

Abstract
Neurodegeneration with brain iron accumulation (NBIA) comprises a clinically and genetically heterogeneous group of disorders affecting children and adults. These rare disorders are often first suspected when increased basal ganglia iron is observed on brain magnetic resonance imaging. For the majority of NBIA disorders the genetic basis has been delineated, and clinical testing is available. The four most common NBIA disorders include pantothenate kinase-associated neurodegeneration (PKAN) due to mutations in PANK2, phospholipase A2-associated neurodegeneration caused by mutation in PLA2G6, mitochondrial membrane protein-associated neurodegeneration from mutations in C19orf12, and beta-propeller protein-associated neurodegeneration due to mutations in WDR45. The ultrarare NBIA disorders are caused by mutations in CoASY, ATP13A2, and FA2H (causing CoA synthase protein-associated neurodegeneration, Kufor–Rakeb disease, and fatty acid hydroxylase-associated neurodegeneration, respectively). Together, these genes account for disease in approximately 85% of patients diagnosed with an NBIA disorder. New NBIA genes are being recognized with increasing frequency as a result of whole-exome sequencing, which is also facilitating early ascertainment of patients whose phenotype is often nonspecific.
INTRODUCTION AND OVERVIEW OF THE NBIA DISORDERS
Neurodegeneration with brain iron accumulation (NBIA) comprises a clinically and genetically heterogeneous group of disorders affecting children and adults. The term NBIA encompasses both single-gene disorders and disorders of unknown etiology that share the feature of high levels of iron in the basal ganglia (Hayflick et al., 2003). A common pathophysiology has been inferred for these disorders based on similar pathologic changes that are observed in selectively vulnerable brain regions. This concept has been reinforced through disease gene discovery with the conspicuous intersection of biochemical pathways (e.g., fatty acid metabolism) and cellular processes (e.g., autophagy/mitophagy) that require normal function of the NBIA genes and their protein products. Most of the NBIA disorders are caused by defects in proteins with no known direct role in iron homeostasis. As new genes have been identified, the phenotypic spectrum has expanded. So, despite their differences, there is value in considering these distinct disorders together from both a biologic perspective as well as from the perspective of clinical care.
A diagnosis of NBIA is often suspected based on brain magnetic resonance imaging (MRI) evidence for increased basal ganglia iron in concert with characteristic clinical features. Widespread use of brain MRI as a diagnostic tool and improved MRI techniques have facilitated early diagnosis of an NBIA disorder, even when the clinical features are not specific. For the majority ofNBIA disorders the genetic basis has been delineated, and targeted diagnostic testing is available. The four most common NBIA disorders are delineated in Table 19.1, along with the minor NBIA disorders that affect smaller numbers of families. Together, these genes account for disease in at least 85% of patients diagnosed with NBIA. However, the path to diagnosis is changing with the increasing use of whole-exome sequencing in clinical care: more patients are diagnosed early in life, sometimes in the absence of increased iron on brain imaging. In addition, new NBIA genes are still being discovered largely as a result of massively parallel sequencing technologies.
Table 19.1.
The neurodegeneration with brain iron accumulation disorders
| Gene | Protein | Disorder |
|---|---|---|
| PANK2 | Pantothenate kinase 2 | Pantothenate kinase-associated neurodegeneration (PKAN) |
| PLA2G6 | Calcium-independent phospholipase A2 group VIa = iPLA2VIa | Phospholipase A2-associated neurodegeneration (PLAN), including infantile neuroaxonal dystrophy (INAD) |
| C19orf12 | C19orf12 | Mitochondrial membrane protein-associated neurodegeneration (MPAN) |
| WDR45 | WD40-repeat protein 45 | Beta-propeller protein-associated neurodegeneration (BPAN) |
| CoASY | Coenzyme A synthase | CoA synthase protein-associated neurodegeneration (CoPAN) |
| FA2H | Fatty acid 2 hydroxylase | Fatty acid-2 hydroxylase-associated neurodegeneration (FAHN) |
| ATP13A2 | Cation-transporting ATPase 13A2 | Kufor–Rakeb disease |
| CPL | Ceruloplasmin | Aceruloplasminemia |
| FTL | Ferritin light chain | Neuroferritinopathy |
The NBIA disorders are rare, though accurate prevalence data are not known. Based on prevalence estimates for pantothenate kinase-associated neurodegeneration (PKAN), which accounts for about half of cases, NBIA occurs in about 1 per 500,000 people worldwide. Certain geographic regions have a high frequency of a specific mutant allele accounting for a relatively high prevalence of a specific NBIA disorder in select populations (e.g., PKAN in Dominican Republic).
History, nosology, and nomenclature
The eponym Hallervorden–Spatz syndrome originally referred to the genetic NBIA disorder now known as PKAN and first described in 1922 by Julius Hallervorden and Hugo Spatz. Since then, inconsistent use of this term in the medical literature has complicated the field. As a result, reports that predate the gene discoveries are difficult to interpret within the context of a specific NBIA disorder.
Because of their deplorable roles in studying brains from institutionalized disabled children and adults who were systematically murdered during the Third Reich, Hallervorden and Spatz have been discredited, and use of the disease eponym has been discontinued (Shevell, 1992, 2012; Zhou et al., 2001). Franz Seitelberger, who described infantile neuroaxonal dystrophy (INAD), trained under Hallervorden and benefited scientifically from his association with Nazi programs (Kondziella, 2009). Therefore use of the eponym Seitelberger disease should be discontinued. A disease nomenclature has now been established that references the gene or protein name in order to connect these disorders to their etiology (Hayflick et al., 2003).
NBIA is an overarching term used to encompass all of the individual disorders, including those for which the specific cause is unknown. The NBIA disorders are categorized by their genetic basis. As a result, a single disease term may include different phenotypes (e.g., phospholipase A2-associated neurodegeneration (PLAN)). As each gene has been found, naming of the disease has followed the convention that was established with the discovery of the first NBIA gene, PANK2 (Hayflick et al., 2003). The protein defect is referenced as part of the name (e.g., pantothenate kinase) and then linked to the term “-associated neurodegeneration,” (e.g., pantothenate kinase-associated neurodegeneration). Finally, a short, easily remembered disease acronym is designated (e.g., PKAN). Secondary nomenclatures lack clear purpose, value, or widespread usage (e.g., NBIA1, 2, 3), and they should probably be discontinued.
APPROACH TO THE PATIENT SUSPECTED TO HAVE AN NBIA DISORDER
Diagnostic evaluation
The combination of clinical features and MRI pattern often leads to suspicion of an NBIA disorder. All children with regression in any functional realm should have a neurologic examination, ophthalmologic examination, and brain MRI. If there is evidence of peripheral nerve disease, then further studies to document the specific changes are indicated. In combination with a detailed family history, this dataset usually enables a high suspicion of an NBIA disorder and prioritizing of disease genes for testing.
The clinical and MRI features may lead to testing for a specific diagnosis or to a categoric diagnosis of an NBIA disorder that then warrants sequential testing to determine the specific disorder. The dominant clinical features of each NBIA disorder are described in the following sections along with the characteristic MRI changes. When these data indicate a specific diagnosis, targeted genetic testing is warranted to identify mutations in the suspected causative gene. However, often the clinical features are nonspecific and the MRI changes, while showing basal ganglia iron, are not sufficiently specific to indicate testing of a single gene. In these cases, clinical testing using a panel of NBIA genes will usually provide the most efficient and cost-effective approach. As technology advances, whole-exome sequencing is likely to provide sufficient coverage and depth of read to entirely replace panels and single-gene testing.
Algorithms to guide the clinician through the diagnostic process for an NBIA disorder are of limited utility because of the broad phenotypic spectra and wide disease variability. However, the MRI patterns for each NBIA disorder typically include characteristic features that are diagnostically valuable when present. Table 19.2 describes the major clinical and brain MRI features of each NBIA disorder. Figure 19.1 compares the main MRI findings in the most common NBIA disorders.
Table 19.2.
Clinical and magnetic resonance imaging (MRI) features of the neurodegeneration with brain iron accumulation disorders
| Disorder | Major clinical features | MRI features |
|---|---|---|
| PKAN | Dystonia, parkinsonism, spasticity, pigmentary retinopathy, acanthocytosis, neuropsychiatric features | T2 hyperintense signal surrounded by hypointense signal in globus pallidus, less involvement of substantia nigra, representing the “eye of the tiger” sign |
| PLAN | Psychomotor regression, ataxia, autism, dystonia, parkinsonism, optic atrophy | Iron seen later in disease, affecting globus pallidus and substantia nigra equally, cerebellar atrophy and gliosis, less often cerebral atrophy |
| MPAN | Spasticity, dystonia, dementia, peripheral nerve involvement | Iron affecting globus pallidus and substantia nigra equally, prominent medial medullary lamina streak on T2 sequences |
| BPAN | Intellectual disability, little to no language, mixed seizure types, juvenile parkinsonism, autism | T2 hypointense signal in substantia nigra even greater than globus pallidus, T1 bright “halo” in substantia nigra/cerebellar peduncles, changes may be seen in early childhood |
| CoPAN | Intellectual disability, dystonia, spasticity, behavioral problems | T2 hypointense signal in globus pallidus, less involvement of substantia nigra based on only 2 cases |
| FAHN | Spasticity, ataxia, dystonia, optic atrophy, dementia and seizures | T2 hypointense signal in globus pallidus, diffuse cerebral atrophy, white-matter changes |
| Kufor–Rakeb disease | Juvenile parkinsonism, dementia | Few have MRI evidence of brain iron, but when present, T2 hypointense signal in striatum, with global atrophy |
| Aceruloplasminemia | Adult-onset retinal degeneration, diabetes mellitus, and chorea or dystonia or ataxia | T2 hypointense signal usually in all of the following: dentate nucleus, substantia nigra, globus pallidus, putamen, caudate and thalamus, without cavitary lesions |
| Neuroferritinopathy | Adult-onset chorea or dystonia with subtle cognitive defects | Cavitary lesions plus T2 hypointense signal in one or more of the following: globus pallidus, substantia nigra, putamen, caudate, thalamus, and dentate |
BPAN, beta-propeller protein-associated neurodegeneration; CoPAN, coenzyme A synthase protein-associated neurodegeneration; FAHN, fatty acid-2 hydroxylase-associated neurodegeneration; MPAN, mitochondrial membrane protein-associated neurodegeneration; PKAN, pantothenate kinase-associated neurodegeneration; PLAN, phospholipase A2-associated neurodegeneration.
Fig. 19.1.
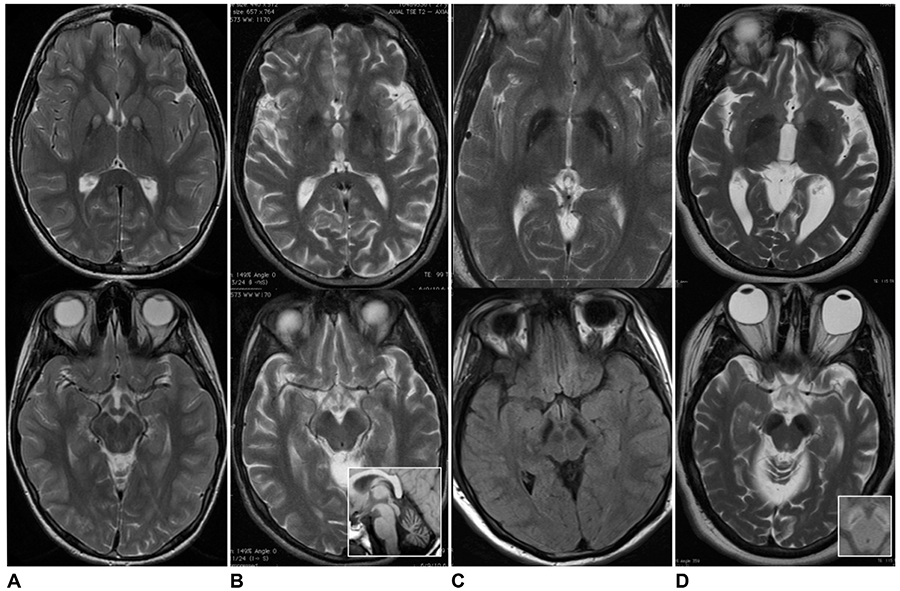
Magnetic resonance imaging of globus pallidus and substantia nigra in four main forms of neurodegeneration with brain iron accumulation. Axial T2-weighted imaging of globus pallidus (top series) and substantia nigra (bottom series) in (A) pantothenate kinase-associated neurodegeneration; (B) phospholipase A2-associated neurodegeneration (inset shows cerebellar atrophy); (C) mitochondrial membrane protein-associated neurodegeneration; and (D) beta-propeller protein-associated neurodegeneration (inset shows T1 hyperintense “halo” in cerebellar peduncles).
Specific MRI sequences for the early detection of iron
The specific sequences that are included in routine MRI studies vary from one institution to another, but they typically do not include sequences that are specifically iron-sensitive. This is true for both adults and children. There are many benefits to adding iron-sensitive sequences to routine MRI studies of some patients, including avoiding the costs of a repeat study, which in children often requires sedation or general anesthetic, and of a prolonged diagnostic odyssey. For children and adults with a movement disorder, extrapyramidal features, or developmental regression, we recommend including iron-sensitive sequences in diagnostic MRI studies of brain. These include gradient echo sequences, susceptibility-weighted imaging, T2*-weighted sequences, and R2* mapping.
A UNIFIED VIEW OF THE NBIA DISORDERS HAS BEEN DRIVEN BY GENETICS
Discovery of the genes underlying the NBIA disorders has enabled significant advances in this field. Beyond providing a diagnostic path and clinical benefits to patients, broader scientific gains have followed from disease gene discovery. New cellular processes and pathways have been identified; selective neuronal and brain region vulnerabilities have been revealed; and our understanding of the pathophysiology of common related neurodegenerative disorders has been enhanced. These achievements would not have been possible without the systematic collection and sharing of biosamples and clinical data from well-phenotyped patients by hundreds of physicians from around the world over the past two decades.
Although the NBIA disorders demonstrate genetic and clinical heterogeneity, they all manifest with degeneration of the globus pallidus and substantia nigra leading to iron accumulation. The vulnerability of these structures occurs despite widespread expression of the NBIA culprit genes, including in brain regions where there is no observed pathology. Globus pallidus and substantia nigra are among the most iron-rich structures in normal brain. This feature may predispose them to secondary iron accumulation; however, other normally iron-rich structures seem not to suffer this result. The basis for the selective vulnerability of globus pallidus and substantia nigra in the NBIA disorders remains unclear.
To date, all of the proteins that are defective in the NBIA disorders are known or suspected to be important for mitochondrial function. In contrast, only two have a known role in iron homeostasis: ferritin light chain and ceruloplasmin. The other NBIA proteins have no known function in iron metabolism, and defects in them seem not to alter systemic iron handling or iron trafficking elsewhere in brain. Although the precise intersection of these pathways remains uncertain, growing attention is being focused on mitochondrial membrane integrity, lipid metabolism, and associated energy and autophagy pathways (Fig. 19.2). As new causative NBIA genes are found, more pieces of this puzzle will be revealed.
Fig. 19.2.

Overview of genes and pathways involved in neurodegeneration with brain iron accumulation (NBIA). Schematic representation of a cell indicating the pathophysiologic mechanisms postulated for NBIA. Nine reported NBIA genes (white box, red outline) and their associated cellular processes are shown. Ceruloplasmin (CP) and ferritin light polypeptide (FTL) are important regulators of cellular iron homeostasis. Phospholipase A2 (PLA2G6) and fatty acid 2-hydroxylase (FA2H) are involved in lipid metabolism and membrane remodeling. Pantothenate kinase 2 (PANK2) and CoA synthase (COASY) are key enzymes in the biosynthesis of coenzyme A (CoA) utilized in a multitude of cellular processes, including the synthesis of fatty acids. WD repeat domain 45 (WDR45) and ATPase type13A2 (ATP13A2) play a role in the degradation process of autophagy. The function of the mitochondrial membrane protein C19orf12 is still unknown, but it is postulated to be associated with fatty acid biogenesis. (Reproduced from Meyer E, Kurian MA, Hayflick SJ (2015) Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genomics Hum Genet 16: 8.1–8.23, with permission.)
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION
General
PKAN is the most common NBIA disorder, accounting for approximately half of NBIA cases. Despite being rare, PKAN is recognizable by its distinctive clinical and radiographic features. No data on prevalence are known; however, estimates are 1 in 1 million (Gregory and Hayflick, 2002, updated 2013).
Clinical features
PKAN may present from early childhood to midadulthood (Hayflick et al., 2003). The age at onset and rate of disease progression generally correlate; early-onset disease portends a more rapid rate of progression, while later-onset disease typically advances in a more insidious pattern. The designation of classic vs. atypical PKAN recognizes that the pediatric phenotype tends to be rather homogeneous while acknowledging that a much broader phenotypic spectrum may occur due to mutations in PANK2. While this binary division serves a purpose, the phenotypes in fact span a continuum.
Classic PKAN
Classic PKAN refers to early-onset, dystonia-dominant disease that is presumably caused by a complete loss of function of the pantothenate kinase 2 protein. This phenotype is fairly consistent across patients; the mean age at onset is 3 years, with the most common presenting feature being gait abnormality and clumsiness due to lower-extremity dystonia. Many children who are eventually diagnosed with classic PKAN have a history of mild global developmental delay and dyspraxia before frank dystonia and spasticity are evident. Seizures are uncommon in PKAN.
Pigmentary retinopathy is common in classic PKAN; however, the limitations from visual impairment typically lag those from impaired motor function caused by dystonia and spasticity. Fundoscopic changes include bone spicule formation, and the pathologic changes reveal loss of photoreceptors, which is also demonstrable by electroretinogram.
While PKAN is considered one of the neuroacanthocytoses, acanthocytes are observed in only a subset of patients. Detection of this erythrocyte morphologic defect is challenging and not part of the routine diagnostic evaluation since genetic testing is highly sensitive and specific and readily available in most developed countries.
Atypical PKAN
Atypical PKAN refers to later-onset, parkinsonism-dominant disease that is presumed to arise as a result of partial loss of function of the pantothenate kinase 2 protein. This functional loss spans a range, so the phenotypic spectrum of atypical PKAN similarly extends from classic disease to adult-onset parkinsonism. Atypical PKAN comprises everything that is not categorized as classic disease.
The early symptoms of atypical PKAN can include neuropsychiatric features, dystonia, or parkinsonism. Speech defects often manifest early in disease, and may include dysarthria, hypophonia, palilalia, or frank stuttering. The later the age of onset, the more likely it is that parkinsonism will dominate the motor phenotype; patients with onset in the second decade usually have mixed dystonia and parkinsonism. Among the neuropsychiatric problems experienced by people with atypical PKAN, the most common are impulsivity and obsessive-compulsive disorder. Some patients eventually diagnosed with PKAN will initially carry a diagnosis of Tourette syndrome or other psychiatric disease.
MRI features
The brain MRI pattern seen in PKAN is distinctive and usually diagnostic (Hayflick et al., 2003). On T2-weighted sequences, the globus pallidus shows a central hyperintense signal region surrounded by hypointense signal. This pattern on coronal section was termed the “eye of the tiger” sign by Sethi et al. (1988). Since that time, the strong association between this specific pattern and the disease has enabled confidence in the diagnosis of PKAN in most patients based on clinical and radiographic data. Clear exceptions have now been well documented, both with other diagnoses leading to a similar pattern on MRI, and with people with a clinical and genetic diagnosis of PKAN lacking this characteristic pattern (Delgado et al., 2012).
Iron-sensitive sequences have improved the MRI diagnosis of PKAN before the classic changes are evident on T2-weighted sequences. Furthermore, it is now clear that pallidal iron levels are increased even in the earliest stages of disease, as demonstrated when these sequences are employed. Involvement of the substantia nigra seems to occur later in disease, with relatively less iron accumulation than is seen in the globus pallidus (Hayflick et al., 2006). In PKAN, the cerebellum is usually normal, in contrast to abnormalities seen in some of the other NBIA disorders.
Genetics
PKAN is an autosomal-recessive disorder caused by defects in the pantothenate kinase 2 protein, which is encoded by the gene PANK2 (Zhou et al., 2001). Only mutations in PANK2 lead to PKAN, and PKAN is the only disorder associated with mutations in PANK2. Two mutant alleles are found in the majority of patients with clinical and radiographic features of PKAN. Very rarely, only one mutation can be found in patients with characteristic features, which likely reflects the current limitations of mutation detection. It is possible that the second PANK2 mutation may be within regions that are not currently sequenced routinely, such as deep intronic or promoter regions. Heterozygous carriers of one PANK2 mutation (i.e., obligate carrier parents and unaffected carrier siblings) are not at increased risk for Parkinson disease or any common neurodegenerative disorder (Hayflick, unpublished data).
Clinical genetic testing is widely available internationally and should include screening for both point mutations and small insertions and deletions. A small number of mutations are common in specific populations, but many different mutations found throughout the PANK2 gene account for the majority of disease.
A clear correlation between phenotype and genotype in PKAN has been elusive. Although nonsense mutations usually lead to classic disease, many missense mutations do as well. In vitro analysis of defective PANK2 protein derived from disease-associated mutations shows subnormal, normal, and supranormal catalytic activity, further challenging genotype–phenotype correlation (Zhang et al., 2006).
Pathophysiology
Many questions remain about how defects in pantothenate kinase 2 lead to this remarkably specific neurodegenerative phenotype. Two other genes encode three additional functional pantothenate kinases in humans. This level of genome redundancy is not surprising for the key regulatory enzyme in the biosynthesis of coenzyme A (CoA), a master regulator of intermediary metabolism.
The current view of the cellular role of PANK2 enzyme, which localizes to the mitochondrial inter-membranous space, is that it serves as a sensor of matrix CoA status (Leonardi et al., 2007). In normal cells, PANK2 is repressed in a state of matrix CoA sufficiency. Therefore in disease, defective or absent PANK2 is predicted to mimic this state, mis-signaling that CoA levels are sufficient and leading to a shutting down of pathways that depend on but lack sufficient quantities of CoA, among them fatty acid oxidation and synthesis.
PKAN pathology is found in brain, retina, erythrocytes, and, provisionally, testis. The dominant brain pathology shows “mummified” neurons, axonal degeneration, and iron accumulation in globus pallidus (Kruer et al., 2011). PKAN is not a synucleinopathy, in contrast to other NBIA disorders, including membrane protein-associated neurodegeneration (MPAN) and PLAN. Tissues in which PANK2 is highly expressed, including many areas of brain, show no abnormalities. How a defect in a ubiquitously expressed gene leads to such a specific phenotype remains an important unanswered question for PKAN. Understanding the basis for the selective vulnerability of large neurons in the globus pallidus will advance our understanding of the pathophysiology of PKAN.
Treatment
Treatment of PKAN is symptomatic at this time (Hogarth, 2015; Hogarth et al., 2017). Several groups are developing rational therapeutics that aim to supply a pathway intermediate as a substitute substrate for CoA synthesis. By bypassing the key regulatory enzyme, this approach would have the potential for flooding the cell with CoA. One of the challenges of this approach is the exquisitely tight regulation under which pantothenate kinase 2 is held, which suggests either that normal cell function requires a delicate titrating of CoA levels or that the cellular “cost” of CoA synthesis is high. As these rational therapeutics are developed and tested, new insights into the disease process, including CoA toxicity, will be revealed.
PHOSPHOLIPASE A2-ASSOCIATED NEURODEGENERATION, INCLUDING INFANTILE NEUROAXONAL DYSTROPHY
General
PLAN comprises a wide phenotypic spectrum that ranges from psychomotor regression beginning in early childhood to dystonia-parkinsonism in adulthood. Both central and peripheral nervous systems are affected, a feature shared with MPAN and beta-propeller protein-associated neurodegeneration (BPAN) but not PKAN. The incidence of PLAN is unknown. The infantile form of this disorder was “lumped” with Hallervorden–Spatz syndrome for many decades based on the common neuropathologic feature of axonal spheroids and the occasional observation of high levels of iron in the basal ganglia.
Clinical features
Infantile-onset PLAN or infantile neuroaxonal dystrophy
Infantile-onset PLAN begins by 6 months to 3 years of age with developmental regression, hypotonia, and spastic tetraparesis (Gregory et al., 2008; Kurian et al., 2008). Visual impairment is common, as are seizures.
The terms ‘infantile neuroaxonal dystrophy’ and ‘INAD’ are entrenched in the medical literature. Use of the term infantile-onset PLAN is gaining favor as the term PLAN, which encompasses the full disease spectrum associated with defective PLA2G6, becomes more commonly used.
Childhood-onset PLAN or atypical neuroaxonal dystrophy
Childhood-onset PLAN encompasses a more slowly progressive psychomotor disorder that is clinically distinct from the infantile- and adult-onset forms of PLAN. These children may initially carry a diagnosis of autism, eventually developing motor features, including ataxia, dystonia, and spasticity. Most have some degree of intellectual disability, as well. In some, iron is seen on brain MRI and can help lead to the diagnosis. For many, the clinical features are nonspecific, and often the diagnosis is delayed in these children. Many have been diagnosed by whole-exome sequencing.
Adult-onset PLAN or dystonia-parkinsonism
The serendipitous finding of mutations in PLA2G6 in individuals with dystonia-parkinsonism led to a marked broadening of the phenotypic spectrum of PLAN (Paisan-Ruiz et al., 2009). These people typically have mild intellectual impairment, with onset in adolescence or early adulthood of dystonia and parkinsonism. At the time of presentation, some patients lack basal ganglia iron accumulation; however, eventually nearly all of these patients do manifest this characteristic as disease progresses.
MRI features
In PLAN, brain iron may accumulate only later in disease regardless of the phenotype (Gregory et al., 2008; Kurian et al., 2008). Therefore, the absence of basal ganglia iron should not lessen clinical suspicion of PLAN. Early MRI features may include cerebellar atrophy often with gliosis, and, less commonly, cerebral atrophy. Optic atrophy may be evident on brain MRI. When iron accumulates in PLAN, it affects both the globus pallidus and the substantia nigra more or less equally and is visualized as hypointense signal on T2-weighted sequences. As with all of the NBIA disorders, iron-sensitive sequences will enable the recognition of this feature earlier in disease.
Genetics
By definition, all cases of PLAN are associated with mutations in PLA2G6 (Morgan et al., 2006). PLAN is autosomal recessive, and patients harbor two mutations. Diagnostic testing includes both sequencing and screening for deletions, insertions, and duplications. Loose genotype–phenotype correlation is observed for PLAN. Individuals with two null alleles of PLA2G6 or a pathogenic variant that impairs catalytic activity of the protein have infantile-onset PLAN. Most people with childhood- or adult-onset disease have missense mutations that do not impair catalytic activity (Engel et al., 2010).
As whole-exome sequencing has become more common in clinical care, many more cases of PLAN have been recognized. The nonspecific early features can make this a challenging diagnosis to suspect. Although there are 18 genes that encode phospholipase proteins, the only human disease associated with defects in any of these genes is PLAN.
Pathophysiology and treatment
This gene encodes a phospholipase A2 enzyme that plays a crucial role in mitochondrial membrane homeostasis. Precisely how this defect leads to the observed pathology is unknown. PLAN is a synucleinopathy with extensive pathologic changes found throughout the central and peripheral nervous systems (Gregory et al., 2008).
Treatment is largely symptomatic in PLAN (Hogarth, 2015). There is some basis to recommend docosahexanoic acid supplements (Rapoport et al., 2011; Cheon et al., 2012), especially given its low toxicity. Gene transfer experiments are underway in a mouse model of INAD to determine whether delivery of functional PLA2G6 to the neonatal brain can prevent or rescue the disease phenotype.
MITOCHONDRIAL MEMBRANE PROTEIN-ASSOCIATED NEURODEGENERATION
Clinical features
This NBIA phenotype was first recognized and described in an Eastern European cohort (Hartig et al., 2011). The features usually begin in childhood or early adulthood and include movement abnormalities, behavioral disturbances, and dementia (Hartig et al., 2011; Hogarth et al., 2013). Dystonia, spasticity, and parkinsonism are common in early disease, as are neuropsychiatric problems, including emotional lability, depression, anxiety, compulsions, and impulsivity. Hallucinations can occur later in the disease. Additional early findings can include cognitive impairment, optic atrophy, motor axonal neuropathy, and bowel/bladder incontinence. Late-onset disease beginning in the third or fourth decade is dominated by neuropsychiatric disturbances, including psychosis, and dementia. This later-onset form of MPAN may follow an aggressive and accelerated rate of progression leading to death usually within 5–10 years (Dogu et al., 2013; Hogarth et al., 2013).
MRI features
MRI evidence of increased basal ganglia iron is usually present early in disease, although rare cases have been reported without brain iron accumulation (Landoure et al., 2013). T2-weighted sequences are characterized by hypointense signal in the globus pallidus and substantia nigra. An MRI feature that can help to distinguish MPAN is T2-hyperintense linear streaking of the medial medullary lamina between the globus pallidus externus and internus (Hogarth et al., 2013).
Genetics
MPAN is caused by mutations in C19orf12, which encodes a mitochondrial membrane protein of unknown function (Hartig et al., 2011). MPAN is an autosomal-recessive disorder. A common Eastern European founder deletion mutation accounts for most cases from that region.
Although the majority of MPAN patients inherit two C19orf12 mutations, one from each parent, possible autosomal-dominant inheritance has been reported (Hogarth et al., 2013). In addition, there are more singletons with an MPAN phenotype and only one identifiable deleterious mutation than is typically observed in rare recessive disorders. Genetic studies looking for deletions, duplications, and insertions have revealed none. These observations have yet to be explained.
Pathophysiology
The function of C19orf12 protein remains unknown, and therefore advances in understanding the MPAN disease process have been mainly descriptive. Putative roles for this mitochondrial membrane protein are in fatty acid biosynthesis and branched-chain amino acid degradation (Hartig et al., 2011). Postmortem brain tissue from affected individuals shows abundant α-synuclein-positive staining and Lewy body formation, in addition to the common NBIA features of iron and axonal spheroids (Hartig et al., 2011). Animal models of MPAN and discovery of the crystal structure of this novel protein will help illuminate its role in the cell.
BETA-PROPELLER PROTEIN-ASSOCIATED NEURODEGENERATION
Clinical features
The early phenotype of BPAN includes neurodevelopmental delay in infancy and childhood, intellectual disability with seizures, and sleep problems as common comorbidities (Haack et al., 2012; Hayflick et al., 2013). Language skills are limited, and expressive language may never be acquired. Midline hand stereotypies are common in BPAN. In time, patients develop a movement disorder, most commonly dystonia or parkinsonism, beginning in adolescence or adulthood. Many children who are eventually diagnosed with BPAN are initially considered to have atypical Rett syndrome or infantile epileptic encephalopathy (Hayflick et al., 2013).
MRI features
The MRI in BPAN may be normal in early childhood, but by the time the movement disorder manifests it shows clear abnormalities that are quite specific. The characteristic changes include T2-weighted hypointense signal in the substantia nigra and globus pallidus, with nigral dominance as a result of iron accumulation. In addition, a unique pattern is observed on T1-weighted sequences of a hyperintense “halo” in the substantia nigra and cerebral peduncles (Hayflick et al., 2013).
Genetics
BPAN is the only NBIA disorder that follows an X-linked dominant pattern of inheritance. Mutations in WDR45, encoding a putative autophagy protein with the structure of a so-called beta-propeller, lead to BPAN (Haack et al., 2012). To date, most cases arise as a result of de novo mutations, and some males have been shown to have evidence for somatic mosaicism, presumably arising from a postzygotic mutation. The occurrence of BPAN in males is not rare, and their phenotype is generally similar to that of females. As whole-exome sequencing has become more common in clinical care, many early cases of BPAN are being recognized, and the spectrum of clinical features is expanding. Several familial cases of BPAN are now documented, as are cases of female monozygotic twins who are discordant for phenotype. Skewing of X-inactivation has been proposed as a mechanism to account for discordant phenotypes, and evidence to support this hypothesis has been published (Haack et al., 2012).
Pathophysiology
The function of WDR45 is currently unknown. It binds or associates with known autophagy proteins, supporting its involvement in recycling of cellular components (Haack et al., 2012; Saitsu et al., 2013). The protein structure suggests a role in regulating multiprotein complexes by orchestrating critical and specific interactions between docking molecules that utilize WDR45. In females, a skewed methylation pattern of the X chromosome might contribute to disease pathogenesis. Studies are needed to determine whether the mutated allele is preferentially inactivated and whether patterns in peripheral blood reflect those in the brain in this neurodegenerative disorder.
COENZYME A SYNTHASE PROTEIN-ASSOCIATED NEURODEGENERATION (CoPAN)
Clinical features
Based on a report of two unrelated Italian patients with CoPAN, this progressive disorder includes childhood-onset dystonia and spasticity with cognitive impairment (Dusi et al., 2014). Later features include parkinsonism, obsessive-compulsive disorder, and peripheral neuropathy.
MRI features
The MRI pattern in CoPAN demonstrates hypointense signal in the globus pallidus and substantia nigra. One patient also had bilateral foci of high signal intensity in the pallidi on T2-weighted imaging. In addition, there was also evidence for swelling of the caudate and putamen in one subject (Dusi et al., 2014).
Genetics
CoPAN is an autosomal-recessive disorder caused by mutations in the gene COASY, encoding CoA synthase (Dusi et al., 2014). This bifunctional protein performs the final two steps in the synthesis of CoA from pantothenate. Though only two families have been reported with CoPAN, this disorder represents an important addition because it further focuses attention on the CoA biosynthetic pathway as essential to the function of neurons of the globus pallidus.
FATTY ACID-2 HYDROXYLASE-ASSOCIATED NEURODEGENERATION (FAHN)
Clinical features
Defects in the gene FA2H were first associated with hereditary spastic paraplegia (SPG35). Since then, mutations in this gene have been associated with a movement disorder and brain iron accumulation to cause a phenotype referred to as FAHN (Kruer et al., 2010). This NBIA disorder has considerable phenotypic overlap with allelic disorders, forming a continuum. The dominant features include spasticity, ataxia, dystonia, and optic atrophy, with progressive intellectual impairment and seizures later in disease.
MRI features
In many children affected with FAHN, brain iron is evident by mid to late childhood. Most individuals with mutations in FA2H eventually develop increased basal ganglia iron. Iron is increased primarily in the globus pallidus, which is observed as hypointense signal on T2-weighted sequences. Other MRI features include white-matter changes and progressive cerebellar and mid-brain atrophy.
Genetics
Mutations in FA2H lead to a range of phenotypes that are now subsumed under the acronym FAHN (Kruer et al., 2010). All are autosomal-recessive disorders caused by two mutations in FA2H. HSP35 was previously considered allelic but now is recognized to be part of FAHN, since many of the original patients eventually developed MRI evidence for increased basal ganglia iron. Single-gene clinical testing is available, as is an NBIA panel of genes. No clear genotype–phenotype correlations are known.
KUFOR–RAKEB DISEASE
Clinical features
Kufor–Rakeb disease is more commonly referred to as PARK9-linked parkinsonism (Ramirez et al., 2006), and it has also been considered a form of ceroid lipofuscinosis. This juvenile form of parkinsonism includes spasticity, dementia, perioral myokymia, and supranuclear gaze palsy. In rare cases, iron accumulates in the basal ganglia, and this had led to its proposed inclusion as an NBIA disorder (Schneider et al., 2010). Most people affected by this autosomal-recessive form of parkinsonism do not accumulate iron in their brain. Peripheral nerve complications are common.
MRI features
When present, iron is evident in the striatum and globus pallidus. Diffuse cortical, subcortical, and spinal cord atrophy are commonly seen as disease progresses.
Genetics
The vast majority of patients with mutations in ATP13A2 have juvenile parkinsonism and do not manifest brain iron accumulation. A few cases in the literature demonstrate basal ganglia iron accumulation. Among cohorts ascertained by high brain iron, mutations are rare (Kruer et al., 2012). Clinical testing is available.
NBIA DISORDERS ASSOCIATED WITH DEFECTS IN IRON HOMEOSTATIC PROTEINS – ACERULOPLASMINEMIA AND NEUROFERRITINOPATHY
Most NBIA disorders are caused by defects in proteins with no known role in iron handling. These include PKAN, PLAN, MPAN, BPAN, Kufor–Rakeb disease, and FAHN, and they lead to iron accumulation, specifically in the globus pallidus and substantia nigra. In contrast, two NBIA disorders, aceruloplasminemia and neuroferritinopathy, are associated with defects in genes that encode iron homeostatic proteins, ceruloplasmin, and ferritin respectively. These disorders are radiographically distinct from the other NBIA disorders largely by the more extensive accumulation of iron in brain involving regions that are normally iron-rich, including caudate, putamen, thalamus, dentate, and red nucleus, in addition to the globus pallidus and substantia nigra.
ACERULOPLASMINEMIA
Clinical features
In aceruloplasminemia iron accumulates in brain and viscera leading to a neurodegenerative disorder as well as retinopathy and diabetes mellitus. The neurologic disease is mainly a movement disorder, including chorea and ataxia, with psychiatric and cognitive changes, as well. Onset is typically in early to mid-adulthood.
MRI features
Aceruloplasminemia and neuroferritinopathy affect many of the normally iron-rich brain structures, a feature that distinguishes them from the other NBIA disorders. These include the globus pallidus, substantia nigra, striatum, red nucleus, thalamus, and dentate. Iron accumulation in liver can also be visualized by MRI.
Genetics
Defects in the gene CP encoding ceruloplasmin are responsible for this autosomal-recessive disorder. Clinical testing is essential in order to establish the diagnosis, which is suspected based on the systemic and neurologic disease with low or absent serum ceruloplasmin levels often accompanied by changes in iron and copper levels. Ceruloplasmin functions as a ferroxidase.
NEUROFERRITINOPATHY
Clinical features
Neurologic features dominate the clinical presentation of neuroferritinopathy, including adult-onset chorea and dystonia and reflecting the brain regions in which iron accumulates. In addition to a movement disorder, patients often manifest cognitive and behavioral defects. Serum ferritin levels may be low, but clinical suspicion should lead to MRI evaluation and genetic testing regardless.
MRI features
Cavitary basal ganglia lesions distinguish neuroferritinopathy from other NBIA disorders. These may be a later feature and are preceded by iron accumulation in the normally iron-rich brain regions, including globus pallidus, striatum, thalamus, dentate, and substantia nigra. A combination of T2 hyperintense and hypointense signal in the basal ganglia with cortical atrophy also helps distinguish this from other NBIA disorders.
Genetics
Mutations in FTL, encoding ferritin light chain, cause neuroferritinopathy, which follows an autosomal-dominant pattern of inheritance with complete penetrance. Most mutations are found in exon 4, including the most common, a single-nucleotide insertion. An allelic but clinically distinct disorder, hyperferritinemia-cataract syndrome, is caused by heterozygous point mutations in the iron response element in the 5-prime noncoding region of FTL. There are no neurologic features in this allelic disorder.
IDIOPATHIC NBIA
The etiology has not yet been identified for a significant portion of people with an NBIA disorder. In different NBIA research cohorts, this group ranges in size from approximately 5% to 15%. However, research cohorts commonly become enriched over time for patients with idiopathic disease and lead to an overestimation of prevalence. Moreover, as new NBIA genes are discovered, the size of this group decreases.
Patients are considered to have an idiopathic NBIA disorder when they manifest a progressive neurologic disorder and their MRI reveals increased iron in the basal ganglia. Since this represents a heterogeneous group by definition, the clinical and radiographic features vary widely. As iron-sensitive MRI sequences and genomewide interrogation methods become more widely used in clinical practice, many of the remaining genetic etiologies will be found. Some will likely result from multi-genic, somatic, or epigenetic variants that will require different testing methods. Nongenetic etiologies accounting for rare NBIA disorders are likely to be identified, as well.
CONCLUSIONS AND FUTURE DEVELOPMENTS
Over the past two decades, the genetic bases of nearly all of the NBIA disorders have been elucidated. Based on their overlapping phenotypes and pathologies, there was reason to expect their disease genes to reveal new pathways critical for neuronal health. Indeed, the intersecting pathways implicate mitochondrial bioenergetics, lipid metabolism, and autophagy/mitophagy as common processes underlying the NBIA disorders. Specific defects in these pathways damage selectively vulnerable cells in the basal ganglia and lead to iron dyshomeostasis.
Despite these rapid genetic advances, there are still no curative or disease-modifying therapies for any of the NBIA disorders. However, rational therapeutics are being developed for several of the NBIA disorders. These efforts include small-molecule product replacement and gene transfer approaches. While iron chelation may provide clinical benefit as an adjunct treatment, its therapeutic value is uncertain. As effective therapeutics advance, they will drive the development of tools for early diagnosis and biomarkers for disease monitoring.
The impact of genetics on the NBIA field is difficult to overstate. Disease gene discovery forms the foundation of knowledge about the biology of all NBIA disorders. Each new gene discovery accelerates the next. Gene transfer and other gene-based therapies hold great promise for the NBIA disorders, and we will ultimately rely on early genetic screening to ascertain the people who will benefit from such treatments. While the era of disease gene discovery may largely be over, genetics will impact the NBIA field for many years to come.
References
- Cheon Y, Kim HW, Igarashi M et al. (2012). Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A (2)-VIA (iPLA (2) beta)-knockout mice. Biochim Biophys Acta 1821: 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado RF, Sanchez PR, Speckter H et al. (2012). Missense PANK2 mutation without “eye of the tiger” sign: MR findings in a large group of patients with pantothenate kinase-associated neurodegeneration (PKAN). J Magn Reson Imaging 35: 788–794. [DOI] [PubMed] [Google Scholar]
- Dogu O, Krebs CE, Kaleagasi H et al. (2013). Rapid disease progression in adult-onset mitochondrial membrane protein-associated neurodegeneration. Clin Genet 84: 350–355. [DOI] [PubMed] [Google Scholar]
- Dusi S, Valletta L, Haack TB et al. (2014). Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 94: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel LA, Jing Z, O’Brien DE et al. (2010). Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 5: e12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A, Hayflick SJ (2002) (updated 2013). Pantothenate kinase-associated neurodegeneration. In: Pagon RA, Adam MP, Ardinger HH et al. (Eds.), GeneReviews (R). University of Washington, Seattle, Seattle, WA. [PubMed] [Google Scholar]
- Gregory A, Westaway SK, Holm IE et al. (2008). Neurodegeneration associated with genetic defects in phospholipase A (2). Neurology 71: 1402–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Hogarth P, Kruer MC et al. (2012). Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 91: 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallervorden J, Spatz H (1922). Eigenartige Erkrankung im extrapyramidalen System mit besonderer Beteiligung des Globus pallidus und der Substantia nigra. Z Ges Neurol Psychiatr 79: 254–302. [Google Scholar]
- Hartig MB, Iuso A, Haack T et al. (2011). Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet 89: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick SJ, Westaway SK, Levinson B et al. (2003). Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348: 33–40. [DOI] [PubMed] [Google Scholar]
- Hayflick SJ, Hartman M, Coryell J et al. (2006). Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. AJNR Am J Neuroradiol 27: 1230–1233. [PMC free article] [PubMed] [Google Scholar]
- Hayflick SJ, Kruer MC, Gregory A et al. (2013). beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 136: 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P (2015). Overview of the NBIA disorders. Mov Disord 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P, Gregory A, Kruer MC et al. (2013). New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 80: 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P, Kurian MA, Gregory A et al. (2017). Consensus Clinical Management Guideline for Pantothenate Kinase-Associated Neurodegeneration (PKAN). Mol Genet Metab 120 (3): 278–287. 10.1016/j.ymgme.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Kondziella D (2009). Thirty neurological eponyms associated with the Nazi era. Eur Neurol 62: 56–64. [DOI] [PubMed] [Google Scholar]
- Kruer MC, Paisan-Ruiz C, Boddaert N et al. (2010). Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 68: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer MC, Hiken M, Gregory A et al. (2011). Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 134: 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer MC, Paudel R, Wagoner W et al. (2012). Analysis of ATP13A2 in large neurodegeneration with brain iron accumulation (NBIA) and dystonia-parkinsonism cohorts. Neurosci Lett 523: 35–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian MA, Morgan NV, Macpherson L et al. (2008). Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70: 1623–1629. [DOI] [PubMed] [Google Scholar]
- Landoure G, Zhu PP, Lourenco CM et al. (2013). Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum Mutat 34: 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R, Rock CO, Jackowski S et al. (2007). Activation of human mitochondrial pantothenate kinase 2 by palmitoylcarnitine. Proc Natl Acad Sci U S A 104: 1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE et al. (2006). PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38: 752–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A et al. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65: 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38: 1184–1191. [DOI] [PubMed] [Google Scholar]
- Rapoport SI, Ramadan E, Basselin M (2011). Docosahexaenoic acid (DHA) incorporation into the brain from plasma, as an in vivo biomarker of brain DHA metabolism and neurotransmission. Prostaglandins Other Lipid Mediat 96: 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitsu H, Nishimura T, Muramatsu K et al. (2013). De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 45 (445–9): 449e1. [DOI] [PubMed] [Google Scholar]
- Schneider SA, Paisan-Ruiz C, Quinn NP et al. (2010). ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 25: 979–984. [DOI] [PubMed] [Google Scholar]
- Sethi KD, Adams RJ, Loring DW et al. (1988). Hallervorden-Spatz syndrome: clinical and magnetic resonance imaging correlations. Ann Neurol 24: 692–694. [DOI] [PubMed] [Google Scholar]
- Shevell M (1992). Racial hygiene, active euthanasia, and Julius Hallervorden. Neurology 42: 2214–2219. [DOI] [PubMed] [Google Scholar]
- Shevell M (2012). The declining use of the Hallervorden-Spatz eponym. J Child Neurol 27: 1308–1309. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Rock CO, Jackowski S (2006). Biochemical properties of human pantothenate kinase 2 isoforms and mutations linked to pantothenate kinase-associated neurodegeneration. J Biol Chem 281: 107–114. [DOI] [PubMed] [Google Scholar]
- Zhou B, Westaway SK, Levinson B et al. (2001). A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28: 345–349. [DOI] [PubMed] [Google Scholar]