

Abstract
Background
Genetic disposition is considered critical for identifying subjects at high risk for disease development. Investigating disease-causing and high and low expressed genes can support finding the root causes of uncertainties in patient care. However, independent and timely high-throughput next-generation sequencing data analysis is still a challenge for non-computational biologists and geneticists.
Results
In this manuscript, we present a findable, accessible, interactive, and reusable (FAIR) bioinformatics platform, i.e., GVViZ (visualizing genes with disease-causing variants). GVViZ is a user-friendly, cross-platform, and database application for RNA-seq-driven variable and complex gene-disease data annotation and expression analysis with a dynamic heat map visualization. GVViZ has the potential to find patterns across millions of features and extract actionable information, which can support the early detection of complex disorders and the development of new therapies for personalized patient care. The execution of GVViZ is based on a set of simple instructions that users without a computational background can follow to design and perform customized data analysis. It can assimilate patients’ transcriptomics data with the public, proprietary, and our in-house developed gene-disease databases to query, easily explore, and access information on gene annotation and classified disease phenotypes with greater visibility and customization. To test its performance and understand the clinical and scientific impact of GVViZ, we present GVViZ analysis for different chronic diseases and conditions, including Alzheimer’s disease, arthritis, asthma, diabetes mellitus, heart failure, hypertension, obesity, osteoporosis, and multiple cancer disorders. The results are visualized using GVViZ and can be exported as image (PNF/TIFF) and text (CSV) files that include gene names, Ensembl (ENSG) IDs, quantified abundances, expressed transcript lengths, and annotated oncology and non-oncology diseases.
Conclusions
We emphasize that automated and interactive visualization should be an indispensable component of modern RNA-seq analysis, which is currently not the case. However, experts in clinics and researchers in life sciences can use GVViZ to visualize and interpret the transcriptomics data, making it a powerful tool to study the dynamics of gene expression and regulation. Furthermore, with successful deployment in clinical settings, GVViZ has the potential to enable high-throughput correlations between patient diagnoses based on clinical and transcriptomics data.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40246-021-00336-1.
Keywords: Annotation, Disease, Gene, Expression, Heat map, RNA-seq
Introduction
Over the past few years, genomic sequencing technologies have improved the clinical diagnosis of genetic disorders and continue to expand the potential of basic sciences in developing insights into human genetic variations and their biological consequences. Gene expression analysis is a widely adopted method to identify abnormalities in normal function and physiologic regulation [1]. It supports expression profiling and transcriptomic analyses to identify, measure, and compare genes and transcripts in multiple conditions and in different tissues and individuals. Several recently published studies have shown that gene expression analysis is a proven method for understanding and discovering novel and sensitive biomarkers among several complex disorders. Two major techniques that are currently being used for gene expression analysis are microarrays and RNA sequencing (RNA-seq) [2]. Microarrays are based on traditional microarray platforms for transcriptional profiling that quantify a set of predetermined whole transcriptome sequences [3], while RNA-seq identifies, characterizes, and quantifies differentially modulated transcriptomes [4]. Due to recent advancements in next-generation sequencing (NGS) technologies and the development of new bioinformatics applications, RNA-seq has become the most widely used method for gene expression analysis [5].
Several RNA-seq data pre-processing pipelines have been developed and published and are freely available [4]. Most of the pipelines follow a similar workflow, which starts with quality checking the sequences, trimming barcodes, sorting sequences, removing duplicates, aligning to reference genome and transcriptome, and calculating different metrics. RNA by expectation maximization (RSEM) is a widely applied and proven algorithm for the quantification and identification of differentially expressed genes (DEGs) that aligns sequences to reference de novo transcriptome assemblies [6]. Its outcomes include quantified gene and isoform abundances with transcripts per million (TPM), fragments per kilobase million (FPKM), reads per kilobase of transcript per million mapped reads (RPKM), and mean expressed transcript lengths. These values are mainly used in case-control studies and gene expression analysis, which requiring bioinformatics expertise to understand the processed RNA-seq data complexities, and computational methods and programming languages to interpret, visualize, and report produced analytic results.
Data visualization is considered essential for RNA-seq interpretation, as it bridges the gap between algorithmic approaches and the cognitive skills of users and investigators. Over the past decade, different data visualization tools have emerged. Some are available as commercial packages (e.g., Tableau, Heatmap.me, Hotja, Crazy Egg, Inspectlet), and others include academic open-source code applications (e.g., BEAVR, NOJAH, Heatmap3, Clustergrammer). However, based on our evaluation, most of these tools are slow; sometimes unable to render large RNA-seq datasets; downloadable, but difficult to install and configure; available only with manual data uploading and management; not freely available and require subscriptions (commercial only); and, lastly, not user-friendly but require good knowledge of programming languages and computational skill sets.
Independent and timely high-throughput NGS data analysis is still a challenge for non-computational biologists and geneticists. In this study, we are focused on supporting RNA-seq-driven gene expression data analysis, annotation with relevant diseases, and heat map visualization without requiring a strong computational background from the user. We present GVViZ (visualizing genes with disease-causing variants), a newly developed bioinformatics application for gene-disease data visualization, annotation, and expression analysis with a dynamic heat map visualization. GVViZ is a findable, accessible, interactive, and reusable (FAIR) platform, based on a set of seven simple instructions that will allow users without computational experience (e.g., bench scientists, non-computational biologists, and geneticists) to analyze data, visualize data, and export data to share results.
Material and methods
GVViZ is a robust bioinformatics, user-friendly, cross-platform, desktop, and database application. Figure 1 explains the workflow and Fig. 2 demonstrates the graphical user interface (GUI) of GVViZ, which includes (1) database connection, (2) data selection, (3) gene selection, (4) querying database, (5) heat map customization, (6) heat map visualization, and (7) exporting of results. The database connection step establishes a link to the SQL server using authenticated user credentials. Data selection allows the user to select among gene types, expression values, and samples. Gene selection offers features to search and select genes and associated diseases for the analysis. Querying database triggers annotation and gene expression analysis based on selected abundances and pruning conditions, samples, genes, and diseases. Heat map visualization provides features to customize and render heat maps, and exporting results allows the user to save outcomes as image (PNG/TIFF) or text (CSV) files.
Fig. 1.

GVViZ workflow design. Overall tasks include the following: (1) database connection, (2) data selection, (3) gene selection, (4) querying database, (5) heat map customization, (6) heat map visualization, and (7) exporting results
Fig. 2.

GVViZ graphical user interface. Sequence of screenshots explaining the overall interactive interfaces
GVViZ simplifies the process of gene expression data analysis, visualization, and exploration of results by using an SQL database, which is divided into two relations: gene expression data and gene-disease annotation. The results of the RNA-seq data processing pipeline (Fig. 3) are automatically parsed and uploaded into gene expression data, which includes TPM, FPKM, expressed transcript lengths, and counts for each of the samples. TPM values are proven to be more accurate measures of the true abundance of RNA molecules from given genes, and TPM counts are more consistent across libraries. Therefore, they potentially allow a more stable statistical analysis. The goal is to perform a gene expression study, where the gene-level TPM estimates, representing the overall transcriptional output of each gene, are compared between conditions.
Fig. 3.
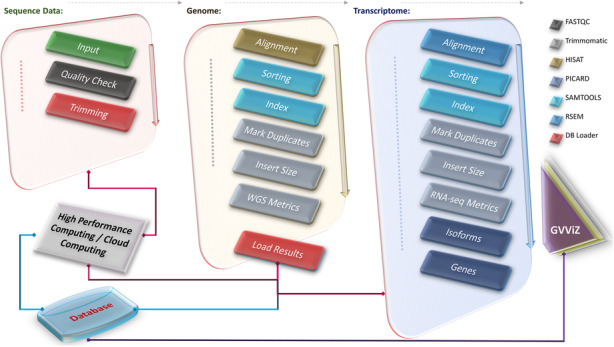
RNA-seq data processing pipeline. We use FastQC for quality checking, Trimmomatic to remove adapters and low-quality sequences, SAMtools to sort and index sequences, MarkDuplicates to remove duplicates, CollectInsertSizeMetrics to compute the size distribution and read orientation of paired-end libraries, HISAT with Bowtie2 to align the sequences to the human reference genome, and RSEM to quantify and identify differentially expressed genes by aligning reads to reference de novo transcriptome assemblies. Furthermore, our RNA-seq pipeline utilizes an in-house developed software application to automatically parse the outcome files of the pipelines and upload the results into a modeled relational database, which are then used by GVViZ for data annotation, analysis, and visualization
The gene-disease annotation relation is populated with published literature-based annotated gene-disease data, collected from different clinical and genomics databases [7], and categorized among gene name/ID, Ensembl ID, category (protein-coding or non-protein-coding, both protein-coding and non-protein-coding, or all available genes in processed RNA-seq samples), and relevant disease (Fig. 1). Our RNA-seq pipeline (Fig. 3) implements FastQC for quality checking [8], Trimmomatic to remove adapters and low-quality sequences [9], SAMtools to sort and index sequences [10], MarkDuplicates to remove duplicates [11], CollectInsertSizeMetrics to compute the size distribution and read orientation of paired-end libraries, and HISAT with Bowtie2 to align the sequences to the human reference genome (hg38) [12, 13]. RSEM is then applied for quantification and identification of differentially expressed genes by aligning the reads to reference de novo transcriptome assemblies [6]. As the final step, our RNA-seq pipeline utilizes an in-house developed scientific software application to efficiently extract and parse information from most of the outcome files (e.g., QC metrics, genes, isoforms) and transfer and load into a modeled relational database. The results based on genes are automatically linked to GVViZ for further annotation, expression analysis, and heat map visualization. Our RNA-seq pipeline starts with mainly the input of short read-based FASTQ files, preferably produced by the Illumina sequencing technology. We recommend the use of paired-end reads, but our pipeline can also work at single-end reads. Users can customize this pipeline based on their needs and can start from any point they would, e.g., instead of starting with FASTQ files, if they have already created SAM/BAM files, they can start directly using HISAT with Bowtie2, and RSEM.
Once GVViZ is successfully connected to the SQL database server, it allows users to design the analysis, select single and multiple sample cohorts, and customize visualization. GVViZ provides SQL-based features to search and select genes and their associated diseases to support gene-disease data annotation. Next, users can select the appropriate gene category (coding, non-coding, both, or all available in samples used for analysis) for the designated analysis. Users need to define the criteria by choosing the right abundance type, setting desired minimum and maximum values, and selecting applicable analytic conditions. Users can select control and diseased samples from the main cohort by picking individual samples and define the range among one or multiple cohorts. GVViZ provides features to customize data visualization, which include titles (header, right y-axis, left y-axis, and x-axis), color schemes (28 gradients), selection and positioning of values (right y-axis and left y-axis), and rendering of heat maps. Finally, users can visualize the results within the GVViZ data visualization panel, as well as export in image (TIFF and PNG) and text (CSV) formats (Fig. 2).
To advance our clinical genomics and precision medicine study, we modeled and implemented an annotated disease-gene-variants database that includes but is not limited to data collected from several genomics databases worldwide [7], including PAS [14, 15], ClinVar [16], GeneCards [17], MalaCard [18], DISEASES [19], HGMD [20], Disease Ontology [21], DiseaseEnhancer [22], DisGeNET [23], eDGAR [24], GTR [25], OMIM [26], miR2Disease [27], DNetDB [28], GTR, CNVD, Ensembl, GenCode, Novoseek, Swiss-Prot, LncRNADisease, Orphanet, and Catalogue Of Somatic Mutations In Cancer (COSMIC) [29]. Our gene datasets consist of 59,293 total genes (19,989 are protein-coding and 39,304 are non-protein-coding) and over 200,000 gene-disease combinations. We have integrated this high-volume and diverse database with GVViZ to support variable and complex gene-disease annotation, visualization, and expression analysis.
GVViZ is based on a product-line architecture, which means each module performs its task independently and its output is used as an input for the next module until the analysis outcome is achieved. It is a multi-platform software application programmed in JAVA, designed following the software engineering principles and the “Butterfly” paradigm [30]. GVViZ has been well tested and can be executed in Microsoft Windows, Linux, Unix, and MacOS operating systems. Along with user guidelines, further GVViZ database design and software development details are available in supplementary material 1.
Results
The performance of GVViZ has been tested and validated in-house with multiple experimental analyses. Here, we report gene-disease annotation, expression mapping, and heat map visualization of different chronic diseases and conditions. We have integrated an annotated gene-disease database with a lab-generated dataset of randomly collected 31 RNA-seq samples. These patients were randomly selected for sequencing to explore variability in expression between individuals. We classified our analysis with an expression cutoff of 100 TPM for any gene in a single sample and mapped them to the physiological conditions: Alzheimer’s disease, arthritis, asthma, diabetes mellitus, obesity, osteoporosis, heart failure, hypertension, and multiple cancer disorders (Fig. 4). The annotation and expression analysis performed by GVViZ produced results linking expression genes to more than one chronic diseases, which included 34 genes linked to Alzheimer’s disease, 51 genes to arthritis, 32 genes to asthma, 43 genes to diabetes mellitus, 2 genes to obesity, 9 genes to osteoporosis, 2 genes to heart failure, and 20 genes to hypertension, and 184 genes were found to be associated to multiple cancer disorders (Fig. 4). GVViZ-produced results (high-resolution figures) are available in supplementary material 2.
Fig. 4.

GVViZ gene-disease annotations, expression analyses, and heat map visualizations of different chronic diseases and conditions. Genes identified for A Alzheimer’s disease, B arthritis, C asthma, D diabetes mellitus, E obesity, F osteoporosis, G heart failure, H hypertension, and I multiple cancer disorders
We observed variability in the expression of Alzheimer’s-related genes CTSB, COX4I1, MT-ND1, CALM3, UBB, MT-CO2, PICALM, FPR2, MT-ND2, CST3, TNFRSF1A, CALM1, CALM2, CTSD, GRN, MT-CO1, ITM2B, and GAPDH between patients (Fig. 4A), having significant expression among most of the samples analyzed. Similarly, we found variable clusters of genes associated with immune-mediated diseases like arthritis and asthma. Arthritis genes included ANXA1, LTB, B2M, TNFRSF1B, HLA-DRA, SLC11A1, S100A12, ITGB2, HLA-DRB1, TNFRSF1A, HLA-C, SERPINA1, HLA-DB1, FCGR3A, CXCR2, HLA-B, TMSB4X, and S100A9 (Fig. 4B) and with significant expression. Asthma-related genes STAT6, IL16, ADAM8, ALOX5, TPT1, CD44, PPBP, HLA-DRB1, FCER1G, HLA-DQB1, ALOX5AP, and SELL had substantial expression (Fig. 4C). Most widely occurring diseases like diabetes mellitus (PIK3CD, IL16, MT-ND1, B2M, HBA1, MT-ND2, SLC11A1, NAMPT, PSMB9, HLA-DRB1, UCP2, HBA2, HLA-DPB1, HLA-A, ADIPOR1, HBB, SOD2, HLA-B) (Fig. 4D) and hypertension (ALOX5, CTS3, RPL5, HBG2, RHOA, HLA-A, HBB, and CYBA) (Fig. 4H) showed variable degree of expression in some genes. We only noticed CTS3 as a significantly regulated gene linked to heart failure (Fig. 4G) and did not find any highly expressed genes among obesity (Fig. 4E) and osteoporosis (Fig. 4F) disorders.
While analyzing multiple cancer disorders (Fig. 4I), we found highly variable expression among genes implicated in cancer: EEF1A1, GNAS, NPM1, PIM1, and RHOA are known oncogenes; H3F3A, PTPRC, SMARCB1, and B2M are possible oncogenes; and CASP8, JAK1, and PRKAR1A are known tumor suppressor genes [31]. While some genes found are known cancer census genes (RAF1, BCL6, STAT3, CCND3, EWSR1, HSP90AB1, LASP1, HSP90AA1, STAT5B, PICALM, NACA, CSF3R, FUS, H3F3B, CALR, MALAT1, LEF1, CXCR4, BIRC3, TPM3, CD79A, HNRNPA2B1, AKT2, SYK, and NUMA1) [32], evidence of aberrated expression of these genes could be a pre-clinical indication for further assessment. A further analysis of age, gender, and clinical history can give a clear idea of why some genes are expressed in nearly all the patients and some are not expressing in any patient. Also, we see many patients expressing most of these disease genes and a few not expressing any at all. Given their old age and no previous diagnosis, these patients should be studied to detect the signs of early-onset diseases.
Discussion
The quest to understand what causes chronic, acute, and infectious diseases has been a central focus of humankind since the beginning of scientific discovery [33, 34]. Our evolving understanding of the complex nature of diseases has led us to realize that to effectively diagnose and treat patients with these conditions, it is essential to utilize a precision medicine approach [35, 36]. By identifying the novel risk factors and disease biomarkers, precision medicine translates scientific discovery into clinically actionable personal healthcare [37–46]. A major barrier to the implementation of precision medicine is the data analysis requirement. Precision medicine requires progressive healthcare IT environments that can efficiently and rapidly integrate data from disparate groups with non-aligned formats to provide decision-making information to healthcare providers without massive amounts of computing time [47]. Despite current progress, there is still no stand-alone platform available to efficiently integrate clinical, multi-omics, environmental, and epidemiological data acquisition [48]. Robust platforms are required in clinical settings to effectively manage, process, integrate, and analyze big data with variable structures [49, 50].
On-demand access and analysis of integrated and individual patient clinical and transcriptomics data can lead to the identification of diagnostic signatures for early dissemination of oncology and non-oncology disorders [51, 52]. It can support better aligning of known disease biomarkers with established treatments necessary for real-time personalized care. However, one of the existing challenges includes timely high-throughput genomics and transcriptomics data interpretation and visualization to support health practitioners in the provision of personalized care [53]. It requires integration and understanding of data with various types, structures, velocity, and magnitude [54]. Visualization of complex and high-volume data in health-related settings will support cognitive work and highly impact time-restricted decision-making [55]. Guidelines for the development of such applied and practical data visualization include but not limited to the implementation of an interactive and friendly user interface [30], efficient mapping of data elements to visual objects [55], use of easy to understand and self-explanatory data visualization techniques, exporting and sharing of produced results, flexible design available with open-source code, and most importantly based on a reproducible approach. The development of academic data visualization approaches will also contribute to improve the collaboration between computational and bench scientists and clinicians to practice precision medicine with impactful scientific discovery and accessible approach at the point of care [56].
In this manuscript, we presented GVViZ, an integrated computational platform to support population and personalized transcriptome analysis with a user-friendly, physician-oriented interface, and essential processes required for RNA-seq-driven gene expression modeling, analysis, integration, management, and visualization. GVViZ is particularly appropriate for demanding clinical settings to facilitate physician’s decision-making. As it offers integrating and using large amounts of transcriptomics generated, and gene-disease annotation data are collected to support the personalized care of individuals with several complex disorders. GVViZ has the potential to bring gene-disease data annotation and analytics to the bedside to facilitate genetic susceptibility for achieving truly personalized treatments for earlier, more effective disease intervention. We emphasize that automated graphical visualization should be an indispensable component of modern RNA-seq analysis, which is currently not the case [57–59]. However, researchers can use our interactive RNA-seq visualization tool to visualize the transcriptomics data making it a powerful tool to study the dynamics of gene expression and regulation. Integration of this tool into clinical settings can help generate a patient’s profile for precision medicine implementation. We used real RNA-seq data to show that our tool can help readily and robustly visualize patterns and problems that may give insight into a patient’s genomic profile, unravel genetic predisposition, and uncover genetic basis of multiple disorders.
The current release of the GVViZ does not support unsupervised gene expression and differential analysis. This is one of the very important aspects that we are looking forward to address in the future. Furthermore, we are planning to account for the potentially varying average transcript length across samples when performing differential gene expression analysis by scaling the TPM matrix (summing the estimated transcript TPMs within genes and multiplying with the total library size in millions). This will transform the underlying abundance measures to incorporate the information provided by the sequencing depth which may considerably improve the false discovery rate.
Conclusion
Here, we introduced GVViZ, a new user-friendly application for RNA-seq-driven gene-disease data annotation, and expression analysis with a dynamic heat map visualization. With successful deployment in clinical settings, GVViZ will enable high-throughput correlations between patient diagnoses based on clinical and transcriptomics data. It will also assess genotype-phenotype associations among multiple complex diseases to find novel highly expressed genes. By mapping known and novel protein-coding and non-coding genes to their respective diseases, GVViZ can efficiently support the interpretation of genetic variants using the American College of Medical Genetics and Genomics (ACMG) guidelines and evaluation of variants in known genes.
Availability and requirements
The software executable (JAR file) is open source and freely available. To execute GVViZ ver.1.0.0, the only requirement is the installation of Java Runtime Environment and MySQL. Once Java and MySQL have been installed, the following two tables need to be created in the MySQL server.
Operating system: Cross-platform (Microsoft Windows, MAC, Unix, Linux)
Programming languages: Java and MySQL
Requirements: The researcher is responsible for MySQL installation and database schema.
License: Freely distributed for global users. Any restrictions to use by non-academics: Copyrights are to the authors.
Download link: GVViZ executable (JAR file) is freely available and can be downloaded through GitHub (https://github.com/drzeeshanahmed/GVViZ-Public).
GVViZ source code and all related material are already uploaded to GitHub and freely available to the community (https://github.com/drzeeshanahmed/GVViZ_SourceCode).
GVViZ online tutorial (video) is available through the following link: https://www.youtube.com/watch?v=x0RroYpk8Nw&ab_channel=Zeeshan.
Supplementary Information
Additional file 1: Supplementary material 1. GVViZ: User guide, database modelling, source code and software configuration.
Additional file 2: Supplementary material 2. GVViZ produced results and high-resolution figures, and quality report by RNA-seq pipeline.
Acknowledgements
We appreciate the great support by the Institute for Health, Health Care Policy and Aging Research (IFH), and Rutgers Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences at the Rutgers, The State University of New Jersey.
We would like to give special thanks to Dr. Christopher Bonin and Dr. Geneva Hargis for the stylistic and native speaker corrections.
We thank the members and collaborators of Ahmed Lab (https://promis.rutgers.edu/) at the Rutgers IFH for their active participation and contribution to this study.
This study was completed in part by research services and/or survey/data resources provided by the Institute for Health Survey/Data Core at Rutgers University, available at http://www.ifhcore.rutgers.edu.
The authors acknowledge the Office of Advanced Research Computing (OARC) at Rutgers, The State University of New Jersey, for providing access to the Amarel cluster and associated research computing resources that have contributed to the results reported here (URL: https://it.rutgers.edu/oarc).
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- COSMIC
Catalogue of Somatic Mutations in Cancer
- DEGs
Differentially expressed genes
- FAIR
Findable, accessible, interactive, and reusable
- FPKM
Fragments per kilobase million
- GUI
Graphical user interface
- NGS
Next-generation sequencing
- RPKM
Reads per kilobase of transcript per million mapped reads
- RSEM
RNA by expectation maximization
- RNA-seq
RNA sequencing
- TPM
Transcripts per million
- GVViZ
Visualizing genes with disease-causing variants
Authors’ contributions
ZA proposed and lead this study. ZA designed and supervised the GVViZ development, and EGR programmed it. ZA implemented the RNA-seq data processing pipeline and applied that to process RNA-seq data used for the analysis. ZA modeled and created the gene-disease annotation and RNA-seq gene expression databases, integrated with GVViZ. SZ participated in the performance evaluation and experiments using GVViZ. ZA and SZ performed the reported analysis. XD guided the study. ZA drafted the manuscript. The authors read and approved the final manuscript.
Authors’ information
ZA is an Assistant Professor of Medicine (Tenure Track) at the Robert Wood Johnson Medical School (RWHMS) and Core Faculty Member at the Institute for Health, Health Care Policy and Aging Research (IFH). EGR is a Postdoctoral Research Associate at the Ahmed Lab, Rutgers IFH. SZ is the Senior Postdoctoral Research Associate at the Rutgers Cancer Institute of New Jersey. XD is the Director of the IFH and the inaugural Henry Rutgers Distinguished Professor of Population Health Sciences. All authors belong to the Rutgers University-New Brunswick.
Funding
This work was supported by the Institute for Health, Health Care Policy and Aging Research, and Robert Wood Johnson Medical School, at Rutgers, the State University of New Jersey.
Availability of data and materials
The data that support the findings of this study are openly available in the following GitHub repository: https://github.com/drzeeshanahmed/GVViZ-Public. The data used in the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Segundo-Val IS, Sanz-Lozano CS. Introduction to the gene expression analysis. Methods Mol Biol. 2016;1434:29–43. doi: 10.1007/978-1-4939-3652-6_3. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405(6788):827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 3.Rao MS, van Vleet TR, Ciurlionis R, Buck WR, Mittelstadt SW, Blomme EAG, Liguori MJ. Comparison of RNA-Seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front Genet. 2019;9:636. doi: 10.3389/fgene.2018.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nature Reviews. Genetics. 2011;12(2):87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeeshan S, Xiong R, Liang BT, Ahmed Z. 100 years of evolving gene-disease complexities and scientific debutants. Brief Bioinformatics. 2020;21(3):885–905. doi: 10.1093/bib/bbz038. [DOI] [PubMed] [Google Scholar]
- 8.Trivedi UH, et al. Quality control of next-generation sequencing data without a reference. Front Genet. 2014;5:111. doi: 10.3389/fgene.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford) 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford) 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebbert MT, et al. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinformatics. 2016;17(Suppl 7):239. doi: 10.1186/s12859-016-1097-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed Z, Zeeshan S, Xiong R, Liang BT. Debutant iOS app and gene-disease complexities in clinical genomics and precision medicine. Clin Transl Med. 2019;8(1):26. doi: 10.1186/s40169-019-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed Z, Zeeshan S, Mendhe D, Dong X. Human gene and disease associations for clinical-genomics and precision medicine research. Clin Transl Med. 2020;10(1):297–318. doi: 10.1002/ctm2.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2015;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards Version 3: the human gene integrator. Database. 2010;2010:baq020. 10.1093/database/baq020. [DOI] [PMC free article] [PubMed]
- 18.Rappaport N, et al. MalaCards: a comprehensive automatically-mined database of human diseases. Curr Protoc Bioinforma. 2014;47:1.24.1–1.24.19. doi: 10.1002/0471250953.bi0124s47. [DOI] [PubMed] [Google Scholar]
- 19.Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX, Jensen LJ. DISEASES: text mining and data integration of disease-gene associations. Methods. 2015;74:83–89. doi: 10.1016/j.ymeth.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 20.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kibbe WA, Arze C, Felix V, Mitraka E, Bolton E, Fu G, Mungall CJ, Binder JX, Malone J, Vasant D, Parkinson H, Schriml LM. Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2014;43(Database issue):D1071–D1078. doi: 10.1093/nar/gku1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang G, et al. DiseaseEnhancer: a resource of human disease-associated enhancer catalog. Nucleic Acids Res. 2017;46(D1):D78–D84. doi: 10.1093/nar/gkx920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, Furlong LI. DisGeNET: a comprehensive platform integrating information on human Sdisease-associated genes and variants. Nucleic Acids Res. 2017;45(D1):D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babbi G, Martelli PL, Profiti G, Bovo S, Savojardo C, Casadio R. eDGAR: a database of disease-gene associations with annotated relationships among genes. BMC Genomics. 2017;18(S5):554. doi: 10.1186/s12864-017-3911-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinstein WS, Maglott DR, Lee JM, Kattman BL, Malheiro AJ, Ovetsky M, Hem V, Gorelenkov V, Song G, Wallin C, Husain N, Chitipiralla S, Katz KS, Hoffman D, Jang W, Johnson M, Karmanov F, Ukrainchik A, Denisenko M, Fomous C, Hudson K, Ostell JM. The NIH genetic testing registry: a new, centralized database of genetic tests to enable access to comprehensive information and improve transparency. Nucleic Acids Res. 2012;41(Database issue):D925–D935. doi: 10.1093/nar/gks1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014;43(Database issue):D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database issue):D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Wu SJ, Yang SY, Peng JW, Wang SN, Wang FY, Song YX, Qi T, Li YX, Li YY. DNetDB: the human disease network database based on dysfunctional regulation mechanism. BMC Syst Biol. 2016;10(1):36. doi: 10.1186/s12918-016-0280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, et al. International Cancer Genome Consortium Data Portal--a one-stop shop for cancer genomics data. Database. 2011;2011:bar026. 10.1093/database/bar026. [DOI] [PMC free article] [PubMed]
- 30.Ahmed Z, Zeeshan S, Dandekar T. Developing sustainable software solutions for bioinformatics by the “Butterfly” paradigm. F1000Res. 2014;3:71. doi: 10.12688/f1000research.3681.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailey MH, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173(2):371–385.e18 [DOI] [PMC free article] [PubMed]
- 32.Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705. [DOI] [PMC free article] [PubMed]
- 33.Ahmed Z. Practicing precision medicine with intelligently integrative clinical and multi-omics data analysis. Human Genomics. 2020;14(1):35. doi: 10.1186/s40246-020-00287-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed Z, Zeeshan S, Foran DJ, Kleinman LC, Wondisford FE, Dong XQ. Integrative clinical, genomics and metabolomics data analysis for mainstream precision medicine to investigate COVID-19. BMJ Innovations. 2021;7(1):6–10. doi: 10.1136/bmjinnov-2020-000444. [DOI] [Google Scholar]
- 35.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khoury MJ, Iademarco MF, Riley WT. Precision public health for the era of precision medicine. Am J Prev Med. 2016;50(3):398–401. doi: 10.1016/j.amepre.2015.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perkins BA, Caskey CT, Brar P, Dec E, Karow DS, Kahn AM, Hou YCC, Shah N, Boeldt D, Coughlin E, Hands G, Lavrenko V, Yu J, Procko A, Appis J, Dale AM, Guo L, Jönsson TJ, Wittmann BM, Bartha I, Ramakrishnan S, Bernal A, Brewer JB, Brewerton S, Biggs WH, Turpaz Y, Venter JC. Precision medicine screening using whole-genome sequencing and advanced imaging to identify disease risk in adults. Proc Nat Acad Sci USA. 2018;115(14):3686–3691. doi: 10.1073/pnas.1706096114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed Z, Mohamed K, Zeeshan S, Dong X. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database. 2020;2020:baaa010. 10.1093/database/baaa010. [DOI] [PMC free article] [PubMed]
- 39.Shieh, Y., et al. (2017). Breast cancer screening in the precision medicine era: risk-based screening in a population-based trial. J Natl Cancer Inst, 109:djw290. [DOI] [PubMed]
- 40.Hou YC, Yu HC, Martin R, Cirulli ET, Schenker-Ahmed NM, Hicks M, et al. Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc Natl Acad Sci U S A. 2020;117(6):3053–62. 10.1073/pnas.1909378117. [DOI] [PMC free article] [PubMed]
- 41.Lee SI, Celik S, Logsdon BA, Lundberg SM, Martins TJ, Oehler VG, Estey EH, Miller CP, Chien S, Dai J, Saxena A, Blau CA, Becker PS. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nature Commun. 2018;9(1):42. doi: 10.1038/s41467-017-02465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beltran H, Eng K, Mosquera JM, Sigaras A, Romanel A, Rennert H, Kossai M, Pauli C, Faltas B, Fontugne J, Park K, Banfelder J, Prandi D, Madhukar N, Zhang T, Padilla J, Greco N, McNary TJ, Herrscher E, Wilkes D, MacDonald TY, Xue H, Vacic V, Emde AK, Oschwald D, Tan AY, Chen Z, Collins C, Gleave ME, Wang Y, Chakravarty D, Schiffman M, Kim R, Campagne F, Robinson BD, Nanus DM, Tagawa ST, Xiang JZ, Smogorzewska A, Demichelis F, Rickman DS, Sboner A, Elemento O, Rubin MA. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1(4):466–474. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo Y, Ahmad FS, Shah SJ. Tensor factorization for precision medicine in heart failure with preserved ejection fraction. J Cardiovasc Transl Res. 2017;10(3):305–312. doi: 10.1007/s12265-016-9727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsanis N. The continuum of causality in human genetic disorders. Genome Biol. 2016;17(1):233. doi: 10.1186/s13059-016-1107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manrai AK, Ioannidis JP, Kohane IS. Clinical genomics: from pathogenicity claims to quantitative risk estimates. JAMA. 2016;315(12):1233–1234. doi: 10.1001/jama.2016.1519. [DOI] [PubMed] [Google Scholar]
- 46.Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169(7):1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazaridis KN, et al. Implementing individualized medicine into the medical practice. American Journal of Medical Genetics. Part C Semin Med Genet. 2014;166C(1):15–23. doi: 10.1002/ajmg.c.31387. [DOI] [PubMed] [Google Scholar]
- 48.van Panhuis WG, Paul P, Emerson C, Grefenstette J, Wilder R, Herbst AJ, Heymann D, Burke DS. A systematic review of barriers to data sharing in public health. BMC Public Health. 2014;14(1):1144. doi: 10.1186/1471-2458-14-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traversi D, Pulliero A, Izzotti A, Franchitti E, Iacoviello L, Gianfagna F, Gialluisi A, Izzi B, Agodi A, Barchitta M, Calabrò GE, Hoxhaj I, Sassano M, Sbrogiò LG, Del Sole A, Marchiori F, Pitini E, Migliara G, Marzuillo C, De Vito C, et al. Precision medicine and public health: new challenges for effective and sustainable health. J Person Med. 2021;11(2):135. doi: 10.3390/jpm11020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mennini M, Arasi S, Fiocchi AG, Assa’ad A. Developing national and international guidelines. Immunol Allergy Clin North Am. 2021;41(2):221–231. doi: 10.1016/j.iac.2021.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Amer B, Baidoo E. Omics-driven biotechnology for industrial applications. Front Bioeng Biotechnol. 2021;9:613307. doi: 10.3389/fbioe.2021.613307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Warner JL. A review of precision oncology knowledgebases for determining the clinical actionability of genetic variants. Front Cell Dev Biol. 2020;8:48. doi: 10.3389/fcell.2020.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Backonja U, Haynes SC, Kim KK. Data visualizations to support health practitioners’ provision of personalized care for patients with cancer and multiple chronic conditions: user-centered design study. JMIR Human Factors. 2018;5(4):e11826. doi: 10.2196/11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.West P, Van Kleek M, Giordano R, Weal M, Shadbolt N. Information quality challenges of patient-generated data in clinical practice. Front Public Health. 2017;5:284. doi: 10.3389/fpubh.2017.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khairat SS, Dukkipati A, Lauria HA, Bice T, Travers D, Carson SS. The impact of visualization dashboards on quality of care and clinician satisfaction: integrative literature review. JMIR Human Factors. 2018;5(2):e22. doi: 10.2196/humanfactors.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonzalez-Hernandez G, Sarker A, O’Connor K, Greene C, Liu H. Advances in text mining and visualization for precision medicine. Pac Symposium Biocomput. 2018;23:559–565. [PMC free article] [PubMed] [Google Scholar]
- 57.Chatterjee A, Ahn A, Rodger EJ, Stockwell PA, Eccles MR. A guide for designing and analyzing RNA-Seq data. Methods Mol Biol. 2018;1783:35–80. doi: 10.1007/978-1-4939-7834-2_3. [DOI] [PubMed] [Google Scholar]
- 58.Liang H, Zeng E. RNA-Seq experiment and data analysis. Methods Mol Biol. 2016;1366:99–114. doi: 10.1007/978-1-4939-3127-9_9. [DOI] [PubMed] [Google Scholar]
- 59.Givan SA, Bottoms CA, Spollen WG. Computational analysis of RNA-seq. Methods Mol Biol. 2012;883:201–219. doi: 10.1007/978-1-61779-839-9_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary material 1. GVViZ: User guide, database modelling, source code and software configuration.
Additional file 2: Supplementary material 2. GVViZ produced results and high-resolution figures, and quality report by RNA-seq pipeline.
Data Availability Statement
The data that support the findings of this study are openly available in the following GitHub repository: https://github.com/drzeeshanahmed/GVViZ-Public. The data used in the current study are available from the corresponding author on reasonable request.