

Abstract
To harness the full potential of human pluripotent stem cells (hPSCs) we combined instrumented stirred tank bioreactor (STBR) technology with the power of in silico process modeling to overcome substantial, hPSC‐specific hurdles toward their mass production. Perfused suspension culture (3D) of matrix‐free hPSC aggregates in STBRs was applied to identify and control process‐limiting parameters including pH, dissolved oxygen, glucose and lactate levels, and the obviation of osmolality peaks provoked by high density culture. Media supplements promoted single cell‐based process inoculation and hydrodynamic aggregate size control. Wet lab‐derived process characteristics enabled predictive in silico modeling as a new rational for hPSC cultivation. Consequently, hPSC line‐independent maintenance of exponential cell proliferation was achieved. The strategy yielded 70‐fold cell expansion in 7 days achieving an unmatched density of 35 × 106 cells/mL equivalent to 5.25 billion hPSC in 150 mL scale while pluripotency, differentiation potential, and karyotype stability was maintained. In parallel, media requirements were reduced by 75% demonstrating the outstanding increase in efficiency. Minimal input to our in silico model accurately predicts all main process parameters; combined with calculation‐controlled hPSC aggregation kinetics, linear process upscaling is also enabled and demonstrated for up to 500 mL scale in an independent bioreactor system. Thus, by merging applied stem cell research with recent knowhow from industrial cell fermentation, a new level of hPSC bioprocessing is revealed fueling their automated production for industrial and therapeutic applications.
Keywords: high density culture, human pluripotent stem cells, in silico process modeling, process scale‐up, stirred tank bioreactor, suspension culture
Wet lab‐derived process characteristics enabled predictive in silico modeling as a rational for suspension‐based human pluripotent stem cells (hPSC) cultivation in stirred bioreactors. Consequently, this hPSC line‐independent strategy yielded 70‐fold cell expansion in 7 days achieving a density of 35 × 106 cells/mL equivalent to 5.25 billion hPSC in 150 mL scale while maintaining a pluripotent phenotype and simultaneously reducing media requirements by 75%.
Significance statement.
Several human pluripotent stem cell (hPSC)‐based therapy concepts have now progressed toward first‐in‐human studies. Recent articles highlighted the urgent need for efficient culture processes to provide the required quantity and quality of hPSCs and then to promote their clinical translation at commercially viable conditions. With the achievements of the present study, not only was the cell production optimized in a way that more cells could be grown within a shorter period of time while using less media, but also the in silico growth model increased knowledge about stem cell growth requirements in suspension, which can also be applied for lineage‐directed differentiations.
1. INTRODUCTION
The application of human pluripotent stem cells (hPSCs; including embryonic and induced; hESC and hiPSC) ranges from advanced drug discovery assays, to in vitro diseases models, and ultimately regenerative therapies. 1 The clinical translation of hPSC progenies has made particular progress in recent years. 2 However, routine cell therapies for solid organs such as the heart, pancreas, liver or brain will require large cell quantities estimated at ~108 to 109 cells per patient. 3 For hPSC‐derived blood cells such as macrophages 4 and erythrocytes 5 even higher numbers ranging beyond 1011 cells per patient are discussed. Moreover, the envisioned 3D printing of whole human organs will depend on the excessive availability of respective hPSC derivatives. To meet such cell demands either by “customized manufacturing” for individual patients (eg, autologous approaches based on patient‐specific hiPSC lines) or by mass produced “of‐the‐shelf” cell products for patient‐independent use (eg, applying HLA‐matched or even HLA‐depleted hPSC lines 6 ) requires robust, efficient, and economically viable hPSCs production processes ultimately compliant with clinical and regulatory standards. 7 , 8
Instrumented stirred‐tank bioreactor (STBR) technology enables the systematic development and upscaling of bioprocesses by equivalent reactor design of incremental dimensions. STBRs ensure the homogeneous distribution of cells, nutrients and gases in suspension culture and enable the continuous online monitoring and, importantly, feedback‐based control of key process parameters such as pH and dissolved oxygen (DO). 8
For hPSC cultivation in three‐dimensional (3D) suspension culture microcarrier have been used for matrix‐based cell attachment equivalent to conventional two‐dimensional (2D) conditions. 9 , 10 , 11 Alternatively, “cell‐only hPSC aggregate” cultivation was demonstrated, 12 , 13 which makes matrix supplementation redundant thereby reducing costs and regulatory hurdles.
Aggregate‐based hPSC cultivation in STBRs recently leveraged process efficiency. A cell density of 2.85 × 106 hPSCs/mL within 7 days was achieved subsequently to single cell‐based process inoculation of 0.5 × 106 hPSCs/mL in 100 mL culture scale. 14 Another study demonstrated a final density of up to 4.7 × 106 hPSCs/mL by the inoculation and passaging of semi‐dissociated clumps. 15 However, this progress seems minor compared to enabled cell densities of >100 × 106 cells/mL reported for optimized high density fermentation of Chinese hamster ovary (CHO) cells typically used for recombinant protein production. 16 , 17 , 18 Such comparison of the (still premature) pluripotent stem cell field to the long‐established industrial fermentation of transformed cell lines may seem misleading; but it highlights the principle gap between the sophisticated field of continuous cell manufacturing in >1000 L scale vs the still adolescent stage of hPSC bioprocessing facing distinct challenges. 7 Notably, the recent progress in 3D hPSC culture was achieved by perfusion feeding, 14 , 15 which is defined as the continuous influx of fresh culture medium paralleled by the equivalent efflux of used medium. Relying on a valid cell retention technology (to avoid cell depletion by the efflux) the strategy aims at promoting process homogeneity by reducing the fluctuation patterns of nutrients and secreted metabolites including the carbon source glucose (Glc) and the cell secreted lactate (Lac) in response to the highly glycolytic metabolism of hPSCs. 14 , 19 , 20
The control of specific process parameters for hPSC cultivation has been very limited yet. Besides the aspired control of pH and DO (crucial to any mammalian cell culture 16 ), the constant increase in the hPSC aggregates' dimensions (subsequent to single cell or semi‐dissociated clump inoculation) 21 may induce physical and physiological limits, 22 suggesting that aggregate size control is a key factor specific to hPSC bioprocessing. 23
Kinetic process modeling aims at the in silico prediction and modulation of feeding strategies to enable rational process optimization alongside the dynamic changes of cells' requirements throughout process progression. 24 In contrast to continuous cell line bioprocessing, very limited kinetic modeling has yet been applied to hPSCs 24 with minor impact on process improvement.
In this study, we take full advantage of the STBR technology to identify and control the complex interplay of proliferation limiting process parameters. Building on an extensive and detailed dataset, the power of in silico modeling is applied to develop novel hPSC culture strategies thereby breaking prior hurdles in hPSC cultivation and aligning it to the field of industrial bioprocessing of continuous mammalian cell lines, while simultaneously promoting the radical reduction of costly media consumption.
2. MATERIALS AND METHODS
2.1. Bioreactor system “DASbox”
The present study was performed using a DASbox Mini Bioreactor System (Eppendorf AG, Hamburg, Germany, https://www.eppendorf.com) for parallel operation of four or more independently controllable DASbox Mini bioreactor vessels for cell culture applications equipped with an eight‐blade impeller (60° pitch) optimized for hPSC expansion. 25 An overhead drive allowed smooth agitation. Sensors for pH and DO monitoring as well as temperature control ensured a tight monitoring and control of critical process parameters. Pumps and sensor were calibrated as described previously. 26 Perfusion was technically established by the incorporation of a porous outflow filter (20‐ to 40‐mm pore size) as a cell retention device similar to that used from Weegman et al.. 27 The culture volume was kept constant by applying the same flow rates for addition of fresh medium (feed) via a head plate port and for the removal of depleted medium (waste) via the outflow filter.
2.2. Cell culture
Experiments were performed utilizing the human induced pluripotent stem cell (hiPSC) line hHSC_1285i_iPS2 (MHHi006‐A) 28 derived from hematopoietic stem cells and for model development and validation the hiPSC line Phoenix (HSC_ADCF_SeV‐iPS2; MHHi001‐A) derived from CD34+ human cord blood hematopoietic stem cells 29 as well as he cell line GMPDU_8 (CD34+hPBHSC_GMPDU_SeV‐iPS8; MHHi008‐A) 30 derived under GMP‐compatible conditions from CD34+cord blood hematopoietic stem cells. Ahead of process inoculation, hiPSCs were expanded in feeder‐free monolayer culture on Geltrex‐coated flasks (250 μL Geltrex per 100 mL DMEM/F‐12 and 1 mL of this Geltrex‐DMEM/F‐12 mix was sufficient for 5 cm2, Invitrogen; Thermo Fisher, Waltham, MA, http://www.thermofisher.com) or to prove the potential GMP compatibility, on CTS Vitronectin coated flasks for GMPDU_8 in E8. The general culturing strategy of hPSCs included cell seeding in E8 + 10 mM Y‐27632 (E8 + RI, synthesized as previously described 31 ) and a media change with E8 after 48 hours for a passaging interval of 3 days and media changes with E8 after 48 and 72 hours for a passaging interval of 4 days. In general, the flasks were cultured in an incubator at 37°C with 5% CO2. For passaging, the cells were washed with PBS without Ca2+/Mg2+ (PBS w/o) and subsequently detached using StemPro Accutase via incubation at 37°C for 3 to 5 minutes or with Versene via incubation at 37°C for 8 to 10 minutes. Afterwards, DMEM/F‐12 was added to dilute the Accutase and the cell suspension was transferred into a conical tube for subsequent centrifugation at 1000g and 4°C for 3 minutes, followed by supernatant removal, resuspension in E8 + RI and cell density determination. Afterwards, cells are seeded in a density of 2‐4 × 104 cells/cm2 into a Geltrex‐ or Vitronectin‐coated flask. Monolayer cultures did not exceed eight passages in feeder‐free culture. The volumes of the respective media and reagents are as follows: E8 is used at 0.2 mL/cm2, Accutase at 0.04 mL/cm2, Versene at 0.04 mL/cm2, and DMEM/F‐12 to dilute Accutase/Versene is used at 0.4 mL/cm2.
2.3. Bioreactor inoculation and process parameters
For bioreactor inoculation, 2D precultures were dissociated using Accutase, or Versene in the case of GMPDU_8 to achieve a single‐cell suspension that was then used to inoculate the bioreactors at a viable cell density of 5 × 105 cells per milliliter in E8 + RI in a final 150 mL culture volume (75 million cells per vessel) as previously described. 26 Cells were cultivated at 37°C, stirred at 60 (or 80, 100, 120) revolutions per minute (rpm), headspace aerated with 3 sL/hour with 21% O2 and 5% CO2. No media changes were performed for 24 hours after single‐cell inoculation. Afterwards the culture medium was constantly replaced at flow rates of 6.25 mL/hour for the processes pUC, p7, p7G, p7GO, pGOS60, pGOS80, pGOS100, and pGOS120, all more advanced processes were conducted at elevated perfusion rates displayed in Supporting information Table 1, while cell aggregates are retained inside the bioreactor by the cell retention device. The media used for perfusion is E8 without the supplementation of RI for the processes pUC and p7, with additional glucose supplementation from day 3 onwards (from 3.15 to 6.15 g/L) for the processes p7G and p7GO, the additional supplementations of 0.1% (final concentration) Pluronic F68 for the processes pGOS60, pGOS80, pGOS100 and pGOS120 and the adapted additional glucose supplementation from day 1 onwards (from 3.15 to 6.15 g/L) and from day 4 onwards (from 6.15 to 7.65 g/L) for the processes pGOS80oF and Stg2M. Control of pH was initiated when the pH was reaching ≤7.0 for the first time and kept constant initially with the reduction of CO2 in the gas stream and afterwards with the addition of 1 M NaHCO3. All the described process were conducted for 7 days in the bioreactor.
2.4. Sampling, aggregate analysis, and cell counting
Sampling from the bioreactors was performed without interruption of stirring as described earlier. 26 To monitor aggregate formation and diameters (d), up to five independent light microscopic images were captured for each sample (Axiovert A1; Zeiss, Thornwood, NY, http://www.zeiss.com) followed by diameter analysis via ImageJ. Mean diameters represent arithmetic averages 45 to 1096 single aggregates.
For cell counting, cell cycle analysis and flow cytometry, aggregates were dissociated into single cells with Accutase as described earlier. 26 , 32 Supernatant was stored at −20°C for subsequent glucose/lactate/amino acid/osmolality analysis.
2.5. Statistical analysis
All bioreactor runs were performed in three or four independent runs. Statistical analyses were performed with GraphPad Prism6. Statistical significance was determined by ordinary one‐way ANOVA or two‐way ANOVA followed by Bonferroni's post‐test. Thereby differences were considered statistically significant at P < .05, P < .01, and P < .001. Results are reported as mean and SE of mean (SEM).
2.6. Cardiac differentiation
Chemically defined cardiac Differentiation in Erlenmeyer flasks (20 mL working volume) was performed as described previously. 26 , 33 In brief, after bioreactor expansion cell density was determined and seeded at 5 × 105 cells/mL for differentiation in CDM3 34 supplemented with 5 μM CHIR99021. Precisely 24 hours later the medium was replaced by CDM3 plus 5 μM IWP2. After precisely 48 hours pure CDM3 was added and replaced every 2 to 3 days thereafter.
2.7. Definitive endoderm and intestinal differentiation
Definitive Endoderm and Intestinal Differentiation was performed as described previously. 35 In brief, after bioreactor expansion cell density was determined and seeded at 10 × 105 cells/mL for differentiation in RPMI 1640 supplemented with 100 ng/mL activin A and 3 μM CHIR99021 in Erlenmeyer flasks (20 mL working volume). Precisely 24 hours later the medium was replaced by 100 ng/mL activin A and 0.8% Knockout Serum Replacement in RPMI 1640. Again 24 hours later the medium was replaced by 100 ng/mL activin A and 8% Knockout Serum Replacement in RPMI 1640. On day 3 of differentiation, aggregates were dissociated with Accutase for 3 minutes in a water bath at 37°C, analyzed for DE markers, and further differentiated into intestinal cells by plating down cells at 2 × 105 cells/cm2 in intestinal medium consisting of DMEM/F12, 2% fetal bovine serum, 500 ng/mL FGF4, 3 μM CHIR99021 and 1% penicillin/streptomycin. Medium was changed every other day until day 7, when cells were analyzed.
2.8. Derivation and differentiation of smNPCs
The derivation of smNPCs from high density hPSC bioreactor cultures was performed following a published protocol 36 with some modifications. In brief, hiPSC aggregates were transferred into low attachment petri dishes and cultivated in N2B27 consisting of 1:1 DMEM‐F12/Neurobasal (both from Invitrogen) supplemented with 1:200 N2 (Invitrogen), 1:100 B27 without vitamin A (Invitrogen) and 1% GlutaMAX (Gibco, Invitrogen). In addition, 3 μM CHIR 99021 (Axon Medchem) and 0.5 μM SAG (Merck Millipore) plus 10 μM SB‐431542 (Axon Medchem) and 1 μM dorsomorphin (Tocris) was added to the N2B27 medium. This medium was replaced after 2 days (day 3). On day 4, medium was changed to expansion medium: N2B27 containing 3 μM CHIR 99021, 0.5 μM SAG and 150 mM ascorbic acid (Sigma). On day 6, aggregates were carefully triturated into smaller pieces and plated in expansion medium on 6‐well plates coated with Matrigel (1:30, growth factor reduced, high concentration; BD Biosciences). Adherent cultures were maintained for additional 3 days with daily medium changes. Three days after plating cultures were treated with 2 mg/mL collagenase IV (Invitrogen) in DMEM/F12 for 30 minutes at 37°C in a cell culture incubator. Expansion medium was added and cells were then dissociated, centrifuged, and plated according to a split ratio of 1:6. From this day on, expansion medium was changed every other day. All subsequent splits were performed using Accutase (Gibco, Invitrogen) prewarmed at 37°C.
For differentiation, smNPCs were plated in expansion medium on Matrigel‐coated 6‐well plates at a density of 1 × 106 cells/well. From the following day on, cells were cultured in differentiation medium: 1:1 DMEM‐F12/Neurobasal medium supplemented with 1:200 N2 (25 μg/mL insulin, 20 nM progesterone, 100 μM putrescine, 30 nM sodium selenite, 10 μM holo‐transferrin; all GE‐Healthcare), 1:100 B27 without vitamin A, 9 μM glucose (Carl Roth) and 1:200 GlutaMAX. Medium was changed twice a week.
3. RESULTS
3.1. pH control and glucose supplementation induces ~2‐fold higher hPSC yield
Following the schematic in Figure 1A, process inoculation by 0.5 × 106 single hPSCs/mL at ~97% cell viability was performed, but resulted into a notable drop down to ~0.3 × 106 viable cells/mL after 24 hours (d1; Figure 1B), in line with previous studies. 14 , 25 Perfusion was automatically started after 24 hours (on d1) while initiation of pH control was process‐triggered by the inherent pH drop below 7.0, typically occurring at ~18 to 24 hours post inoculation (Figure 1C). On d2, cell counts were by tendency higher at pH 7.0 controlled (p7) vs pH uncontrolled (pUC; Figure 1B) conditions. To anticipate Glc limitation, Glc was supplemented from d3 onward to the feed medium in those processes designated “pH 7.0 con + Glc” (p7G).
FIGURE 1.
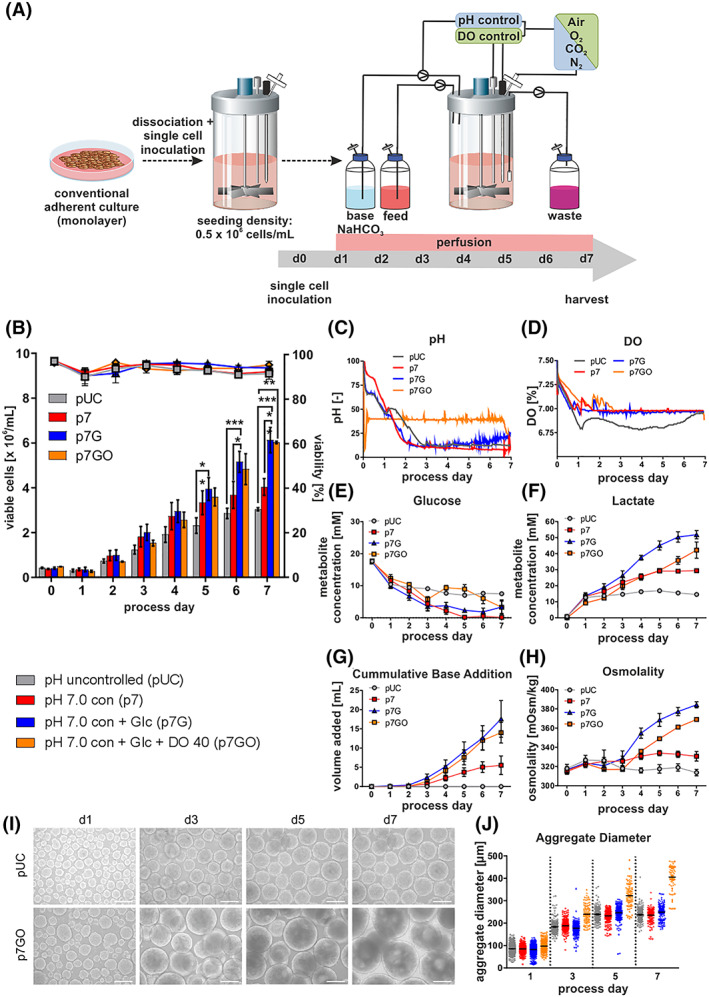
Impact of pH/DO control and Glc supplementation on process performance. A, Human induced pluripotent stem cells (hHSC_1285i_iPS2) were detached from monolayer cultures and seeded as single‐cell suspensions at a seeding density of 0.5 × 106 cells/mL on day 0 to stirred tank bioreactors at 60 rpm. During the first 24 hours cultures were maintained without any medium exchange. After 24 hours, perfusion was started. For the pH controlled processes, a triggered clock was starting the regulation when pH set‐point was reached for the first time and keeping it constant afterwards with first the reduction of CO2 in the gas stream and afterwards the addition of 1 M NaHCO3. The dissolved oxygen concentration (DO) was kept at 40% (of air saturation) by adjusting the O2 concentration in the gas stream. B, Viable cell densities (bar chart) and viability (line graphs) for processes using the cell line hHSC_F1285T_iPS2. Processes conditions: pH uncontrolled (pUC; gray) or controlled at pH 7.0 without (p7; red) or with (p7G; blue) additional Glc supplementation from day 3 onwards (n = 4) or additional DO control (p7GO; orange). All processes were performed using a MPR of D = 1/d. Metabolic activity in pH controlled and uncontrolled perfusion processes. C,D, Online process parameter measurement of pH and DO. To ensure clarity of illustration, representative single pH and DO are shown only. Respective process‐dependent patterns of Glc (E), Lac (F) concentrations and osmolality (H) in cell culture supernatants as well as cumulative base addition over the cultivation process (G) (n = 4). I, Impact of process conditions on hiPSC aggregate formation and size. Exemplified light microscopy pictures of process derived aggregate samples on days 1, 3, 5 and 7 (scale bars = 200 μm). J, Distribution of aggregate diameters over the cultivation time. As noted before, process perfusion was initiated on day 1 and additional Glc supplementation via addition of 3 g/L Glc to culture medium used for the perfusion was started from day 3 onwards in respective processes (blue lines). All Data displayed in this figure was generated with the cell line hHSC_1285_iPS2. Results are presented as mean ± SEM. Differences were considered statistically significant at *P < .05, **P < .01, and ***P < .001
Figure 1C shows typical pH patterns, demonstrating that feedback‐based, automated base supplementation was efficient for maintaining pH values closely around the defined set point of pH 7.0. In contrast, at uncontrolled conditions, the pH dropped toward pH ~6.8 on d1 despite the compensatory effect of the initiated medium perfusion.
At p7 conditions, Glc rapidly dropped to depletion on ~d5 (Figure 1E), which was prevented in p7G. Notably, confirming our previous observations, 14 Glc and Lac levels stagnated in pUC on ~d1‐2. (Figure 1E,F).
In accordance with established glycolytic metabolism of hPSCs, 37 progressive cell proliferation at p7 control resulted in elevated Lac accumulation (Figure 1F). Even more drastic differences became apparent in response to Glc supplementation in p7G, leading into Lac concentrations of ~50 mM on d6‐7; such Lac levels were previously suggested as proliferation inhibiting or toxic in 2D hPSC culture. 38 Glc feeding combined with the increased base requirements for pH control at increasing Lac concentrations (Figure 1G) together resulted into drastically increased osmolality values (Figure 1H).
On d7 (7 days of process duration was chosen for proper comparison to prior studies 14 , 25 , 39 , 40 ), the previously established pUC “control process” resulted in an average viable cell density of 3.04 × 106/mL closely recapitulating 3.0 × 106 cells/mL observed at equivalent conditions. 14 At p7, the average cell density was 4.03 × 106 cells/mL equivalent to an increase of >30% over pUC. Moreover, p7G yielded 6.13 × 106 cells/mL representing a ~12‐fold expansion of inoculation and a significant increase of >50% over p7 and >100% over pUC, strongly suggesting that pH control and Glc supplementation is a favorable direction for the desired progress.
3.2. DO control results in larger aggregates but no increase in cell yield
pH control—by trend—induced a faster DO decrease (Figure 1D). Subsequently, DO levels typically stagnated at ~8% to 20% from d2‐3 onwards. Lack of DO control may (equivalent to other parameters) impact on both the cell yield and the process reproducibility, particularly upon transition to different STBR systems and process upscaling. Thus, in addition to pH control and Glc supplementation, DO was set at 40% of air saturation reflecting a common set‐point in mammalian cell bioprocessing. 41 , 42 In contrast to triggered pH control, DO was directly set to 40% at process inoculation and subsequently maintained at this level (Figure 1D; established by automated mixing of air, oxygen and nitrogen), while keeping the overall gas flow rate (into the reactor vessel headspace) constant.
Notably, immediate DO control (p7GO) resulted—by tendency—in lower cell counts on d1‐4 vs p7 and p7G (Figure 1B). Since 2D hPSC pre‐cultures (used for process inoculation; Figure 1A) were cultured at higher oxygen concentration conditions, 43 the immediate transition to 40% DO in suspension may underlie the transient growth retardation observed here. However, p7GO ultimately resulted in ~6 × 106 cells/mL on average, that is, highly similar but not advantageous to p7G (Figure 1B). Interestingly, substantially larger aggregate dimensions were observed at p7GO compared to any other condition (Figure 1I,J; S1a). This became apparent already 24 hours after inoculation and was accompanied by lower cell counts on d1 (Figure 1B) as indicated above. Together, this suggests that the increased loss of viable cells (apparently triggered by the immediate transition to 40% DO, in combination with stirring‐induced shear) promotes the (undesired) agglomeration of remaining viable cells into larger aggregates, likely catalyzed by dead cell‐born debris, a known mechanism in bioprocessing. 44 However, the sensitivity of the single cell‐based inoculation strategy (as indicated by our above observations), and the importance of the first 24 hours of the “2D/3D transition phase” for the entire process outcome (as suggested by previous studies 32 ), directed the next steps of investigations.
3.3. Modulated inoculation overcomes early cell loss and boosts cell yields
Single cell‐based inoculation, in contrast to applying semi‐dissociated clumps, enables exact cell number determination thus supporting process comparability and reproducibility. 15 On the down side, the strategy results in a previously described viable cell loss of ~30% to 50% within the first ~24 hours post inoculation (Figure 1B). However, we here find that a shorter passaging interval of the 2D pre‐culture (former 96 hours, resulting in a 90%‐95% cell layer confluence) reduced to 72 hours (schematic in Figure 2A; resulting in a confluence of only 65%‐75%) is favorable for suspension culture inoculation. To reflect the hypothesized growth retardation by lowered DO (set to 40%; see above) the process was now inoculated at 100% DO and while control was initiated after DO levels dropped down to 40% (typically occurring after ~24 hours of culture; Figure 2C) and stabilized at this DO thereafter. Moreover, the Poloxamer Pluronic F68 (an established shear protectant in stirred CHO culture 17 , 45 ) was supplemented at inoculation. These combined measures (Figure 2A, left) significantly prevented the critical early cell loss (Figure 2A, bar graph) now resulting in a typical post‐inoculation lag‐phase for the first 24 hours, well known in bioprocessing. 46
FIGURE 2.
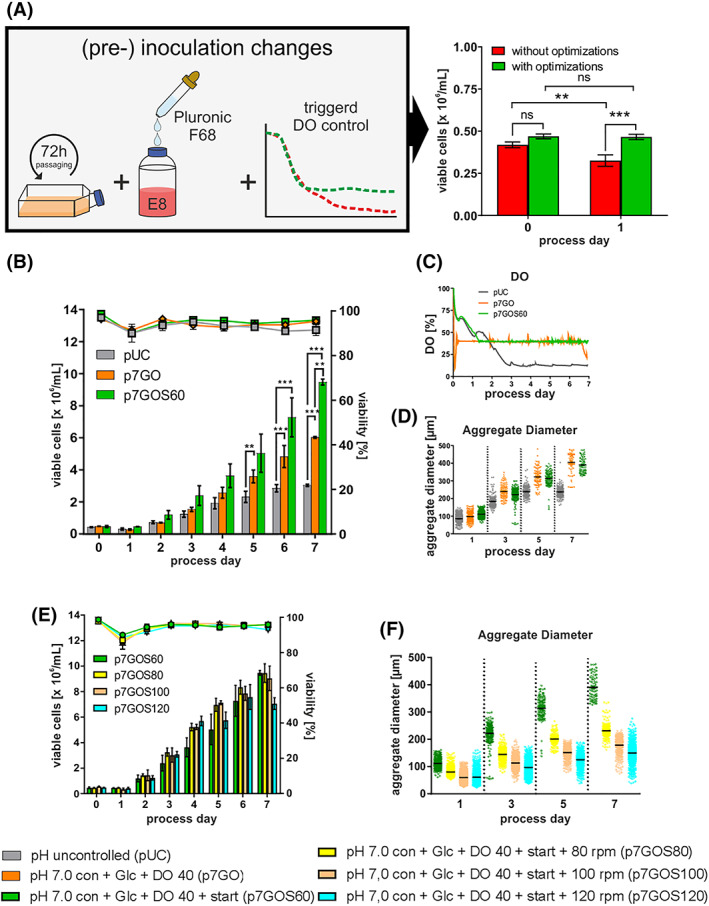
Impact of optimized single cell inoculation on process performance. A, Schematic outline of changes performed that resulted in the increased cell densities (green bars; n = 11) and control conditions (red bars; n = 15). B, Viable cell densities (bar chart) and viability (line graphs) for processes using the cell line hHSC_F1285T_iPS2. Processes conditions: pH uncontrolled (pUC; gray) or controlled at pH 7.0 with additional Glc supplementation from day 3 onwards and additional DO control (p7GO; orange) or optimized inoculation (p7GOS60; green). All processes were performed using a MPR of D = 1/d. C, Online process parameter measurement of dissolved oxygen (DO). D, Distribution of aggregate diameters over the cultivation time. Impact of stirring rates on process performance and aggregate size. E, Viable cell densities (bar chart) and viability (line graphs) for perfusion processes using the cell line hHSC_F1285T_iPS2. Processes conditions: controlled at pH 7.0, Glc supplementation, DO 40% control and optimized inoculation at 60 rpm (gray; n = 2), 80 rpm (red; n = 5), 100 rpm (blue; n = 3) and 120 rpm (green; n = 3). All processes were performed using a MPR of D = 1/d. F, Distribution of aggregate diameters over the cultivation time. All Data displayed in this figure was generated with the cell line hHSC_1285_iPS2. Results are presented as mean ± SEM. Differences were considered statistically significant at *P < .05, **P < .01, and ***P < .001
The advanced strategy designated p7GO + start (p7GOS60; Figure 2B) resulted in a significant increase in growth kinetics enabling 9.5 × 106 cells/mL on average thereby accounting for another >50% increase in cell yield compared to p7G and p7GO, respectively. Strikingly, both DO controlled process p7GO and p7GOS60 promoted the formation of relative large aggregates at later process stages ranging at ~210 to 440 μm on d5 and ~320 to 480 μm on d7 (Figure 2D, S1a), suggesting the induction of potential growth limitations due to diffusion limits expected at >300 μm of aggregate diameter. 47
3.4. Stirring rate increase enables aggregate size reduction without limiting hPSC viability
Due to the significant increase of aggregate size above 300 μm in consequence to our process optimization measures, the following experiments were focused on aggregate size control and overall size reduction. It has been suggested that hPSCs are relatively sensitive to mechanical/hydrodynamic stress. 25 , 48 However, in the presence of the shear protectant Pluronic F68, we found that aggregate dimensions can be systematically reduced by increasing the stirring rate from the above established 60 rpm (rpm) up to 120 rpm. For example, at 80 rpm, the average aggregate diameter was ~250 μm on d7 with only a minor population of >300 μm (yellow in Figure 2F). However, despite this stirring‐controlled reduction in aggregate diameters (ie, by higher stirring rates without reducing the cell viability, as confirmed by lactate dehydrogenase [LDH] measurement Figure S2c), the final cell yield did not increase (as compared to established 60 rpm processes) but rather dropped, particularly at 120 rpm conditions (Figure 2E). This in mind, however, it should be noted that growth kinetics did indicate improved proliferation on d2‐5 at 80 and 100 rpm (Figure 2E), suggesting advantageous effects, which may have been masked at later stages such as on process d6‐7 by other bottlenecks. Under these assumptions, we consequently applied 80 rpm conditions (p7GOS80 in Figure 2E,F) further on.
3.5. Manually adapted perfusion feeding results in >6‐fold higher cell yields
Besides the aggregate dimensions, two other bottlenecks were suggested at this stage of the study: Low Glc and high Lac concentrations from ~d3 onwards (Figure S2a,b). To simultaneously increase the Glc supply and dilute Lac, the daily medium perfusion rate (dMPR) was increased from 1 culture volume/day (throughout process duration; applied for all processes above) stepwise to 2 culture volume/day on day 5 (Figure 3B; Table S1) aiming at accommodating the cell density‐triggered process requirements. Such “optimized feeding” (oF) resulted in an additional doubling of the final cell density that is, 18 × 106 cells/mL on average in p7GOS80oF (Figure 3A). The specific growth rate μ was also highest at p7GOS80oF (Figure 3C) and the mean aggregate diameter increased at p7GOS80oF compared to p7GOS80 but remained at ~280 μm on average on d7 (Figure S2d) thus below 300 μm. Interestingly, no major changes in respective Glc and Lac patterns where observed (Figure 3D,E), suggesting that—in consequence to the significant boost in cell density—these parameter may still be growth limiting, despite the applied increase in medium perfusion.
FIGURE 3.

Impact of optimized feeding strategy on process performance. A, Viable cell densities (bar chart) and viability (line graphs) for perfusion processes using the cell line hHSC_F1285T_iPS2. Processes conditions: pH uncontrolled (pUC; gray; n = 4) or controlled at pH 7.0 and Glc supplementation (p7G; blue; n = 4) or with additional DO 40%, optimized inoculation and 80 rpm (p7GOS80; yellow; n = 5) or with additional optimized feeding strategy (p7GOS80oF; purple; n = 4). All processes (except for purple) were performed using a MPR of D = 1/d. B, Perfusion rate applied for the respective processes. C, Based on viable cell densities calculated values for the specific growth rate μ. Respective process‐dependent patterns of Glc (D) and Lac (E) concentrations in cell culture supernatants. Calculated values for the specific Glc consumption rate are depicted in (F). All Data displayed in this figure was generated with the cell line hHSC_1285_iPS2. Results are presented as mean ± SEM. Differences were considered statistically significant at *P < .05, **P < .01, and ***P < .001
3.6. In silico‐directed process modeling and modulation enables hPSC line independent high density culture
Building on the above described wet lab results, we next aimed at establishing a process‐reflecting and ultimately a process‐predictive in silico model for enabling: (a) more mechanistic process understanding and optimization, (b) reducing resource consuming trial‐and‐error experiments, (c) providing a universal blueprint for process transition to other culture systems and for up‐scaling, (d) and tailoring the nutrient demands closely aligned to process requirements, thus systematically increasing the efficiency.
Using the established Berkeley Madonna software 49 we postulated that hPSC growth is following Monod‐kinetics, established for typical production cell lines such as CHO or HEK 50 , 51 ; while such modeling approach is common to single cell‐adapted suspension cultures of continuous cell lines, it has not yet been enabled for the multi‐parametric complexity and specific requirements of aggregate‐based hPSC cultivation.
We first defined Monod‐variables for each parameter hypothesized to impact on growth kinetics, initially including: Glc‐, Lac‐, Glutamine (Gln)‐concentrations and the dMPR; for each variable respective Monod half‐saturation constants (KS) were defined (summarized in Table S2).
The resulting Stage 1 model (Stg1M) allowed for the comprehensive in silico prediction of the experimental kinetics for p7GOS80 and p7GOS80oF just by the input of process‐specific dMPR, Glc‐ and Gln‐concentration data (Figure 4A).
FIGURE 4.
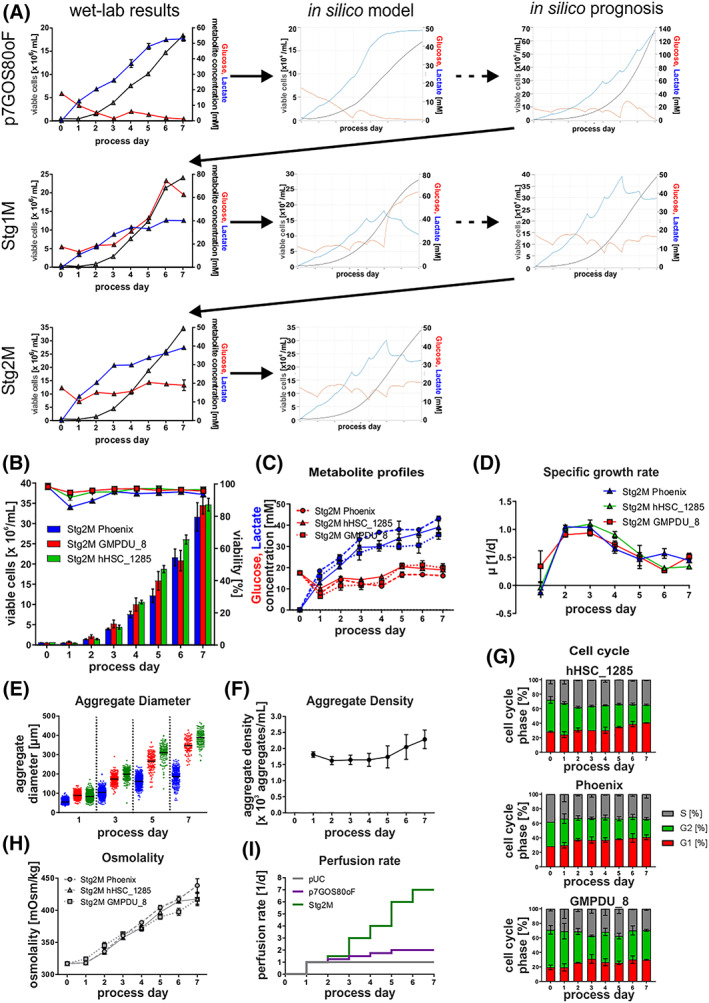
Model‐based in silico process optimization. A, Outline of the iterative model building process, including in silico results of each optimization step. B, Viable cell densities (bar chart) and viability (line graphs) for processes using the cell lines Phoenix (blue; n = 3), hHSC_F1285T_iPS2 (green, n = 3), and GMPDU_8 (red, n = 2). Process strategy followed the results of the stage 2 model. C, Respective process‐dependent patterns of glucose (red) and lactate (blue) concentrations in cell culture supernatants using the cell lines Phoenix (dashed lines), hHSC_F1285T_iPS2 (solid lines), and GMPDU_8 (dotted lines). D, Based on viable cell densities calculated values for the specific growth rate μ. E, Distribution of aggregate diameters over the cultivation time. F, Aggregate density of GMPDU_8 during the cultivation. G, Cell cycle analysis of cultures of the cell lines hHSC_1285T_iPS2 (n = 3), Phoenix (n = 3) and GMPDU_8 (n = 2) cultured under Stg2M conditions. H, Respective process‐dependent patterns of osmolality in cell culture supernatants using the cell lines Phoenix (dashed lines), hHSC_F1285T_iPS2 (solid lines), and GMPDU_8 (dotted lines). I, Perfusion rate applied for the respective processes. All Data displayed in this figure was generated with the three cell lines hHSC_1285_iPS2, Phoenix, and GMPDU_8
To apply Stg1M for process optimization, the Glc feed concentration and dMPR were shifted in silico toward the maximal cell yield (predicted at 70 × 106 cells/mL on d7; Figure 4A) while keeping the virtual Glc‐ and Lac‐patterns in a relative narrow range, to avoid detrimental overshoots. However, corresponding wet‐lab runs reached 23 × 106 cells/mL on d7 while revealing an unpredicted Glc peak pattern on d6 (Figure 4A, S4b‐d), suggesting improper modeling and/or lack of relevant parameters (Figure 4A). Consequently Monod‐like constants for osmolality and the aggregate diameter were added to the models and the KS for Lac was reduced.
The emerging Stage 2 model (Stg2M) did still tally with p7GOS80 and p7GOS80oF but now also correctly predicted the wet‐lab patterns (particular the observed Glc peak), not reflected by the Stg1M model (Figure 4A, Figure S3c,d).
Subsequently, Stg2M‐directed in silico modifications of the Glc‐feed concentration and the dMPR (Figure 4F,I) was applied, whereby dMPR was substantially elevated to avoid both overshooting Glc and osmolality peaks. Stg2M modeling predicted up to 40 × 106 cells/mL (Figure 4A) and the corresponding experiments reproducibly achieved 33 × 106 cells/mL, which was confirmed by three genetically independent hiPSC lines (Figure 4B); the respective Glc‐, Lac‐, and osmolality‐patterns notably closely reflected the in silico predictions (Figure 4C,H). Moreover, comprehensive assessment of the aggregate density (ie, the number of aggregates per culture volume) was found to be remaining nearly constant throughout the entire process duration (Figure 4F). This important finding suggests, that hPSC aggregates, which are primarily formed by single cell aggregation during first day after process inoculation, constantly increase in aggregate size (and thus result into increasing cell counts) along the process, but without any net increase in aggregate numbers over the entire process duration.
However, in order to address the 3D structure of aggregates, and to test whether the advanced process condition at Stg2M might impact on the endogenous structure of aggregates as compared to p7GOS80oF described above, native aggregates were harvested at the process endpoint and stained via the live cell marker Calcein‐AM. Applying 2‐photon microscopy, we found the presence of cavities, which were more dominant at p7GOS80oF conditions, whereas fewer and smaller cavity structures were found in aggregates derived from Stg2M conditions (Figure S2f). Notably, since Stg2M‐aggregates have a larger average diameter compared to p7GOS80oF‐samples (Figure 4E vs S2d) it seems unlikely that the aggregate dimensions (suggestive of hypoxic conditions inside the spheres) is underlying the observed cavitation. However, since glucose limitations were observed at later stages of p7GOS80oF cultures (Figure 3D), but were eliminated at Stg2M conditions, it is tempting to speculate that such suboptimal glucose supply (or related metabolic conditions) maybe an underlying trigger of process‐dependent cavity formation.
Final adaptations of process modeling (noted in Figure S3a), led to the ultimate Stage 3 model (Stg3M; Table S6), now reflecting a close overlap of wet‐lab results for all cell lines tested (ie, hHSC_1285, Phoenix and GMPDU_8) with the predicted in silico patterns (Figure S4a) with small prediction errors only (Table 1).
TABLE 1.
Prediction relative errors associated with the Monod‐type kinetic model for the six monitored variables
| Variable | Relative errors for set of experiments (%) [e r ± SD] | ||||
|---|---|---|---|---|---|
| p7GOS80 | p7GOS80oF | Stg2M hHSC | Stg2M Phoenix | Stg2M GMPDU_8 | |
| Cell density | 2.53 ± 3.93 | 4.48 ± 4.18 | 2.20 ± 1.88 | 2.48 ± 2.39 | 3.86 ± 3.41 |
| Glucose | 9.34 ± 8.04 | 16.43 ± 10.17 | 11.68 ± 8.06 | 18.51 ± 11.85 | 18.09 ± 15.71 |
| Lactate | 8.09 ± 5.77 | 9.08 ± 6.47 | 12.94 ± 9.56 | 12.32 ± 6.92 | 12.20 ± 10.18 |
| Glutamine | 12.37 ± 9.07 | 16.02 ± 9.91 | 7.95 ± 5.37 | 18.18 ± 9.22 | 12.96 ± 8.54 |
| Aggregate diameter | 4.21 ± 3.42 | 2.08 ± 1.45 | 2.49 ± 2.37 | 3.00 ± 5.27 | 2.29 ± 1.53 |
| Osmolality | 0.87 ± 0.64 | 2.62 ± 2.09 | 2.49 ± 2.37 | 3.75 ± 2.85 | 1.94 ± 1.58 |
| Cumulative base addition | 17.31 ± 16.98 | 3.05 ± 2.37 | 2.60 ± 2.76 | 3.33 ± 4.06 | 3.45 ± 5.50 |
Notably, our Stg3M also reflects (and is adaptable to) cell line‐specific differences such as different growth rates (μ; Figure 4D) and formation of smaller aggregates (Figure 4E) by Phoenix and GMPDU_8, respectively (see Supporting information Table S2). Moreover, by the degree of system control enabled here, differential DO patterns provide valuable hints toward the cell line‐dependent metabolic differences such as higher oxygen demands observed for GMPDU_8 and Phoenix (Figure S2e).
Cell cycle profiling revealed the highest amount of dividing cells (those in G2/S‐phase) on d0‐d1. Subsequently the number of resting cells in G1 continuously increased from ~25% on d0 to ~40% on d7 nicely reflecting patterns for μ calculated from entirely independent analytical methods and process data (Figure 4D,G). Remarkably, however, the amount of cells in S‐phase remained relative high and stable at ~35% at our most advanced process conditions which may support hPSCs differentiation potential as recently suggested in monolayer cultures. 52 , 53
3.7. High density cultivation is compatible with the hPSCs' pluripotency and differentiation potential
Since pluripotency is a key feature of hPSCs, maintenance of pluripotency‐associated markers expression at the protein level—as we have observed for all conditions tested (Figure 5A)—is of key importance; this finding was further corroborated by IF staining for seeded cells (Figure 5B). Moreover, no karyotype abnormalities where identified as exemplarily shown for hHSC_1285 and Phoenix derived for endpoint analysis on 7 days at Stg2M conditions (Figure 5c).
FIGURE 5.
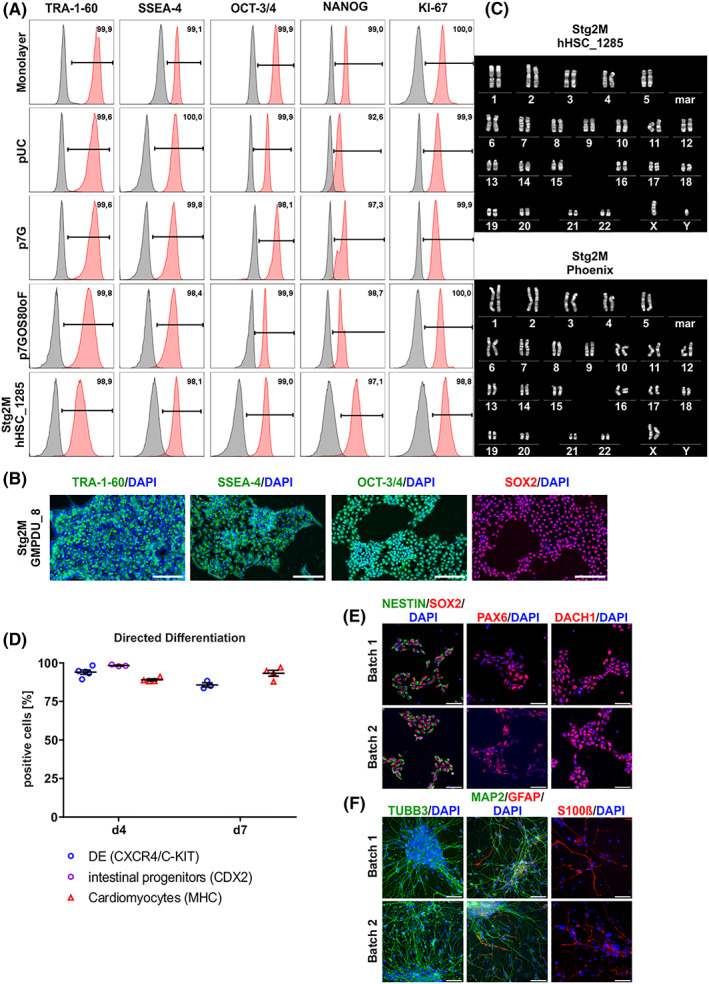
A, Pluripotency marker expression of cells harvested at process endpoints. Exemplified flow cytometry analysis plots showed that the majority of cells harvested at process endpoint (day 7) expressed pluripotency associated surface markers SSEA‐4 and TRA‐1‐60 as well as transcription factors OCT‐3/4 and NANOG at levels equivalent to monolayer pre‐cultures used for bioreactor inoculation (isotype controls shown in gray). Additionally, no decrease in proliferation marker KI‐67 could be measured. All Data displayed in (A) was generated with the cell line hHSC_1285_iPS2. B, Process‐derived hPS cell aggregates were dissociated and plated down. Representative pictures of immunofluorescence staining of day 7 derived cells stained for pluripotency markers TRA‐1‐60, SSEA‐4, OCT‐3/4, and SOX2. Positive staining is shown in green/red. Isotype controls confirmed staining specificity (not shown). DAPI stained nuclei in blue. Scale bars = 200 μm. All data displayed in (b) was generated with the cell line GMPDU_8. C, Karyotype of cells cultured for 7 days under the Stg2M conditions displayed for the cell lines hHSC_1285_iPS2 and Phoenix. D, Process‐derived hPS cell aggregates of GMPDU_8 cultured under Stg2M conditions for 4 and 7 days were directed differentiated into definitive endoderm (DE; based on double positive CXCR4/C‐KIT) and intestine (based on CDX2, only on day 4) as well as cardiomyocytes (based on MHC). Characterization of small molecule neural precursor cells (smNPCs) derived from high density hPSC bioreactor cultures. E, Samples from two independent high density hPSC bioreactor batches were differentiated into smNPCs and subsequently immunostained for markers of neural stem cells/progenitors including SOX2, NESTIN, and PAX6 as well as the neural rosette‐associated transcription factor DACH1. F, Upon growth factor withdrawal for 4 weeks differentiated smNPC cultures were stained for the neuronal markers TUBB3 and MAP2 as well as the glial markers GFAP and S100beta. Nuclei were counterstained with DAPI. Scale bars: 50 μm. All data displayed in (D)‐(F) was generated with the cell line GMPDU_8
Furthermore, the undirected differentiation of d7‐derived aggregates resulted in the formation of cell lineages representative of endoderm (SOX17), mesoderm (Desmin), and ectoderm (TUJ1) shown in Figure S6a. Applying recent differentiation strategies, GMPDU_8 cells cultured at Stg2M for 4 or 7 days where further directed into definitive endoderm (DE), intestinal progenitors 35 and cardiomyocytes 33 resulting into the highly reproducible induction of ~85% to 95% of respective progenies (Figure 5D). Moreover, neural induction into “small molecule neural precursor cells” (smNPCs) 36 was highly efficient, as confirmed by SOX2, NESTIN, PAX6 and DACH1 staining of respective samples (Figure 5E). Subsequently, after 4 weeks of growth factor withdrawal, smNPCs spontaneously differentiated into TUBB3‐ and MAP2‐positve neurons as well as glial cells expressing GFAP and S100beta (Figure 5F). Together, this highlights that our advanced culture strategy is fully compliant with maintaining hPSCs' pluripotency and differentiation potential.
3.8. Targeting aggregate diameter kinetics enables process scale‐up
Upscaling aggregates‐based cultures is another distinct challenge. Process upscaling of single cell‐adapted, continuous cell lines in industry is typically achieved by maintenance of the impeller tip speed or volumetric power input in relation to the increase in process volume and bioreactor dimensions. 54 , 55 But tip speed adaptation to an increase in process volume failed in our hands (data not shown) to maintain the above established hPSC aggregate patterns (shown in Figure 4B). In contrast, adaptation of the volumetric power input to the process scale by means of stirring rate adaptation (Figure 6A) allowed for the close reproduction of aggregate size kinetics (Figure 6B). The hereto required calculations where simplified (see Equation 34, methods section) due to the unchanged “impeller‐diameter‐to‐vessel‐ratio” and power number within the same bioreactor platform (DasBox; applied for 150, 200, 250 mL process volume) and in a larger bioreactor system as well (BioBlock; applied for 500 mL process volume; Figure 6A; note adaptations in Equation 35). This “volumetric power input‐based upscaling strategy” enabled, in addition to the aggregate pattern (Figure 6B), also the reproduction of growth kinetics and metabolic patterns of Glc (Figure 6D) and Lac (Figure 6E).
FIGURE 6.

A, Schematic outline of process volume and bioreactor systems used to evaluate the up‐scaling capability. B, Distribution of aggregate diameters over the cultivation time. C, Viable cell densities (bar chart) and viability (line graphs) for processes using culture volumes of 150 mL (gray; n = 3), 200 mL (red; n = 3), 250 mL (blue; n = 3), and 500 mL (green, n = 3). Respective process‐dependent patterns of Glc (D) and Lac (E) concentrations in cell culture supernatants. Online process parameter measurement of DO (F). To ensure clarity of illustration, representative single DO are shown only. G, Overlay of online capacitance measurement (black line) and offline measured viable cell densities (green points) in 500 mL process. All data displayed in this figure was generated with the cell line hHSC_1285_iPS2
Importantly, the offline cell density assessment was closely matched by the online capacitance measurement enabled in the 500 mL process scale (Figure 6F), thereby revealing that this online method which is typically applied for the growth kinetic assessment of single cell‐adapted mammalian cell cultivation, 56 is fully applicable to pluripotent cell aggregate cultures as well. However, whereas the pH control was accomplished at all stages of process upscaling (Figure S5c), DO control was only partially enabled that is, typically failed after 2 to 3 days at our conditions (Figure 6F). This suggests that the applied head space gassing, due to the bioreactor system‐inherent change of the surface‐to‐volume ratio, was insufficient to comply with the oxygen requirements of the high cell density conditions achieved by us. Interestingly, this DO reduction had now obvious impact on cell yields: Only a minor reduction in the 250 mL scale approach was observed (Figure 6C), which was most drastically affected by the due to headspace gassing‐induced DO drop (due to the most prominent change in surface‐to‐volume ratio).
However, in summary, these observations are calling for the future implementation of submerse gassing, to avoid such limits and keep all parameters controlled along the envisioned increase in process dimensions.
4. DISCUSSION
In this study, the controlled bioprocessing of hPS cells to high densities is revealed; in essence, a 10‐fold improvement of cell yields was achieved, as compared to best current practice in STBR‐based expansion strategies. 14 , 15 , 25 , 40
By overleaping the “virtual mark” of >107 cells/mL and ultimately attaining >30 × 106 cells/mL, our study, for the first time, promotes hPSC bioprocessing to standards previously established for continuous cell lines only. 16 , 17 , 18 Our approach was indeed inspired by translating strategies established for the fermentation of continuous cell line to the unique requirements and challenges imposed by pluripotent stem cells.
The application of instrumented STBRs to hPSC culture was previously restricted to the monitoring of individual process parameters such as DO and pH. 14 , 15 , 57 In contrast, we here concerted the multi monitoring modalities of STBRs for directing feedback‐based process regulation. We first demonstrate the overarching importance of pH control for maintaining hPSC proliferation in line with the central role of this parameter for process development of conventional cell lines. 16 , 17 , 18 While no systematic studies on pH control in hPSC have been published, it is well established that pH is a dominant trigger of metabolic activity and proliferation. Tumor cells can develop mechanisms of selective growth advantage by tolerating local tissue pH gradient induced by their own metabolism. 58 But the in vitro proliferation of continuous cell lines is readily diminished below pH 7.0, 59 , 60 whereby CHO and BHK cells are able to consume lactate 61 , 62 (the main driver of media acidification), in clear contrast to hPSCs.
Having the “pH roadblock” identified and removed/controlled, we were confronted with the combinatorial challenge of rising Glc demands, consequently boosting Lac secretion and subsequently driving base consumption required for maintaining pH stability. Together, this “metabolic feedback loop” accounted for rising osmolality levels, reflecting the steep increase in growth kinetics.
In our process, the stage‐dependent Glc consumption rate qGlc ranged at ~7.5 to 20.5 pmol/(cell × d) (Figure 3F) tallying with published values for hPS cells (Silva et al. 19 ). This highlights that the hPSC‐related qGlc is about 5‐ to 20‐fold higher compared to the typical qGlc values of 1.1 to 4.5 pmol/(cell × d) reported for CHO cells. 63 , 64 , 65 This notion reflects the glycolysis‐dependent metabolism of hPSCs 19 , 38 and pronounces the hPSC‐induced challenges for maintaining Glc, Lac, pH, and osmolality at stable, proliferation‐supporting levels; these challenges are potentiated by high density cultivation.
Interestingly, in contrast to growth inhibiting effects described for >25 mM Lac in 2D hESC culture, 38 our data suggest maintenance of robust hPSC proliferation at up to 50 mM Lac (Figure 3A,E). This discrepancy may suggest that stagnation‐inducing effects (eg, of the Lac concentration) become effective much earlier in static 2D culture, potentially due to the local accumulation of metabolites in the cell proximity and the gradient formation of metabolites in consequence of static conditions, which is prevented in our dynamic 3D suspension. Notably, regarding the impact of osmolality, we found that up to ~500 mOsm is tolerated by hPSC, in accordance with Nagashima et al. 66
However, our study underscores that perfusion feeding is “the gate‐keeper approach” for enabling controlled and progressive hPSC cultivation. In contrast to conventional batch feeding, perfusion feeding—including the process specific adaptation of the medium perfusion rate—enables the required control of pH, base, Glc, osmolality etc. in parallel to the dilution of growth‐inhibiting metabolites, as exemplified by Lac. Perfusion feeding thus provides a “holistic solution” to the metabolism‐induced challenges of hPSC bioprocessing toward high density.
For more rational process development, we have established in silico modeling. This was enabled by adapting the Monod approach formally established for prokaryotes 50 and conventional mammalian cell lines. 51
A first attempt for in silico modeling of hPSCs cultures was recently described by Galvanauskas, 24 but this approach was limited to the reflection of some wet‐lab result, only. In contrast, we here applied in silico modeling for accurate process simulation, prediction of advanced conditions, followed by the reiterative feedback of wet‐lab analysis for further model adaptation. Our final in silico algorithm developed in this study is capable of accurately predicting the growth kinetics of an aggregate‐based suspension culture of hPSC only by the input of: the inoculation density, the Glc and Gln feed concentration, and the respective medium perfusion rate. On the other hand, our model predicts patterns of Glc‐, Lac‐ and Gln‐concentrations, the osmolality, the base amount required for pH stabilization, and finally the important aspect of aggregate size patterns.
Beyond the metabolism‐related challenges, cell aggregation and the control of aggregate growth dynamics is “the other gate‐keeper” to advanced hPSC culture in suspension. Therefore, in addition to monitoring the process‐dependent increase in aggregate diameters, we have also accurately assessed the aggregate density (Figure 4F). Notably, we found that after aggregate formation (within 24 hours post single cells inoculation) their density (ie, the number of aggregates per volume) remains essentially unchanged along the entire process.
This has two consequences: First, this suggests that—despite the stirring‐induced culture dynamics—no substantial (if any) net formation (eg, by “aggregate division” or break) or net loss (eg, by fusion) of aggregates occurs. Thus, second, an increase of the overall cell density can only result from an increase in cell numbers per aggregate, thus triggering the constant increase in aggregate dimensions. This later assumption is underpinned by both the linear correlation of the average aggregate volume and the respective cell density as well as the calculated number of cells per aggregate and respective the cell density at any process stage (Figure S5a,b).
Vice versa, these findings indicate that the initial aggregate formation is a process limiting step. This assumption is highlighted by our optimization of the inoculation step: Obviating the former loss of 30% to 50% viable cell counts in the first 24 hours, 14 , 25 is subsequently boosting the proliferation kinetics (Figure 2B).
Having achieved a high degree of advancement via the Stg2M conditions, the process was applied to three genetically independent hiPSC lines. This revealed that our comprehensive process control enables achieving highly comparable growth kinetics and high density yields for all cell lines tested (Figure 4B,D) despite the notion of line‐specific properties such as different aggregate size kinetics (Figure 4E). This outcome is of utmost importance since it underscores the universal applicability of our strategy and highlights its “unifying properties,” promoting the use of any established hPSC line at hand including, for example, patient‐specific hiPSCs.
In this study, we have applied 7 days of process duration to, first, ensure relevant comparison to published benchmarks 11 , 12 , 19 , 23 and, second, to explore the possibility for maintaining long‐term exponential hPSC proliferation. However, after the 24 hours post inoculation lag‐phase, our in silico optimized final process Stg2M supports cell proliferation at the maximal cell specific growth rate μ of 1.0 equivalent to an exponential growth at a population doubling time (PDT) of ~16.6 hours (this is notably faster than published maximal values for hPSCs showing ~24 to 48 hours PDT in feeder‐based culture 67 , 68 , 69 as well as for 27.5 hours PDT for culture on the defined and xeno‐free matrix CELLstart 70 ) until day ~4 of culture, followed by a stepwise decrease of μ (Figure 4D). Under the assumption, that our optimized and controlled culture environment is not raising substantial limitations after day 4, we hypothesize that the aggregate‐inherent milieu may induce this proliferation decrease at later stages, even if the proposed diffusion‐limiting average aggregate size remains below 300 μm (Figure 4E).
While focusing on improving the growth kinetics, preserving the quality of the yielded cells is of utmost importance. For hPSCs, the mandatory quality criteria include: retaining the pluripotency, maintenance of the unrestricted differentiation potential into derivatives of all three germ layers, and karyotype stability. By comprehensive analysis along all process optimization steps, we have observed the expected high expression of all relevant pluripotency markers tested (ie, TRA‐1‐60, SSEA‐4, OCT‐3/4, and NANOG) at the protein level. Importantly, high homogeneous expression patterns of pluripotency‐associated markers was also detected in all cell samples collected from the highest cell densities generated at the process endpoint at optimized conditions (Figure 5A). From a functional perspective, we have observed the efficient, lineage specific differentiation of straight STBR‐derived hPSC aggregates into neuro‐ectodermal progenies, mesoderm‐derived cardiomyocytes, and intestinal progenitors representing endodermal derivatives (Figure 5D,E). Finally, maintenance of the normal karyotype was found across all hiPSC lines when tested after high density cultivation. Together, this strongly suggests that our progressive bioprocess conditions are fully compatible with the relevant quality criteria applied to pluripotent stem cells.
For future process logistics, we suggest a culture duration limited to 4 days (ie, when ~10 × 106 cells/mL is readily achieved by our conditions) followed by the inoculation of the next larger STBR system in a seed‐train approach. 15 This in mind, we have adapted an established equation for volumetric power input to our aggregate‐based suspension culture. Applying this calculation, we show the practical Proof‐of‐Concept for rational process upscaling from 150 to 500 mL culture scale (including transition to a larger STBR system) while fully maintaining all essential process characteristics established in 150 mL scale (Figure 6B‐E).
Our advanced high density culture also promotes economic aspects. As outlined in Table S3, process optimization induced >75% reduction of media consumption, which (irrespective of labor costs that are also reduced by process upscaling and automation) is the key cost component in mass cell production.
Our progress, combined with the non‐requirement of costly matrices and the future potential of process automation, opens a new perspective for developing hPSC culture toward industry scale processes that is, at ~10 to 1.000 L process scale and beyond, which is also urgently aspired in the “field of in vitro meat production” using for example, porcine or bovine pluripotent stem cells as the starting cell source for muscle and fat differentiation. 71
Regarding regenerative medicine, high density cultivation of hPSC is an important asset for fueling the envisioned large scale production of functional cell lineages. 7 , 33 , 34 , 35 , 53 , 72 , 73 , 74 , 75 , 76 , 77 This is, on the one hand, absolutely necessary for the commercially viable mass production of universal, non‐patient specific “off the shelf” cell products envisioned for routine clinical applications. 5 , 6 , 7 On the other hand, patient‐specific manufacturing of hiPSCs, the ultimate goal for autologous therapies, will benefit from the technology as well: It supports the customized generation of high cell numbers in small process scale at rigorously reduced costs.
By demonstrating that hPSC aggregates derived from our novel process can be directly applied for both spontaneous and the efficient directed differentiation into specific lineages, we reveal that the technology fully supports the enormous potential of hPSCs.
5. CONCLUSION
Taken together, this study provides novel solutions to yet unsolved challenges for hPSC culture at fully controlled conditions. The work enables to merge the enormous potential of the still infant field of pluripotent stem cell culture with mayor advancements achieved in the long‐standing field of mass cell culture of conventional cell lines in biotechnology.
CONFLICT OF INTEREST
O.B. declared leadership position and owner of LIFE & BRAIN GmbH, Bonn, Germany, Inventor on several patents and patent applications of LIFE & BRAIN GmbH and University of Bonn unrelated to the content of this manuscript. All of the other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
F.M.: conception and design, collection and assembly of data, data analysis and interpretation, article writing, ImageJ codes writing, Berkley Madonna model development; K.U. and C.K.: collection and assembly of data, data analysis and interpretation; C.H.: ImageJ codes writing, collection of data; W.T., A.F., C.M.F., A.H., Y.B.: collection of data; A.S.: collection and assembly of data, data analysis; M.P. and O.B.: conception and design, data analysis and interpretation; S.K.: multiphoton microscopy imaging; U.M. and R.O.: conception and design, financial support; R.Z.: conception and design, data analysis and interpretation, article writing, financial support.
Supporting information
Appendix S1: Supporting information.
ACKNOWLEDGMENTS
We would like to thank U. Rinas and T. Scheper from the Institute for Technical Chemistry, Leibniz University, Germany for providing FGF‐2; A. Kirschning and G. Dräger from the Institute of Organic Chemistry, Leibniz University, Germany for providing Y‐27632; T. Schlaeger from Boston Children's Hospital, Harvard Medical School, MA, United States for the idea of measuring aggregate density in a practical way; C. Thiele from the Institute of Reconstructive Neurobiology, University of Bonn Medical Faculty & University Hospital Bonn, Germany for the excellent technical support in neuroectodermal differentiations; G. Göhring and coworkers from the Institute for Human Genetics, Hannover Medical School, Germany for karyotyping; M. Weiß from the Institute for Technical Chemistry, Leibniz University, Germany for amino acid analysis. R.Z. received funding from: the German Research Foundation (DFG; Cluster of Excellence REBIRTH EXC 62/2, ZW64/4‐1), the German Ministry for Education and Science (BMBF, grants: 13N14086, 01EK1601A, 01EK1602A), and the European Union H2020 program to the project TECHNOBEAT (grant 668724).
Manstein F, Ullmann K, Kropp C, et al. High density bioprocessing of human pluripotent stem cells by metabolic control and in silico modeling. STEM CELLS Transl Med. 2021;10:1063–1080. 10.1002/sctm.20-0453
Funding information European Union H2020 program to the project TECHNOBEAT, Grant/Award Number: 668724; German Ministry for Education and Science, Grant/Award Numbers: 01EK1602A, 01EK1601A, 13N14086; German Research Foundation; Cluster of Excellence REBIRTH, Grant/Award Numbers: ZW64/4‐1, EXC 62/2
Contributor Information
Felix Manstein, Email: manstein.felix@mh-hannover.de.
Robert Zweigerdt, Email: zweigerdt.robert@mh-hannover.de.
DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33:409‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ludwig TE, Kujak A, Rauti A, et al. 20 years of human pluripotent stem cell research: it all started with five lines. Cell Stem Cell. 2018;23:644‐648. [DOI] [PubMed] [Google Scholar]
- 3. Chen AK, Reuveny S, Oh SK. Application of human mesenchymal and pluripotent stem cell microcarrier cultures in cellular therapy: achievements and future direction. Biotechnol Adv. 2013;31:1032‐1046. [DOI] [PubMed] [Google Scholar]
- 4. Ackermann M, Kempf H, Hetzel M, et al. Bioreactor‐based mass production of human iPSC‐derived macrophages enables immunotherapies against bacterial airway infections. Nat Commun. 2018;9:5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah SN, Gelderman MP, Lewis EMA, et al. Evaluation of stem cell‐derived red blood cells as a transfusion product using a novel animal model. PLoS One. 2016;11:e0166657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jang Y, Choi J, Park N, et al. Development of immunocompatible pluripotent stem cells via CRISPR‐based human leukocyte antigen engineering. Exp Mol Med. 2019;51:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kempf H, Andree B, Zweigerdt R. Large‐scale production of human pluripotent stem cell derived cardiomyocytes. Adv Drug Deliv Rev. 2016;96:18‐30. [DOI] [PubMed] [Google Scholar]
- 8. Zweigerdt R. Large scale production of stem cells and their derivatives. Adv Biochem Eng Biotechnol. 2009;114:201‐235. [DOI] [PubMed] [Google Scholar]
- 9. Bardy J, Chen AK, Lim YM, et al. Microcarrier suspension cultures for high‐density expansion and differentiation of human pluripotent stem cells to neural progenitor cells. Tissue Eng Part C Methods. 2013;19:166‐180. [DOI] [PubMed] [Google Scholar]
- 10. Badenes SM, Fernandes TG, Rodrigues CAV, Diogo MM, Cabral JMS. Microcarrier‐based platforms for in vitro expansion and differentiation of human pluripotent stem cells in bioreactor culture systems. J Biotechnol. 2016;234:71‐82. [DOI] [PubMed] [Google Scholar]
- 11. Badenes SM, Fernandes TG, Miranda CC, et al. Long‐term expansion of human induced pluripotent stem cells in a microcarrier‐based dynamic system. J Chem Technol Biotechnol. 2017;92:492‐503. [Google Scholar]
- 12. Olmer R, Martin U, Zweigerdt R. Expansion and differentiation of human iPS and ES cells in stirred tank bioreactors. J Stem Cells Regen Med. 2010;6:119. [PubMed] [Google Scholar]
- 13. Elanzew A, Sommer A, Pusch‐Klein A, Brüstle O, Haupt S. A reproducible and versatile system for the dynamic expansion of human pluripotent stem cells in suspension. Biotechnol J. 2015;10:1589‐1599. [DOI] [PubMed] [Google Scholar]
- 14. Kropp C, Kempf H, Halloin C, et al. Impact of feeding strategies on the scalable expansion of human pluripotent stem cells in single‐use stirred tank bioreactors. Stem Cells Translational Medicine. 2016;5:1289‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abecasis B, Aguiar T, Arnault É, et al. Expansion of 3D human induced pluripotent stem cell aggregates in bioreactors: bioprocess intensification and scaling‐up approaches. J Biotechnol. 2017;246:81‐93. [DOI] [PubMed] [Google Scholar]
- 16. Xu S, Gavin J, Jiang R, Chen H. Bioreactor productivity and media cost comparison for different intensified cell culture processes. Biotechnol Prog. 2017;33:867‐878. [DOI] [PubMed] [Google Scholar]
- 17. Xu S, Jiang R, Chen Y, Wang F, Chen H. Impact of Pluronic([R]) F68 on hollow fiber filter‐based perfusion culture performance. Bioprocess Biosyst Eng. 2017;40:1317‐1326. [DOI] [PubMed] [Google Scholar]
- 18. Yang WC, Minkler DF, Kshirsagar R, Ryll T, Huang YM. Concentrated fed‐batch cell culture increases manufacturing capacity without additional volumetric capacity. J Biotechnol. 2016;217:1‐11. [DOI] [PubMed] [Google Scholar]
- 19. Silva MM, Rodrigues AF, Correia C, et al. Robust expansion of human pluripotent stem cells: integration of bioprocess design with transcriptomic and metabolomic characterization. Stem Cells Translational Medicine. 2015;4:731‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwedhelm I, Zdzieblo D, Appelt‐Menzel A, et al. Automated real‐time monitoring of human pluripotent stem cell aggregation in stirred tank reactors. Sci Rep. 2019;9:12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rowley J, Abraham E, Campbell A, Brandwein H, Oh S. Meeting lot‐size challenges of manufacturing adherent cells for therapy. BioProcess Int. 2012;10:16‐22. [Google Scholar]
- 23. Kropp C, Massai D, Zweigerdt R. Progress and challenges in large‐scale expansion of human pluripotent stem cells. Process Biochem. 2017;59:244‐254. [Google Scholar]
- 24. Galvanauskas V, Simutis R, Nath S, Kino‐oka M. Kinetic modeling of human induced pluripotent stem cell expansion in suspension culture. Regener Ther. 2019;12:88‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olmer R, Lange A, Selzer S, et al. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng Part C Methods. 2012;18:772‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kempf H, Kropp C, Olmer R, Martin U, Zweigerdt R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat Protoc. 2015;10:1345‐1361. [DOI] [PubMed] [Google Scholar]
- 27. Weegman BP, Nash P, Carlson AL, et al. Nutrient regulation by continuous feeding removes limitations on cell yield in the large‐scale expansion of mammalian cell spheroids. PLoS One. 2013;8:e76611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hartung S, Schwanke K, Haase A, et al. Directing cardiomyogenic differentiation of human pluripotent stem cells by plasmid‐based transient overexpression of cardiac transcription factors. Stem Cells Dev. 2013;22:1112‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haase A, Göhring G, Martin U. Generation of non‐transgenic iPS cells from human cord blood CD34+ cells under animal component‐free conditions. Stem Cell Res. 2017;21:71‐73. [DOI] [PubMed] [Google Scholar]
- 30. Haase A, Glienke W, Engels L, et al. GMP‐compatible manufacturing of three iPS cell lines from human peripheral blood. Stem Cell Res. 2019;35:101394. [DOI] [PubMed] [Google Scholar]
- 31. Palecek J, Zweigerdt R, Olmer R, et al. A practical synthesis of rho‐kinase inhibitor Y‐27632 and fluoro derivatives and their evaluation in human pluripotent stem cells. Org Biomol Chem. 2011;9:5503‐5510. [DOI] [PubMed] [Google Scholar]
- 32. Zweigerdt R, Olmer R, Singh H, Haverich A, Martin U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat Protoc. 2011;6:689‐700. [DOI] [PubMed] [Google Scholar]
- 33. Halloin C, Schwanke K, Löbel W, et al. Continuous WNT control enables advanced hPSC cardiac processing and prognostic surface marker identification in chemically defined suspension culture. Stem Cell Rep. 2019;13:366‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burridge PW, Holmström A, Wu JC. Chemically defined culture and Cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet. 2015;87:21.23.21‐21.23.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sahabian A, Sgodda M, Naujok O, et al. Chemically‐defined, Xeno‐free, scalable production of hPSC‐derived definitive endoderm aggregates with multi‐lineage differentiation potential. Cells. 2019;8(12):1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reinhardt P, Glatza M, Hemmer K, et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One. 2013;8:e59252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Folmes CD, Terzic A. Energy metabolism in the acquisition and maintenance of stemness. Semin Cell Dev Biol. 2016;52:68‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen X, Chen A, Woo TL, Choo ABH, Reuveny S, Oh SKW. Investigations into the metabolism of two‐dimensional colony and suspended microcarrier cultures of human embryonic stem cells in serum‐free media. Stem Cells Dev. 2010;19:1781‐1792. [DOI] [PubMed] [Google Scholar]
- 39. Nampe D, Joshi R, Keller K, Zur Nieden NI, Tsutsui H. Impact of fluidic agitation on human pluripotent stem cells in stirred suspension culture. Biotechnol Bioeng. 2017;114:2109‐2120. [DOI] [PubMed] [Google Scholar]
- 40. Kwok CK, Ueda Y, Kadari A, et al. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single‐use bioreactors. J Tissue Eng Regen Med. 2018;12:e1076‐e1087. [DOI] [PubMed] [Google Scholar]
- 41. Heidemann R, Lutkemeyer D, Buntemeyer H, Lehmann J. Effects of dissolved oxygen levels and the role of extra‐ and intracellular amino acid concentrations upon the metabolism of mammalian cell lines during batch and continuous cultures. Cytotechnology. 1998;26:185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Link T, Bäckström M, Graham R, et al. Bioprocess development for the production of a recombinant MUC1 fusion protein expressed by CHO‐K1 cells in protein‐free medium. J Biotechnol. 2004;110:51‐62. [DOI] [PubMed] [Google Scholar]
- 43. Lees JG, Cliff TS, Gammilonghi A, et al. Oxygen regulates human pluripotent stem cell metabolic flux. Stem Cells Int. 2019;2019:8195614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Renner WA, Jordan M, Eppenberger HM, Leist C. Cell‐cell adhesion and aggregation: influence on the growth behavior of CHO cells. Biotechnol Bioeng. 1993;41:188‐193. [DOI] [PubMed] [Google Scholar]
- 45. Mauritz C, Schwanke K, Reppel M, et al. Generation of functional murine cardiac myocytes from induced pluripotent stem cells. Circulation. 2008;118:507‐517. [DOI] [PubMed] [Google Scholar]
- 46. Lloyd DR, Leelavatcharamas V, Emery AN, Al‐Rubeai M. The role of the cell cycle in determining gene expression and productivity in CHO cells. Cytotechnology. 1999;30:49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sen A, Kallos MS, Behie LA. Effects of hydrodynamics on cultures of mammalian neural stem cell aggregates in suspension bioreactors. Ind Eng Chem Res. 2001;40:5350‐5357. [Google Scholar]
- 48. Koch L, Deiwick A, Franke A, et al. Laser bioprinting of human induced pluripotent stem cells‐the effect of printing and biomaterials on cell survival, pluripotency, and differentiation. Biofabrication. 2018;10:035005. [DOI] [PubMed] [Google Scholar]
- 49. Macey R, Oster G, Zahnley T. Berkeley Madonna User's Guide. Berkeley, California: University of California, Department of Molecular and Cellular Biology; 2009. [Google Scholar]
- 50. Monod J. The Growth of Bacterial Cultures. Annual Review of Microbiology. 1949;3 (1):371–394. 10.1146/annurev.mi.03.100149.002103. [DOI] [Google Scholar]
- 51. Kyriakopoulos S. Kinetic modeling of mammalian cell culture bioprocessing: the quest to advance biomanufacturing. Biotechnol J. 2018;13:e1700229. [DOI] [PubMed] [Google Scholar]
- 52. Jacobs K, Zambelli F, Mertzanidou A, et al. Higher‐density culture in human embryonic stem cells results in DNA damage and genome instability. Stem Cell Rep. 2016;6:330‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laco F, Woo TL, Zhong Q, et al. Unraveling the inconsistencies of cardiac differentiation efficiency induced by the GSK3beta inhibitor CHIR99021 in human pluripotent stem cells. Stem Cell Rep. 2018;10:1851‐1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kern S, Platas‐Barradas O, Portner R, Frahm B. Model‐based strategy for cell culture seed train layout verified at lab scale. Cytotechnology. 2016;68:1019‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu S, Hoshan L, Jiang R, et al. A practical approach in bioreactor scale‐up and process transfer using a combination of constant P/V and vvm as the criterion. Biotechnol Prog. 2017;33:1146‐1159. [DOI] [PubMed] [Google Scholar]
- 56. Metze S, Ruhl S, Greller G, Grimm C, Scholz J. Monitoring online biomass with a capacitance sensor during scale‐up of industrially relevant CHO cell culture fed‐batch processes in single‐use bioreactors. Bioprocess Biosyst Eng. 2020;43:193‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Serra M, Brito C, Correia C, Alves PM. Process engineering of human pluripotent stem cells for clinical application. Trends Biotechnol. 2012;30:350‐359. [DOI] [PubMed] [Google Scholar]
- 58. Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786‐795. [DOI] [PubMed] [Google Scholar]
- 59. Kim HS, Lee GM. Differences in optimal pH and temperature for cell growth and antibody production between two Chinese hamster ovary clones derived from the same parental clone. J Microbiol Biotechnol. 2007;17:712‐720. [PubMed] [Google Scholar]
- 60. Núñez EG, Leme J, De Almeida Parizotto L, et al. Influence of aeration‐homogenization system in stirred tank bioreactors, dissolved oxygen concentration and pH control mode on BHK‐21 cell growth and metabolism. Cytotechnology. 2014;66:605‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wilkens CA, Altamirano C, Gerdtzen ZP. Comparative metabolic analysis of lactate for CHO cells in glucose and galactose. Biotechnol Bioprocess Eng. 2011;16:714‐724. [Google Scholar]
- 62. Cruz HJ, Freitas CM, Alves PM, Moreira JL, Carrondo MJT. Effects of ammonia and lactate on growth, metabolism, and productivity of BHK cells. Enzyme Microb Technol. 2000;27:43‐52. [DOI] [PubMed] [Google Scholar]
- 63. Gupta SK, Srivastava SK, Sharma A, et al. Metabolic engineering of CHO cells for the development of a robust protein production platform. PLoS One. 2017;12:e0181455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ogata M, Wakita KI, Sakano K, et al. Characteristics of CHO‐K1 cell culture producing two types of recombinant soluble thrombomodulin. J Ferment Bioeng. 1993;75:293‐298. [Google Scholar]
- 65. Pan X, Dalm C, Wijffels RH, Martens DE. Metabolic characterization of a CHO cell size increase phase in fed‐batch cultures. Appl Microbiol Biotechnol. 2017;101:8101‐8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nagashima T, Shimizu K, Matsumoto R, Honda H. Selective elimination of human induced pluripotent stem cells using medium with high concentration of L‐alanine. Sci Rep. 2018;8:12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cowan CA, Klimanskaya I, McMahon J, et al. Derivation of embryonic stem‐cell lines from human blastocysts. New Engl J Med. 2004;350:1353‐1356. [DOI] [PubMed] [Google Scholar]
- 68. Park YB, Kim YY, Oh SK, et al. Alterations of proliferative and differentiation potentials of human embryonic stem cells during long‐term culture. Exp Mol Med. 2008;40:98‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu Z, Pu J, Jiang H, et al. Generation of naivetropic induced pluripotent stem cells from Parkinson's disease patients for high‐efficiency genetic manipulation and disease modeling. Stem Cells Dev. 2015;24:2591‐2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen VC, Couture SM, Ye J, et al. Scalable GMP compliant suspension culture system for human ES cells. Stem Cell Res. 2012;8:388‐402. [DOI] [PubMed] [Google Scholar]
- 71. Ben‐Arye T, Levenberg S. Tissue engineering for clean meat production. Front Sustain Food Syst. 2019;3 . [Google Scholar]
- 72. Lachmann N, Ackermann M, Frenzel E, et al. Large‐scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep. 2015;4:282‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Olmer R, Engels L, Usman A, et al. Differentiation of human pluripotent stem cells into functional endothelial cells in scalable suspension culture. Stem Cell Rep. 2018;10:1657‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Correia C, Serra M, Espinha N, et al. Combining hypoxia and bioreactor hydrodynamics boosts induced pluripotent stem cell differentiation towards cardiomyocytes. Stem Cell Rev. 2014;10:786‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fischer B, Meier A, Dehne A, et al. A complete workflow for the differentiation and the dissociation of hiPSC‐derived cardiospheres. Stem Cell Res. 2018;32:65‐72. [DOI] [PubMed] [Google Scholar]
- 76. Stevens KR, Murry CE. Human pluripotent stem cell‐derived engineered tissues: clinical considerations. Cell Stem Cell. 2018;22:294‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Protze SI, Lee JH, Keller GM. Human pluripotent stem cell‐derived cardiovascular cells: from developmental biology to therapeutic applications. Cell Stem Cell. 2019;25:311‐327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.