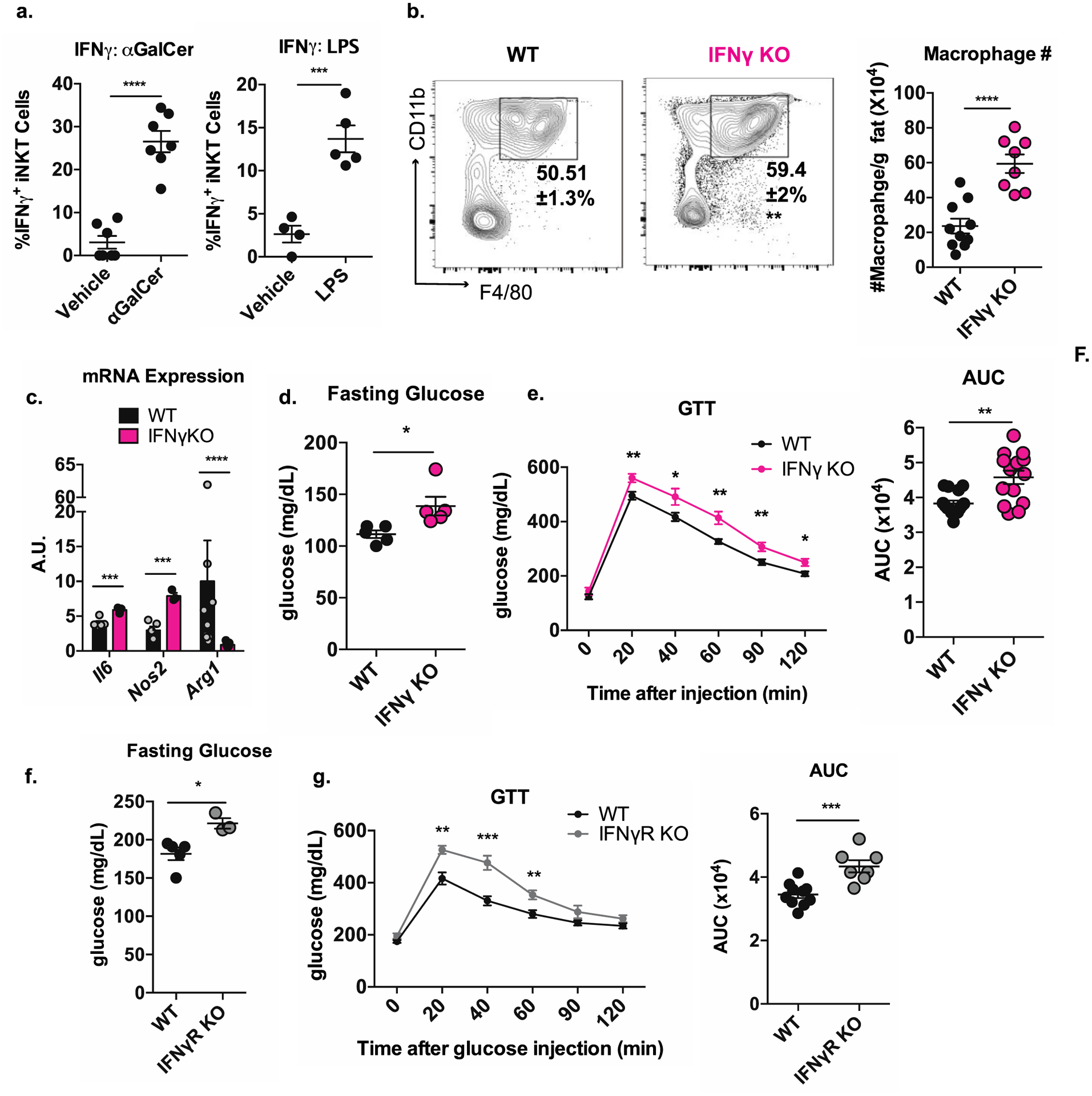
(a) Percentage of IFNγ+ adipose tissue iNKT cells from mice injected with vehicle or αGalCer (left) or vehicle or LPS (right) four hours prior (n = 4–7 mice per group).
(b) Percentage among CD45+ and absolute number of ATMs in WT versus IFNγ KO mice (n = 8–10 mice per group).
(c) Transcript levels Il6, Nos2, and Arg1 in adipose tissue of WT and IFNγ KO mice (n = 3–7 mice per group).
(d-e) Fasting glucose (d) and glucose tolerance test (e) of WT versus IFNγ KO mice (n = 14–15 mice per group).
(f-g) Fasting glucose (f) and glucose tolerance test (g) of WT versus IFNγR KO mice (n = 7–10 mice/group).
NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001. Two tailed Student’s t-test. Mann Whitney U test was use to assess significance for Arg1 in Fig. 6c, as the data were not normally distributed. Error bars indicate mean (± S.E.M.). Data representative of (a, b, c, e, f, g, i) or combined from (d, h) two independent experiments.
Unexpectedly, we found that IFNγ KO mice had a significant expansion of ATMs, containing over three times as many per gram of adipose tissue as WT mice (Figure 6b). The ratio of macrophages expressing M1-like versus M2-like markers was not altered in IFNγ KO mice (Figure S5e). Macrophage accumulation is a driver of inflammation in obese adipose tissue and systemic metabolic dysfunction (Weisberg et al., 2003), and we previously showed that iNKT-deficient mice exhibit macrophage accumulation in adipose tissue (Lynch et al., 2012) (Figure S5f). In fact, previous reports have shown that both ATMs with M1 and M2 markers have similar capacity to produce inflammatory cytokines when they expand uncontrolled (Kratz et al., 2014; Xu et al., 2013). Therefore, while the balance of “M1” and “M2” macrophages is important in adipose tissue, the total number of macrophages is the major determinant of inflammation. Consistent with this, we found a higher level of transcripts for the macrophage-derived inflammatory mediators Il6 and Nos2 and a marked reduction in the anti-inflammatory macrophage marker Arg1 in the adipose tissue of IFNγ KO mice (Figure 6c).
Given the greater macrophage accumulation, we examined the metabolic state of lean IFNγ KO mice. We found that IFNγ signaling was essential for glucose homeostasis in lean mice: IFNγ KO mice had significantly higher fasting glucose compared to WT counterparts (25% increase) (Figure 6d) and had impaired glucose handling (Figure 6e). These results were phenocopied in mice lacking the IFNγ receptor (Figure 6f, g). Of note, we observed no difference in body mass between WT and IFNγ KO mice (Figure S5g). Thus, iNKT cells are a significant source of IFNγ in adipose tissue, and in the absence of IFNγ signaling, lean mice experience ATM accumulation, greater adipose inflammation, and systemic metabolic dysfunction.
iNKT cell-derived IFNγ licenses NK cell cytotoxicity against ATMs
To define a mechanism by which iNKT cells and IFNγ limit macrophage numbers in adipose tissue, we first characterized the kinetics of ATMs in IFNγ KO mice. We found no differences in macrophage proliferation rates between WT and IFNγ KO mice (Figure 7a, left). In contrast, when we analyzed macrophage death with 7-AAD, we found that there was a 50% reduction in 7-AAD+ macrophages in IFNγ KO adipose tissue compared to WT (Figure 7a, right), suggesting that macrophages may be accumulating due to a lower rate of death.
Figure 7. iNKT cell-derived IFNγ licenses NK cell cytotoxicity against ATMs in lean mice.
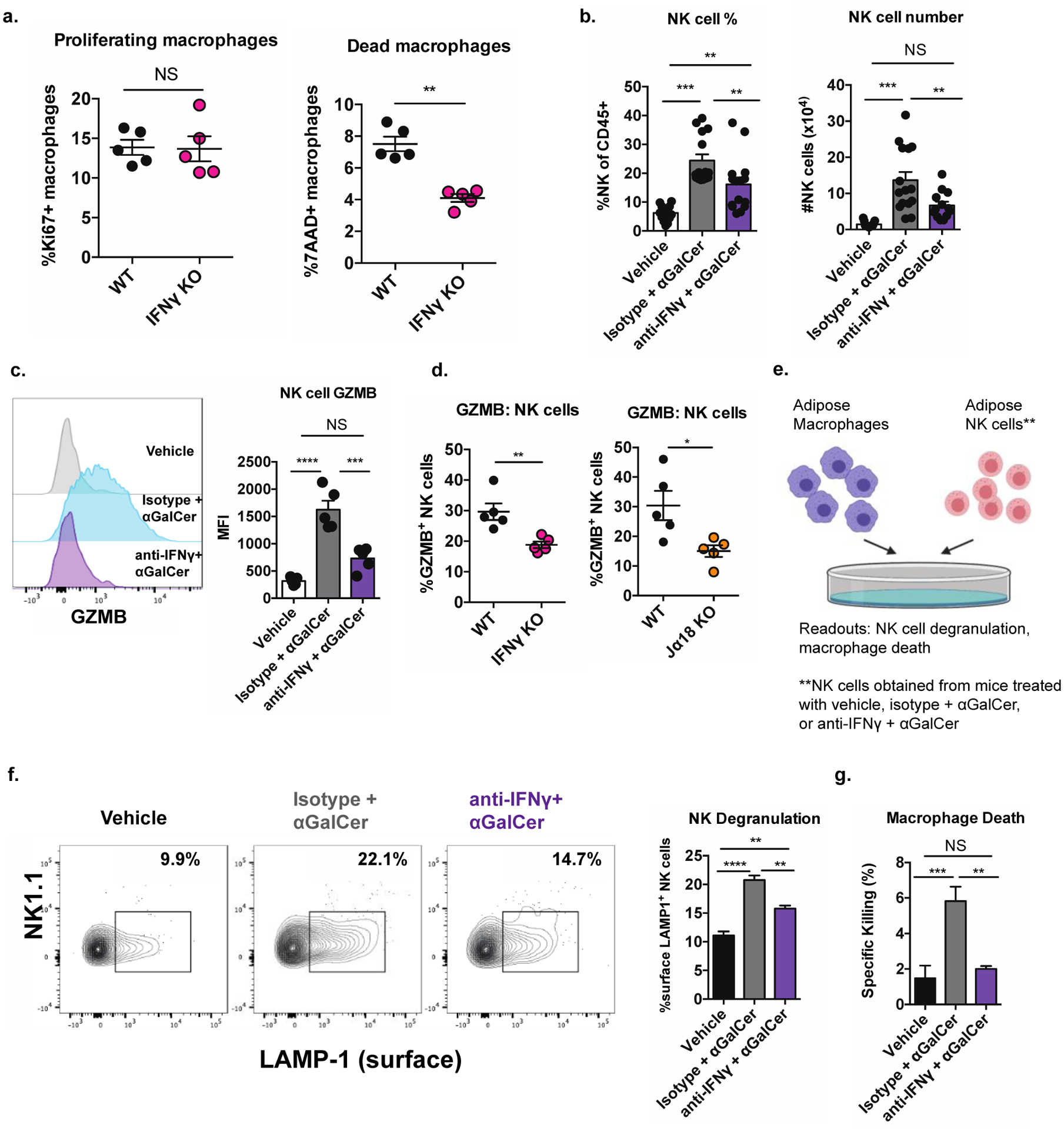
(a) Percentage of Ki67+ (left) and 7-AAD+ (right) ATMs WT and IFNγ KO mice (n = 5 mice/group).
(b) Percentage (left) and number (right) of adipose tissue NK cells from mice given the indicated treatments three days prior to analysis (n= 14–15 mice per group).
(c) Expression of GZMB in adipose NK cells of mice treated as in (b) (n = 5 mice per group).
(d) Expression of GZMB in adipose NK cells of Jα18KO and IFNγ KO mice compared to WT controls (n = 5–6 mice per group).
(e) Diagram of cytotoxicity assay.
(f-g) Percentage of surface LAMP-1+ NK cells (reflecting degranulation) (f) and percentage of dead ATMs (g) resulting from coculture of ATMs with adipose NK cells from mice treated as in (b) (n = two biological replicates, plated in triplicate).
NS, not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001. ANOVA with post-hoc Tukey test (b, c, f, g) or Two tailed Student’s t-test (a, d). Error bars indicate mean (± S.E.M.). All data representative of two independent experiments (c-f) or combined from three independent experiments (b) or one experiment (a). For panel b, two outliers were eliminated using a ROUT test with Q = 0.1% or Grubb’s test with alpha = 0.05
Given the lower macrophage death rate, we hypothesized a role for adipose tissue NK cells in this process as they have been shown recently to induce ATM death at steady state (Boulenouar et al., 2017). While NK cells are known to be enriched in obesity, where they are reported to contribute to inflammation by producing inflammatory cytokines (Lee et al., 2016; O’Sullivan et al., 2016; Wensveen et al., 2015), we recently uncovered a homeostatic role for NK cells in lean adipose tissue. In the lean state, NK cells are cytotoxic against all populations of ATMs in a perforin-granzyme B dependent manner. This “physiological killing” restrains macrophage accumulation and pathologic inflammation, and depletion of NK cells in mice results in ATM expansion and metabolic dysfunction (Boulenouar et al., 2017). During obesity, the cytotoxic ability of adipose NK cells is lost, largely due to lipid accumulation and alteration of cell-intrinsic metabolism (Michelet et al., 2018).
Given that iNKT cell-derived IFNγ has been demonstrated to regulate NK cell functions in several other organs (Carnaud et al., 1999; Fujii et al., 2004; Fujii et al., 2003), we asked if this axis contributed to macrophage cytolysis in lean adipose tissue. To explore this possibility, we specifically activated iNKT cells in lean mice in vivo with αGalCer in the presence or absence of IFNγ blocking antibodies. Three days post αGalCer injection, the percentage and number of adipose NK cells markedly expanded, and this was significantly abrogated by blockade of IFNγ (Figure 7b). In the spleen, iNKT cell-derived IFNγ rapidly transactivates IFNγ production in NK cells, largely by inducing IL-12 production by splenic myeloid cells (Carnaud et al., 1999; Fujii et al., 2004; Fujii et al., 2003). However, activation of adipose tissue iNKT cells did not transactivate NK cells to produce IFNγ (Figure S6a). Instead, we found that iNKT cell activation significantly enhanced the cytotoxic capacity of adipose tissue NK cells; αGalCer stimulation induced a five-fold increase in granzyme B expression in adipose tissue NK cells, and this was completely abrogated by blockade of IFNγ (Figure 7c). Consistent with this, adipose tissue NK cells in both iNKT-deficient and IFNγ-deficient mice exhibited a two-to-three-fold reduction in granzyme B expression (Figure 7d). Interestingly, NK cells from iNKT-deficient mice also produced less IFNγ themselves after PMA/I stimulation (Figure S6b), and had reduced expression of Tbet (Figure S6c).
Finally, we directly tested if iNKT-derived IFNγ drove adipose NK cell killing of macrophages with a cytotoxicity assay. Specifically, we co-cultured ATMs with adipose tissue NK cells obtained from mice treated with a vehicle control or with αGalCer in the presence or absence of IFNγ blocking antibodies (Figure 7e). Confirming our hypothesis, iNKT cell activation enhanced the ability of adipose NK cells to directly kill macrophages as measured by NK cell degranulation/surface LAMP-1 (Figure 7f) and macrophage death (Figure 7g), and these effects were abrogated by IFNγ blockade. Thus, in the lean state, iNKT cells direct NK cell physiological killing of ATMs in an IFNγ-dependent manner, which restrains physiologic inflammation and promotes metabolic health.
Discussion
Here, we sought to gain insight into the biology and functions of adipose tissue iNKT cells by utilizing scRNA-seq and discovered unexpected transcriptional and functional heterogeneity. While NK1.1POS adipose tissue iNKT cells dominantly produce IFNγ, almost all of the previously described regulatory properties belong only to the NK1.1NEG population. Chief among these properties is the production of IL-10, which has been considered a defining characteristic of the adipose tissue iNKT compartment. Mechanistically, we found that adipose tissue FFA accumulation within adipose tissue iNKT cells drives expression of E4BP4 and IL-10 via IRE1α-XBP1 signaling and that this pathway was primarily active within NK1.1NEG iNKT cells. Therefore, these two iNKT cell populations exist at opposite ends of a spectrum, with NK1.1POS iNKT cells being dominantly instructed by developmental signals to produce IFNγ and NK1.1NEG iNKT cells being dominantly instructed by the tissue microenvironment to produce IL-10.
These findings in normal physiology may be relevant to iNKT cells in the context of disease, particularly cancer. IL-10-producing iNKT cells were recently discovered in intestinal polyps and colorectal cancer, where there is a high level of ER stress (Wang et al., 2018). Additionally, splenic iNKT cells can be induced to produce IL-10 by strong stimulation with αGalCer, limiting the ability of these cells to be targeted in anti-tumor responses (Sag et al., 2014). If the IRE1α-XBP1s pathway governs the generation of these cells, then this becomes a potential therapeutic target. Whether this pathway is relevant for other populations of innate T cells also remains an open area for investigation.
Our study also uncovered a metabolically protective role for iNKT cell-derived IFNγ lean animals. At steady state, IFNγ produced by NK1.1POS iNKT cells transactivates adipose NK cells and facilitates their cytotoxicity against tissue-resident macrophages. In the absence of iNKT cells or IFNγ signaling adipose NK cells lose their cytotoxic capacity, and this results in macrophage expansion, local inflammation and metabolic dysfunction. In this way, two divergent populations of adipose iNKT cells collaborate to maintain metabolic homeostasis through non-redundant mechanisms – NK1.1NEG iNKT cells by directly producing regulatory cytokines and NK1.1POS iNKT cells by indirectly limiting the accumulation of pathogenic macrophages.
While many reports have described adipose tissue iNKT cells as anti-inflammatory and protective against metabolic disease, several groups have reported the opposite finding- that adipose iNKT cells contribute to obesity-related metabolic disease by producing TNF and IFNγ (Satoh et al., 2016; Wu et al., 2012). Several theories for this discrepancy have been proposed, including differences in diet, gut microbiome, and analytical methods. However, a mechanistic explanation for these divergent findings has not been demonstrated. It is tempting to speculate that these contrasting results may be driven by differences in the iNKT cell subsets we have identified in this report. Indeed, differences in either the activation state or the relative abundance of these two subsets could influence whether the bulk iNKT cell population is interpreted as protective or pathogenic during obesity. Supporting this, the NK1.1NEG population was expanded by fasting, which is well known to drive anti-inflammatory signaling pathways in adipose tissue. Our results provide a new framework for future studies of adipose tissue iNKT cells in different mouse cohorts.
Interestingly, two studies focusing on the role of IFNγ in the obese state observed no differences in glucose tolerance between lean WT and IFNγ KO mice, although they did not analyze adipose tissue inflammation or immune cell compartments (Rocha et al., 2008; Wong et al., 2011). While we do not know the reason for this discrepancy, one significant difference between these studies and ours is the ages of the mouse cohorts used. The previous studies used aged mice (28 weeks and older) for metabolic analysis. Our studies, in contrast, almost exclusively used younger mice (ages 6–12 weeks) due to the higher number and important role of iNKT cells in adipose physiology during that time window (Kohlgruber et al., 2018; LaMarche et al., 2019; Lynch et al., 2015). Given that the immunological environment in adipose tissue is well documented to change with age, particularly around 16 weeks of age (Feuerer et al., 2009; Kohlgruber et al., 2018; Kolodin et al., 2015; LaMarche et al., 2019), it is possible that IFNγ plays its most essential metabolic role in adipose tissue at younger ages. Another study found that IFNγ deficiency enhanced glucose tolerance in lean mice, but demonstrated that this effect was completely dependent on the gut microbiome (Greer et al., 2016). Altering or reducing the microbiota with antibiotics abrogated the metabolic differences between WT and IFNγ KO mice (Greer et al., 2016). It serves reason that specific vivarium microbiomes may underscore different contributions of IFNγ signaling to metabolic homeostasis. Other factors including environment, food, and technical differences also cannot be discounted.
That IFNγ serves a metabolically protective role in lean adipose tissue seems paradoxical, especially given the importance of preserving an anti-inflammatory microenvironment in this organ. However, a growing body of research has shown that IFNγ signaling can have markedly anti-inflammatory effects depending on context. For example, during influenza infection excessive IFNγ signaling suppresses phagocytic ability of alveolar macrophages by inducing scavenger receptor downregulation, and this suppression results in enhanced susceptibility for secondary pneumococcal infection (Sun and Metzger, 2008). In allergic asthma, IFNγ produced by a unique iNKT cell population protects against disease by antagonizing type 2 inflammation in the lung (Chuang et al., 2019). IFNγ also has several anti-inflammatory effects on myeloid cells, including downregulation of IL-1β and IL-8 and induction of caspase-dependent apoptosis (Mühl and Pfeilschifter, 2003). Our finding that IFNγ serves an anti-inflammatory role in lean adipose tissue highlights the importance of this phenomenon in the maintenance of a metabolic organ.
It appears that adipose tissue has a unique ability to utilize stereotypically proinflammatory cytokines to drive homeostatic outcomes. Indeed, IL-17A (Kohlgruber et al., 2018), TNF (Wernstedt Asterholm et al., 2014) and IL-18 (Murphy et al., 2016) all maintain metabolic fitness in adipose tissue when they are produced in a controlled manner in low quantities. Here we have shown that IFNγ, a metabolically deleterious cytokine in obesity, acts in a beneficial way when produced by NK1.1POS adipose tissue iNKT cells in the lean state. Thus, adipose tissue contains two populations of iNKT cells that, when activated in the correct physiological context, cooperate to subdue local inflammation and preserve systemic metabolic integrity.
Limitations of Study
Our study leaves open several questions suitable for future study. It is unknown if PLZF and E4BP4 expression are regulated by a common pathway, or if these two transcription factors counter-regulate each other.
Additionally, it is unknown if expression of NK1.1 is simply a distinguishing marker between adipose iNKT cell subsets, or if this NK receptor has a functional role in their biology. As stated above, we acknowledge that our results vis-à-vis the metabolic effect of IFNγ-deficiency differs from those of two previous studies. While we speculate that this may be due to significant differences in the ages of the mouse cohorts, we cannot discount well-known regulators of metabolism such as diet and facility-specific microbiomes, as well as more nebulous environmental factors such as ambient noise and frequency of mouse handling. These factors may also help explain differing results on the protective versus pathogenic role of iNKT cells in adipose tissue. Besides these variables, our new insights into adipose iNKT cell heterogeneity and function provide the opportunity to study the proportions of distinct subsets and their activation states at a more refined level which may offer insight into the results in different laboratories.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael B. Brenner (mbrenner@research.bwh.harvard.edu).
Materials Availability
No new materials were created for this study.
Data and Code Availability
ScRNA-seq data from this manuscript have been deposited in the Gene Expression Omnibus under accession code GSE142845. Non-RNA-seq data have been deposited in Mendeley Data (doi: 10.17632/2d6ywgb8bj.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Jα18 KO / CD1d KO, Vα14 TN (Clancy-Thompson et al., 2017), and CD1d1floxed (Olszak et al., 2014) mice were kindly provided by Dr. Mark Exley, Dr. Stephanie Dougan and Dr. Richard Blumberg, respectively. C57BL/6 (WT), AdipoQ Cre (RRID:IMSR_JAX:010803), IL-10 eGFP (TIGER) (RRID:IMSR_JAX:008379) , IFNγ KO (RRID:IMSR_JAX:002287), and IFNγR KO (RRID:IMSR_JAX:003288) mice were obtained from Jackson Laboratories. Animals were randomly assigned to experimental and control groups. In almost all experiments, male mice between 6–14 weeks of age were used. Both male and female Vα14 TN mice were used as a source of adipose tissue iNKT cells for sorting and adoptive transfer experiments. Mice were bred and housed at 25°C under specific pathogen-free conditions at Brigham and Women’s Hospital’s Center for Comparative Medicine. Mice were in good general health, were subject to a 12:12 hour light:dark schedule, and had free access to food and water. Where indicated, mice were fed either standard fat diet (Pico Labs 5053) or HFD (Research Diets, 60 kcal% fat) beginning at six weeks of age. HFD was for a duration of 8 weeks and induced weight gain of roughly 15 grams. All animal work was approved by, and in compliance with, the Institutional Animal Care and Use Committee of Brigham and Women’s Hospital.
METHOD DETAILS
Metabolic Testing
All metabolic testing was performed on mice fasted for 6–12 hours. For glucose tolerance tests, mice were injected I.P. with 2 g/kg (lean mice) or 1g/kg (obese mice) dextrose and blood glucose levels were measured at the indicated time points post injection.
Tissue Processing
Adipose tissue was excised, minced with a razor, and digested in 1mg/ml Collagenase Type II (Worthington) in RPMI shaking for 25 minutes at 37°C. Digested cells were filtered through a 70μM nylon mesh and centrifuged at 15,000 rpm for 7 minutes to pellet the stromovascular fraction (SVF). Spleen was disrupted through a 70μM filter and pelleted. Red blood cells in the spleen and SVF were lysed with ACK Lysing Buffer (VWR) prior to further analysis.
Histology
Visceral adipose tissue was fixed in 4% paraformaldehyde in PBS overnight and then transferred to 70% ethanol for storage. Samples were processed, paraffin embedded, and stained with H&E by the Dana Farber Rodent Histopathology Core. TIF images of sections were collected at 20X magnification, and adipocyte size was analyzed on ImageJ using the Adiposoft automated software package. Size cutoffs were set between 10μM-100μM and manually adjusted as needed.
Flow Cytometry and Cell Sorting
All antibody staining of live cells was performed in Biolegend Cell Staining Buffer. Single cell suspensions were incubated in Fc-receptor blocking antibody (Clone 93, Thermo Fisher) on ice for 15 minutes prior to cell surface antigen staining. Dead cells were excluded with 7-aminoactinomycin D or Fixable Viability dyes (UV or eFluor 780, Thermo Fisher). For intracellular antigen staining, cells were fixed with either BD Pharmingen’s Cytofix/Cytoperm kit or Thermo Fisher’s FoxP3/Transcription Factor Fixation/Permeabilization kit for 30 minutes at room temperature. iNKT cells were identified as lymphocytes binding to anti-TCRβ antibodies and αGalCer analog PBS57-loaded CD1d tetramer (NIH Tetramer Core Facility/Emory Vaccine Center). For analysis of iNKT cells and NK cells, a “dump” channel with antibodies against CD19 and F4/80 was used to eliminate nonspecific staining. For neutral lipid staining, cells were stained with the LipidTox reagent (Thermo Fisher) per the manufacturer’s instructions. Gating strategies are provided in Supplemental Information (Figure S7).
Generation of splenic iNKT cell lines
Lines were generated from C57BL/6 mouse spleens as previously described (Chiba et al., 2009). Briefly, iNKT cells were isolated from bulk splenocytes by tetramer-based MACS enrichment followed by FACS sorting, and then expanded by exposure of to irradiated CD11c+ splenocytes pulsed with αGalCer. Stimulated cells were maintained in a cocktail of IL-2 and IL-7, and frozen for use in experiments 21 days later. Cells were used in experiments one day after thawing.
In vitro stimulations
Where indicated, iNKT cells were incubated for 6–12 hours with 0.1g epidydimal fat, 20μM 4μ8c, 10–1mg/ml 4-PBA, or 1μg/ml tunicamycin. In other experiments, tissues were digested as described above, and cultured for 2–4 hours in the presence of phorbol 12-myristate 13-acetate and Ionomycin (PMA/I) (eBioscience Cell Stimulation Cocktail, Thermo Fisher) and Brefeldin A (Thermo Fisher). Cultures were in complete RPMI media [RPMI supplemented with HEPES (Invitrogen), L-glutamine, penicillin/streptomycin, 2-Mercapto Ethanol, and 10% FBS (Gemini)]. For palmitate cultures, dry palmitic acid (Sigma) was dissolved in methanol and then diluted in culture to 200μM. A vehicle control was used for all cultures.
Adipose-conditioned media and liquid-liquid extractions
To generate adipose-conditioned media, 1g of epididymal adipose tissue was cultured in 1ml of complete RPMI overnight, and centrifuged to collect cell-free supernatant. Conditioned media was fractionating using a liquid-liquid extraction with 2:1 chloroform:methanol to collect aqueous (protein) and organic (lipid) phases for biological testing. Fractions were dried, resuspended in equivalent volumes of complete RPMI, and sonicated before being tested on iNKT cells.
In vivo stimulations
For BrdU labeling, mice were injected I.P. with 1 mg BrdU daily for six days. One day later, tissues were harvested and BrdU incorporation in iNKT cells was evaluated with the BrdU Flow Kit (BD Pharmingen) per the manufacturer’s instructions. As a cytokine-driven model of iNKT cell activation, mice were injected I.V. with 2mg ultra-pure LPS from E. coli O111:B4 (InvivoGen) per kg body weight. As an antigen-driven model of iNKT cell activation, mice were injected I.P. with 1 μg αGalCer analog KRN7000 (Avanti Polar Lipids). Both αGalCer and LPS were prepared in DMSO and diluted 1:10 (v/v) in sterile saline immediately prior to injection. Mice were sacrificed and tissues harvested after either 4 or 72 hours. In some experiments, mice were injected I.P. with 200μg ultra-low endotoxin azide-free anti-IFNγ (XMG1.2) or isotype control antibodies (Biolegend) 8 hours prior to αGalCer injection. For in vivo analysis of apoptosis mice were injected I.V. with the fluorescent poly-caspase binding reagent FAM-FLIVO (Immunochemistry Technologies) and analyzed per the manufacturer’s instructions. For ex-vivo intracellular cytokine staining, suspensions of SVF cells were cultured for two hours at 37°C in complete media in the presence of Brefeldin A prior to staining with flow cytometry antibodies.
Adoptive Transfer
In Figures 1 and 3, 1 × 106 CD45.2 splenic iNKT cells were injected I.P. into congenically marked CD45.1 mice. 1–2 weeks later, transferred cells were harvested from recipient spleen and adipose tissue. In Figure 5, obese Jα18KO mice were I.P. injected with vehicle control or with 20,000 NK1.1POS or NK1.1NEG iNKT cells sorted from the adipose tissue of lean Vα14 TN mice. Two weeks later, mice were subjected to metabolic testing and adipose tissue analysis as described above.
Cytotoxicity assays
Adipose NK cells from indicated mice were sorted and cultured in 1:1 ratios with ATMs from lean, unmanipulated mice. Macrophages were labeled with 0.01μM of CellTracker Green (Thermo Fisher) prior to co-incubation. Cells were centrifuged in round bottom plates to force conjugation and incubated for 2–4 hours in the presence of monensin (Biolegend) and anti-CD107a antibodies. 7-aminoactinomycin D was added to the cells immediately prior to analysis on a flow cytometer.
RT-PCR Analysis
iNKT cells or adipose SVF were suspended in a 5:1 ratio of TRIzol Reagent (Life Technologies) : chloroform. After centrifugation, the upper aqueous phase was extracted and mixed in a 1:1 ratio with 70% ethanol. RNA was then isolated with RNeasy Mini Kits (Qiagen). cDNA was prepared using Quantitect RT-PCR kit (Qiagen), and qPCR was performed on a Stratagene Mx3000 instrument using Brilliant III SYBRGreen (Agilent). Relative expression values were normalized to expression of 18s or Hprt. A list of primers can be found in Table S3.
scRNA-seq sequencing and data pre-processing
Single-cell RNA-seq was performed was performed using the 10x Genomics platform. Single cell suspensions from the spleens of mice treated with saline or αGalCer were tagged by TotalSeq-A Mouse hashtag antibody #1 and #2 (Biolegend) respectively, and a total of 7,500 cells from each labeled suspension were combined and loaded onto a 10x Chromium Controller to generate single cell Gel Beads-in-emulsion (GEMS). Similarly, cells from the visceral adipose tissue of mice treated with saline were separately loaded onto a 10x Chromium Controller to generate GEMS. GEMs were then processed to generate UMI-based libraries according to the 10X Genomics Chromium Single Cell 3’ protocol. Libraries were sequenced using a NextSeq 500 sequencer (Illumina). Cell Ranger v3.0.2 was used to process the raw BCL files and produce a final feature by barcode UMI count matrix. First, raw BCL files were demultiplexed using Cell Ranger mkfastq to generate fastq files with default parameters. Fastq files were then aligned to the mm10 genome (v1.2.0) and feature reads were quantified simultaneously using Cell Ranger count for feature barcoding. The resulting filtered feature-barcode UMI count matrices containing quantification of gene expression in each cell, and, in the case of the splenic iNKT UMI matrix, quantification of hashtag antibody binding, were then utilized for downstream analysis of the scRNA-seq data.
Downstream analysis of scRNA-seq data
A total of 15,702 cells expressing a median of 1,685 genes per cell were loaded from the pre-processed single cell data files. For analysis of the adipose iNKT cells only, cells expressing less than 1000 genes or more than 6000 genes in total were excluded from the analysis to remove possible doublets, empty droplets and cells with poor quality transcripts. Genes which were expressed in less than 3 cells in total were removed from the analysis to prevent false positive identification of transcripts. Cells expressing less than 1% or greater than 10% of mitochondrial genes as a % of total gene counts were considered to be apoptotic or dying and were also removed from the analysis. Following quality control, UMI counts were normalized using regularized negative binomial regression with the sctransform package (Hafemeister and Satija, 2019).
For dimensional reduction of the normalized adipose iNKT cell data, principal component analysis (PCA) was performed using n = 50 dimensions and PCA variability was determined using an Elbow plot. A total of 40 dimensions were selected for calculating the initial UMAP using a minimum distance of 0.3 and a spreading factor of 1. An initial SNN graph was constructed (k = 20 nearest neighbors, n = 40 dimensions) and graph-based clustering was performed using the leidenalg algorithm at a resolution of 0.22 (Traag et al., 2019). At this stage a small number of outlier cells expressing macrophage markers such as Cd14 (CD14) and Adgre1 (F4/80) were identified and were removed from the analysis. A total of 3000 variable features were then used to recalculate a new PCA and a total of 40 dimensions were selected for calculating the final UMAP using a minimum distance of 0.3 and a spreading factor of 1. A new SNN graph was then constructed (k = 20 nearest neighbors, n = 25 dimensions) and graph-based clustering was performed using the leidenalg algorithm using a resolution of 0.075 to generate the final clusters for analysis. Differentially expressed genes (DEGs) and cluster markers were identified using the Wilcoxon Rank Sum test and DEGs were defined as genes which had FDR-corrected p-values of 1E-20 or less, were expressed in at least 30% of all cells in a given cluster (min.pct = 0.3) and had log2(Fold Change) (Log2FC) threshold of at least 0.25. Sub-clustering of NKT1-B cells was performed by identifying n = 3000 new variable features, computing a new PCA and then calculating a new UMAP using a total of 40 dimensions (minimum distance = 0.3, spreading factor = 1).
For analysis of splenic and adipose tissue iNKT cells together, splenic cells iNKT cells first underwent quality control filtering similar to the filtering steps previously described for adipose tissue iNKT cells. Briefly, cells expressing less than 500 genes or more than 4000 genes in total and genes expressed in less than 3 cells in total were removed from the analysis. Cells expressing less than 2.5% or greater than 10% of mitochondrial genes as a % of total gene counts were then also filtered out. Antibody hashtag data was demultiplexed using the Seurat HTODemux function (Butler et al., 2018; Stuart et al., 2019) with a positive quantile of 0.99 and doublets identified as positive for both antibodies were removed from the analysis. HTO data was normalized using centered log ratio transformation and all cells which displayed a value greater than 0.5 for hashtag antibody #1 (corresponding to splenic iNKT cells isolated from saline-injected mice) were selected for further analysis. The raw UMI counts of these selected splenic iNKT cells were then merged with the raw UMI counts of the previously analyzed adipose tissue iNKT cells to generate a new merged data matrix including both splenic and adipose tissue iNKT cells. UMI counts of cells in the merged matrix were normalized using regularized negative binomial regression with the sctransform package and PCA was performed using n = 100 dimensions. PCA variability was determined using an Elbow plot and an initial UMAP was generated using a total of 100 dimensions, a minimum distance of 0.3 and a spreading factor of 1. Following initial graph-based clustering (k = 20 nearest neighbors, n = 25 dimensions) two small clusters of splenic cells expressing macrophage associated genes (Cd14, Adgre1 etc.) and Cd8a were identified, and these contaminating cells were removed from the analysis. A total of 3000 variable features were then used to calculate a new PCA and a total of 50 dimensions were selected for calculating the final UMAP using a minimum distance of 0.3 and a spreading factor of 1. Differential expression testing was performed using the MAST package (Finak et al., 2015) with a min.pct and a log2FC threshold of zero.
All downstream analysis was performed using R v.3.6.1 and RStudio Desktop 1.2.5001 on a Windows 10 (64 bit) system using the following R packages and libraries: dplyr v.0.8.3, ggplot2 v.3.2.1, Seurat v.3.1.1, sctransform v.0.2.0, uwot 0.1.4, reticulate v.1.13, leiden v.0.3.1, viridis v.0.5.1, MAST v.1.12.0 and R tools v.35; and the following python libraries: umap-learn v.0.3.9, leidenalg v.0.7.0, pycairo v.1.18.1 and python-igraph v.0.7.1
QUANTIFICATION AND STATISTICAL ANALYSIS
Pilot studies were performed to estimate required sample size for adequate power and control and experimental groups were randomly assigned. Experiments were repeated two to three times to ensure reproducibility unless otherwise indicated. Data represent mean ± S.E.M. unless otherwise indicated. Significance was determined by Student’s two-tailed t-test with Bonferroni correction, Mann-Whitney U Test, or one-way ANOVA with post-hoc Tukey test, where indicated. Significance is presented as *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001 with P > 0.05 considered non-significant. Details on specific statistical tests can be found in each figure legend. All statistical analyses were performed on GraphPad Prism 6. Investigators were not blinded during this study. In figure 7b, 2 out of 43 samples were excluded using a ROUT test with Q = 0.1% or Grubb’s test with alpha = 0.05.
Supplementary Material
Table S2, related to Figure 4. Differentially expressed genes between adipose and splenic iNKT cells by scRNAseq
Table S1, related to Figure 2. Differentially expressed genes in each scRNAseq cluster
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq™-A0301 anti-mouse Hashtag 1 Antibody | Biolegend | Ca#155801 |
| TotalSeq™-A0302 anti-mouse Hashtag 2 Antibody | Biolegend | Ca#155803 |
| Ultra-LEAF™ Purified anti-mouse IFN-γ Antibody | Biolegend | Ca#505834 |
| anti-mouse/human CD11b (M1/70) | Biolegend | Ca#101242 |
| anti-mouse CD45 (30-F11) | Biolegend | Ca#103138 |
| anti-mouse TCRβ (H57–597) | Biolegend | Ca#109224 |
| anti-mouse NKp46 (29A1.4) | Biolegend | Ca#137612 |
| anti-mouse PLZF (Mags.21F7) | Thermofisher | Ca#53-9320-82 |
| anti-mouse T-bet (4B10) | Thermofisher | Ca#45-5825-82 |
| anti-mouse RORγT (B2D) | Thermofisher | Ca#25-6981-82 |
| anti-mouse E4BP4 (S2M-E19) | Thermofisher | Ca#12-5927-82 |
| anti-mouse CD69 (FN50) | Thermofisher | Ca#25-0699-41 |
| anti-mouse Nur77 (12.14) | Thermofisher | Ca#53-5965-82 |
| anti-mouse TNF (MP6-XT22) | Thermofisher | Ca#25-7321-82 |
| anti-mouse IL-17A (eBio17B7) | Thermofisher | Ca#45-7177-82 |
| anti-mouse IFN-γ (XMG1.2) | Thermofisher | Ca#53-7311-82 |
| anti-mouse IL-2 (JES6–5H4) | Thermofisher | Ca#25-7021-82 |
| anti-mouse IL-10 (JES5–16E3) | Thermofisher | Ca#12-7101-41 |
| anti-mouse IL-4 (11B11) | Thermofisher | Ca#25-7041-82 |
| anti-mouse IL-13 (eBio13A) | Thermofisher | Ca#12-7133-82 |
| anti-mouse F4/80 (BM8) | Thermofisher | Ca#17-4801-82 |
| anti-mouse CD19 (ID3) | Thermofisher | Ca#47-0193-82 |
| anti-mouse NK1.1 (PK136) | Thermofisher | Ca#25-5941-82 |
| anti-mouse CD3e (500A2) | Thermofisher | Ca#HM3420 |
| anti-mouse CD11c (N418) | Thermofisher | Ca#47-0114-82 |
| anti-mouse CD301 (ER-MP23) | Thermofisher | Discontinued |
| anti-mouse CD107a (eBio1D4B) | Thermofisher | Ca#12-1071-82 |
| anti-mouse Granzyme B (NGZB) | Thermofisher | Ca#25-8898-82 |
| anti-mouse CD4 (RM4–5) | Thermofisher | Ca#48-0042-82 |
| anti-mouse CD8a (53–6.7) | Thermofisher | Ca#11-0081-82 |
| anti-mouse ST2 (RMSST2–2) | Thermofisher | Ca#17-9335-82 |
| anti-mouse Foxp3 (FJK-16s) | Thermofisher | Ca#11-5773-82 |
| anti-mouse CD45.2 (104) | Thermofisher | Ca#45-0454-82 |
| anti-mouse CD45.1 (A20) | Thermofisher | Ca#25-0453-82 |
| anti-mouse Ki67 (SolA15) | Thermofisher | Ca#11-5698-82 |
| anti-mouse Siglec-F (E50–2440) | BD Biosciences | Ca#565526 |
| PE Mouse anti-XBP-1S | BD Biosciences | Ca#562642 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alpha-galactosylceramide (KRN7000) | Avanti | Ca#867000 |
| Recombinant murine IL-2 | Peprotech | Ca#212–12 |
| Recombinant murine IL-7 | Peprotech | Ca#217–17 |
| Palmitic acid | Sigma | Ca#P0500 |
| 4μ8c | Sigma | Ca#SML0949 |
| Sodium phenylbutyrate | Sigma | Ca#SML0309 |
| Tunicamycin | Sigma | Ca#T7765 |
| Chloroform | Sigma | Ca#288306 |
| Methanol | Sigma | Ca#34860 |
| Monensin solution, 1000X | Biolegend | Ca#420701 |
| CellTracker™ Green CMFDA Dye | Thermofisher | Ca#C2925 |
| Annexin V | Thermofisher | Ca#88-8007-72 |
| Brefeldin A | Thermofisher | Ca#00-4506-51 |
| LipidTox Green | Thermofisher | Ca#H34475 |
| Trizol Reagent | Thermofisher | Ca#15596026 |
| FoxP3 staining kit | Thermofisher | Discontinued |
| Cell stimulation cocktail | Thermofisher | Ca#00-4970-03 |
| 7AAD | BD Biosciences | Ca#559925 |
| Cytofix/Cytoperm | BD Biosciences | Ca#554714 |
| Collagenase Type 2 | Worthington | Ca#LS004176 |
| ACK Lysing Buffer | VWR | Ca#10128–802 |
| Ultrapure LPS, E. coli 0111:B4 | InvivoGen | Ca#tlrl-3pelps |
| Critical Commercial Assays | ||
| FAM-FLIVO Kit | ImmunoChemistry Technologies | Ca#980 |
| BRDU Flow Kit | BD BioSciences | Ca#559619 |
| RNeasy Mini Kit | Qiagen | Ca#74104 |
| Brilliant III SYBR Green | Agilent | Ca#600883 |
| Deposited Data | ||
| Raw scRNAseq Data | GEO | GSE142845 |
| Raw data (non-RNAseq) | Mendeley Data | doi: 10.17632/2d6ywgb8bj.1 |
| Experimental Models: Cell Lines | ||
| Primary murine iNKT cell line | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Vα14 TN | Stephanie Dougan Lab (Clancy-Thompson, et al., 2018) | N/A |
| Mouse: CD1d1floxed | Richard Blumberg Lab (Olszak, et al., 2014) | N/A |
| Mouse: B6.129S6-Tcra-Jtm1Tgi/J (Jα18 KO) | Mark Exley Lab | N/A |
| Mouse: B6.129S6-Cd1d1/Cd1d2tm1Spb/J (CD1d KO) | Mark Exley Lab | N/A |
| Mouse: C57BL/6J (WT) | Jackson Laboratories | Ca#000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) | Jackson Laboratories | Ca#002014 |
| Mouse: B6.129S7-Ifngtm1Ts/J (IFNγ KO) | Jackson Laboratories | Ca#002287 |
| Mouse: B6.129S6-Il10tm1Flv/J (TIGER) | Jackson Laboratories | Ca#008379 |
| Mouse: B6;FVB-Tg(Adipoq-cre)1Evdr/J (AdipoQ Cre) | Jackson Laboratories | Ca#010803 |
| Oligonucleotides | ||
| qPCR Primers (18s, Nfil3, Il10, Xbp1 total, Xbp1s, Hprt, Nos2, Il6, Arg1, Il13, Il4 | Integrated DNA Technologies | See Table S3 |
| Software and Algorithms | ||
| ImageJ | ImageJ | www.imagej.net |
| Adiposoft package | ImageJ | www.imagej.net/Adiposoft |
| FlowJo | Treestar | https://www.flowjo.com/ |
| R | The R Project for Statistical Computing | www.r-project.org |
| Graphpad Prism 6 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Mouse PBS57:CD1d Tetramer | NIH Tetramer Core Facility/Emory Vaccine Center | N/A |
| Standard Fat (Chow) Diet | Pico Labs | Ca#0007688 |
| High Fat Diet (60% kcal) | Research Diets, Inc | Ca#D12492 |
| Blood Glucose Meter | McKesson | Ca#960302 |
| Blood Glucose Test Strips | McKesson | Ca#960298 |
Acknowledgments:
The authors thank the Brigham and Women’s Hospital Single Cell Genomics Core for assistance with sequencing and pre-processing of the scRNASeq data, the NIH Tetramer Core Facility for recombinant CD1d PBS57 tetramers, A.T. Chicoine for cell sorting, R.S. Blumberg for CD1dfloxed mice, and S.K. Dougan for Vα14 TN mice.
Funding:
This work was supported by NIH grants F31 AI138353 (to N.M.L.), R01 AI113046 (to M.B.B.) and R01 AI134861 (to L.L.) and ERC Stg 679173 and American Diabetes Association 1-16-JDF-061 (to L.L.).
Footnotes
Declaration of Interests: The authors declare no competing interests.
References
- Beach JA, Nary LJ, Hirakawa Y, Holland E, Hovanessian R, and Medh RD (2011). E4BP4 facilitates glucocorticoid-evoked apoptosis of human leukemic CEM cells via upregulation of Bim. J Mol Signal 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O’Connor D, Stutte S, Tavakkoli A, Winters D, et al. (2017). Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 46, 273–286. [DOI] [PubMed] [Google Scholar]
- Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, et al. (2015). Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, and Bendelac A (1999). Cutting Edge: Cross-Talk Between Cells of the Innate Immune System: NKT Cells Rapidly Activate NK Cells. J Immunol 163, 4647–4650. [PubMed] [Google Scholar]
- Chiba A, Cohen N, Brigl M, Brennan PJ, Besra GS, and Brenner MB (2009). Rapid and reliable generation of invariant natural killer T-cell lines in vitro. Immunology 128, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YT, Leung K, Chang YJ, DeKruyff RH, Savage PB, Cruse R, Benoit C, Elewaut D, Baumgarth N, and Umetsu DT (2019). A natural killer T-cell subset that protects against airway hyperreactivity. J Allergy Clin Immunol 143, 565–576 e567. [DOI] [PubMed] [Google Scholar]
- Clancy-Thompson E, Chen GZ, Tyler PM, Servos MM, Barisa M, Brennan PJ, Ploegh HL, and Dougan SK (2017). Monoclonal Invariant NKT (iNKT) Cell Mice Reveal a Role for Both Tissue of Origin and the TCR in Development of iNKT Functional Subsets. J Immunol 199, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSpirito JR, and Mathis D (2015). Immunological contributions to adipose tissue homeostasis. Semin Immunol 27, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel I, Seumois G, Chavez L, Samaniego-Castruita D, White B, Chawla A, Mock D, Vijayanand P, and Kronenberg M (2016). Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol 17, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Liu K, Smith C, Bonito AJ, and Steinman RM (2004). The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med 199, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Shimizu K, Smith C, Bonifaz L, and Steinman RM (2003). Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med 198, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Stankovic S, and Baxter AG (2010). Raising the NKT cell family. Nat Immunol 11, 197–206. [DOI] [PubMed] [Google Scholar]
- Govindarajan S, Gaublomme D, Van der Cruyssen R, Verheugen E, Van Gassen S, Saeys Y, Tavernier S, Iwawaki T, Bloch Y, Savvides SN, et al. (2018). Stabilization of cytokine mRNAs in iNKT cells requires the serine-threonine kinase IRE1alpha. Nat Commun 9, 5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer RL, Dong X, Moraes AC, Zielke RA, Fernandes GR, Peremyslova E, Vasquez-Perez S, Schoenborn AA, Gomes EP, Pereira AC, et al. (2016). Akkermansia muciniphila mediates negative effects of IFNgamma on glucose metabolism. Nat Commun 7, 13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, and Hotamisligil GS (2011). Inflammatory mechanisms in obesity. Annu Rev Immunol 29, 415–445. [DOI] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv, 576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, Lee SK, Alfadda AA, Kim SS, Choi SH, et al. (2013). A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol 33, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JY, Park J, Kim JI, Park YJ, Lee YK, and Kim JB (2017). Deletion of CD1d in Adipocytes Aggravates Adipose Tissue Inflammation and Insulin Resistance in Obesity. Diabetes 66, 835–847. [DOI] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, and Qi L (2012). Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem 287, 13561–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JS, Koh JM, So JS, Jeon YK, Kim HY, and Chung DH (2017). Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1alpha-dependent decay. Sci Rep 7, 14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlgruber A, and Lynch L (2015). Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Curr Diab Rep 15, 92. [DOI] [PubMed] [Google Scholar]
- Kohlgruber AC, Gal-Oz ST, LaMarche NM, Shimazaki M, Duquette D, Koay HF, Nguyen HN, Mina AI, Paras T, Tavakkoli A, et al. (2018). gammadelta T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol 19, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, and Mathis D (2015). Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 21, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMarche NM, Kohlgruber A, and Brenner MB (2019). Innate T cells govern adipose tissue biology. J Immunol 201, 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberle D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, et al. (2016). Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab 23, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Holzapfel KL, Zhu J, Jameson SC, and Hogquist KA (2013). Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol 14, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, Cohen DE, Ghosh A, Lu B, Corrigan M, et al. (2016). iNKT Cells Induce FGF21 for Thermogenesis and Are Required for Maximal Weight Loss in GLP1 Therapy. Cell Metab 24, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, Besra G, Vomhof-Dekrey EE, Tighe M, Koay HF, et al. (2015). Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol 16, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, Balk SP, O’Shea D, O’Farrelly C, and Exley MA (2012). Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 37, 574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG, and O’Farrelly C (2009). Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol 39, 1893–1901. [DOI] [PubMed] [Google Scholar]
- McNab FW, Pellicci DG, Field K, Besra G, Smyth MJ, Godfrey DI, and Berzins SP (2007). Peripheral NK1.1 NKT cells are mature and functionally distinct from their thymic counterparts. J Immunol 179, 6630–6637. [DOI] [PubMed] [Google Scholar]
- Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. (2018). Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nature Immunology 19, 1330–1340. [DOI] [PubMed] [Google Scholar]
- Motomura Y, Kitamura H, Hijikata A, Matsunaga Y, Matsumoto K, Inoue H, Atarashi K, Hori S, Watarai H, Zhu J, et al. (2011). The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4+ T cells. Nat Immunol 12, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühl H, and Pfeilschifter J (2003). Anti-inflammatory properties of pro-inflammatory interferon-γ. International Immunopharmacology 3, 1247–1255. [DOI] [PubMed] [Google Scholar]
- Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, Wentworth JM, Vasanthakumar A, Gerlic M, Whitehead LW, et al. (2016). IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab 23, 155–164. [DOI] [PubMed] [Google Scholar]
- O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, and Sun JC (2016). Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity 45, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, Lin CS, Jobin C, Brand S, Sotlar K, et al. (2014). Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature 509, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicci DG, Hammond KJ, Uldrich AP, Baxter AG, Smyth MJ, and Godfrey DI (2002). A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J Exp Med 195, 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramanik J, Chen X, Kar G, Henriksson J, Gomes T, Park JE, Natarajan K, Meyer KB, Miao Z, McKenzie ANJ, et al. (2018). Genome-wide analyses reveal the IRE1a-XBP1 pathway promotes T helper cell differentiation by resolving secretory stress and accelerating proliferation. Genome Med 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, and Libby P (2008). Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 103, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag D, Krause P, Hedrick CC, Kronenberg M, and Wingender G (2014). IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest 124, 3725–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Hoshino M, Fujita K, Iizuka M, Fujii S, Clingan CS, Van Kaer L, and Iwabuchi K (2016). Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep 6, 28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, Prop S, Meerding J, Hamers N, Besra G, et al. (2012). Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest 122, 3343–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, and Metzger DW (2008). Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med 14, 558–564. [DOI] [PubMed] [Google Scholar]
- Tan CY, Virtue S, Murfitt S, Roberts LD, Phua YH, Dale M, Griffin JL, Tinahones F, Scherer PE, and Vidal-Puig A (2015). Adipose tissue fatty acid chain length and mono-unsaturation increases with obesity and insulin resistance. Scientific Reports 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sedimbi S, Lofbom L, Singh AK, Porcelli SA, and Cardell SL (2018). Unique invariant natural killer T cells promote intestinal polyps by suppressing TH1 immunity and promoting regulatory T cells. Mucosal Immunol 11, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW (2003). Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, et al. (2015). NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16, 376–385. [DOI] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, and Scherer PE (2014). Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MT, Johansson C, Olivares-Villagomez D, Singh AK, Stanic AK, Wang CR, Joyce S, Wick MJ, and Van Kaer L (2003). The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc Natl Acad Sci U S A 100, 10913–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, Fam BC, Cempako GR, Steinberg GR, Walder K, Kay TW, Proietto J, and Andrikopoulos S (2011). Deficiency in interferon-gamma results in reduced body weight and better glucose tolerance in mice. Endocrinology 152, 3690–3699. [DOI] [PubMed] [Google Scholar]
- Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, and Locksley RM (2011). Eosinophils Sustain Adipose Alternatively Activated Macrophages Associated with Glucose Homeostasis. Science 332, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, et al. (2012). Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A 109, E1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, and Ferrante AW Jr. (2013). Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18, 816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YL, Chiang YJ, and Yen JJ (2002). GATA factors are essential for transcription of the survival gene E4bp4 and the viability response of interleukin-3 in Ba/F3 hematopoietic cells. J Biol Chem 277, 27144–27153. [DOI] [PubMed] [Google Scholar]
- Zhao M, Liu Q, Liang G, Wang L, Luo S, Tang Q, Zhao H, Su Y, Yung S, Chan TM, et al. (2013). E4BP4 overexpression: a protective mechanism in CD4+ T cells from SLE patients. J Autoimmun 41, 152–160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2, related to Figure 4. Differentially expressed genes between adipose and splenic iNKT cells by scRNAseq
Table S1, related to Figure 2. Differentially expressed genes in each scRNAseq cluster
Data Availability Statement
ScRNA-seq data from this manuscript have been deposited in the Gene Expression Omnibus under accession code GSE142845. Non-RNA-seq data have been deposited in Mendeley Data (doi: 10.17632/2d6ywgb8bj.1).