

Abstract
Gene editing blood‐derived cells is an attractive approach to cure selected monogenic diseases but remains experimental. A systematic search of preclinical controlled studies is needed to determine the persistence of edited cells following reinfusion. All studies identified in our systematic search (to 20 October 2020) examining the use of CRISPR/Cas9 gene editing in blood‐derived cells for transplantation were included. Meta‐analysis was performed to determine the engraftment and persistence of gene edited cells. A total of 3538 preclinical studies were identified with 15 published articles meeting eligibility for meta‐analysis. These in vivo animal studies examined editing of hemoglobin to correct sickle cell disease (eight studies), inducing resistance to acquired immunodeficiency syndrome (two studies), and six other monogenic disorders (single studies). CRISPR‐Cas9 edited hematopoietic stem and progenitor cells demonstrated equivalent early engraftment compared to controls in meta‐analysis but persistence of gene‐edited cells was reduced at later time points and in secondary transplant recipients. Subgroup analysis in studies targeting the hemoglobin gene revealed a significant reduction in the persistence of gene‐edited cells whether homology‐directed repair or nonhomologous end‐joining were used. No adverse side effects were reported. Significant heterogeneity in study design and outcome reporting was observed and the potential for bias was identified in all studies. CRISPR‐Cas9 gene edited cells engraft similarly to unedited hematopoietic cells. Persistence of gene edited cells, however, remains a challenge and improved methods of targeting hematopoietic stem cells are needed. Reducing heterogeneity and potential risk of bias will hasten the development of informative clinical trials.
Keywords: CRISPR/Cas9, gene editing, gene therapy, genetic disease, hematopoietic stem cells, monogenic disorders, sickle cell disease, transplantation
Among 15 studies identified in our systematic search of the literature, meta‐analysis of 21 animal cohorts that examined long‐term persistence of transplanted hematopoietic stem cells that underwent CRISPR/Cas9 gene editing revealed a decline over time.

Significance statement.
Gene editing of blood‐derived cells is a promising strategy to correct genetic disorders but remains experimental. This study conducted a systematic review of the literature and identified 15 animal studies that transplanted blood‐forming stem cells that were edited using CRISPR/Cas9. Although edited cells engrafted similarly to unedited cells, this meta‐analysis revealed that edited cells declined over time after transplantation. New approaches that better target blood stem cells are needed to overcome this limitation. Also, future studies should reduce potential sources of bias by blinding outcome assessors and randomizing animals to accelerate translation to clinical studies.
1. INTRODUCTION
Recent advances in high‐throughput sequencing technologies have played a pivotal role in our understanding of genetic components that drive human diseases. We now know that alterations in over 4000 genes are associated with diseases (www.omim.org/statistics/geneMap; accessed November 15, 2020). Although some illnesses such as acute myeloid leukemia and acute lymphoid leukemia are driven by a complex interplay between various genetic and epigenetic factors, 1 , 2 others such as thalassemia, sickle cell disease (SCD), Friedreich's ataxia (FRDA), severe combined immunodeficiency (SCID‐X1), and x‐linked hyper‐immunoglobulin M syndrome (XHIM) are associated with monogenic mutations. Hematopoietic stem cell (HSC) transplantation using HSCs obtained from human leukocyte antigen (HLA) matched donors can be curative. HLA‐matched donors, however, are not available for many patients 3 and transplant‐related complications such as infection and graft‐vs‐host disease introduce significant morbidity and risk of mortality that complicates allogeneic transplantation. Taken together, new approaches such as gene therapy and/or gene editing of ex vivo expanded autologous HSCs have attracted significant attention.
Gene therapy involves correction of a mutated gene either by replacing the gene (gene replacement therapy) using exogenous DNA or editing the gene at its native location using endogenous DNA repair machinery. Although gene replacement therapy appears straight forward, this strategy has limitations and involves multiple risks that include unwanted immune system reactions to foreign DNA, and insertional mutagenesis like oncogene activation caused by regulatory elements in the gene delivery vectors that can contribute to leukemia. 4 Furthermore, in some monogenic illnesses, gene replacement therapy might not be feasible because of tightly regulated transgene expression. For example, gene replacement therapy in preclinical models of XHIM has led to lymphoproliferative disorder because of upregulation of the CD40 ligand. 5 For these reasons, gene editing strategies may be more appealing and feasible and offer the potential to cure a broader range of monogenic diseases with reduced risks.
Targeted gene editing strategies rely on endogenous cell repair machinery that are activated once double‐stranded breaks (DSBs) in DNA are induced at the intended gene locus. The two canonical pathways involved in the repair of DSBs in DNA are homology‐directed repair (HDR) or nonhomologous end joining (NHEJ). 6 , 7 In NHEJ, the two DNA ends are ligated in an error‐prone method, thereby creating insertions and deletions (INDELs) that lead to functional gene knockouts (KOs). Unfortunately, this process cannot be controlled and often creates a heterogenous population of gene‐edited cells. 8 In contrast, the HDR repair process is more precise and efficient. In HDR, the DNA repair machinery uses the sister chromatid or externally supplied homologous donor template as a scaffold to repair the DSB. 7 Therefore, by designing specific homologous donor templates, one can efficiently edit disease causing missense or nonsense mutations present within the human genome. The three popular platforms that currently exist to induce site specific DSBs within mammalian cells are (a) transcription activator‐like effector nucleases (TALENs), (b) zinc‐finger nucleases (ZFNs), and (c) the CRISPR‐Cas9 system. CRISPR‐Cas9 is the most commonly used and efficient platform that allows multiplexed gene editing and studies have progressed to the preclinical stage.
Clustered regularly interspaced short palindromic repeats (CRISPR) are short repeat sequences separated by spacers found within microbes. 9 In prokaryotes, CRISPR‐associated genes (Cas) encode proteins that can complex with phage or plasmid DNA, cut the DNA into short sequences, and integrate the short DNA sequences into the host genomic CRISPR locus as spacers. These spacers are expressed as mobile surveillance RNAs that direct Cas proteins to protect against subsequent attack by the same phages or plasmids. In essence, CRISPR serve as a prokaryotic adaptive immune‐system and protects bacteria from invading phages and plasmids. 10 Cas9 is derived from Streptococcus pyogenes and is the most commonly used endonuclease as part of the CRISPR platform. In 2008, Brouns et al 11 discovered that the Cas9 protein forms a ribonucleoprotein (RNP) complex with a RNA duplex consisting of CRISPR RNA sequences (crRNA transcribed from the acquired phage sequences) and a second RNA molecule (tracrRNA). The duplex is known to guide the Cas9 protein to the DNA target site upon which the Cas9 induces DSBs. 12 In 2012, Gasiunas et al 13 and Jinek et al 14 demonstrated that the first 20 nucleotides of the crRNA could be designed against a desired DNA target site. Jinek et al 14 further simplified the CRISPR‐Cas9 platform into a two‐system platform by fusing the tracrRNA and crRNA into a single 100 nucleotide guide RNA (sgRNA). Subsequently in 2013, Cong et al 15 demonstrated for the first time that the CRISPR‐Cas9 system can be used to edit mouse and human cells.
Given the rapid increase in publications reporting the preclinical use of CRISPR/Cas9 gene editing for transplantation and cellular therapy, a knowledge synthesis study of preclinical data is needed to gain an objective understanding of what approaches are most effective for the persistence of gene‐edited cells upon reinfusion. Important questions remain in relation to the ability of CRISPR‐Cas9 gene edited human cells to engraft and their ability to persist for long term. Furthermore, insight regarding safety and the identification of barriers that hinder progression into clinical trials is also needed. In this manuscript, we performed a systematic review and meta‐analysis of preclinical controlled animal studies where CRISPR/Cas9 gene edited blood‐derived human cells were administered in a transplantation approach. Of note, our preliminary search confirmed that clinical trials examining the use of CRISPR/Cas9 gene edited blood‐derived cells have not yet been published, even though some trials may be ongoing or recently completed.
2. METHODS
2.1. Search strategy
The systematic search of the published literature was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) guidelines. 16 Our search strategy was developed to identify studies in Medline, Embase, and Pubmed. The following terms: (hematopoietic stem cell transplantation) AND (transplantation) AND (bone marrow [BM] transplantation) AND (cord blood transplantation) AND (peripheral blood progenitor cell transplantation) AND (CRISPR) AND (gene editing) were used as part of our search strategy. Databases were searched from 1947 up to 20 October 2020. The complete electronic search strategy is presented in Supplementary Table 1 and was prepared with the assistance of an information specialist (Risa Shorr). The search strategy was peer reviewed by a second information specialist. Our research protocol was registered with the International Prospective Register of Systematic Reviews (PROSPERO; registration number CRD42020207607 registered 12 October 2020; https://www.crd.york.ac.uk/prospero/).
2.2. Study selection and data extraction
We included all controlled interventional preclinical studies that tested the in vivo use of CRISPR‐Cas9 gene edited blood‐derived cells for transplantation. Review articles, editorials, preclinical in vitro or ex vivo studies, and conference abstracts were excluded. Studies that specifically addressed the treatment of cancer, including studies that reported the use of CRISPR/Cas9 gene editing as a means to manufacture chimeric antigen receptor T cells, were excluded from further analysis in this study since these studies were primarily focused on cancer‐related outcomes with less focus on persistence of gene‐edited cells following infusion and will be analyzed and reported separately. Studies that reported on blood‐derived cells for applications other than restoration of the blood system were also excluded 17 (ie, marrow‐derived mesenchymal stromal cells) as persistence of these cells after infusion may not have been a primary concern.
All records from the search were imported into Rayyan (https://rayyan.qcri.org/; accessed October 12, 2020). Duplicates were excluded. Abstracts of all the identified studies were independently screened by three investigators (H. Maganti, A. Bailey, A. Kirkham), and all potentially relevant articles were retrieved for further review based on the eligibility criteria (see below). Data were extracted independently by H. Maganti and A. Bailey using standardized forms (created using Excel, Microsoft, Seattle, Washington). Consensus was achieved through discussion with a senior team member (D.S. Allan) as needed to resolve discrepancies. Data were estimated from published graphs using Digitizelt (Version 2.2; Braunschweig, Germany) when raw data were not reported.
2.3. Data analysis
For the meta‐analysis involving preclinical in vivo studies, raw data relating to engraftment percentages and/or percentage edited cells within the BM at input and endpoint were abstracted from the manuscripts or from published supplementary data. In studies where the raw data were not presented, the data were estimated from the presented graphs using Digitizelt (Version 2.2). To minimize bias, raw data were extracted by two individuals (H. Maganti and A. Bailey) and all discrepancies were resolved through consensus. Control (unedited) and edited arms of individual pre‐clinical studies were compared using Student's t test. Bias and significance in pooled analysis was done using DerSimonian and Laird random effects model for long‐term persistence of gene‐edited cells and fixed effects model for cell engraftment. Key parameters such as the number of animals which contributed to specific results reported in the study, cells used for transplantation in mice, randomization, and reporting of investigator and/or lab personnel blinding were also noted and summarized. These key parameters have been previously identified as potential sources of bias in systematic reviews and their reporting is crucial in order to strengthen the conclusions of the review. 18 Potential sources of bias were examined in each study using the Systematic Review Centre for Laboratory Animal Experimental (SYRCLE) tool. 19
3. RESULTS
3.1. Identification of relevant published studies
A total of 3538 studies were identified in our systematic literature search. After excluding duplicates and screening for potential relevance, 26 studies underwent comprehensive review for assessment of eligibility. Eleven studies were subsequently removed for the following reasons: in vitro studies only (three reports), nonhematopoietic gene edited cells (two report), case reports (two reports), and CAR‐T (four studies), which will be analyzed and reported separately. The summary of the study selection process is provided in Figure 1. A total of 15 studies 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 met the eligibility and underwent further analysis. No published clinical studies were identified.
FIGURE 1.
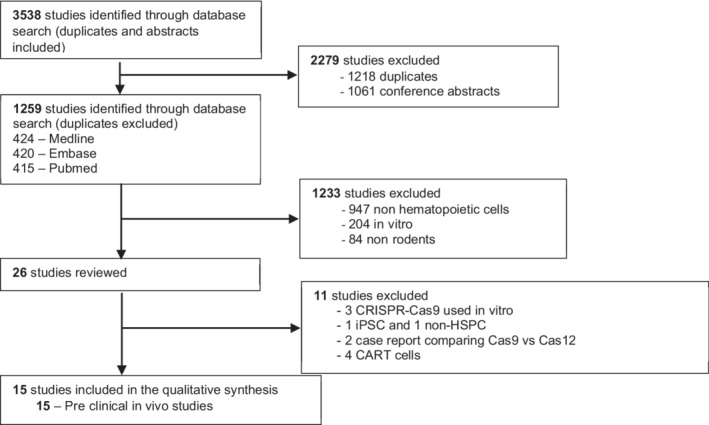
Result of systematic search of the literature
3.2. Characteristics of published preclinical studies
The included studies addressed gene editing of several gene loci implicated in monogenic diseases, and/or monogenic approaches to disease amelioration, including hemoglobin disorders such as SCD (eight studies), resistance to human immunodeficiency virus (HIV) infection through deletion of C‐C chemokine receptor five (CCR5) (two studies), XHIM (one study), X‐linked chronic granulomatous disease (X‐CGD) (one study), FRDA (one study), immunodysregulation polyendocrinopathy entropathy X‐linked syndrome (IPEX) (one study), and SCID‐X1 (one study). All studies used mouse models (Table 1).
TABLE 1.
Summary of preclinical studies. Unless specified the tissue source of all studies were from healthy donors. In Charlesworth et al, cells were transduced with AAV6 24 hours post electroporation
| Study | Targeted gene (disease) | Cells edited | Tissue source | Cas9 delivery (electroporation) | Repair template | In vivo follow‐up (weeks) | Goal of gene editing |
|---|---|---|---|---|---|---|---|
| Xiao et al 2019 17 | CCR5 (AIDS) | CD4+ T | Peripheral Blood | Lentivirus | None | 6.5 | Gene knock‐out |
| Xu et al 2017 18 | CCR5 (AIDS) | CD34+ | Fetal Liver | Plasmid | None | 12 | Gene knock‐out |
| Kuo et al 2018 19 | CD40L (XHIM) | CD34+ | mPB | mRNA/RNP | AAV6 | 12 | Gene knock‐in |
| De Ravin et al 2017 20 | CYBB (X‐CGD) | CD34+ | X‐CGD patient mPB | mRNA | ssODN | 20 | Targeting a point mutation and correcting it |
| Goodwin et al 2020 21 | FOXP3 (IPEX) | CD34+ | Umbilical Cord Blood | RNP | AAV6 | 14 | cDNA knock‐in |
| Rocca et al 2020 22 | FXN (FRDA) | CD34+ | mPB | RNP | None | 12 | Deleting GAA expansions (intron 1) |
| Charlesworth et al 2018 23 | HBB (SCD) | CD34+ | Umbilical Cord Blood | RNP | *AAV6 |
1*: 16 2*: 16 |
Targeting a point mutation and correcting it |
| Dever et al 2016 24 | HBB (SCD) | CD34+ | mPB | RNP | AAV6 | 16 | Targeting a point mutation and correcting it |
| Dewitt et al 2016 25 | HBB (SCD) | CD34+ | mPB | RNP | ssODN | 16 | Targeting a point mutation and correcting it |
| Park et al 2019 26 | HBB (SCD) | CD34+ |
P1: mPB P2: BM |
RNP | ssODN |
P1: 19 P2: 16 |
Targeting a point mutation and correcting it |
| Pattabhi et al 2019 27 | HBB (SCD) | CD34+ | mPB | RNP |
AAV6 ssODN |
12 | Targeting a point mutation and correcting it |
| Wu et al 2019 28 | BCL11A (SCD) | CD34+ | mPB | RNP | None |
1*: 16 2*: 16 |
Disruption of GATA1 binding sequences within the promoter |
| Metais et al 2019 29 | HBG1/HBG2 (SCD) | CD34+ | SCD patient mPB | mRNA/RNP | None | 17 | Disruption of BCL11a binding site within the HBG1/2 promoter |
| Weber et al 2020 30 | HBG1/HBG2 (SCD) | CD34+ | mPB | RNP | None | 16 | Disruption of LRF binding site within the HBG1/2 promoter |
| Pavel‐Dinu et al 2019 32 | IL‐2RG (SCID‐X1) | CD34+ | Umbilical Cord Blood | RNP | AAV6 |
1*: 16 2*: 16 |
cDNA knock‐in |
Abbreviations: 1*, primary transplant; 2*, secondary transplant; AAV, adeno‐associated virus; AIDS, acquired immunodeficiency syndrome; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; FRDA, Friedreich's ataxia; IPEX, immunodysregulation polyendocrinopathy entropathy X‐linked syndrome; mPB, peripheral blood mobilized with GCSF or plerixafor; P1, patient 1; P2, patient 2; RNP, ribonucleo‐protein complex; SCD, sickle cell disease; SCID‐X1, X‐linked severe combined immunodeficiency; ssODN, single stranded donor oligonucleotides; X‐CGD, X‐linked chronic granulomatous disease; XHIM, X‐linked hyper IgM syndrome.
All studies provided information regarding the number of animals used for the control and experimental conditions. Information regarding animal allocation to treatment groups was provided in the Results section (3 studies) or in the figure legends where they contributed data (12 studies). None of the preclinical studies reported on randomization, allocation concealment, or provided information regarding physiological baseline characteristics to ensure the treatment and control groups were balanced. All included studies, however, provided information about the mice (strain and source) and method of transplantation (Table 2). No information pertaining to power calculations and sample size determination was provided in any of the studies.
TABLE 2.
Assessment of long‐term engraftment of CRISPR‐Cas9 edited cells in preclinical studies. In Charlesworth et al, AAV is added 24 hours post electroporation. Unless specified the same number of gene‐edited and mock treated cells were transplanted. Results from secondary transplants (2*) are in italics
| Study | Animal (strain) | Route of delivery (cells) | Cells delivered (culture time), groups | In vitro culture media | PB engraftment (human CD45+) | % edited cells in BM (ddPCR) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control, % (n) | Edited (n)% | Time point (weeks) | Input % (n) | Endpoint % (n) | Time point (weeks) | |||||
| Xiao et al 2019 | Mice (NSG) | Tail vein (CD4+ T) | 1 × 105 (5d) | RPMI, IL2 | 317 ± 258 cells (8) | 186 ± 76 cells (7) | 2 | 40 ± 2.3 (8) | NR | NR |
| Xu et al 2017 | Mice (NSG) | Intrahepatic (CD34+) | 1 × 106 (4d) | Stem span SFEM II media, FLT3L, TPO, IL6 | 13.0 ± 9.5 (9) | 9.5 ± 5.1 (9) | 6 | 42.2 ± 3.6 (5) | 25.2 ± 17.0 (12) | 12 |
| Kuo et al 2018 | Mice (NSG) | Intrahepatic (CD34+) | 1 × 106 (3d) | X‐VIVO15, Glutamine, TPO, FLT3, SCF | 12.8 ± 19.9 (7) | 12.2 ± 15.1 (16) | 12 | 27.6 ± 4.0 (6) | 10.3 ± 11.2 (15) | 12 |
| De Ravin et al 2017 | Mice (NSG) | Tail vein (CD34+) | 1‐3 × 106 (3d) | Stem span SFEM II media, FLT3L, TPO |
P1: 38.7 ± 16.3% (3) P2: NR |
P1: 32 ± 12.9% (3) P2: NR |
8 |
P1: 18.3 ± 2.7 (2) P2: 20.1 ± 2.7 (3) |
P1: 12.1 ± 3.7 (6) P2: 10.2% ± 3.2 (5) |
20 |
| Goodwin et al 2020 | Mice (NSG‐SGM3) | Intrahepatic (CD34+) | 1 × 106 (4d) | Stem span SFEM II media, FLT3L, TPO, IL6, UM171, SR1 | 92.5 ± 12.8 (6) | 91.8 ± 5.3 (5) | 4 | 70.1 ± 4.5 (2) | 57.7 ± 17.16 (4) | 14 |
| Rocca et al 2020 | Mice (NSG) | Intrahepatic (CD34+) | 1 × 106 (3d) | IMDM, IL3, IL6, SCF | 24.9 ± 21.2 (8) | 28 ± 24.6 (8) | 6 | 37.5 ± 12.3 (8) | 5.4 ± 6.8% (8) | 12 |
| Charlesworth et al, 2018 | Mice (NSG) | Intrafemoral (CD34+) |
A: 5 × 104 (4d) B: 5 × 104 (12d) |
Stem span SFEM II media, FLT3L, TPO, IL6, UM171, SR1 |
A: 1*: NR A: 2*: NR B: 1*: NR B: 2*: NR |
A: 1*: 3.1 ± 4.5 (10) A: 2*: 0.43 ± 0.60 (2) B: 1*: 17.2 ± 16.1 (9) B: 2*: 0.43 ± 0.46 (2) |
8 |
A: 36.2 ± 14.4 (11) B: 81% ± 14.4 (2) |
A: 1*: 3.1 ± 0.1 (8) A: 2*: 0.21 ± 0.29 (2) B: 1*: 16.6 ± 0.7 (7) B: 2*: 0.41 ± 0.36 (2) |
A: 1*: 16 A: 2*: 16 B: 1*: 16 B: 2*: 16 |
| Dever et al 2016 | Mice (NSG) | Tail vein (CD34+) | 4‐7 × 105 (4d) | Stem span SFEM II media, FLT3L, TPO, IL6, SR1 | 29.3 ± 14.6 (10) | 21.7 ± 2.6 (6) | NR | 16 ± 5.2 (10) | 9.5 ± 2.2 (6) | 16 |
| Dewitt et al 2016 | Mice (NSG) | Tail vein (CD34+) | 0.75‐1 × 106 (5d) | Stem span SFEM II media, FLT3L, TPO, SCF | NR | 37 ± 21 (6) | 8 | 66.8 ± 15 (6) | 48 ± 3.5 (6) | 16 |
| Park et al 2019 | Mice (NSG) | Intrafemoral (CD34+) |
0.5 × 106 (P1: PB: 4d) (P2: BM: 5d) |
GMP SCGM, SCF, FLT3L, TPO, IL3 |
P1: 37 ± 5 (3) P2: 16.8 ± 5.5 (3) |
P1: 42 ± 6 (3) P2: 7.5 ± 9 (3) |
4 |
P1: 43 ± 5 (3) P2: 60% ± 8 (3) |
P1: 36 ± 8 (3) P2: 54 ± 4 (3) |
P1: 19 P2: 16 |
| Pattabhi et al 2019 | Mice (NBSGW) | Tail vein (CD34+) |
2 × 106 (3d) A: rAAV6 B: ssODN |
GMP SCGM, SCF, FLT3L, TPO, IL6 | 83.98 ± 8.59 (8) |
A: 64.3 ± 19.2 (17) B: 83.8 ± 13% (18) |
4 |
A: 24.3 ± 7.5 (17) B: 17.5 ± 6 (18) |
A: 0.66 ± 0.66 (17) B: 4.136 ± 2.1% (17) |
A: 12 B: 12 |
| Wu et al 2019 | Mice (NBSGW) | Retro‐orbital (NR) | 0.5 × 106 (3d) | X‐VIVO, FLT3L, SCF, TPO |
P1: 1*: 21.6 ± 8.6 (3) P1: 2*: 0.28 ± 0.17 (3) P2: 1*: 88.4 ± 3.4 (3) |
P1: 1*: 16.0 ± 13.5 (5) P1: 2*: 0.26 ± 0.18 (4) P2: 1*: 88.8 ± 2.8 (3) |
4 |
P1: 1*: 95.6 ± 1.5 (2) P2: 1*: NR |
P1: 1*: 84.5 ± 2.6 (11) P1: 2*: 78.6 ± 0.6 (4) P2: 1*: NR |
P1: 1*: 16 P1: 2*: 16 |
| Metais et al 2019 | Mice (NBSGW) | Tail vein (CD34+) | 1 × 106 (6d) | Stem span SFEM II media, FLT3L, TPO, SCF |
P1: 82.3 ± 11.5 (3) P2: 73.7 ± 2.3 (3) |
P1: 79.8 ± 9.6 (4) P2: 74.2 ± 3.5 (5) |
6 |
P1: 79.7 (1) P2: 54.9 (1) |
P1: 60.7 ± 2.5 (3) P2: 18.5 ± 2.8 (5) |
P1: 17 P2: 17 |
| Weber et al 2020 | Mice (NSG) | Retro‐orbital (CD34+) | 1 × 106 (7d) | Stem span SFEM II media, FLT3L, TPO, IL3, | 45.6 ± 29.5 (4) |
C197: 39.6 ± 20.7 (4) C196: 44.0 ± 19.2 (4) C115: 34.8 ± 15.7 (4) |
NR |
C197: 77.3 ± 3.7 (4) C196: 87.4 ± 4.6 (4) C115: 89.6 ± 2.8 (4) |
C197: 43.0 ± 9.3 (4) C196: 60.3 ± 6.1 (4) C115: 47.6 ± 4.2 (4) |
C197: 16 C196: 16 C115: 16 |
| Pavel‐Dinu et al 2019 | Mice (NSG) |
A: Intrahepatic B: Intrafemoral (CD34+) |
Control: 5.7 × 106 Edited: 9.7 × 106 (2d) |
Stem span SFEM II media, FLT3L, TPO, UM171, Stem Regenin I |
1*: 61 ± 22.7 (7) 2*: A: 6.6 ± 3.7 (7) 2*: B: 0.6 ± 2.8 (4) |
1*: 45.6 ± 15.3 (10) 2*: A: 7.7 ± 3.5 (6) 2*: B: 11.4 ± 3.2 (10) |
4 | 45% ± 15% (n = 3) |
1*: 25.5 ± 13.2 (19) 2*: A: 20.0 ± 12.7% (9) 2*: B: 9.5 ± 14.9% (13) |
1*: 16 2*: A: 16 2*: B: 16 |
Notes: All the data presented in this table has been extrapolated from the graphs presented in the various studies. C197, C196, C115: These are different sgRNA clones that target the promoter region of HBG1/2 genes where LRF1 binds.
Abbreviations: 1*, primary transplant; 2*, secondary transplant; AAV, adeno‐associated virus; BM, bone marrow; HDR, homology‐directed repair; IF, intrafemoral; IH, intrahepatic; NHEJ, nonhomologous end Joining; P1, patient 1; P2, patient 2; PB, peripheral blood; RNP, ribonucleo‐protein complex; ssODN, single‐stranded donor oligonucleotides.
3.3. Characteristics of cells types targeted
Blood‐derived cells were isolated from frozen samples using density gradient centrifugation methods. None of the studies utilized freshly collected cells. The tissue sources were all human‐derived and included umbilical cord blood (four studies), BM (one study), peripheral blood (one study), mobilized peripheral blood (mPB) following administration of granulocyte colony‐stimulating factor and/or plerixafor (eight studies), and fetal liver (one study). One study 29 used mPB or BM in their transplant experiments. Fourteen studies isolated and/or targeted HSPCs from the tissue source for gene editing by selecting CD34+ cells prior to gene editing and cell expansion in culture. T lymphocytes were targeted in one study (Table 1). Two studies derived their HSPCs from mobilized PB of patients with SCD and one study derived their HSPCs from mobilized PB of patients with X‐CGD (Table 1). All other cells used were derived from healthy subjects.
3.4. CRISPR‐Cas9 gene editing
Cas9 protein was transfected via electroporation (14 studies) or transduced using lentivirus (1 study). Cas9 was delivered either in a ribonucleoprotein complex with sgRNA (10 studies) or as separate protein and RNA components (2 studies). Two studies delivered Cas9 as mRNA and one study delivered Cas9 as a plasmid (Table 1). The density at which the cells were plated for CRISPR‐Cas9 editing varied between studies. All studies cultured cells in media that promoted active proliferation and minimal differentiation (Table 2). The number of days the cells were expanded post gene‐editing varied between studies (Table 2). Two studies created functional gene KOs. Ten studies used the CRISPR‐Ca9 system to either induce or edit specific mutations (Table 1). Three studies created cells with gene knock‐ins. Nine studies used donor templates to facilitate HDR. The donor template was delivered as either Adeno‐associated virus type 6 (AAV6) (six studies) or single stranded donor oligonucleotides (ssODN) (four studies). One study 30 used both AAV6 and ssODN as donor templates. Only three studies attempted to correct the abnormal gene and disease phenotype in animal models (two studies of SCD and one study of X‐CGD) while the remainder demonstrated their ability to target gene loci implicated in disease by introducing marker gene expression such as green florescent protein (GFP) or to knock‐in or knock‐out a gene at a specific locus such as CCR5 (Table 2). Correction of abnormal genes from patients and/or cell lines, and gene expression levels and protein function assays were performed in complementary in vitro testing and are not summarized or analyzed further in this report.
FIGURE 2.
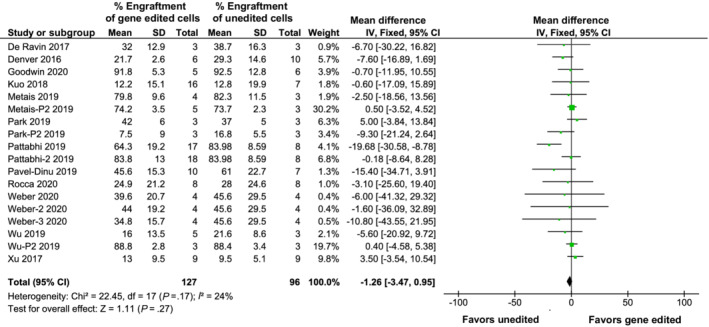
CRISPR‐Cas9 gene editing does not negatively impact the ability of hematopoietic stem and progenitor cells to engraft in vivo, as measured by fluorescent‐activated cell sorting
3.5. Transplantation and long‐term engraftment of gene‐edited cells
CRISPR‐Cas9 edited cells were transplanted via tail vein (six studies), retro‐orbital (two studies), intrahepatic (five studies), or via intrafemoral injection (three studies) (Table 2). Fourteen studies performed long‐term (≥12 weeks) primary transplant experiments (Table 2). Eleven studies reported a decline in the percentage of gene‐edited human cells within the BM at the endpoint of the study compared to the time of transplantation (Table 2). Gene edited cells were typically sorted by flow cytometric cell sorting, gating on expression of a marker gene such as GFP. Some cells appear to have manifested transient expression of GFP which was later lost prior to infusion, which likely accounts for the percentage of edited cells at the time of transplant that was less than 100% in most studies.
A total of 12 studies reported in vivo engraftment data of gene edited hematopoietic cells based on samples of peripheral blood or BM from the transplanted mice. Following a pooled‐fixed effects analysis, the mean difference in engraftment of gene edited cells showed no significant difference when compared to unedited cells (−1.26%; 95% confidence interval [CI], −3.47% to 0.95%; I 2 24%; P = .17) with overall human engraftment ranging from 7.5% to 92% (see Figure 2).
A total of 14 studies (21 individual cohorts) reported the long‐term persistence (≥12 weeks) of gene edited cells within the BM or peripheral blood of transplanted mice. One study did not report the end‐point data and hence was precluded from meta‐analysis. Following a pooled random effects analysis, the mean reduction of gene‐edited cells at endpoint compared to input was 18.79% (95% CI, −25.11 to −12.46; P < .00001; Figure 3) with the percentage of edited cells at endpoint ranging from 3.1% to 95%, although significant heterogeneity was observed between studies (I 2 = 96%). Only three studies 26 , 31 , 34 performed secondary transplants to evaluate the ability of gene‐edited HSPCs harvested from the BM of transplanted mice and used to engraft and repopulate the BM of a second set of mice. The percentage of engrafted cells following secondary transplants was reduced compared to primary animals (from 84% to 0.26%) in all cases.
FIGURE 3.
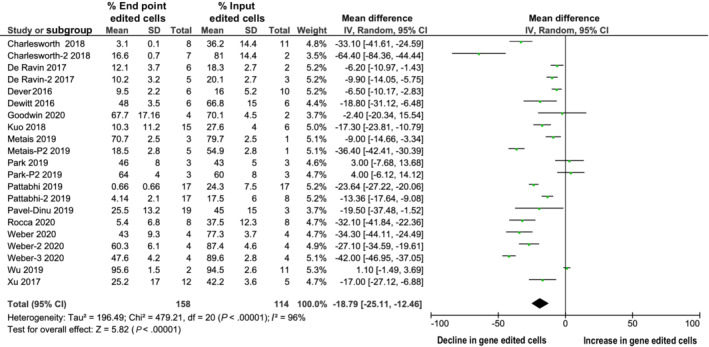
The percentage of gene edited hematopoietic cells decline over long‐term, as measured by digital droplet PCR at input and endpoint
3.6. Impact of the CRISPR‐Cas9 gene editing vector: Analysis of studies editing the hemoglobin gene
A total of eight studies addressed editing HSPCs at the hemoglobin B locus (HBB). Analyzing this group of homogenous studies provided an opportunity to compare the impact of different gene editing platforms. Although two studies used the AAV6 platform to deliver the donor vector, two studies used ssODN to deliver the donor vector. One study used both ssODN and AAV6 independently in separate experiments. Three studies did not use a donor vector. In subgroup‐analysis regarding the reduction in long‐term persistence of gene‐edited cells when using ssODN (−12.16% compared to baseline levels of engraftment, 95% CI: −16.45 to −7.86; Figure 4A) compared to studies using AAV6 as the transduction vector (−28.94%; 95% CI: −43.78 to −14.11; Figure 4B), there was no significant difference observed between the two methods.
FIGURE 4.
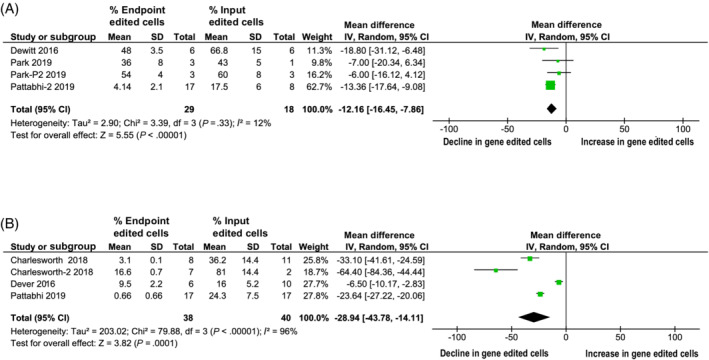
The percentage of hematopoietic cells within sickle cell disease (SCD) studies edited using single stranded donor oligonucleotides (ssODN) decline slower over long‐term compared to hematopoietic cells edited using AAV6, as measured by digital droplet PCR at input and endpoint. A, The percentage of hematopoietic cells within SCD studies using ssODN as measured by digital droplet PCR at input and endpoint. B, The percentage of hematopoietic cells within SCD studies using AAV6 as measured by digital droplet PCR at input and endpoint
3.7. CRISPR‐Cas9 gene edited cells for treatment of AIDS
Two studies 20 , 21 investigated the use of CRISPR‐Cas9 gene edited T cells and CD34‐positive cells for the management of the acquired immunodeficiency syndrome (AIDS). The in vivo results from one study 20 demonstrated that KO of CCR5 did not affect T‐cell function. The in vivo data from the second study 21 demonstrated that HSPCs devoid of CCR5 could differentiate into T cells that lacked CCR5. Interestingly, mice transplanted with either CCR5 KO T cells or HSPCs showed reduced viral burden and resistance to HIV infection.
4. RISK OF BIAS ANALYSIS
Two independent reviewers (H. Maganti, A. Bailey) assessed the risk of bias of each study included study according to the Systematic Review Centre for Laboratory animal Experimentation (SYRCLE) risk of bias tool for animal studies. 19 Consistent with most preclinical studies, we found the included preclinical studies had potential risk of bias in several areas such as outcome reporting, use of randomization methods, blinding, and a priori protocols. None of the studies mentioned whether the experiments were done in a double‐blinded manner, whether randomization was performed among the control and treatment cohorts or how the animal cohort size was determined. Future studies should strive to reduce bias by following reporting guidelines outlined by the SYRCLE tool.
5. DISCUSSION
The CRISPR/Cas9 gene editing system offers an efficient way of making precise genetic changes to the human genome. CRISPR‐Cas9 can therefore be employed for disruption, addition, and correction of genes, thereby enabling a new class of genetic/cellular therapies. The preclinical data summarized in this review confirm that CRISPR/Cas9 edited hematopoietic cells can be used as cellular therapy for the treatment of monogenic disease and induce resistance to infectious diseases. We identified that preclinical studies mostly addressed hemoglobin disorders such as SCD (50%) followed by inducing resistance to HIV infection (12.5%). Meta‐analysis of transplant data from studies included in our analysis revealed that gene edited HSPCs engraft similarly to unedited cells. The percentage of gene‐edited cells was shown to decline over time in these studies, however, highlighting the challenge of improved targeting of HSCs. Improved targeting of HSCs through isolation of more homogenous stem cell populations and/or more effective methods of transducing quiescent stem cells will improve outcomes and accelerate progress toward clinical application. Moreover, reducing sources of potential bias in future studies, through greater use of randomization and blinding of outcome assessors will accelerate the development of studies that can be translated to the clinical domain.
Re‐establishing hematopoiesis in transplant recipients involves early repopulation with short‐term hematopoietic progenitor cells (ST‐HPCs), which are responsible for initial engraftment after transplantation, and long‐term hematopoietic stem cells (LT‐HSCs). LT‐HSCs and ST‐HPCs differ markedly in their in vivo repopulation capacity. Although primary transplantation experiments in mice measure the ability of ST‐HPCs and progenitors to support initial hematopoiesis, secondary transplantation experiments directly measure the ability of LT‐HSCs to engraft to the BM and support longer‐term and sustained hematopoiesis. Engraftment is the process by which transplanted cells home to their niche, start to repopulate, and give rise to progeny. Several seminal studies have reported that human HSPCs when transplanted into mice home to the BM and their differentiated progeny make their way into the peripheral blood of mice. Within the studies included in our systematic review, engraftment was assessed using peripheral blood of mice approximately 2 to 8 weeks after transplantation when human engraftment typically emerges and BM samples at the endpoint of the study, typically at 12 to 16 weeks after transplant. The long‐term persistence of the engrafted gene‐edited HSCs and their progeny are critical for gene‐therapy to be a viable treatment option for monogenic illnesses. Interestingly, while our meta‐analysis revealed that gene‐edited HSPCs have similar initial engraftment potential compared to mock‐treated controls, we observed that the percentage of gene‐edited cells declines significantly over time. Surprisingly, only three studies performed secondary transplantation experiments using gene edited cells derived from primary transplant recipients. Nevertheless, all the three studies did report a further decline in the percentage of gene edited cells compared to the proportion of edited cells that were recovered from initial animals. While the heterogeneity between studies reporting on ST‐HPC engraftment in primary animals was low (I 2 = 24%), the heterogeneity between the few studies reporting on LT‐HSC engraftment in secondary animals was high (I 2 = 96%) and limits confidence in the pooled estimates from these studies. Taken together, these analyses suggest that (a) insufficient numbers of gene‐edited LT‐HSCs may have been transplanted into mice and (b) that the population of LT‐HSCs transplanted into the mice may have varied significantly in terms of gene editing efficiency between the different studies. One strategy that could help overcome this limitation is by enriching for gene‐edited LT‐HSCs prior to transplantation. Most studies used CD34‐expression as a means of HSC enrichment prior to transplantation. Several seminal studies 35 , 36 , 37 have reported that CD34 is not a robust marker for HSC enrichment from cultured HSPCs as cell surface expression may diminish during cell culture. Combinations of additional cell surface markers (eg, CD34+CD90+, CD34+CD45RA‐) or novel markers like EPCR, ITGA3, and AC133 have enabled improved enrichment of HSCs from ex vivo cultured HSPCs isolated from UCB. Studying the isolation of HPCs from other sources such as BM and mPB will also be needed.
Interestingly, another model that has aided researchers to study the long‐term persistence of gene edited cells is the nonhuman primate (NHP) autologous transplantation model. 38 , 39 , 40 Unfortunately, because the NHP model does not use human hematopoietic cells, it did not fit our inclusion criteria and hence we did not include NHP studies within our analysis. Nevertheless, it is important to note that the NHP system has several advantages and has provided valuable preclinical data. The NHP HSCs express common cell surface markers that are homologous to human surface markers therefore reagents such as growth factors or selection reagents such as MGMT/P140K platform that are used to select gene‐edited NHP HSCs are easily transferable to the human HSCs. 38 Furthermore, the scale of cell populations collected, edited, and transplanted into NHPs is similar to human patients therefore this model allows to better predict the number of gene edited cells required for transplantation and the cost associated with this process. Seminal work by the Hans‐Peter Kiem's group has shown that CRISPR‐Cas9 gene edited cells persist long‐term within the NHP model. 40 Moreover, the data from the NHP models have been consistent with the early human SCD clinical trial (NCT03655678 and NCT03745287) data that report the persistence (1 year post transplantation) of CRISPR‐Cas9 gene edited hematopoietic cells within the peripheral blood and BM of SCD patients at high levels. 41
Hemoglobinopathies have been previously treated successfully using ex vivo lentiviral therapy where a functional HBB gene was delivered into autologous HSCs. 42 Our systematic review also identified hemoglobin disorders such as SCD to be an appealing target for CRISPR‐Cas9 mediated precision gene therapy, comprising 50% of the articles included in our systematic review. Although studies previously attempted to correct mutations associated with SCD using ZFNs and TALENS, 43 , 44 it was done with limited success and many off‐target effects. All eight studies identified in our systematic review were able to partially rescue the SCD phenotype with minimal off‐target effects. Interestingly, although five studies targeted the HBB gene, two studies targeted the HBG1/2 promoter region to disrupt the binding sites of LRF and BCL11a. In one study, the promoter region of the BCL11A gene was targeted to disrupt the binding site of GATA1. 32 Within the studies that targeted the HBB gene directly, two used AAV6 as the donor vector and two studies used ssODN. One study used both AAV6 and ssODN as donor vectors. When we compared the long‐term persistence of gene‐edited cells using the two different methodologies, we did not detect a significant difference and we were limited in our analysis by heterogeneity between studies and the small sample size. Further studies that examine the methodology for donor template delivery are needed to understand any potential impact on long‐term persistence of gene edited cells.
Data from preclinical in vivo studies included in our analysis revealed the majority of the CRISPR‐Cas9 gene edited HSPCs underwent NHEJ repair, which induces new INDELs. These data are consistent with other observations from DNA repair studies that show HDR machinery to be enriched within actively dividing (cells in S and G2 phases) but not quiescent (G0) cells. 45 However, unlike HDR, NHEJ machinery is found to be active throughout all stages of cell cycle. Therefore, it is likely that within uncultured LT‐HSCs (CD34+CD38−CD90+CD45RA−CD49f+ and present in G0 stage), 46 , 47 the NHEJ repair machinery is more abundant relative to HDR machinery and that LT‐HSCs readily undergo NHEJ compared to HDR. Several studies have attempted to resolve this potential limitation by shifting the NHEJ/HDR equilibrium toward HDR with the help of NHEJ inhibitors. 48 , 49 , 50 Although data from these studies are promising, this approach has mostly been restricted to studies using murine pluripotent cells. The relative success of this approach using human LT‐HSCs remains unknown.
Traditional antiretroviral therapy does not appear capable of eradicating the entire HIV‐1 reservoir. In patients that have undergone antiretroviral therapy, the HIV‐1 provirus often remains hidden and can reactivate after cessation of therapy, which is followed by productive infection and disease progression. 51 , 52 Therefore, it seems impossible to completely cure HIV with drug treatment alone at this juncture. Furthermore, antiretroviral therapy is often associated with side effects and is expensive for people in developing countries. Interestingly, a subset of people that have deletions of CCR5 have demonstrated resistance to HIV and allogeneic transplantation of BM from CCR5‐negative donors into patients with HIV has been curative, although can be associated with significant toxicity and is resource intensive. 53 , 54 Autologous gene therapy could be a viable option to cure patients with HIV and avoid the risks of allogeneic transplantation. Two studies 20 , 21 included in our systematic review investigated the use of CRISPR‐Cas9 gene edited T cells and CD34 cells for the management of AIDS. Both the studies knocked out CCR5 gene using CRISPR‐Cas9. Interestingly, KO of CCR5 did not significantly affect T‐cell function. Furthermore, the CCR5 KO HSPCs showed healthy long‐term engraftment and gave rise to functional T cells that were also devoid of CCR5. These CCR5 KO T cells showed resistance to HIV. Together, these studies demonstrated that CRISPR‐Cas9 provides another avenue to develop gene therapy products for the treatment of HIV.
Our analysis has several limitations worthy of mention. First, included studies had potential risk of bias in at least some categories of the SYRCLE analysis. 19 Future studies can address these limitations through the consistent reporting of randomization methods, blinding, and registering protocols a priori. Second, significant heterogeneity was detected in our analysis focusing on the long‐term persistence of gene‐edited cells. This heterogeneity was likely attributed to several factors including the HSPC tissue source (ie, umbilical cord blood vs mobilized PB vs BM), the method used to deliver the HSPCs (intrahepatic vs intra‐femoral vs retro‐orbital vs tail vein injection), the number of HSPCs transplanted, the ex vivo HSPC culture conditions, and/or the donor delivery vectors used (ssODNs vs AAV6). Moreover, in some of our pooled estimates of effect size, some studies appeared to garner more significant weight than others, related to the method for calculating weighting based on the variance reported for each study. The inclusion of more studies with similar variance would avoid unbalanced weighting of studies that influence meta‐analysis. Standardization of gene editing, cell culture, and transplantation protocols would enable more impactful knowledge synthesis efforts that should further accelerate translation to clinical studies.
To conclude, CRISPR‐Cas9 gene edited hematopoietic cells demonstrate initial engraftment similar to unedited hematopoietic cells within preclinical animal models of monogenic diseases. Data from long term preclinical in vivo studies suggest that the overall percentage of gene edited cells decline significantly over time perhaps because of low targeting efficiencies of LT‐HSCs or deleterious impact on their engrafting potential. 55 Future studies should leverage insights gained from our systematic review and meta‐analysis by reducing heterogeneity between studies, limiting potential risk of bias, and improving the targeting efficiency of HSCs. This will hasten the translation of this novel therapeutic technology toward clinical trials that may benefit patients.
CONFLICT OF INTEREST
D.S. Allan is Medical Director, Stem Cells, Canadian Blood Services. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
H.M.: conceptualization, methodology, validation, formal analysis and investigation, data curation, writing ‐ original draft preparation, writing ‐ review and editing, visualization; A.B., A.K.: methodology, validation, formal analysis and investigation, data curation, writing ‐ review and editing, visualization; R.S.: methodology; N.P.: resources, writing ‐ review and editing, visualization, supervision, funding acquisition; D.S.A.: conceptualization, methodology, validation, formal analysis and investigation, resources, data curation, writing ‐ original draft preparation, writing ‐ review and editing, visualization, supervision, project administration, funding acquisition.
Supporting information
Supplementary Figure 1 Supplementary Figure
ACKNOWLEDGMENT
Funding for trainee stipends (A. Kirkham, A. Bailey) from Canadian Blood Services Foundation, and Postdoctoral Fellowship from CBS (H. Maganti).
Maganti HB, Bailey AJM, Kirkham AM, Shorr R, Pineault N, Allan DS. Persistence of CRISPR/Cas9 gene edited hematopoietic stem cells following transplantation: A systematic review and meta‐analysis of preclinical studies. STEM CELLS Transl Med. 2021;10:996–1007. 10.1002/sctm.20-0520
Authored by a member of CBA.
Funding information Canadian Blood Services Foundation
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Goldman SL, Hassan C, Khunte M, et al. Epigenetic modifications in acute myeloid leukemia: prognosis, treatment, and heterogeneity. Front Genet. 2019;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maganti HB, Jrade H, Cafariello C, et al. Targeting the MTF2‐MDM2 Axis sensitizes refractory acute myeloid leukemia to chemotherapy. Cancer Discov. 2018;8(11):1376‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem‐cell grafts in the U.S. registry. N Engl J Med. 2014;371(4):339‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cavazzana‐Calvo M, Hacein‐Bey S, de Saint BG, et al. Gene therapy of human severe combined immunodeficiency (SCID)‐X1 disease. Science. 2000;288(5466):669‐672. [DOI] [PubMed] [Google Scholar]
- 5. Brown MP, Topham DJ, Sangster MY, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998;4(11):1253‐1260. [DOI] [PubMed] [Google Scholar]
- 6. Komor AC, Badran AH, Liu DR. Editing the genome without double‐stranded DNA breaks. ACS Chem Biol. 2018;13(2):383‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeggo PA. DNA breakage and repair. Adv Genet. 1998;38:185‐218. [DOI] [PubMed] [Google Scholar]
- 8. Rodgers K, McVey M. Error‐prone repair of DNA double‐strand breaks. J Cell Physiol. 2016;231(1):15‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mojica FJ, Juez G, Rodriguez‐Valera F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol. 1993;9(3):613‐621. [DOI] [PubMed] [Google Scholar]
- 10. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709‐1712. [DOI] [PubMed] [Google Scholar]
- 11. Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9‐crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109(39):E2579‐E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho HM, Lee KH, Shen YM, et al. Transplantation of hMSCs genome edited with LEF1 improves cardio‐protective effects in myocardial infarction. Mol Ther Nucleic Acids. 2020;19:1186‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henderson VC, Kimmelman J, Fergusson D, Grimshaw JM, Hackam DG. Threats to validity in the design and conduct of preclinical efficacy studies: a systematic review of guidelines for in vivo animal experiments. PLoS Med. 2013;10(7):e1001489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hooijmans CR, Rovers MM, de Vries RB, et al. SYRCLE's risk of bias tool for animal studies. BMC Med Res Methodol. 2014;14:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao Q, Chen S, Wang Q, et al. CCR5 editing by Staphylococcus aureus Cas9 in human primary CD4(+) T cells and hematopoietic stem/progenitor cells promotes HIV‐1 resistance and CD4(+) T cell enrichment in humanized mice. Retrovirology. 2019;16(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu L, Yang H, Gao Y, et al. CRISPR/Cas9‐mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV‐1 resistance in vivo. Mol Ther. 2017;25(8):1782‐1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuo CY, Long JD, Campo‐Fernandez B, et al. Site‐specific gene editing of human hematopoietic stem cells for X‐linked hyper‐IgM syndrome. Cell Rep. 2018;23(9):2606‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Ravin SS, Li L, Wu X, et al. CRISPR‐Cas9 gene repair of hematopoietic stem cells from patients with X‐linked chronic granulomatous disease. Sci Transl Med. 2017;9(372):eaah3480. [DOI] [PubMed] [Google Scholar]
- 24. Goodwin M, Lee E, Lakshmanan U, et al. CRISPR‐based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv. 2020;6(19):eaaz0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rocca CJ, Rainaldi JN, Sharma J, et al. CRISPR‐Cas9 gene editing of hematopoietic stem cells from patients with Friedreich's ataxia. Mol Ther Methods Clin Dev. 2020;17:1026‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Charlesworth CT, Camarena J, Cromer MK, et al. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/sgRNA gene targeting. Mol Ther Nucleic Acids. 2018;12:89‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 beta‐globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeWitt MA, Magis W, Bray NL, et al. Selection‐free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8(360):360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park SH, Lee CM, Dever DP, et al. Highly efficient editing of the beta‐globin gene in patient‐derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47(15):7955‐7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pattabhi S, Lotti SN, Berger MP, et al. In vivo outcome of homology‐directed repair at the HBB gene in HSC using alternative donor template delivery methods. Mol Ther Nucleic Acids. 2019;17:277‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu Y, Zeng J, Roscoe BP, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25(5):776‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Metais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3(21):3379‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weber L, Frati G, Felix T, et al. Editing a gamma‐globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020;6(7):eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pavel‐Dinu M, Wiebking V, Dejene BT, et al. Gene correction for SCID‐X1 in long‐term hematopoietic stem cells. Nat Commun. 2019;10(1):1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito CY, Kirouac DC, Madlambayan GJ, Yu M, Rogers I, Zandstra PW. The AC133+CD38‐, but not the rhodamine‐low, phenotype tracks LTC‐IC and SRC function in human cord blood ex vivo expansion cultures. Blood. 2010;115(2):257‐260. [DOI] [PubMed] [Google Scholar]
- 36. Tomellini E, Fares I, Lehnertz B, et al. Integrin‐alpha3 is a functional marker of ex vivo expanded human Long‐term hematopoietic stem cells. Cell Rep. 2019;28(4):1063‐1073.e1065. [DOI] [PubMed] [Google Scholar]
- 37. Fares I, Chagraoui J, Lehnertz B, et al. EPCR expression marks UM171‐expanded CD34(+) cord blood stem cells. Blood. 2017;129(25):3344‐3351. [DOI] [PubMed] [Google Scholar]
- 38. Beard BC, Trobridge GD, Ironside C, McCune JS, Adair JE, Kiem HP. Efficient and stable MGMT‐mediated selection of long‐term repopulating stem cells in nonhuman primates. J Clin Invest. 2010;120(7):2345‐2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Humbert O, Peterson CW, Norgaard ZK, Radtke S, Kiem HP. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta‐hemoglobinopathies. Mol Ther Methods Clin Dev. 2018;8:75‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Humbert O, Radtke S, Samuelson C, et al. Therapeutically relevant engraftment of a CRISPR‐Cas9‐edited HSC‐enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. 2019;11(503):eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR‐Cas9 gene editing for sickle cell disease and beta‐thalassemia. N Engl J Med. 2021;384(3):252‐260. [DOI] [PubMed] [Google Scholar]
- 42. Ribeil JA, Hacein‐Bey‐Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848‐855. [DOI] [PubMed] [Google Scholar]
- 43. Hoban MD, Cost GJ, Mendel MC, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125(17):2597‐2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang X, Wang Y, Yan W, et al. Production of gene‐corrected adult Beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells. 2015;33(5):1470‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9(4):297‐308. [DOI] [PubMed] [Google Scholar]
- 46. Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long‐term multilineage engraftment. Science. 2011;333(6039):218‐221. [DOI] [PubMed] [Google Scholar]
- 47. Sumide K, Matsuoka Y, Kawamura H, et al. A revised road map for the commitment of human cord blood CD34‐negative hematopoietic stem cells. Nat Commun. 2018;9(1):2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS‐1 enhances CRISPR/Cas9‐ and TALEN‐mediated knock‐in efficiency. Nat Commun. 2016;7:10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR‐Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Riesenberg S, Maricic T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat Commun. 2018;9(1):2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853‐860. [DOI] [PubMed] [Google Scholar]
- 52. Katlama C, Deeks SG, Autran B, et al. Barriers to a cure for HIV: new ways to target and eradicate HIV‐1 reservoirs. Lancet. 2013;381(9883):2109‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV‐1 coreceptor accounts for resistance of some multiply‐exposed individuals to HIV‐1 infection. Cell. 1996;86(3):367‐377. [DOI] [PubMed] [Google Scholar]
- 54. Samson M, Libert F, Doranz BJ, et al. Resistance to HIV‐1 infection in caucasian individuals bearing mutant alleles of the CCR‐5 chemokine receptor gene. Nature. 1996;382(6593):722‐725. [DOI] [PubMed] [Google Scholar]
- 55. Schiroli G, Conti A, Ferrari S, et al. Precise gene editing preserves hematopoietic stem cell function following transient p53‐mediated DNA damage response. Cell Stem Cell. 2019;24(4):551‐565.e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Supplementary Figure
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.