

Abstract
Remarkable progress in ageing research has been achieved over the past decades. General perceptions and experimental evidence pinpoint that the decline of physical function often initiates by cell senescence and organ ageing. Epigenetic dynamics and immunometabolic reprogramming link to the alterations of cellular response to intrinsic and extrinsic stimuli, representing current hotspots as they not only (re-)shape the individual cell identity, but also involve in cell fate decision. This review focuses on the present findings and emerging concepts in epigenetic, inflammatory, and metabolic regulations and the consequences of the ageing process. Potential therapeutic interventions targeting cell senescence and regulatory mechanisms, using state-of-the-art techniques are also discussed.
Subject terms: Medical research, Cell biology
Introduction
Individual cells face three major cell fate choices: to survive, to senesce, or to commit suicide. The balance between these processes ensures that cell turnover in an organism remains essentially in functional equilibrium (homeostasis). While cell survival and death have been intensively investigated for long, cell senescence is relatively less understood due to the complexity of the ageing process and heterogeneity of ageing phenotypes.1 Past investigations of cell senescence have focused on its role in tumor suppression, cell cycle arrest, tissue repair, and DNA replicative stress/damage response. Many mediators are linking to intrinsic (e.g., organelle homeostasis, chronic inflammation, and epigenetic alterations) and extrinsic factors (e.g., UV exposure, drug toxicity, and lifestyle) that deteriorate cell physiology.2 We now know many pathways involve in the regulation of cell senescence, such as AMP-activated protein kinase (AMPK) energy-sensing,3 histone/protein (de-)acetylation,4,5 cyclic GMP–AMP synthase (cGAS)–the cyclic GMP–AMP receptor stimulator of interferon genes (STING) signaling pathways,6 which impact the rate and extent of cell senescence. Of note, existing studies define that senescence as a relatively inert, non-proliferating, irreversible cell state.1 Although the removal of senescent cells serves as an attractive option to mitigate age-related functional decline and extend health span,7 researchers are trying to reprogram senescent cells back to functionally healthy cells, especially for cardiomyocytes and neurons that are hardly proliferative. Can cell senescence be reversed? Are there different forms or stages of senescent cells that exert distinct physiological effects? Does senescence have any yet new functions to the cells or tissues? To our knowledge, there are still many open questions that await answers.
As the biological techniques advance, many new targets and signaling pathways participating in cell senescence regulation have been identified, although some are still controversial. Nonetheless, the common signals and mechanisms converge upon dysregulated inflammation, alteration of epigenetic modifications, and metabolic imbalance.8–10 Here in this review, we highlight those known and yet unexploited key molecular and signaling pathways, particularly the inflammatory, epigenetic, and metabolic aspects that link cell senescence and organismal ageing (Fig. 1). Therapeutic targets and novel techniques established in recent cell senescence studies are also discussed.
Fig. 1.

Dysregulated inflammation, alteration of epigenetic modifications, and metabolic imbalance converge to cell senescence and ageing. Cross talks among epigenetic modifiers (writers, readers, and erasers), inflammatory gene expression and immune cell response, and metabolic alterations contribute to senescent phenotypes and organ degeneration. MiDAS mitochondrial dysfunction-associated senescence, SASP senescence-associated secretory phenotype, OXPHOS oxidative phosphorylation, ROS reactive oxygen species, TCA tricarboxylic acid
Definition of cell senescence
Although research on cell senescence lasts for decades, until recently, the field reached a consensus on the definition of cell senescence, that is, a type of cell state that can be stimulated by multiple stress signals throughout the life cycle and is characterized by cell cycle arrest, senescence-associated secretory phenotype (SASP), and dysregulated metabolism and macromolecular damage1 (Fig. 2). Consequently, many types of cell senescence have been proposed, including replicative senescence, programmed developmental senescence, and stress-induced senescence.11 Such diverse senescent pathways represent active and passive modes to establish a delicate balance between different cell populations—as abstract as it sounds. It is worth noting that ageing and cell senescence are different but closely related. Organismal ageing emphasizes the degeneration of tissues or organs caused by accumulated damages upon a period.12 In contrast, cell senescence can occur whenever under a specific stress condition, and it may also play a positive role in wound healing and tumor inhibition.13 The determinant that matters favorable or lousy cell senescence largely relies on the duration: long-term type of cell senescence is prone to inflammation and disease, while short-term type sometimes seems beneficial because the immune system can quickly scavenge the senescent cells.14
Fig. 2.
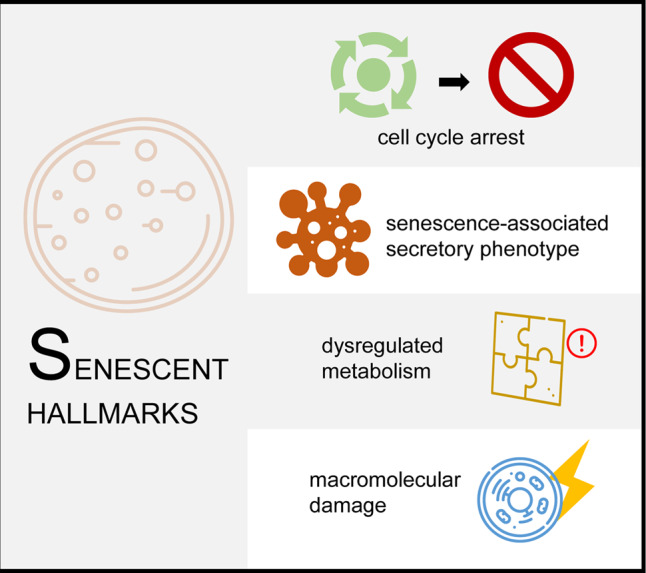
The hallmarks of cell senescence
The hallmarks of cell senescence
As described above, senescent cells are categorized by differences in stress stimuli, thereby exhibiting various phenotypes and hallmarks. At present, the most commonly used senescent marker is senescence-associated beta-galactosidase (SA-β-gal), closely related to the lysosomal stress response, but not necessarily dependent on senescence.15 Activation of p53, p21CIP1, p16INK4a, ataxia telangiectasia mutated (ATM)/ATM and RAD3-related (ATR), and retinoblastoma (RB) can be used as auxiliary markers apart from the occurrence of morphological changes, permanent cell cycle arrest, cell secretion, and metabolic and chromatin remodeling.16 As none of those mentioned above markers can solely determine the specific type of cell senescence, various methods and hallmarks are required to clarify the exact category of a senescent cell.17 A summary of known hallmarks of cell senescence is shown in Table 1.
Table 1.
Selected modulators of cell senescence
| Senescent modulators | Circumstances | References |
|---|---|---|
| SA-β-gal-positive staining | Development, ageing, all stages | 15 |
| p21CIP1 | Cell cycle arrest, ageing, all stages | 568 |
| p16INK4a | Cell cycle arrest, ageing, all stages | 569 |
| p53 | Cell cycle arrest, ageing, DNA damage | 570 |
| Rb | Cell cycle arrest, ageing, DNA damage | 571 |
| Lamin B1 | Decrease in ageing, genome instability | 572 |
| TAFs/TIFs | DNA damage, telomere attrition | 573,574 |
| Phospho-γH2AX | DNA damage | 575 |
| SASP components | Ageing, DAMPs, all stages | 18 |
| Mitochondrial dysfunction | Decrease respiration and ATP production | 576 |
| Protein aggregates | UPR, ageing, loss of proteostasis | 577 |
| Oxidative stress | Increase ROS/RNS | 576 |
| Autophagy malfunction | Ageing, protein toxicity | 578 |
| Macromolecular condensates | Various stress, ageing | 19 |
| HP1 | Various stress, SAHF | 579 |
| DNA methylation | Various stress, SAHF | 580 |
| Histone methylation | Various stress, SAHF | 581 |
| Morphology | Ageing | 582 |
| Multi-omics alteration | Various stress, ageing | 583 |
Permanent cell cycle arrest
Typically, senescent cells exhibit permanent cell cycle arrest that differs from quiescent or differentiated cells. The current definition of cell senescence emphasizes a state of cell cycle withdrawal upon external stimuli.1 In contrast, quiescent cells can reenter the cell cycle, and differentiated cells can be dedifferentiated under certain circumstances. Besides, senescence typically accompanies augmented p21CIP1 or p16INK4a expression, while quiescence is p27KIP1 dependent, and differentiation can involve multiple signaling pathways (e.g., Wnt/Notch/Hedgehog/p16INK4a).
Senescence-associated secretory phenotype
First discovered by Jean-Philippe Coppe and colleagues in 2008, SASP refers to a state when senescent cells release certain substances that mediate a series of (patho-)physiological effects, including pro-inflammatory cytokines, chemokines, growth hormones, angiogenic factors, and matrix metalloproteinases.18 The different biological activities induced by the components of SASP suggest that it may interact with local and surrounding cells, and constitutes a mechanism to regulate microenvironment, which could be either beneficial or deleterious, depending on the secretion factors, site (cell types), duration (acute or chronic), and secretion-induced stimuli.
Dysregulated metabolism
Metabolic disturbance during cell senescence manifests the loss of molecular and protein homeostasis. Several processes, such as DNA damage response (DDR) induced by telomere attrition, decreased tricarboxylic acid cycle activity, and ATP production by mitochondrial dysfunction, declined degradation of the proteasome and autophagolysosome, the changes in SASP and epigenetic alteration, all lead to the remodeling of metabolic signals and metabolites in the cells.
Macromolecular damage
Biomacromolecular damage is another typical phenotype of cell senescence. Stimuli-like ionizing radiation, chemotherapeutics, and other drug damage, oxidative stress, and senescence-induced telomere dysfunction can mediate such damage, subsequently leading to the aggregation of aberrant macromolecular substances.19 Of note, the emerging concept of liquid–liquid phase separation in cell senescence has recently made the research of biomacromolecule condensate in the ascendant.
Effects of cell senescence on organ function and diseases
Accumulating studies have proven the relationship between senescent cells and organismal ageing. Meanwhile, the concept of eliminating senescent cells to counteract ageing-related conditions has emerged and succeeded in rodent models. Baker and colleagues have found a large number of p16INK4a-positive senescent cells in various tissues that cause a range of ageing symptoms, including sarcopenia, cataracts, and lipodystrophy.7 Accordingly, targeted clearance of p16INK4a senescent cells alleviates the adverse symptoms and successfully extend the health span in many diseased models.7
The field began to look for traces of senescent cells in common ageing diseases in humans, and successfully established a causal relationship between pathogenesis of ageing-related diseases and cell senescence. Take atherosclerosis as an example, we have known that plaques composed of fat and protein gradually accumulate on the inner arterial wall, which is prone to cause coronary atherosclerotic disease, stroke, or other ischemic severe diseases. Next, senescence-associated macrophages were recruited to the arterial wall, where the plaque initially formed. As time elapsed, other senescent cell types appeared near these sites. Compared with other control cells, these senescent cells expressed abundant secretory factors and metabolites that promoted the pathogenesis of atherosclerosis, concurrent with significant alterations in epigenetic imprints.20 Using a variety of approaches to remove these senescent cells attenuated the lesions, and thus alleviating the progress of atherosclerosis.20 Consequently, focusing on the epigenetic and immunometabolic regulation of cell senescence may shed light on managing ageing-related diseases and therapeutic interventions.21
In this review, we highlight the recent advances in the understanding of the inflammatory, epigenetic, and metabolic basis of cell senescence, a comprehensive overview of relevant molecules and signaling pathways associated with cell senescence and organismal ageing are discussed. Finally, novel techniques and strategies intervening in the ageing process are briefly summarized.
Ageing and inflammation
Infection-related diseases are responsible for many deaths globally, and aged people are more vulnerable to severe and life-threatening infections.22,23 Chronic inflammation represents an essential phenomenon in both murine and human ageing.24 Recent studies from different aspects investigations conclude that inflammation is a commonly shared mark of ageing tissues. Gene comparisons of young and old tissues from mice, rats, and humans revealed that age-related gene expression changes most remarkably involve a strong induction of inflammation and immune response genes.25 The combination of high levels of inflammatory signals, increased comorbid conditions, and reduced function of the immune system increases the vulnerability of aged individuals to infection.26
Despite its crucial role in defending infections during life, inflammation may turn into a hazardous factor to health for the aged individuals, when it gets into chronic and persistent, such chronic increases in low-grade inflammation during ageing, also known as “inflammageing”, is a hallmark of ageing.16,27 Unlike acute inflammation, inflammageing is characterized by maintaining a low-grade, sustained background of inflammation even in the absence of acute infection and clinically diagnostic disease.27,28 Increasing evidence has been shown that inflammageing is a risk factor that leads to reduced tissue repair and generative capacity, which is associated with many ageing-related diseases.29–31
Consequently, extending the understanding of the underlying mechanism of inflammageing and ageing-associated diseases helps realize healthy ageing among the growing senile population. In this section, we summarize the current findings illustrating the cause and effect of inflammageing, and how inflammation gives rise to the evolution of ageing-related diseases. Novel approaches to attenuate pathophysiological conditions are also discussed, aiming at combating adverse inflammageing.
Source of chronic inflammation in ageing
High levels of pro-inflammatory cytokines shape the ageing-associated pro-inflammatory status, although the source of ageing-related chronic low-grade inflammation remains incompletely understood.24,32–34 Nevertheless, a variety of source that contributes to the pro-inflammatory microenvironment has been identified.35
Senescent cells
Cells are driven into a senescent, nondividing state by many factors, such as telomere shortening, DNA damage, oxidative stress, genotoxic stress, and altered chromatin structure.36–39 The immune system efficiently clears away the senescent cells to maintain systemic homeostasis. However, the removal capacity declines with age (partially due to the immunosenescence), resulting in the increased SASP. Therefore, it becomes more explicit that SASP builds a relationship connecting cellular senescent with various biological processes, where SASP represents a potential pharmaceutical target to manipulate the development of ageing and ageing-related diseases (Fig. 3). In agreement, mounting evidence suggests that therapeutic removal of senescent cells could delay or even prevent various age-related diseases, such as atherosclerosis and osteoarthritis.7,20,40,41
Fig. 3.

Inflammatory sources lead to senescence-associated secretory phenotype (SASP). Several pro-inflammatory sources have been identified to trigger the chronic inflammation during cellular senescence and organismal ageing, featured by the activation of a group of SASP-related genes, and subsequently the release of SASP components
Cell debris
Cell debris, including damage-associated molecular patterns (DAMPs), damaged organelles, and macromolecules, are recognized and removed by the immune system. With age, cell debris accumulates due to impaired clearance and overproduction, inducing augmented inflammation and impairs tissue regeneration.42,43 The ageing-associated mitochondrial compromise can lead to a release of DAMPs, namely the secretory phenotype by mitochondrial dysfunction-associated senescence (MiDAS), which causes particular attention in recent years.44–47 Congruently, ageing-associated mitochondrial stress can also lead to aberrant activation of inflammasomes and result in a functional decline in aged hematopoietic stem cells (HSCs), further stressing the immune system.48 Intriguingly, a study found that the level of circulating mitochondrial DNA (mtDNA) is significantly increased in elderly individuals and contributes to the increased systemic inflammation, although the exact source for circulating mtDNA remains undefined.49
Immunosenescence
The innate immune system gradually overtakes the adaptive immune system during ageing.50 In general, the function of the immune system declines with age, collectively termed immunosenescence, featured by the reduced output of natural killer cells and thymic T cells, decreased phagocytic capacity of macrophages, as well as the impaired activation of neutrophils and maldevelopment of B cells in the aged.50,51 Immunosenescence has been considered a dominating problem in the aged population and is associated with inappropriate immune responses, resulting in the declined removal capability of senescent cells and DAMPs.52 Reciprocally, inflammageing leads to chronic, continuous generation of inflammatory factors that exhausts the adaptive immune responses, culminating with immunosenescence.10 Of note, due to the suppression of the adaptive immune response, the innate immune response could be reinforced as a compensatory means to protect the organism from infections. Thus, the immunosenescence and inflammageing could operate in parallel and form a vicious feedback loop.
Gut dysbiosis
The gut mucosa barrier plays a vital role in defending against bacterial invasion. However, the integrity of the gut is impaired with age. The permeability of the epithelial cells is damaged, allowing the bacteria and other toxins to enter the blood, called “leaky gut”.53–55 The gut microbiota of older people also exhibits a biased diversity.53–55 For instance, reduced anti-inflammatory bacteria like Bifidobacterium spp., while increased pro-inflammatory bacteria like Streptococcus spp. was found in the aged gut.56 These changes lead to an increase in the susceptibility to infections in aged people.57 Another study also shows that gut microbiota links to longevity, in which healthier seniors showed microbiome signatures that are similar to young people.58 In line with this, the long-living (>90 years old) individuals favor increased gut microbiota diversity than that in young controls, with several beneficial bacteria identified in the gut.59
Obesity
Obesity is featured by excessive fat accumulation that secret many inflammatory adipokines.60,61 During ageing, immune cells infiltrate into the fat tissues that can be activated upon various stimuli. Bernier and colleagues recently demonstrated that anti-inflammatory Disulfiram, an FDA-approved drug treating chronic alcohol addiction, reversed established diet-induced obesity and metabolic dysfunctions in middle-aged mice.62 Thus, bodyweight control or calorie restriction (CR) that eliminates pro-inflammatory fat deposition would reduce inflammation during ageing.
Sex hormones
Existing evidence shows that sex steroids regulate the immune system by expressing their specific receptors in different immune cells.63 With age, the levels of sex hormones, such as estrogen and progesterone in females and testosterone in males are downregulated.64–66 Interestingly, after menopause, the number of lymphoid cells decreases, accompanied by a strong induction of pro-inflammatory cytokines.67–69 In contrast, postmenopausal females receiving hormone replacement therapies (HRT) showed increased B cells and reduced concentration of pro-inflammatory cytokines compared with that without HRT.69,70 Despite that testosterone replacement therapy has not been reported with aged male individuals, one study using old nonhuman primates clearly showed that supplementation of androgens in aged male rhesus macaques partially reverted the reduced number of naive T cells via enhancing thymic output, implicating a possible connection between age-related hormone dysregulation and immune dysfunction.71
Other sources
Apart from the sources discussed above, several lifestyle-related factors affect the secretory phenotypes of inflammageing.72 First, long-term smoking has been associated with the increased susceptibility of respiratory diseases, and especially lung cancer in the elderly, with a significantly elevated production of pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6, TNF-alpha, and acute phase proteins.73,74 Second, a sedentary lifestyle among the aged individuals also accelerates fat accumulation and myeloid-biased hematopoiesis, siding with the pro-inflammatory microenvironment. In agreement with that, a recent study found that regular exercise activity results in the reduced inflammatory cell production, limiting the secretion of the inflammatory cytokines via modulating hematopoietic and progenitor cell proliferation in both murine and humans.75 Similarly, sleep problem perplexes aged individuals that aids mental stresses with elevated circulating inflammatory cytokines.
Mechanisms involve in inflammageing
Although the mechanism of inflammageing has not been thoroughly studied, many factors include oxidative stress, pro-inflammatory cytokines, DNA damage, dysfunction of cellular organelles, defects in autophagy, and stem cell ageing are involved in regulating inflammageing at both transcriptional and posttranscriptional levels.76
Cytokines induction
Pathogen-associated molecular pattern receptors, such as the toll-like receptors (TLRs) expressed on immune cells, are the principal receptors that sense pathological stimuli and lead to cytokine induction. TLRs are the first to be affected by invading pathogens and mediate a series of physiological reactions, such as inflammation, cell survival, proliferation, and apoptosis.77 During ageing, the activation of TLRs downstream signaling pathways is altered.78,79 Among the transcription factors that regulate chronic inflammation across multiple diseases and tissues, NF-kB (nuclear factor kappa-light-chain enhancer of activated B cells) and STAT (signal transducer and activator of transcription) are the two well studied.80 NF-kB positively regulates many genes that encode pro-inflammatory cytokines, therefore acting as a master regulator of SASP.81–83 Moreover, NF-kB drives several ageing phenotypes, particularly in the skin, spine, brain, and blood system.84–87 Notably, mTOR controls the translation of IL-1a and thus regulates SASP, indicative of its role in the regulation of SASP.88,89 mTOR also has been manifested to control the translation of MK-2 kinase, which phosphorylates the specific RNA-binding protein ZFP36L1, preventing the degradation of the transcripts of many SASP factors.89 These findings lead to the assumption that mTOR accumulation helps accelerate the synthesis of SASP factors. Moreover, the surroundings of senescent cells and their communications also contribute to the SASP, for instance, the NOTCH/JAG1 signaling controls the interaction between senescent cells with their microenvironment.90,91
Oxidative stress-induced inflammageing
Based on the close relationships between oxidative stress, inflammation, and ageing, De La Fuente and Miquel proposed an oxidation-inflammatory theory of ageing (oxi-inflammageing).92 That is, oxidative stress leads to inflammageing and influences the homeostasis of the body. The redox state and the function of immune cells affect the velocity of ageing and life span.92 Therefore, antioxidants treatment may improve immune function. In line with this, resveratrol and metformin supplementation could extend life span via reducing oxidative stress.93,94
DNA damage response
DNA damage induces several signaling transductions that result in damage repair, cell cycle arrest, apoptosis, and cell death.95,96 Apart from the responses mentioned above, DNA damage also triggers cellular senescent and ultimately induces SASP.97 p38 is the primary regulator of DDR, and its activation could induce NF-kB signaling, causing the SASP-related gene expression.98,99 Studies showed that p38 inhibition prevents the secretion of various inflammatory factors involved in SASP.100,101 DNA damage also leads to the imbalance of systemic metabolism via inducing tissue inflammation.102 It should be noted that DNA damage accumulated during ageing, potentially contributing to the increase in chronic inflammation with age.
Cytosolic double-strand DNA-induced inflammageing
Viral DNA in cytoplasm triggers the cGAS–STING pathway, leading to the interferon (IFN) production and subsequently activates inflammatory response.103–105 Cytoplasmic DNA released from stressed mitochondria or damaged nuclei can also lead to innate immune signaling response via inflammasome or cGAS signaling.106,107 The cGAS–STING signaling also connects genomic instability and DNA damage to inflammation.108 Accumulated evidence suggests that endogenous retroelements, such as short interspersed nuclear elements (SINEs; including Alu) and long interspersed nuclear elements (LINEs; including LINE1), play an essential role in initiating inflammation.109–111 De Cecco and colleagues demonstrated that LINE1 was transcriptionally activated in senescent cells, thereby leading to type-I interferon (IFN-I) induction and promoting SASP.112
Micro-RNAs
The intracellular signaling cascades that regulate inflammageing are subject to numerous layers of regulation, including the regulation by micro-RNAs.113,114 For example, miRNAs participate in modulating TLR, retinoic acid-inducible gene I (RIG-I), and NF-kB signaling pathways.115–118 miRNA can directly bind on TLR signaling or activate the RNA-sensing TLRs. In turn, the expression of miRNA can also be regulated by TLRs, RIG-I, and NF-kB activation, revealing a feedback loop controlling the immune response.117,119–121
Stem cell ageing
Stem cells underlie tissue homeostasis, while ageing causes a functional decline in the stem cells, compromising tissue regeneration and contributing to age-related degenerative diseases.122 Chronic inflammation is one of the main factors that induce stem cell ageing.123 During ageing, aberrant activation of the NLRP3 inflammasome restraints the function of HSCs.48 Likewise, inflammageing is the main culprit of skeletal stem and progenitor cell dysfunction.124 The chronic inflammatory process accompanied by ageing leads to dysfunctional differentiation of stem cells, loss of self-renewal capacity, and results in stem cell ageing.123 Conversely, stem cell ageing is also responsible for systematic inflammageing. An increase in NF-kB activity has been reported in aged HSCs, leading to enhanced sensitivity in aged HSC to inflammatory stimuli, which result in the higher production of IL-6 and a myeloid-biased differentiation.87,125 Mesenchymal stromal cells (MSCs) ageing leads to adipocytes accumulation in old bones and dysregulates hematopoiesis.126,127 A detailed summary of epigenetics and ageing, particularly in stem cell ageing, is reviewed in an independent section.
Together, the possible mechanisms discussed above help understand how chronic inflammation accumulate and persist during ageing. These factors also provide potential drug targets for therapeutic interventions to delay the ageing process and prevent inflammageing-associated diseases.
Inflammageing-associated chronic diseases
Inflammatory signaling has beneficial functions in many physiological processes, such as embryo development and wound healing. However, excessive and persistent inflammatory responses are detrimental, as reflected by increased morbidity and mortality, leading to a decline in life quality. Indeed, several studies have shown experimental evidence linking inflammation to chronic age-related diseases29(Fig. 4). For instance, old mice were about 6.5-fold and fourfold more sensitive to the lethal toxicity of lipopolysaccharide and exogenous TNF than young controls, respectively.128 The enhanced sensitivity of old mice to inflammatory stimuli is possibly due to the already existing higher basal level of inflammation signal in aged animals. Therefore, the resolution phase is much more extended than young mice.27,129
Fig. 4.
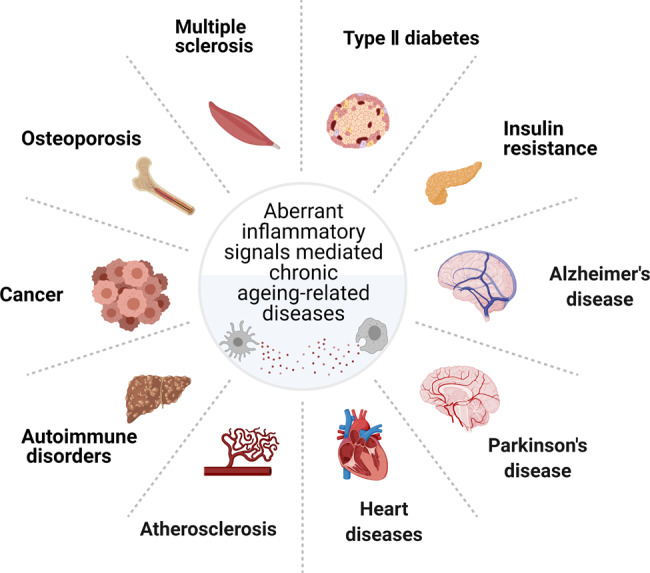
Inflammageing and its associated diseases. Multiple inflammatory signaling pathways, such as the NF-kB, NOTCH/JAG1, toll-like receptor, DNA damage response, cytosolic DNA sensing, autophagy, mRNA stabilization, and mTOR signaling pathways have been linked to age-related chronic diseases in various organs
It is worth noting that inflammation has been well established as a significant component of neurodegenerative disorders, yet it is unclear if this is a direct cause of the disease or a consequence of progressive neurodegeneration.130,131 Over the past decade, there has been a revolution in understanding how cytokines contribute to the etiology of the leading neurodegenerative disorders, including Alzheimer’s (AD) and Parkinson’s disease (PD). Inflammation also involves in many cardiovascular diseases, although whether inflammation causes a heart attack or other cardiovascular disorders require further investigation, inflammation serves as a universal sign for the atherogenic response. Atherosclerosis is a chronic inflammatory condition where atherosclerotic plaques show cellular senescence.132,133 Cytokines are involved in all stages of the pathogenesis of atherosclerosis, having both pro- or anti-atherogenic effects.134,135 Chronic tissue inflammation has a vital role in the etiology and immunopathogenesis of rheumatoid arthritis, with genetic and environmental factors contributing to a predilection to develop the disease.136,137 Osteoporosis is a disease in which bone loses calcium and become fragile. Young people maintain a balance between bone loss and bone formation. However, with ageing, the balance is disturbed toward bone loss due to the increases in chronic inflammation.138 Sustained low-grade inflammation can be found in type 2 diabetes due to a high concentration of circulatory inflammatory cytokines.139 The inflammageing also alters the function of the blood system, leading to decreases in lymphopoiesis, overproduction of myeloid cells, cytopenias, and anemia.140–142 Besides, the persistence of inflammageing intensively involves in hematological diseases, such as myelodysplastic syndrome and acute myelogenous leukemia.143–145 Other studies also link that inflammageing to hypertension, frailty, dementia, and chronic kidney disease.146–148
On the other hand, cell senescence inhibits aberrant cell proliferation and tumorigenesis, yet ageing is considered the most significant risk factor for cancer development. Nearly 60% of people suffering from various kinds of cancers are 65 years old or older. Paradoxically, although cellular senescence function as an anticancer program, the secreted SASP factors are associated with malignant tumor progression.149 The mechanism that links SASP and cancer have been extensively studied.1 For instance, IL-6 has been shown to activate WNT signaling and promotes cell proliferation.150,151 MPP3 (stromelysin) and vascular endothelial growth factor drive cancer cell invasion or tumor angiogenesis.152,153 Besides, SASP factors have been demonstrated to facilitate epithelial–mesenchymal transitions in the nearby premalignant epithelial cells, and resulted in cancer cell invasion and metastasis.154,155 Furthermore, many SASP factors can deteriorate the surroundings and remodel the tissue microenvironment, promoting cancer progression.156–159 Although the “seed and soil” theory has established for >100 years, the internal relationship between tumor microenvironment and cancer development becomes a research hotspot in recent years.160 The most abundant cells that compose the cancer microenvironment are cancer-associated fibroblasts (CAFs).161,162 Apart from the positive effect of CAFs on enhancing cancer proliferation and invasion, CAFs also contribute to tumor-associated inflammation.163,164 A study has shown that NF-kB signaling inhibition can abolish the effect of CAFs on promoting immune cell recruitment, neovascularization, and tumor growth in a mouse model of squamous skin carcinogenesis.163 Targeting on CAFs favors a positive role in extending life span and delaying cancer proliferation.165
Therapeutic strategies to attenuate inflammageing
The pathways discussed above, which drive age-related inflammation, are potential therapeutic targets to modulate inflammageing and consequently, beneficial for the aged. One study indicates that switching off the immune machinery via mediating NLRP3 inflammasome activity could halt or even reverse these age-related diseases.166 Congruently, inhibition of NF-kB signaling could reduce the sensitivity of aged HSCs to inflammatory stimuli, leading to better maintenance of the hematopoietic system.87 Pharmaceutical or genetic removal of p16INK4a-positive senescent cells delays the ageing onset and tumorigenesis in the mouse model, although very recently p16+ liver sinusoid endothelial cells are found to be indispensable for the health span in mice.7,167,168 Furthermore, given the evidence that obesity causes increased inflammation, bodyweight control, and healthy diet consumption will be beneficial for reducing inflammageing.61 Similarly, exercise helps decrease inflammatory factors, which play an anti-inflammation effect across multiple systems, including cardiac, blood, and muscle.75,169 Likewise, CR lower inflammation and protect against age-related diseases. A recent study explored the effect of CR on multiple tissues at the single-cell level and found that genes related to immunity, inflammation, and lipid metabolism are most affected by the CR.170 Intake of reverse transcriptase inhibitor Lamivudine (3TC),112 or natural compounds represent a safe and effective option that helps ameliorate many age-associated disabilities and diseases.93,112 Resveratrol supplement could reduce ovarian inflammation, attenuated spinal cord injury, and suppressed tumorigenesis by targeting NF-kB and mTOR signaling in a SIRT1-dependent manner.171–174 Similarly, metformin supplementation can also reduce SASP by blocking NF-kB activity,175 although the sex-dependent effects on life span remain controversial upon metformin treatment.176–180 Other interventions, such as sleeping modulation, thymic replacement to increase adaptive immune function, maintain gut integrity, and to improve environmental quality, could be potentially helpful in altering the dynamic of inflammation and preventing the inflammageing related disease.5
Although these attempts have significant impacts on treating ageing and inflammation-associated diseases, the spatiotemporal regulation of pro-inflammatory cytokine release and its landscape have not been completely understood. Besides, due to the limited sensitivity of the current technique, many unknown age-associated pro-inflammatory cytokines in blood await to be detected. Nevertheless, single-cell omics and lineage tracing would surely empower a deeper understanding of inflammageing, and provide better solutions to counteract age-related inflammatory diseases.
Ageing and epigenetics
Epigenetic regulation is used to classify heritable changes in gene expression that are not attributable to changes in DNA sequences.181 Mounting evidence suggests that epigenetic dysregulation is also an essential driver for cellular senescence and stem cell ageing.182–186 This part highlights the functional importance of epigenetic regulation in terminally differentiated cells and stem cells, in the context of altered DNA methylation, changes in histone modifications, and synergistic relationships between epigenetics and metabolism in ageing.
DNA methylation
In mammalian cells, DNA methylation occurs predominantly at CpG dinucleotides. Methylated cytosine (mC) is found throughout the genome at high frequency, predominantly located at promoter regions of genes,187 which plays a critical role in transcriptional silencing.188 DNA methyltransferases (DNMTs) DNMT3A and DNMT3B establish genome-wide de novo methylated nucleotides, DNMT1 maintain methylated nucleotides, and TET protein family-regulated DNA demethylation.187,189
DNA methylation during cellular senescence
During cellular senescence, the landscapes of DNA methylation are changed in a context-dependent manner. For instance, local hypermethylation could be induced by senescence-associated heterochromatin foci (SAHF), which recruit DNMT1 to focal sites,190,191 while oncogene-induced senescence fails to exhibit such alterations in DNA methylation,192 reinforcing the diverse characteristics of DNA epigenetic alterations during senescence. Interestingly, mtDNA methylation has also been changed in replicative senescent cells. One study reveals that 76% of mtDNA noncoding regions are hypomethylated in senescent cells,193 where p53-induced downregulation of mitochondrial DNMT represents a possible mechanism for the hypomethylation of mtDNA.194 However, no significant global DNA methylation changes are observed in multiple forms of stress-induced premature senescence, including doxorubicin-induced senescence, irradiation-induced senescence, oncogene-induced senescence, and nonpermissive temperature-induced senescence.195,196 The different types of cell senescence-associated DNA methylation also lead to distinct gene expression patterns and cell phenotypes. Further investigations are required to explore the differences among the epigenetic mechanisms underlying replicative senescence and stress-induced premature senescence.
Of note, DNA methylation changes usually lead to a decline in the number and function of stem cells, like self-renew ability defect and differentiation bias, which are often similar to those observed in the ageing process.1 The effect of ageing on the DNA methylome of purified adult stem cells from young and old mice was detected by global methylated DNA immunoprecipitation sequencing.197,198 These studies show that HSCs display global DNA hypermethylation during ageing197,198 concomitant with decreased 5-hmC levels.198 Furthermore, ageing muscle stem cells (MuSCs) showed a slight increase in their DNA methylation age at the single-cell level.199
Ageing-associated gains of DNA methylation were also over-occupied at loci associated with polycomb gene (PcG) binding and some transcription factors binding in old HSCs.197,198,200 Similarly, PcG targets were also hypermethylated in MSCs during ageing, though a predominance of ageing-associated hypomethylation as reported.201,202 The correlation between age-related changes in DNA methylation and age-related changes in transcription was also examined in these studies, suggesting that the ageing process could disrupt these PcG proteins or transcription factors to bind DNA and regulate transcription.201,202
Sub-telomere region DNA methylation and ageing
Senescence-associated DNA methylation alterations are engaged in the regulation of telomere dysfunction. Telomere damage is not only determined by the telomere length, but also controlled by the epigenetic conditions in telomeric/sub-telomeric regions.203 In young wild-type cells, sub-telomeric regions are hypermethylation in CpG islands, and enriched by HP1a protein and repressive histone modification marks (H3K9me3 and H4K20me3), and lack of permissive histone modification marks (H3K9ac and H4K20ac).203 However, MEF cells from telomerase-deficient mice exhibit more “open” state of telomeric/sub-telomeric chromatin, as indicated by loss of CpG island DNA methylation, loss of repressive histone modifications (H3K9me3 and H4K20me3), decreased CBX3 binding accompanied by increased H3 and H4 acetylation, and increase the level of H3K9ac and H4K20ac.203,204
Our work discovered that the deletion of DDR factor Gadd45a rescued the heterochromatin remodeling via base excision repair-mediated active DNA demethylation in sub-telomeric regions in telomere-deficient cells, which generates an uncondensed chromatin structure to promote DDR signaling.204 Although Gadd45a has been linked to the global DNA methylation and transcriptional regulation, Gadd45a loss does not change the global DNA methylation pattern in our experimental setting,204 indicating that manipulation of the Gadd45a gene could delay organ ageing, and prolong the health span and life span of premature ageing mice.
DNA methylation and human ageing clocks
Age predictors based on a small set of CpG sites DNA methylation levels have been developed for humans and several other species.205 Early studies have found that age-related DNA hypomethylation patterns occur in many body tissues of the elderly.187,205–209 However, detailed analyses of several studies uncovered CpG islands site-specific DNA hypermethylation associated with ageing tissues,200,206,210–213 and these hypermethylation changes are generally related to age rather than the tissue type,211,214,215 suggesting some level of synergetic control of DNA methylome during ageing.
The DNA methylome of different organs or tissues can be used to predict the biological age.216 For example, DNA methylation in human peripheral blood has been manifested to correlate with ageing.217,218 A recent analysis of human blood samples does confirm that, with age, most hypermethylation is not related to changes in cell composition, but directly related to ageing.213 The increase in DNA methylation age of blood over 5 years was associated with a 16% higher mortality rate than age.219,220 Several research groups have observed an acceleration in DNA methylation age in some age-associated diseases, including AD, cardiovascular disease, and cancer.221,222 DNA methylation has been reported to regulate neuronal differentiation in early CNS development. A global methylome reconfiguration was associated with synaptogenesis ranging from mammalian fetal to adult brain development.187 In human, 353 CpG sites were identified to form an epigenetic age clock.223 The DNA methylation levels change with normal ageing in many tissues, including the brain, peripheral blood.223 Gene-specific DNA methylation changes are involved in rewarding in a context-dependent manner and are essential for memory formation, neurogenesis, and neuronal plasticity.224,225 Lower levels of DNA methylation on the promoter of target genes in peripheral blood samples have been reported to contribute to AD.226,227 The expression of DNMT1 and global 5mC and 5hmC were also shown to be decreased in AD neurons and hippocampus.228,229 Marioni et al.230 showed that greater DNA methylation acceleration is correlated with a lower cognitive score, weaker grip strength, and poorer lung function in humans during later life. Horvath et al.231 found accelerated DNA methylation age in Down syndrome patients with clinical signatures of “accelerated ageing.” There is also evidence that frailty, a syndrome with a pronounced association with age-related phenotypes, has a significant association with DNA methylation age, but not with telomere length.232 Zheng et al.233 pointed out that DNA methylation age estimated from blood tissue can also be used to predict cancer incidence and mortality. The apparent genetic clock derived from DNA methylation is better at estimating actual age than transcriptome and proteomic data or telomere length.209 In conclusion, the age-related DNA methylation changes may reflect the biological age to some extent, therefore constituting the biological age clock.
Histone modifications
Histone modification is an additional epigenetic regulatory layer that is more complicated than DNA methylation. The unstructured N-terminal of histones can be used for posttranslational modification, including acetylation, methylation, phosphorylation, sumoylation, ubiquitination, and other modifications that change chromatin structure and accessibility. These modifications can regulate transcriptional activity. Here, we focus on histone acetylation and methylation, which are the two most well-studied markers in cellular senescence and ageing.
Histone (de-)acetylation and (de-)methylation during cellular senescence
Global decreases in H4K16Ac, H3K4me3, H3K9me3, and H3K27me3, while increases in the level of H3K9Ac and H4K20me3 occur in replicative senescent cells.234–236 Such histone modifications have also been found in stress-induced premature senescence cells.237 However, the histone marker alteration patterns differ in different stress-induced premature senescence cells based on various stress factors.237 Thus, the diversity of histone modifications in senescence cells may cause diverse gene expression patterns and senescence phenotype.
The role of senescence-associated histone modification changes in the senescence regulatory mechanisms has been broadly explored. The histone methyltransferases (HMTs) complex, polycomb repressive complex (PRC), was investigated in repressing the p16 gene expression.238 PRCs binds directly to the p16 locus and induces H3K27me3 occurrence, which leads to transcriptional suppression of p16.239 Besides, cell senescence can be delayed via inhibition of histone acetyltransferases (HATs) and induced by inhibiting histone deacetylases (HDACs).240–242 A recent study revealed that HAT p300 is a primary driver of the replicative senescence phenotype via a high-throughput screen.242 The depletion of p300 suppresses senescence-related gene expression, ensuing delayed senescence.242 Therefore, p300 is a candidate target for anti-ageing therapeutics.
However, certain histone modifications in senescence and ageing may be contrasting and even paradoxical. Tissue or cells from ageing organisms show increased H4K16ac, H4K20me3, or H3K4me3, along with decreased H3K9me3 and H3K27me3, which are quite different from cellular senescence.243 The difference in histone modification between cellular senescence and organismal ageing may attribute to multiple sources for ageing-associated damage, such as mutation, reactive oxygen species (ROS), and environmental stress, that change the epigenetic pattern of ageing and differ from cell senescence.244,245 In turn, epigenetic therapies based on histone modification that target cell senescence may inhibit the accumulation of senescent cells. Naturally occurring activators of SIRT1, including resveratrol, nicotinamide riboside, and nicotinamide mononucleotide, limit the accumulation of senescent cells.246–249
Histone acetylation and methylation during stem cells ageing
Histone associated epigenetic changes in adult stem cell ageing have been reported for numerous stem cell populations, remarkably HSCs and MuSCs.125,250 An increase in the level of the repressive histone modification H3K27me3 has been observed in both HSCs and MuSCs.251 However, H3K4me3, an active histone modification mark, shows an increase in HSCs but decreases marginally in MuSCs with age.250
Many genetic studies have revealed the critical role of HDACs and HATs activity in stem cell function. In the hematopoietic system, the significant phenotypes related to CREB-binding protein suggest the vital role of HATs in HSC function.252–254 Mononuclear leukemia zinc finger protein Moz is a kind of HATs translocation protein in human acute myeloid leukemia. During embryo development, the gene is eliminated, resulting in the severe loss of HSC and other progenitors with limited lineage.255 These results strongly suggest that histone acetylation is necessary for HSC self-renewal. HAT activity also plays an essential role in the homeostasis and function of HSCs and precursor cells. Eighteen mammalian HDACs have been identified and divided into four families. Class I HDAC plays a role in differentiation and seems to have a high degree of functional redundancy.256,257 In the hematopoietic system, loss of HDAC class I leads to a decrease in bone marrow cells,258 and in some cases, causes loss of stem cells and progenitor cells.259 In addition, the simultaneous knockout of HDAC3, HDAC5, and HDAC7 (class I and class II HDACs) resulted in the CDKN1A (p21) upregulation and inhibition of cell proliferation,260 similar to p21 induction and cell cycle arrest in human mesenchymal stem cells after drug-induced inhibition of HDAC activity.261
Class III HDAC includes the NAD+-dependent sirtuin family, while other HDAC families need Zn2+ as a cofactor. In mesenchymal stem cells, SIRT1 is related to differentiation into bone and cartilage by deacetylation of β-catenin.262 In adult neural stem cells, the loss of SIRT1 leads to increased self-renewal and proliferation with the increase of oligodendrocytes.263 Similarly, SIRT2 also hinders the differentiation of oligodendrocytes.264 In adult HSCs, loss of SIRT6 leads to enhanced Wnt signaling, decreased self-renewal, and over-proliferation.265 Similarly, SIRT1 guides the differentiation of epidermal stem cells by promoting the production of keratinocytes.266 In HSCs and MuSCs, SIRT1 loss leads to premature cell differentiation, implicating it is a regulator of self-renewal of these cells.267–269 Robust SIRT1 activity is also related to maintaining the quiescence of MuSCs, while the decrease of SIRT1 activity measured by increasing H4K16ac is related to the decline in NAD+ level in activated MuSCs.267 The increase of the H4K16ac level in these activated stem cells is due to the transformation of metabolism from fatty acid oxidation to glycolysis. Interestingly, although HSCs utilize glycolysis rather than oxidative phosphorylation (OXPHOS) and therefore have low levels of available NAD+, SIRT1 activity seems to be needed to regulate histone acetylation to maintain proper HSC function in ageing.268
The level of H4K16ac of aged HSCs was increased by immunostaining.270 Interestingly, compared with the young HSCs with high-level polarized H4K16ac expression, the H4K16ac level in the old HSCs decreased, concurrent with significantly changed cell distribution.270 The drug inhibition of Cdc42 and partial recovery HSC function reversed the change of H4K16ac in aged HSCs.270 Although the exact role of altered H4K16ac in aged HSC remains elusive, H4K16 deacetylation has been shown to hinder DDR and repair of double-strand breaks.271 Therefore, H4K16 deacetylation in aged HSC may contribute to the accumulation of DNA damage.272
In contrast to histone acetylation, histone methylation can be used as a context-dependent inhibitor or permissive marker, which indirectly regulates gene expression. Although histone methylation occurs on lysine and arginine residues, most stem cell studies detect methylation catalyzed by HMT on lysine residues.251 Changes in some other chromatin features, most of which are also altered with age, have been shown to regulate stem cell function.273 For instance, the H3K27me3 demethylase UTX is essential for MuSC-mediated muscle regeneration.274 The increase of histone inhibitory markers, such as H3k9me3 and H3K27me3, were observed in the aged MuSCs and HSCs, which indicated that the heterochromatin increased gradually in the ageing process.251 H3K4me3, an active chromatin marker, was enriched in the old HSCs, suggesting that epigenetic enhancement was observed in the transcriptional activation of stemness related genes.198,251 Contrary to what was observed in HSCs, the detection of H3k4me3 in MuSCs showed little difference between the cells isolated from young and old mice.250
Furthermore, H3K27me3 was added by polyclonal inhibition complex 2 (PRC2). When compared with the young HSCs, the level of H3K27me3 signals in aged HSC is essentially the same, but the coverage and intensity of H3K27me3 signals in aged HSC are expanded.275 However, unlike the young MuSCs, the aged MuSCs showed a transition to the euchromatin state after activation, with increased histone acetylation while decreased in H3K27me3.250,276 How the changes of H3K27me3 in the ageing process affect the function of stem cells in muscle and blood remain unanswered, yet this inhibitory marker may limit the regeneration potential of these stem cells, which will decrease with age.
Epigenetic defects in progeroid laminopathies
The reconstruction of the chromosomal domain is also a feature of senescent cells. Senescent cells undergo substantial changes in three-dimensional chromatin organization globally, as evidenced by the combination of whole-genome chromosome conformation capture (Hi-C), fluorescence in situ hybridization, and in silico modeling.277 Among them, lamin A/C represents an epigenetic regulator of ageing partially due to its direct interaction with chromatin in a specific DNA sequence termed the lamin A-associated domains (LADs).278,279 In the process of cell senescence induced by an oncogene, the remodeling of LADs results in the unexpected recruitment of the decompressive sequence to the nuclear plate.280,281 Lamin A/C also promotes epigenetic changes by interacting with epigenetic enzymes.282 Under physiological conditions and in young cells, lamin A/C interacts with SIRT1 and enhances its deacetylase activity. It also promotes SIRT6 function during DNA repair and is found to recruit HDAC2.283–285 Importantly, the interaction of laminin with SIRT1, HDAC2, and SIRT6 decreased when protein A or progerin accumulated.283,285
The level of H3k9me3 in Hutchinson-Gilford progeria syndrome (HGPS) and mandibulofacial dysplasia type A (MADA) cells were decreased.286,287 HGPS cells lose heterochromatin protein HP1 and other heterochromatin markers, including H3K27me3 and H4K20me3.287,288 In addition, increased H4K16ac and H3K9ac were reported in HGPS cells and MADA cells, respectively.285 Congruently, many changes in DNA methylation levels at specific CpG sites were observed in the immortalized B cells of HGPS patients.289 Interestingly, according to the epigenetic clock,231 the same cells are older than expected. LADs were also involved in the epigenetic landscape remodeling of HGPS cells.290 In HGPS cells, progerin destroys the interaction of lamin A/C with SIRT1 and SIRT6, affecting chromatin localization and deacetylase function.283,284
Epigenetic regulation of retrotransposable elements during ageing
Two subtypes of non-LTR retrotransposons, LINEs and SINEs, together make up nearly half of the human genome.291–293 Heterochromatin region in young cells and organisms silences reversible transposable factors, but due to the lack of regulation of higher-order chromatin structure, they are activated in the context of cell senescence and tissue ageing.294 Interventions to prolong life, such as CR, decreased retrotransposon expression in elderly mice.295 In the liver and muscle cells of old mice, CR delayed the loss of constitutive heterochromatin and inhibited the expression of repetitive components, including LINE1 and satellite components, which were in the centromeric, pericentromeric, and telomeric region. CR also inhibited the interaction between microRNA and chromodomain helicase DNA-binding protein 1, thus preventing the activation of retrotransposons induced by ageing and poor diet.296 Therefore, blocking the transcription of endogenous retrotransposon factors through diet restriction can improve the age-related phenotype, and support the view that retrotransposon leads to ageing and age-related diseases.
SIRT6 mono-ADP ribosylates KAP1 and promotes KAP1 interaction with HP1α, packaged as inhibitory heterochromatin in the LINE1 element. SIRT1 also binds to and inhibits major satellite repeats in yeast and mammalian cells.297 Another known heterochromatin regulator, retinoblastoma protein (Rb), antagonizes the activation of LINE1 in senescent cells and decreases the percentage of Rb on the LINE1 promoter in senescent human cells and senescent mouse tissues.298 Homologous protein transcription factors inhibit the expression of LINE1 in adult dopaminergic neurons299 and prevent its degeneration. In addition, partial overexpression of histone H3 and H4 reversed the transcription defects observed during ageing and reduced the reverse transcriptional transposition, which indicated that the increased reverse transcriptional transposition in old yeast was the result of histone loss during ageing.291 In addition, CRISPR-Cas9 screening also provided a genome-wide gene investigation related to the retrotransposition control of LINE1, revealing that the vertebrate-specific chromatin-modifying complex human silencing hub and the subunit of MORC family CW zinc finger protein 2 promoted the deposition of H3K9me3 to silence the transcription of LINE1 element.300,301 Recent studies have further shown that inhibition of the transposition of LINE1 by nucleoside reverse transcriptase inhibitors can inhibit the secretion of SASP and IFN by senescent human fibroblasts, and prolong the life span of the D. melanocyte model without DICR2, a heterochromatin structure regulating gene.112 These findings collectively suggest that epigenetic remodeling plays a vital role in the anti-ageing process, preventing the activation, and mobilization of retrotransposons by increasing heterochromatin stability. Genome-wide quantitative analysis will provide new insights into the frequency, structure, and location of retrotransposons during ageing, and clarify their overall contribution to ageing and rejuvenation.
Sirtuin-mediated epigenetic regulation in stem cell ageing
Metabolism and epigenetics are closely linked, which together affect the ageing of the body. The availability of key nutrients, such as glucose, fatty acids, and amino acids, directly affects organisms’ life span. Glycolysis disorders have been shown to prolong life span302 and supplement D-glucosamine, an antagonist of glucose, which can damage glucose metabolism and prolong the life span of nematodes and mice.303 In addition, amino acid and lipid composition are closely related to age. They can be used as indicators of health span, as shown by the metabonomics analysis of plasma of healthy young and old individuals.304 However, mitochondrial metabolism is the most correlative with epigenetic regulation. Small molecules, such as NAD+, alpha-ketoglutarate (α-KG), and coenzyme A derived from mitochondria, have changes in the content of these metabolites during the ageing process and affect the activity of enzymes that use these metabolites as substrates for epi-modification.
The sirtuin protein family is one of the first known epigenetic enzymes and a key regulator of ageing and CR.305,306 In mammals, the sirtuin family contains seven Sir2 homologs, Sirt1–Sirt7, whose expression or enzyme activity increases after CR. It is worth noting that CR can prolong the life span of mice by inducing SIRT1 expression.4 Congruently, SIRT1 overexpression mimics the beneficial effects of CR. SIRT6 deficiency resulted in a shortened life span in mice and early death in nonhuman primates.110,307 In contrast, SIRT6 overexpression and CR induced SIRT6 activation delayed ageing.282 In addition, sirtuin activators, such as SRT1720 or SRT2140, can increase the health span of obese mice and the life span of mice on a standard diet.308,309 These longevity-extending effects of sirtuins are realized mainly by their enzyme functions, such as deacetylase and single ADP ribosyltransferase, especially when histone is used as the substrate. Recent findings of sirtuins in epigenetic regulation of adult stem cells are summarized below.
SIRT1
SIRT1 is very important to maintain the static and regeneration ability of HSCs under environmental stress and ageing conditions.310 Under normal circumstances, no abnormality was seen in the hematopoietic cells of SIRT1-KO mice.311 However, the hematopoietic commitment of SIRT1-deficient HSC is impaired in vitro, concurrent with a reduced survival rate of hematopoietic progenitor cells, especially in hypoxia or delayed addition of growth factor.312 Moreover, conditional ablation of SIRT1 in adult hematopoietic stem/progenitor cells (HSPCs) autonomously induces HSPCs expansion and loss of long-term repopulation under stress.310 This stress-induced loss of HSC function is associated with genomic instability, p53 activation, and increased DNA damage in SIRT1-deficient HSPCs.310 SIRT1 deficiency also resulted in a significant increase in H4K16ac and upregulated the expression of HOXA9, a key regulator of HSPC function and proliferation.268,310,312 In general, SIRT1 is essential to maintain different ASC pools by maintaining quiescence, self-renewal, and regenerative capacity, especially in response to stress and injury.
SIRT2
Sirt2 is a mammalian sirtuin, which primarily exists in the cytosol and has deacetylase activity.313 Luo et al. suggested that mitochondrial stress and SIRT2 inactivation lead to the activation of inflammatory corpuscles in NLRP3 of HSC and the decline of HSC ageing function.48 Specifically, diminished SIRT2 expression augmented mitochondrial stress, while SIRT2 overexpression or NLRP3/caspase-1 inactivation attenuated the impaired regenerative capacity of aged HSCs.48
Mitochondrial sirtuins (Sirt3, 4, and 5)
In addition to the three nuclear sirtuins (Sirt1, 6, and 7), mitochondrial sirtuins (mtSIRT) include three members, i.e., SIRT3, SIRT4, and SIRT5, which are all involved in regulating stem cell metabolism.314 Among them, mitochondrial SIRT3 was found to be highly enriched in HSCs.315 Brown et al. suggested that SIRT3 deficiency reduced the HSC pool of old mice and impaired the self-renewal of HSC during continuous transplantation stress, partly due to the hyperacetylation of superoxide dismutase (SOD2) and subsequent increase in oxidative stress.315 SIRT3 decreased with age and its overexpression in HSC reduced oxidative stress and maintained reconstructive ability.315 SIRT4 expression is upregulated during cell senescence of different types.316 SIRT4 overexpression can induce trophoblast stem cell senescence.317 SIRT5 has demalonylase, deglutarylase, and desuccinylase activities318,319 that regulates ammonia detoxification.320 Although not well studied in stem cells, SIRT5 was thought to desuccinylate and activate SOD1 to maintain a low ROS level in stem cells.321
SIRT6
Specific deletion of Sirt6 in HSCs of adult mice resulted in the amplification of HSPC, which is associated with acetylation of H3K56 and the increase of transcriptional factor in the Wnt signaling pathway.265 The increased proliferation further hampered HSC quiescence and led to HSC depletion. As a result, the long-term regeneration capacity of Sirt6-defective HSCs was severely impaired.265 Furthermore, Sirt6-mediated stress resistance also helps maintain the in vitro function of MSCs.322 Human bone marrow mesenchymal stem cells derived from SIRT6-deficient mice were susceptible to oxidative damage due to the increased level of ROS.322 Mechanistically, the SIRT6-mediated antioxidant effect by H3K56ac deacetylation activated the Nrf2-mediated antioxidant gene.322 Although it is not clear how the deacetylase acts as a coactivator to promote anti-oxidation genes in MSC, this study shows that SIRT6 is crucial to mediate the anti-stress and anti-ageing effect of MSC.
SIRT7
Sirt7 is also highly expressed in HSC, which exerts its regenerative ability by regulating the unfolded protein response in mitochondria.323 Quiescent HSCs are maintained in a state of inactive metabolism that can be easily activated via boosting mitochondrial content. Mohrin et al. showed that mitochondrial protein folding stress (PFSmt) induced the interaction between Sirt7 and Nrf1, and inhibited the expression of mitochondrial ribosomal protein and mitochondrial translation factor. Sirt7-deficient HSCs exited quiescence and exhibited an ageing phenotype, including the increase of PFSmt, apoptosis, a decrease of reproductivity, and biased bone marrow differentiation. On the contrary, the upregulation of Sirt7 improved the regenerative ability of HSC in the elderly.323
Together, posttranscriptional and posttranslational regulation via sirtuins maintains stem cell function to cope with various stress stimuli and ageing. Although mtSIRT is less studied in stem cell function, three nuclear sirtuins (Sirt1, 6, and 7) participate in the static control of stem cells, which is of considerable significance to maintain the regenerative ability of stem cells and prevent premature ageing. Pharmacological interventions targeting sirtuins may hold great promises to counteract stem cell ageing and, therefore, tissue homeostasis.
Approaches in epigenetic control of rejuvenation
Somatic cells are induced to restore pluripotency through various reprogramming strategies, the most common of which is the overexpression of four transcription factors Oct4, Sox2, Klf4, and Myc (referred to as OSKM).324 The mouse nucleus reprogrammed by OSKM can produce viable embryos and further develop into fertile adults without showing premature ageing, indicating that the time sequence of the donor nucleus has been reset. Consequently, core reprogramming appears to be able to reset ageing clocks. Partial reprogramming of the adeno-associated virus vector expressing OSKM can significantly improve axon regeneration after injury.325 The expression of OSKM leads to extensive remodeling of chromatin, accompanied by alterations of epigenetic enzymes and other transcription factors. For example, as a pioneer factor, Oct4 can loosen heterochromatin and reduce the global levels of inhibitory H3K9me2, H3K9me3, and 5-methylcytosine,326 which are obstacles in reprogramming.327,328 In addition, during OSKM-mediated pluripotent reprogramming, the epigenetic memory of the primitive cells is largely eliminated and rewritten in the subsequent differentiation process.329
Many drugs, compounds, and supplements with anti-ageing properties have also been identified and attracted considerable attention by pharmaceuticals, which can prolong the life span and healthy ageing of model organisms (such as mice, Drosophila melanogaster, and Caenorhabditis elegans).330 For instance, metformin regulates the activation of AMPK, which directly governs the activities of several epigenetic enzymes, such as HATs, HDACs, and DNMTs.331,332 In addition, metformin restores AMPK-mediated phosphorylation and stabilizes Tet2, thereby preventing changes in 5-hydroxymethylcytosine levels.333 Aspirin supplementation has also been shown to generalize the anti-ageing effect of CR.334 The accumulation of senescent cells is one of the signs of ageing. Senolytics selectively eliminate senescent cells, representing a new anti-ageing drug335 that may delay the ageing process. Consistently, eliminating p16INK4a-positive senescent cells was able to prolong the life span of early ageing model mice and wild-type mice.167,336
Metabolic intermediates and by-products of the tricarboxylic acid cycle act as cofactors and substrates of various epigenetic enzymes, including acetyl CoA for acetylation and S-adenosylmethionine (SAM) for methylation.337,338 In addition, α-KG, an intermediate product of the tricarboxylic acid cycle, induces DNA and histone demethylation by activating the jumoniji C domain-containing demethylase and lysine demethylase. A recent study showed that the increase of α-KG activated JMJD3 (histone H3K27 demethylase) and PHF8 (histone lysine demethylase H3K9me1/2 specificity), leading to the removal of inhibitory markers and the induction of mitochondrial unfolded protein response gene expression. These changes are enough to prolong the life span of nematodes.339,340
Another important metabolite is NAD+, which is the cofactor of sirtuins. It connects the gene regulation of epigenetic with mitochondria. High levels of NAD+ can improve mitochondrial function, supplement the stem cell pool, and prolong the life span in mice. Supplementation of NAD+ precursor to aged mice can delay the decline of mitochondrial function, improve muscle, nerve, and melanocyte stem cell performance, alleviate age-related physiological decline (for example, type 2 diabetes and cognitive impairment), and prolong life span.341
Apart from the investigations of novel epigenetic interventions in pursuit of rejuvenation, the study of epigenetic regulation at the single-cell resolution has deepened our understanding of the diversity of the single-cell state and the process of cell state maintenance.342–344 Specific single-cell DNA sequencing provides epigenetic information of DNA modification, DNA accessibility, and chromosome conformation, thus deepening the understanding of the impact of epigenome on the transcriptome. A variety of single-cell epigenome-sequencing techniques have been developed, such as single-cell sodium bisulfite sequencing for DNA methylation detection,345 single-cell chromatin immunoprecipitation sequencing for the identification of histone modification and protein–DNA interaction, single-cell transposable accessible chromatin sequencing, and Hi-C sequencing for the evaluation of chromatin accessibility and chromosome conformational information.346–348 These techniques have been combined with single-cell transcriptome to study gene regulatory profiles and analyze cell heterogeneity. Recently, a new method has been established to record transcriptome synchronized with chromatin accessibility, thus enabling the analyses of the functional relationship between these two characteristics in the same cell.349
The intensity of the regulatory link between epigenetic modification and transcription may vary in different developmental stages and cell types, adding a layer of complexity and uncertainty in delineating the specific spatiotemporal transcriptional regulation. The application of these single-cell and multi-omics techniques enables us to understand the regulation of epigenetic factors on gene expression under physiological and pathological conditions.350
In sum, stem cells residing in different tissues accumulate defects during ageing, preventing stem cells from repairing damage, and maintaining tissue homeostasis, with altered epigenetics a potential hallmark (Fig. 5). Existing evidence has suggested that the developmental pathway in embryogenesis is the key to epigenetic regulation that contributes to stem cell ageing. These observations raise important issues for future research. While it is unclear whether epigenomic changes are an essential element of ageing and how these changes occur during ageing remain elusive, many attempts are on the way to depict the full landscape of acute and chronic changes in the epigenetic modification during stem cell ageing. Given that mutations in epigenetic modifiers have become a marker of the ageing hematopoietic system, understanding the ageing-related clonal dominance mechanism of stem cells with mutations in epigenetic modifiers is of great interest. Finally, epigenetic integration of damage signals, as a hardly neglected cause of stem cell and organismal ageing, has brought new hope for the translational pathway. Since the epigenome changes are largely reversible in principle, manipulating epigenetic imprints holds great prospects in improving tissue maintenance, regeneration ability, and, ultimately, extending health span.
Fig. 5.

Epigenetic regulators and interventions in ageing. Age-dependent epigenetic remodeling by various (de-)methylases and (de-)acetylases affects chromatin accessibility, thereby regulating gene transcription and expression. Interventions, such as calorie restriction and drug treatment, may reverse the age-dependent degeneration in an epigenetic-modifying manner. HDACs histone deacetylases, DNMTs DNA methyltransferases, TET ten–eleven translocation, HMT histone methyltransferase, TSS transcriptional start site, SAM S-adenosylmethionine, α-KG alpha-ketoglutarate, NAD nicotinamide adenine dinucleotide
Ageing and metabolism
The unique metabolic signature of senescent cells shapes the distinct senescent phenotype; for instance, augmented glycolysis represents one of the metabolic hallmarks during replicative senescence. Changes in intracellular and extracellular metabolites may lead to the consequence of senescence in adjacent cells, aka a bystander effect of the senescence-associated metabolic pattern.351 This section will discuss the metabolic regulation of cell senescence that interacts with many aspects of cellular physiology, including redox balance, genomic integrity, immunometabolism, proteostasis, organelle homeostasis, and metabolic signaling pathways and interventions (Fig. 6).
Fig. 6.

Intrinsic cues to metabolic reprogramming during ageing. Six aspects, namely protein homeostasis, genetic and epigenetic instability, organelle dysfunction, redox imbalance, immune response, and altered signaling pathways, lead to the metabolic regulation of ageing
Metabolic regulation of redox balance in ageing
Redox reactions occur throughout the cellular metabolism with the production of a small number of reactive oxygen radicals.352 Studies using model organisms, such as yeast, nematode, drosophila, and mouse have shown that cell senescence is closely related to dysregulation of redox balance.353–356 With ageing, increased oxidative stress featured by increased oxidized glutathione (GSSG), while lowered levels of glutathione (GSH) and reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) have been observed, which causes lipid, protein, and DNA damages (e.g., DNA single- or double-stranded breaks, fatty acid chain breakage, increased membrane fluidity, and protein hydrolysis, inactivation of proteases, etc.). All of the above injuries can lead to aberrant cellular metabolism and signal transduction, culminating by altering cell fate.357 Consequently, systemic clearance of the excess free radicals through enzyme-based, such as SOD2, catalase, glutathione peroxidase, coenzyme Q10, etc.), and nonenzyme-based (e.g., vitamins, β-carotene, selenium, GSH/GSSG, cysteine, etc.) defense systems are required to maintain physiological balance. It has been generally accepted that redox imbalance caused by boosting of ROS and reactive nitrogen species (RNS) production, or “oxidative stress”, and elevated intracellular NADH:NAD+ ratio, or “reductive stress”, reduced OXPHOS lead to the disorders of the mitochondrial electron transport chain (ETC), therefore, decreased ATP synthesis and cell respiration.358
ATP synthesis utilizing various substrates is fundamental to cell viability. NAD+/NADH and FAD/FADH2 redox couples synthesize most ATP; in particular, NAD+ involves regulating both redox and metabolic homeostasis.359 Accumulation of mitochondrial proton donors (NADH and FADH2) attenuates OXPHOS through the ETC, thereby causing reductive stress and increased ROS production. ROS exerts multiple functions in various pathophysiological responses closely related to augmented NADH/NAD+ ratio and accumulation of L-2-hydroxyglutarate, a reductive metabolite that buffers reductive stress via inhibiting glycolysis and the Kreb’s cycle.360 Increased intracellular NADH concentration caused by hypoxia will generate a reverse electron transfer, resulting in higher succinate levels and increasing oxidation, and subsequently higher ROS levels.360 A similar process has been seen in the mechanically overloaded heart. Thus, a stable ratio of NAD+/NADH is essential to maintain cellular homeostasis. Any alteration in the ratio of NAD+/NADH will cause oxidative or reductive stress, which may lead to accelerated cell senescence. Accordingly, restoration of NAD+ via precursors may reestablish the NADH/NAD+ ratio, thereby reducing the cardiovascular injuries and attenuating cell senescence.361–363 Indeed, mounting studies have shown the influence of exogenous NAD+ repletion in the regulation and homeostasis in different models (gut, heart, muscle, etc.).364–369 A recent study shows augmented circulating α-hydroxybutyrate levels associated with increased NADH/NAD+ ratio and impaired glucose metabolism,370 while a normalized NADH/NAD+ ratio can achieve by constructing LOXCAT-mediated conversion of lactic acid to pyruvate.371
Although targeted regulation of the NADH/NAD+ ratio or NAD+ level has great potential as an intervention for cell senescence and organ ageing,372 there are still some critical questions about the relationship between NADH/NAD+ and cell senescence. For instance, in vivo NADH/NAD+ ratio and NAD+ level (particularly in different subcellular organelles) cannot be determined by conventional biochemical analysis. The exact mechanisms by which exogenous NAD+ and NADH work remain elusive. Furthermore, under the condition of ageing-related metabolic remodeling, the spatiotemporal distribution of NADH/NAD+ or NAD+ in different cellular compartments is still unclear. Given the crucial roles of NADH/NAD+ and NAD+ in metabolic regulations of redox balance and ageing, new technologies are urgently needed to provide a landscape of precise dynamics of NADH/NAD+ or NAD+ at subcellular organelle, cellular, and tissue levels. Some attempts have successfully detected NADH/NAD+ ratio or NAD+ concentration in vivo by constructing genetically encoded fluorescent probes and reporter mice,373,374 which hold high potentials to delineate the compartmentalized distribution of NADH/NAD+ ratio or NAD+ concentration upon various ageing-related stress stimuli.
Another vital player in redox balancing is the pentose phosphate pathway (PPP). PPP in the cytoplasm converts glucose into 5-phosphate ribose and produces NADPH.375 The reductive NADPH functions as an antioxidative mechanism produced during the oxidation phase, starting with the conversion of glucose-6-phosphate to 6-gluconolactone and NADPH. This is an irreversible reaction, catalyzed by glucose-6-phosphate dehydrogenase (G6PDH, a rate-limiting enzyme of this pathway), while inhibited by NADPH feedback. Next, the lactone hydrolyzes to 6-phosphogluconic acid that is further dehydrodecarboxylated to form ribulose 5-phosphate, concomitant with another molecule of NADPH. NADPH is used for the reductive reactions in the synthesis of biomolecules, such as fatty acids, cholesterol, deoxyribose, tetrahydrofolic acid, etc. NADPH is also used to reduce GSSG, thus maintaining redox balance in cells.375 The flux of PPP is mainly determined by the amount of reduction force. Besides, high ROS levels in senescent cells also require more NADPH to maintain redox balance.376
The causal role of oxidative damage in the ageing process remains controversial, partly because of the absence of a clear correlation between the efficacy of antioxidant defenses and extended cellular function or longevity.377 The lack of human studies on toxic oxidative metabolites in tissue ageing also makes scientists tiptoe cautiously at the crossroad. Nevertheless, existing evidence demonstrates that ageing-related pathological fibrosis can be attenuated via the Nox-4–Nrf2 antioxidant axis.378 Other interventions that promote cysteine metabolism and hydrogen sulfide production also exhibit therapeutic benefits and delay tissue ageing.357,379 Mitochondria represent the central platform of cellular metabolism and contain their own genome. The accumulation of mutations in mtDNA during ageing has been validated in many cell systems,380 with ROS as a principal cause.381 Despite several studies implicating the role of ROS in cell senescence, others also suggest that it may not necessarily be the case. One study using an empirical mathematical model (stochastic step model of replicative senescence) suggests that increased mitochondrial ROS production in replicative senescent cells is a consequence of the senescence phenotype rather than the reverse.382 Another report shows that overexpression of the mitochondrial localized antioxidant SOD2 and the mitochondrially targeted catalase are insufficient to inhibit the senescence phenotype in hyperoxia-induced senescent cells.383 Because mitochondrial and non-mitochondrial enzymes produce ROS during hyperoxia (70% O2), the inability of mitochondrial antioxidants to reverse growth arrest in hyperoxia-induced senescence suggests that cytosolic ROS may assist growth arrest. Hence, the mechanisms involved in linking mitochondrial ROS and cell senescence still need to be further studied. Nonetheless, studies on genetically manipulated mouse model suggested that many metabolic pathways have been found to involve oxidative stress management, and thus regulating cell function and maintenance, including the Akt/mTOR,384 FoxO,385 AMPK,386,387 the ATM-BID,388 Nrf2/Keap1,389 and the sirtuins,315 and so on. Together, these studies suggest that cell senescence is closely correlated with the metabolic and redox state, influencing the intracellular homeostasis of ROS and its functionalities.
Metabolic regulation of DNA damage in ageing
Cells have evolved various systems to regulate nutrient availability to maintain homeostasis, and have also developed active DNA repair machinery to avoid detrimental genomic instability during ageing, with two distinct cellular activities highly coordinated.102,390 For example, premature ageing phenotypes in excision repair cross-complementing group 1 knockout mice and progeroid Xpg mice can be attenuated after feeding a restricted diet.391 During the neonatal, a high oxygen environment induces cardiomyocyte cell cycle arrest through DDR and directs perinatal cardiac metabolic switch.392,393 To date, many key regulatory molecules exerting dual roles in regulating DNA repair and cellular metabolism have been identified, including p53,394 sirtuins,395,396 poly(ADP-ribose) polymerases (PARPs),397 and ATM.398 All these factors converge to the telomere at the end of the chromosome in eukaryotes. Of note, telomere shortening represents one of the common mechanisms for organ ageing.399 Telomerase inactivation in early life accelerates ageing phenotypes regardless of telomere length.400 Gene mutations in the telomerase complex cause accelerated telomere erosions, which leads to heritable syndromes in multiple systems, takes congenital keratosis, for example, the main cause of death is DNA damage-related bone marrow failure.401 Consistently, telomerase gene mutations and accelerated telomere shortening have also been found in patients with aplastic anemia, suggesting that telomere shortening is closely related to the decline in adult stem cell regeneration, and premature ageing of tissues and organs.402 In high-turnover tissues, such as the hematopoietic system, we also found that telomere shortening triggers cell senescence or apoptosis by activating DDR,403–405 suggesting a close connection between DDR and telomere maintenance. Intriguingly, other studies have shown that telomere-related proteins can be localized on mitochondria, thus affecting mitochondrial metabolism.406 For instance, in low-turnover tissues, such as the heart, telomerase dysfunction perturbs cellular energy metabolism thus promoting ageing. The seminal study from DePinho’s lab demonstrated a direct link between telomere dysfunction-induced DDR to mitochondrial metabolic compromise, where critical shortening of telomere length induces p53 activation, thereby lowering PGC1 expression.407 Recently, another study also indicates that telomere attrition can lead to p53-dependent sirtuin repression,408 in turn triggering a metabolic remodeling. Since telomere attrition occurs in each cell division, at some time point, it will trigger a DDR (e.g., through PARP and ATM), thereby reducing the stability of the genome and eventually leading to cell senescence. Congruently, mice with hyper-long telomeres show less metabolic ageing and longer life spans.409 We also demonstrated that mild elevation of mitochondrial biogenesis regulator PGC1α in late-generation telomerase-deficient mice was sufficient to attenuate metabolic compromise and extending health span.362
p53
p53 represents a sophisticated molecule that wires the nuclear-metabolic axis during ageing; that is, p53 involves glycolysis, OXPHOS, and regulation of the PPP and other pathways via regulating a plethora of target genes.410 Under the ageing-related pseudo-hypoxic state, a reduced glycolytic rate can be achieved through the downregulation of monocarboxylate transporter 1 expression by p53-mediated prevention of lactate efflux.411 In addition, p53 activates the transcription of cytochrome c oxidase 2 synthesis, a core component of OXPHOS, and plays a key role in the regulation of cytochrome c oxidase complex assembly.412
p53 affects several physiological and metabolic pathways, all heavily involved in modulating ageing and the establishment of senescence.413 There is growing interest in dissecting how metabolism affects DDR and vice versa. One hallmark of aged cells is the deregulation of genes involving in DDR. The cell cycle, as well as senescence, can be regulated by metabolism and mitochondrial activity through p53. For instance, vascular smooth muscle cell senescence-induced atherosclerosis was associated with p53-dependent degradation of telomere repeat-binding factor-2.414 Besides, mitochondrial stress in the form of increased ROS levels also induces cell cycle arrest using mediators, such as p53 and p27.415 Meanwhile, phosphorylation of Ser-15 by ATM activates p53 in response to DNA damage.416 Our study also reveals a mechanism of cell senescence, in which distinct effects of p53 and mTORC1 pathways on HSC ageing are governed by Wild-type p53-induced phosphatase 1 (Wip1), which negatively regulates several tumor suppressors and DDR pathways,417 but also B lymphocyte maturation and tissue regeneration.418,419
ATM
First identified in 1995, the ATM kinase contains a PI3K-like kinase domain at the C-terminal and represents the core regulator for DNA double-strand break and repair. During ageing, ATM/ATR-dependent DDR, rather than the p53/p16INK4a axis, was required for the GATA-binding protein 4-mediated senescence-associate secretory phenotype.420 Cells isolated from AT patients were sensitive to radiation, indicating that ATM involves DNA repair. ATM is a relatively upstream kinase that phosphorylates hundreds of substrates in response to DNA damage. The MRE11–RAD50–NBS1 complex, Tip60, ATR, protein phosphatase 2A, and Wip1 regulate ATM activation. In turn, the activated ATM can activate checkpoint kinase 2, structural maintenance of chromosomes-1, Fanconi anemia, complementation group D2, breast cancer 1, early onset, and H2A.X variant histone. Studies have shown that oxidative stress can also activate ATM by phosphorylating ATM at Ser1981, resulting in phosphorylation of p53 and AMPKα, therefore regulating gene expression and energy metabolism. ATM deficiency in mice causes insulin resistance, increased adiposity, atherosclerosis, and a variety of metabolic syndromes,421 while ATM activation is required in insulin-induced glucose transport in slow and fast muscle fibers utilizing glucose oxidation.422 Intriguingly, the repletion of NAD also attenuated the severity of AT-induced neuropathology, suggesting that accumulated DNA damage links to dysfunctional mitochondrial metabolism.423 ATM is the sensor for ROS in human fibroblasts, which also mediates mitochondrial ROS signaling and extends the life span of yeast. A more recent study demonstrated that ATM promotes antioxidant defenses and the repair of double-strand DNA breaks by activating the PPP, representing a link between DNA repair processes and cellular metabolism.
Sirtuins
Sirtuins are a cluster of NAD+-dependent enzymes involving the regulation of metabolism and genome integrity. Many proteins related to DNA repair have been identified as direct substrates of sirtuins, e.g., RB-binding protein 8, endonuclease (CtIP), PARPs, and p53.424–426 Conversely, p53 can posttranscriptionally regulate non-mtSIRT via miRNAs, while transcriptionally regulating mtSIRT through PGC1α/β.408 Existing evidence manifests that nuclear DNA damage signal to the mitochondria during CR with age, where sirtuin network bridges such nucleus–mitochondria communication linking genome instability to altered mitochondrial metabolism.427 Another evidence shows that sirt3 deacetylates and activates IDH2 to boost NADPH, thus mediating reduced oxidative stress and DNA damage.428 In addition, sirt7 deacetylates p53 to inactivate its pro-apoptotic effect, thereby attenuating cardiac hypertrophy and inflammatory cardiomyopathy.429 These observations lead to attempts to boost sirtuin as an anti-ageing strategy. Indeed, sirtuin overexpression exerts a salutary effect in healthy ageing by enhancing the resistance to DNA damage and metabolic insult.430
PARP
PARP represents another NAD+-consuming enzyme that catalyzes the formation of PAR polymers. As a base excision repair protein, PARP overactivation has been linked to mitochondrial dysfunction in xeroderma pigmentosum group A, as well as ataxia telangiectasia and Cockayne syndrome with similar pathogenesis involving nuclear–mitochondrial communication.426 Prolonged PARP activation also couples NAD+ and ATP depletion, thereby triggering cellular energy crisis and metabolic reprogramming.431,432 Some metabolic transcriptional factors (e.g., HIF-1a, NRF-1, and NRF-2) and nuclear receptors (e.g., estrogen receptor, retinoid X receptor, and peroxisome proliferator-activated receptors) have been identified to interact with PARP, affecting glucose and lipid metabolism and related metabolic syndromes.433 Collectively, these data suggest a link between DNA damage-induced poly(ADP-ribosyl)ation and mammalian longevity.434
In sum, the accumulation of DNA damage can promote metabolic dysfunction in two ways: cell-autonomous and non-cell-autonomous mechanisms. Tissue regeneration is impaired by DNA damage-induced senescence and/or apoptosis of stem cells and somatic cells, leading to metabolic dysfunction. DNA damage induces non-cell-autonomous tissue inflammation through the upregulation of cytokines and chemokines, thereby hampering systemic insulin signaling. DNA damage can also affect systemic metabolic homeostasis through influences on cellular metabolism and the endocrine system. Activation of the DDR in specific tissues could influence the function of vital metabolic organs, thereby provoking systemic insulin resistance. Thus, a better understanding of the systemic DDR may help us develop novel therapeutic strategies for metabolic disorders. Additional factors await identification to shed further light on the role of DNA damage in metabolic homeostasis.
Metabolic regulation of immune response in ageing
Inflammation is one of the most described ageing-related phenotypes in ageing research. Senescent cells activate innate and adaptive immune responses that can be beneficial and detrimental.435 Meanwhile, immune organs senesce rapidly after advanced age, causing diseases such as infection, cancer, and other many chronic inflammatory diseases.436,437 In response to ageing-related intrinsic and extrinsic factors, induction into pro-inflammatory cell senescence represents a conserved antitumor and pro-survival mechanism that promotes the clearance of damaged components, restriction of fibrosis and tissue repair, thereby maintaining the relative homeostasis.438 However, persistently activated immunosurveillance can accelerate immune senescence, featured by a blunted immune response, bias to myeloid hematopoiesis, and impaired immunometabolism, possibly stem from the ageing of haematopoietic stem cells.403,439 Many investigations reveal that different stimuli can lead to age-related inflammatory manifestations, such as DDRs, telomere attrition, oncogene activation, oxidative stress, chemotherapy drugs, or radiation damage, all of which contribute to the SASP.440–443 In turn, SASP causes an inflammatory microenvironment that dampens tissue regeneration.444 Notably, the metabolic signatures of senescent cells have been intensively investigated, with some typical changes in immunometabolism during ageing discussed below.
Altered secretome
The altered secretory phenotype in inflammatory cell senescence includes (1) altered nucleic acid secretion, such as micronucleus formation and disruption, leakage of mtDNA, release of LINE1, which may activate RIG-I-MAVS or cGAS–STING signaling pathways to initiate an inflammatory response;110,445 (2) altered protein secretion, such as inflammatory chemokines, cytokines, and other factors, which may exert tissue repairing or degrading effect to eliminate danger signals;446 (3) altered metabolite secretion, such as carnitine and low density lipoprotein, which may correlate with risk of ageing-related oncogenic diseases, cardiovascular, and cerebrovascular diseases.447 Of note, such DAMPs can activate the cellular immune response that differs from the pro-inflammatory response of immune cells induced by pathogens (PAMP), indicating the secretome signature to be specific and stimulus-dependent.448 Moreover, energy demand changes dramatically when immune cells shift from a resting state to an active state, which often causes energy deficits when cells become senescent and, consequently, contributes to the metabolic remodeling accompanied by the altered secretion of cytokines, chemokines, and inflammatory mediators.
Extracellular vesicle secretion
Extracellular vesicles (EVs) are bilayer vesicles released from cells into the extracellular matrix.449 For many years, EVs have been regarded as a mechanism for excreting intracellular components as waste products, and are important mediators for exchanging proteins and lipids between secretory cells and target cells. Two major types of EVs, microvesicles, and exosomes contain a variety of components (e.g., enzymes, miRNAs, transcription factors, membrane-bound and soluble receptors, lipids, and DNA, etc.) and involve in cell communication, migration, angiogenesis, and tumor cell growth. Mounting evidence has been suggested that EV secretion plays a crucial role in age-related inflammatory and metabolic abnormalities.450 For instance, one study demonstrates that the levels of extracellular nicotinamide phosphoribosyltransferase (eNAMPT) decline with age in mice and humans. Increasing eNAMPT promotes NAD+, counteracting ageing, and extending health span.451 Other studies reveal that EVs participate in metabolic reprogramming during macrophage polarization,452 wound healing,453 and fibrotic replacement.454,455 EVs derived from immune cells contain antigen peptide–MHC complex and various antigens, which can control the exchange of antigen information between immune cells, thus regulating activation or inhibition of immune cells and other immune responses.
Metabolic reprogramming
Throughout the life of an organism, the immune system continually senses and responds to environmental threats, which is a relatively energy-consuming process.456 Altered energy substrate (e.g., glucose, lipid, and glutamate) metabolism is commonly found in the stressed cells.339 Metabolic pathways control the duration and strength of innate and adaptive immune responses and the generation of memory cells.457,458 Immune cells can switch between different metabolic states, preferentially using different substrates (glucose, amino acids, and fatty acids) to maintain various functions of specific effectors.456 Cellular metabolism controls the function of immune cells by controlling key metabolic nodes, bringing new paradigms to immunology and prospects to treat inflammatory diseases and autoimmune diseases through metabolism. Mills et al. demonstrated that activated macrophages experience substrate alteration for ATP production from OXPHOS to glycolysis, concurrent with elevated ROS and succinate levels that triggers a pro-inflammatory cell state.459 Tomas et al. demonstrated that the risk stratification of low- and high-risk plaque in atherosclerosis links the macrophage infiltration to altered mitochondrial substrate oxidation, that is, low-risk plaque exhibits fatty acid oxidation-prone and downregulated glycolysis or anaplerosis by amino acids, while opposing energy preference in high-risk plaques.460 In line with this, a similar finding was found by different investigators461 and further evidence associates the regulation of immunometabolism of macrophages with other ageing-related diseases, such as type 2 diabetes462 and macular degeneration.463 Altogether, these studies suggest that the metabolic reprogramming of immune cells might be a promising therapeutic approach to treat ageing-related diseases.
Mitochondrial dysfunction
In addition to their roles in cellular energy metabolism and apoptosis, mitochondria are thought to be the central hub of the innate immune response,464 that is, mitochondria can not only act as the platform of immune response adaptors (such as MAVS and NLRP3), but also participate in the immune response by producing ROS. During ageing, mtDNA, Tfam, ROS, ATP, cardiolipin, and N-formyl peptide can be released as DAMPs and are recognized by pattern recognition receptors upon mitochondrial stress or compromise, leading to the activation of MiDAS and SASP.44 A recent study showed that T cells with dysfunctional mitochondria promoted ageing in mice via inducing circulating cytokines. Blocking TNF-α, a critical cytokine released during the T cell metabolic failure, partially rescued the premature ageing phenotype.465 Interestingly, He et al. demonstrated age-associated acetylation of NLRP3 in macrophages might cause inflammatory microenvironment and insulin resistance, while enhancing Sirt2 expression diminished inflammation and related disease phenotype by directly deacetylation of NLRP3,5 indicating a possible interplay between epigenetic regulation of inflammageing and mitochondrial metabolic reprogramming. Of note, abnormalities between ER–mitochondrial communication (i.e., ER–mitochondrial dysfunction) could be another crucial player mediating the ageing-related inflammatory phenotype in macrophages.466 Another study has demonstrated that de novo NAD biosynthesis in macrophages is the key to the homeostasis of the immune response during ageing,467 while α-KG repletion could maintain the α-KG pool in aged mice, lower the bodyweight and fat mass, and improve glucose tolerance in mice fed a high-fat diet.468 Collectively, these studies implicate that targeting mitochondrial homeostasis in the immune cells may represent a novel avenue to counteract inflammageing and normalize immunometabolism.
Serum phosphate
Studies performed over the past few years make it clear that changes in serum phosphate levels have profound effects in mice and human. Changes in extracellular and intracellular phosphate concentration affect glucose metabolism, insulin sensitivity, and oxidative stress in vivo and in vitro. High concentrations of extracellular phosphate are toxic to cells, which induce cell damage and inflammation when precipitated with calcium (to form calciprotein particles that comprised calcium–phosphate crystals and fetuin-A), thus potentially affect ageing processes.468,469 Indeed, impaired urinary phosphate excretion increases serum phosphate level and induces an inflammageing phenotype in mice, and calciprotein particles are found in patients with chronic kidney disease associated with a (mal)adaptation of the FGF-23-Klotho endocrine system.469 Direct evidence shows that knocking out fibroblast growth factor-23, a regulator of phosphate homeostasis, causes hyperphosphatemia and vascular calcification in mice, concurrent with multiple ageing-like phenotypes.470 Deletion of Klotho exhibits similar abnormalities, both could be alleviated by resolving phosphate retention (e.g., on a low-phosphate diet), indicating an underlying link between phosphate and inflammageing. In line with this, augmented circulating levels of calciprotein particles are detected in concert with an increase in serum phosphate and age, which correlate positively with vascular stiffness and chronic sterile inflammation, suggesting that calciprotein particles may be an endogenous pro-ageing factor.471 Furthermore, old mice showed muscular degeneration correlated with high serum phosphate concentration and increased levels of integrin-linked kinase and p53, indicative of a possible mechanism in developing sarcopenia.472 Another study confirms that hyperphosphatemia induces endothelin-1-mediated senescence in human endothelial cells.473 In contrast, Klotho deficiency-induced heart ageing is independent of phosphate metabolism since Klotho-mutated mice exhibit a normal range of serum phosphate.474
One-carbon metabolism
One-carbon (1C) metabolism formed by three reactions, namely the folate cycle, the methionine cycle, and the trans-sulfuration pathway, is of great importance to nucleotide biosynthesis, amino acid homeostasis, methylation modifications, and redox balance.475 Recent advances reveal that 1C metabolism is also associated with inflammageing. Perturbations in 1C metabolism have been linked to a biased pro-inflammatory state and may also increase the risk of age-related diseases.476 Indeed, a specific deficit in the induction of enzymes of 1C metabolism leads to the accumulation of impaired naive T cells in aged mice, which could be rescued by the addition of products of 1C metabolism (formate and glycine) to the cells.477 1C units in the methionine cycle support the generation of SAM thereby a high SAM:S-adenosylhomocysteine (SAH) ratio, leading to enhanced histone H3 lysine 36 trimethylation and IL-1β expression in macrophages.478 Conversely, serine deprivation lowers IL-1β production and inflammasome activation, and alters the transcriptomic and metabolic profile in M1 macrophages via inhibited mTOR signaling.479 Congruently, elevated homocysteine, SAM, and SAH levels in plasma are more susceptible to cardiometabolic syndromes.480 Moreover, defect of complex I decreases mitochondrial 1C NADPH production, which is associated with increased inflammation and cell death.481 These findings highlight a potential regulatory mechanism for 1C metabolism to modulate the inflammatory response. Notably, anti-ageing mechanisms by which metformin can activate the gerosuppressor/tumor suppressor AMPK. One such mechanism may be 1C metabolism that drives the de novo synthesis of purine nucleotides (e.g., AMP), suggesting that de novo biosynthesis of purine nucleotides, which is based on the metabolism of 1C compounds, is a new target for metformin’s actions in ageing.482 Therefore, quantification and intervention of 1C metabolism metabolites may open a new avenue to further our understanding of inflammageing.
Microbial burden
Ageing is correlated with a reduced abundance of salutary commensal microbes that maintain the intact intestinal barrier and limit the amplification of pathogenic commensals by secretion of specific metabolites, for instance, the short-chain fatty acids.483 Increased attention has been paid to microbial homeostasis that connects microbiota to the functionality of multiple organs during ageing. Indeed, gut microbial dysbiosis links to aberrant immune responses and miscommunication between gut microbes and innate immune response may cause various diseases.484,485 One study implicates that microbiota-induced IFN-Is instruct a poised basal state of dendritic cells.486 Lloyd and Marsland propose that microbes in the host are crucial to establishing lung homeostasis and are affected by ageing to reshape the immune network during chronic inflammation.487 Ang et al. found that the ketogenic diet changed the landscape of gut microbiota by reducing intestinal pro-inflammatory Th17 cells, suggesting diet interventions could be a possible means to attenuate ageing and related metabolic diseases.488 Similarly, other studies have testified interactions between gut microbiota with cerebral and cardiovascular diseases.489,490 In addition, based on deep learning techniques such as neural networks, Galkin et al. established a mathematical model that can reflect the relationship between the microbiological profiles of gut microbiota and the biological age of host, using >3000 microbiome samples from 1165 healthy individuals. They concluded that 39 microbiota taxa were extracted as intestinal markers to best correlate physiological age.491 In sum, these studies highlight the importance of microbes in regulating organ function and potential effects on immunosenescence.
Metabolic regulation of protein homeostasis in ageing
Protein quality control in living organisms is essential for the maintenance of cell viability that maintains a relatively balanced state of the proteome in regards to synthesis, folding and unfolding, modification, trafficking, and degradation of specific proteins.492 Protein homeostasis in cells is mainly coordinated by the molecular chaperones, and two proteolytic systems, namely the ubiquitin–proteasome system and autophagy–lysosome system. The imbalance of protein homeostasis is one of the crucial factors causing ageing and the development of ageing-related diseases.493 Cell senescence can be accelerated when various organelles meet overloaded stress or proteotoxicity, such as misfolding of nuclear, ribosomal, peroxisomal, and mitochondrial proteins,494–497 leading to aberrant protein aggregates that dampen the cross talk of organelles. Accordingly, several cellular hallmarks of proteotoxic stress have been identified, including dysregulated mTOR signaling and autophagy, compartment-specific unfolded protein response in mitochondria (UPRmt), and in the endoplasmic reticulum (UPRER).
mTOR
The evolutionarily conserved mTOR is a serine/threonine kinase that participates in the central regulation of nutrient sensing, stress response, and longevity.498 Two mTOR complexes, namely mTORC1 and mTORC2, have been extensively studied, and both exert their functions by other kinases, such as S6 kinase and Akt.499 Park et al. demonstrated that mTORC1 could influence integrated stress response via augmenting activating transcription factor 4 (ATF4) at the posttranscriptional level by 4E-BP1.500 Zhang et al. revealed that mTORC1 enhances the protein synthesis capacity, while concordantly inhibiting autophagy, promoting the production of proteosomes via the activation of Nrf1.501 Conversely, other studies suggested mTORC1 inhibition induced an increase in proteasome abundance and protein degradation.502,503 mTORC2 has been demonstrated to inhibit chaperone-mediated autophagy during ageing that can be restored by phosphatase PHLPP1, thereby representing a lysosomal pathway to counteract age-associated dysfunction of protein degradation.504 Moreover, rapamycin-induced insulin resistance is triggered by the inhibition of mTORC2, although it is irrelevant with longevity.505
Autophagy
Impairment in autophagy has linked to the derailment of proteostasis that is closely associated with ageing and age-related diseases, including macroautophagy, microautophagy, and chaperone-mediated autophagy. A significant effort has been made to elaborate on the role of autophagy in the field of cerebral proteinopathies, including Huntington’s disease (HD), AD, PD, and amyotrophic lateral sclerosis (ALS).506–508 Sorrentino et al. reported that enhancing mitophagy helped resolve amyloid-β proteotoxicity in mouse and C. elegans, providing a promising strategy targeting mitochondrial proteostasis to delay AD.509 Poewe et al. showed that α-synuclein aggregation and clearance of undegraded proteins were therapeutic targets for PD.510 Similarly, enhancement of cytosolic proteostasis pathways (through autophagy or proteasome) shows promise for HD and ALS treatments.511,512
Moreover, identifying novel mitophagy receptors or booster of mitophagy further enhances our understanding of the complexity of mitochondrial quality control. Li et al. showed that mitophagy receptor protein FUNDC1 located at the mitochondrial outer membrane that is associated with cytoplasmic chaperone protein HSC70, leading to the recruitment of damaged or misfolded cytosolic proteins to mitochondria, and then shuttle to the mitochondrial matrix through the TOM–TIM complex, culminating with the degradation by matrix-localized mitochondrial protease LONP1.513 Ryu et al. identified the natural compound urolithin A as a mitophagy inducer, which enhanced mitochondrial and muscle function.514 We and others have found the potential therapeutic benefits by PGC1α and TFEB upregulation in counteracting declined autophagy capacity and reduced proteostasis.362,515 Together, these findings indicate a regulatory effect of autophagy in protein homeostasis necessary for proper organ function, especially in the brain and other high energy-demanding organs.
UPRmt
UPRmt is one key mechanism in mitochondrial quality control involving multiple components, such as chaperones, proteases, and other stress sensors and effectors.516 Apart from cell respiration and ATP production, mitochondria also participate in immune response, cell senescence, and apoptosis. Notably, most proteins required by the processes above are translated from nuclear-encoded genes; thus, it is crucial to orchestrate mitochondrial–nuclear communication that ensures a precise transcriptional response by the UPRmt. Indeed, a plethora of defects within aged mitochondria that activate the UPRmt implicates intrinsic surveillance of mitochondrial function being reliant on mitochondrial stress response.517 Zhao et al. discovered that the accumulation of unfolded proteins in the mitochondrial matrix resulted in transcriptional upregulation of nuclear-encoded mitochondrial stress genes, including hsp60, hsp10, mtDnaJ, and ClpP, without interfering gene expression that was related to endoplasmic reticulum (ER) stress. Teske et al. demonstrated that CHOP-induced ATF5 to trigger cell death in response to perturbations in protein homeostasis.518 Notably, a CHOP element, coupled with the C/EBP domain, was found in the gene promoter regions.519 Further analysis revealed that c-JNK could bind to an AP1-binding site within the promoter regions of CHOP and C/EBP, underscoring an indispensable role of the JNK signaling pathway in UPRmt.520 Furthermore, using the nematode model, transcriptional factors DVE-1 and ATFS-1, along with H3K27 demethylases jmjd-1.2 and jmjd-3.1 and other factors, have been identified as UPRmt activators.521–523 Although ample studies provide a framework for the signal transduction of UPRmt, the exact and complete molecular mechanism that cells (especially in mammals) sense mitochondrial stress signals and transmit the signals to the nucleus await further elucidation.
UPRER
Response to ER stress caused by unfolded or misfolded proteins (UPRER) represents another intrinsic mechanism to counteract multiple stress stimuli that leads to perturbed proteostasis. The UPRER contains three upstream stress sensors in the ER membrane, i.e., inositol-requiring enzyme-1, PKR-like ER kinase, and ATF6, which is activated by the presence of unfolded proteins within the ER, ensuring signal transduction to the well-known downstream effector molecules.524 UPRER compromise lies at the nexus of inflammageing and metabolic dysfunction, while its constitutive activation attenuates multiple age-related diseases and extends longevity.525 Most of the pioneering findings regarding UPRER are derived from the studies using worms. Taylor and Dillin revealed how to rescue declined UPRER in the ageing of C. elegans and found that age-onset loss of ER proteostasis could be restored by neural expression of a constitutively active form of X-box-binding protein 1.526 Similarly, Daniele et al. facilitated UPRER specifically in neurons to promote ER function by lipid depletion through lipophagy. Interestingly, this beneficial metabolic alteration is independent of chaperone induction, but is regulated by EH domain-binding protein 1-mediated lipophagy, thereby enhancing organismal stress resistance and extending the life span.527 Schinzel et al. utilized a whole-genome CRISPR-based knockout screen to identify transmembrane protein 2 (TMEM2), a hyaluronidase that degraded hyaluronan in the extracellular matrix, was capable of modulating ER stress through CD44, ERK, and p38. Ectopic expression of human TMEM2 promoted ER homeostasis and extended longevity in C. elegans, indicating a conserved health-promoting pathway via elevating the TMEM2–UPRER axis.528 Although more efforts are required to illustrate the orthologs of human UPRER targets and pathways, these findings advance our understanding of how the UPRER changes with age and how this impacts disease development, thus opening new therapeutic avenues to treat age-associated diseases.
Metabolic regulation of organelle homeostasis in ageing
Imbalance of cellular metabolism and organelle homeostasis during ageing is one of the major drivers causing ageing-related diseases.16 Several aspects that aim to delineate the changes in organelle ageing, in particular, to clarify the causal relationship between organelle dysfunction and ageing and/or degenerative diseases, and to explore new strategies targeting organelle homeostasis to intervene ageing and related disorders at varying levels of metabolism, have been investigated for long and some critical findings are of great importance.529,530
Mitochondria
For decades, mitochondrial biology lies at the center of mechanistic exploration in metabolic regulation of cell senescence and organismal ageing.531 Mitochondrial dysfunction has been linked with mtDNA mutations, aberrant mitochondrial respiration, dynamics, biogenesis, autophagy, and other quality control machinery.532 Apart from their roles above in redox balance and immune response, mitochondria also involve in proteostasis and other cellular functions that intensively interact with ER and nucleus.
Guo et al. developed grazing incidence structured illumination microscopy and found most of the mitochondrial fission events, while ~60% of the mitochondrial fusion events actively occurred at the contact sites between mitochondria and ER.533 Consistently, Gӧbel et al. demonstrated that mitofusin 2-mediated mitochondria–ER contacts in astrocytes promote repairing the injured brain.534 Moreover, reduced mitochondrial fusion and Huntingtin levels were observed in impaired dendritic maturation, thereby leading to behavioral deficits,535 indicative of a fundamental role of mitodynamics in tissue homeostasis. Another critical player, mitochondrial permeability, has been manifested as an indispensable regulator of mitochondrial function. VDAC oligomerization warranted an increased mitochondrial outer membrane permeability, which was essential to trigger released mtDNA-induced inflammatory response.536 Ying et al. demonstrated that short-term mitochondrial permeability transition pore opening (aka mitoflash) induced a nuclear reprogramming by histone methylation, suggesting a novel mitochondrial regulation of nuclear epigenetics.537 Besides, MacVicar et al. identified that the mTORC1–LIPIN1–YME1L regulatory axis was a posttranslational regulator of mitochondrial proteostasis at the interface between metabolism and mitochondrial dynamics.538
Lysosome
Lysosomes are crucial cellular organelles for human health that function in digesting and recycling of extracellular and intracellular macromolecules. Given that many neurodegenerative diseases (e.g., PD and AD), as well as reduced life span, are linked to lysosomal dysfunction, enhancing lysosomal function becomes an option to promote health span in certain circumstances, such as reduced ability to clear protein aggregates during neural stem cells ageing. Leeman et al. demonstrated that aged neural stem cells exhibited decreased lysosomal genes, thereby a slower clearance of protein aggregates. Normalization of lysosomal function in these cells could restore the defects and enhance the quiescent neural stem cell activation.539 Castellano et al. investigated the metabolic cue that activated mTORC1 at lysosome and found that SLC38A9-mediated import and Niemann–Pick C1-mediated export of lysosomal cholesterol were the drivers for mTORC1 activation.540 Further analysis found that arginine was essential for mTORC1 activation and represented as the lysosomal messenger sensed by SLC38A9.541 Via developing a method for rapid lysosomal isolation and metabolomics, Abu-Remaileh et al. manifested that mTOR inhibition decreased the lysosomal efflux of most essential amino acids in a V-ATPase-dependent manner.542 Moreover, overexpression of the lysosomal biogenesis regulator TFEB homolog (HLH-30) in worms has been proven beneficial. It may prolong life span through repression of mTOR signaling and engagement of autophagic processing.543 Collectively, these results reinforce the concept that regulation of lysosomal function is one of the most efficient anti-ageing interventions.
Peroxisome
Peroxisomes are dynamically and metabolically active that signal various intracellular events, therefore exhibiting a high degree of plasticity, linking redox balance, and nutrient digestion to immune metabolism. They are capable of autonomous replication via fission. Recent studies have revealed that beyond the essential roles in fatty acid oxidation, anaplerotic metabolism, and production and scavenging of ROS, peroxisomes also participate in the metabolic regulation of cell senescence.544 Evidence shows the significant role of weakened peroxisomal functions, including diminished peroxisomal autophagy (pexophagy), loss of communication with ER and mitochondria, and dysregulation of peroxisomal proteostasis in accelerated ageing and related diseases.496 Peroxisomes not only eliminate (mainly detoxify by peroxiredoxins, glutathione peroxidases, and catalases), but also produce (mainly oxidize by xanthine oxidases and acyl-CoA oxidases) H2O2. Consequently, peroxisomes may serve as a direct source of oxidative stress, especially during ageing, by decreased peroxisome turnover and downregulated detoxifying enzymes.545 Dixit et al. revealed that the well-known mitochondrial antiviral signaling protein, MAVS, was also a peroxisome-localized adaptor that functioned in innate antiviral immunity.546 The tuberous sclerosis complex has also been found on the peroxisome to inhibit mTORC1 in response to endogenous ROS.547 Similar findings were reported that a portion of extranuclear ATM was localized to peroxisomes and mediated ROS-induced pexophagy,548 while AMPK and dietary restriction extended the life span of C. elegans via coordinating mitochondrial and peroxisomal homeostasis.3 Of note, the peroxisomes remain the least understood of all organelles, yet accumulating studies uncover their roles in regulating metabolism and ageing in concert with mitochondria, ER, and other organelles. Further investigations on peroxisomal quality control warrant a deeper understanding of their roles in cellular homeostasis.
Together, comprehensive mapping of metabolic networks should help dissect the interaction and cross talk among different organelles, furthering our understanding of the molecular and cellular basis of organelle ageing.
Novel interventions and techniques in metabolic ageing research
Increasing attention has been focused on depicting the metabolic signatures of cells from single-cell resolution, as well as the whole metabolic landscape of organisms in a dynamic manner. In particular, identifications of age-related metabolic phenotypes or biomarkers that are closely correlated with overall fitness are increasingly being recognized as therapeutic targets. The discovery of senescence-related genes and metabolites by combining transcriptomics, proteomics, epimicromics, secretomics, metabolomics, phenomics, and large population cohort studies sheds light on the complex senescence signaling networks. Among the multi-omics, single-cell RNA sequencing is the hotspot to analyze the molecular mechanism of organ ageing.549 For example, Wang et al. compared young and aged nonhuman primate ovaries using single-cell transcriptomics. In-depth analyses of gene expression dynamics of oocytes revealed oxidative damage was an important determinant in ovarian ageing.550 Bian et al. characterized a comprehensive map of human macrophage development at single-cell resolution, providing a deeper understanding of the functions of tissue-resident macrophages.551 In addition, the advances in metabolomics, especially single-cell metabolomics and their applications, further add strength to the field of clinical pharmacology.552,553
Continuous focus on the clearance of intracellular senescence-inducing factors is a relevant direction for treating age-associated disorders. Many reports manifest that removal of senescent cells (e.g., via senolytics) or abnormally active transposons (e.g., via inhibitors to LINE1 transposons) from ageing animal models is salutary.7,40,112,554 Conversely, the repletion of rejuvenating factors (NAD+) also improves various organ functions, which have been experimentally proven through genetic manipulation or diet/supplement intervention, although detecting NAD+ levels in vivo remains challenging. Genetically encoded fluorescent probes represent promising tools to realize high-dynamic, high-resolution, high-throughput NADH/NAD+ detection in vivo, empowering a real-time visualization of compartmentalized NAD+ that facilitate screening for NAD-associated drug design or gene candidates identification.363,374 Furthermore, manipulation of melatonin,555 supplementation of compounds, such as spermidine,556 acarbose,557 and urolithin A514 may also exert an anti-ageing effect through metabolic remodeling. Intriguingly, a controversial study using parabiosis demonstrated that the enhanced level or administration of GDF11 could correct DNA damage accumulated in ageing mouse satellite cells,558 while its paralog myostatin regulates energy homeostasis in the heart and prevents heart failure.559 Although heterochronic parabiosis of young and old mice implies the potential to attenuate the ageing process and improve cell function, how to rejuvenate senescent cells via autologous rather than allogeneic resources remains a central question to avoid issues regarding immunity and ethics.
CR, drug intervention, microbial regulation, and sleep/exercise represent simple but also noninvasive approaches to extend health span and ameliorate age-related pathologies, at least, via enhancing the tissue function across a broad spectrum of animals (Fig. 7). The level of CR is indeterminate from individual-by-individual, and long-term CR sounds impracticable to the general public. In contrast, resveratrol, a naturally occurring small molecule that induces metabolic benefits resembling CR, increases mitochondrial biogenesis, partly through activation of SIRT1 and PGC1α.560 Similarly, metformin, a hypoglycemic agent widely applied in type II diabetes mellitus, has been found to extend longevity and health span in animal models.178,561 Rapamycin also exerts a similar protective response in C. elegans and mice.562 Such an endeavor is worth trying that directs strategies to mimic the life-extending effect using known compounds and pathways. In addition, emerging evidence suggests that sleep modulation links haematopoiesis to atherosclerosis,563 while the development of microbiota-based interventions, such as probiotics and prebiotics extended health span.564 Physical exercise also gives considerable benefits in improving energy metabolism, yet this is unsuitable to the population with severe conditions, such as cardiovascular diseases, asthma, osteoarthrosis, diabetes, etc., which predominantly happen among the aged.
Fig. 7.

Approaches to alter local and systemic metabolism. Six aspects, including environmental stimuli, sleep modulation, exercise, intestinal microbes, diet, and drug interventions, may reprogram the metabolism thereby benefiting to healthy ageing
Conclusive remarks
Many conventional drugs have been found to exert an anti-ageing effect. Thus, developing methods aiding high-throughput screening of anti-ageing drugs from currently approved drug libraries is cost-efficient to promote pharmacological interventions of ageing, which required state-of-the-art techniques to delineate the dynamics and interactions of key molecules/metabolites involving the ageing process (Fig. 8). Further elucidation of regulatory mechanisms of organ homeostasis in senescent cells will be informative to anti-ageing agent design.
Fig. 8.

Novel techniques accelerate the discovery of anti-ageing mechanisms and therapies. Illustrations of three advanced approaches, i.e., single-cell sequencing, 3D-genome analysis, and genetically encoded fluorescent probes in dissecting genetic, epigenetic, and metabolic alterations in ageing and anti-ageing research
Apart from practices in assisting medical image diagnosis, pathological classification, and treatment decision-making, machine learning, such as convolution neural network, has been integrated into ageing biology to calculate or predict cell ageing.565–567 The algorithm models are continually optimized to define the best cell senescence markers. It is thought the extent of cell senescence or organismal ageing could be readily predicted by certain factors (e.g., inflammatory cytokines or metabolites) in a blood test soon. Such artificial intelligence can also be utilized to pick candidate anti-ageing compounds or drug targets in high-throughput screening, which could lead to disruptive innovation of the pharmaceutical industry and finalizing our goal to health ageing.
In sum, the inflammatory, epigenetic, and metabolic regulation of cellular components and activities is the core event of environmental and genetic manipulations that link cell senescence to organismal ageing. Owing to the significant progress in understanding the aspects above, incremental approaches and concepts that help healthy ageing have been identified, and it will continue to be a prosperous field of research for the next decades to come.
Acknowledgements
This work was supported by Grants 2016YFA0100602, 2017YFA0103302, and 2018YFA0109300 from the National Key Research and Development Program of China; Grants 81525010, 81770155, 91749117, 91749203, 81901403, 82030039, 82022026, and 82071572 from the National Natural Science Foundation of China; Grant 2C32003 from Opening Project of Key Laboratory of Integrative Chinese and Western Medicine for the Diagnosis and Treatment of Circulatory Diseases of Zhejiang Province; Grant 2019B151502008 from the Science Foundation for Distinguished Young Scholars of Guangdong Province; Grant 2018GZR110103002 from Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory; and Grant 2017ZT07S347 from the Program for Guangdong Introducing Innovative and Enterpreneurial Teams. Figures 3–5 are created with BioRender.com.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Xudong Zhu, Zhiyang Chen
These authors jointly supervised this work: Hu Wang, Zhenyu Ju
Contributor Information
Hu Wang, Email: wanghu19860315@163.com.
Zhenyu Ju, Email: zhenyuju@163.com.
References
- 1.Gorgoulis V, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–827. doi: 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Rodier F, Campisi J. Four faces of cellular senescence. J. Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weir HJ, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017;26:884–896 e885. doi: 10.1016/j.cmet.2017.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen HY, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 5.He M, et al. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 2020;31:580–591 e585. doi: 10.1016/j.cmet.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H, et al. cGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang N, Sen P. The senescent cell epigenome. Aging. 2018;10:3590–3609. doi: 10.18632/aging.101617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018;2018:9076485. doi: 10.1155/2018/9076485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fulop T, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 2017;8:1960. doi: 10.3389/fimmu.2017.01960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmeer C, et al. Dissecting aging and senescence-current concepts and open lessons. Cells. 2019;8:1446. doi: 10.3390/cells8111446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018;128:1238–1246. doi: 10.1172/JCI95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee BY, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 16.Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardson AG, Schadt EE. The role of macromolecular damage in aging and age-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:S28–S32. doi: 10.1093/gerona/glu056. [DOI] [PubMed] [Google Scholar]
- 20.Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020;16:263–275. doi: 10.1038/s41574-020-0335-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavazzi G, Krause KH. Ageing and infection. Lancet Infect. Dis. 2002;2:659–666. doi: 10.1016/S1473-3099(02)00437-1. [DOI] [PubMed] [Google Scholar]
- 23.Yoshikawa TT. Epidemiology and unique aspects of aging and infectious diseases. Clin. Infect. Dis. 2000;30:931–933. doi: 10.1086/313792. [DOI] [PubMed] [Google Scholar]
- 24.Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vukadinov J, et al. [Aging and infection] Med. Pregl. 2003;56:243–246. doi: 10.2298/MPNS0306243V. [DOI] [PubMed] [Google Scholar]
- 27.Franceschi C, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 28.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 29.Straub RH, Schradin C. Chronic inflammatory systemic diseases: an evolutionary trade-off between acutely beneficial but chronically harmful programs. Evol. Med. Public Health. 2016;2016:37–51. doi: 10.1093/emph/eow001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzaferro S, et al. Bone, inflammation and chronic kidney disease. Clin. Chim. Acta. 2020;506:236–240. doi: 10.1016/j.cca.2020.03.040. [DOI] [PubMed] [Google Scholar]
- 31.Neves J, Sousa-Victor P. Regulation of inflammation as an anti-aging intervention. FEBS J. 2020;287:43–52. doi: 10.1111/febs.15061. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez-Rodriguez L, Lopez-Hoyos M, Munoz-Cacho P, Martinez-Taboada VM. Aging is associated with circulating cytokine dysregulation. Cell. Immunol. 2012;273:124–132. doi: 10.1016/j.cellimm.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Morley JE, Baumgartner RN. Cytokine-related aging process. J. Gerontol. A Biol. Sci. Med. Sci. 2004;59:M924–M929. doi: 10.1093/gerona/59.9.M924. [DOI] [PubMed] [Google Scholar]
- 34.Germolec DR, Shipkowski KA, Frawley RP, Evans E. Markers of inflammation. Methods Mol. Biol. 2018;1803:57–79. doi: 10.1007/978-1-4939-8549-4_5. [DOI] [PubMed] [Google Scholar]
- 35.Sanada F, et al. Source of chronic inflammation in aging. Front. Cardiovasc. Med. 2018;5:12. doi: 10.3389/fcvm.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.d’Adda di Fagagna F, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 37.Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodier F, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welch D, Dawes PJ. Childhood hearing is associated with growth rates in infancy and adolescence. Pediatr. Res. 2007;62:495–498. doi: 10.1203/PDR.0b013e3181425869. [DOI] [PubMed] [Google Scholar]
- 40.Jeon OH, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017;23:775–781. doi: 10.1038/nm.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collison J. Osteoarthritis: removing old chondrocytes to combat disease. Nat. Rev. Rheumatol. 2017;13:388. doi: 10.1038/nrrheum.2017.73. [DOI] [PubMed] [Google Scholar]
- 42.Lindborg JA, Mack M, Zigmond RE. Neutrophils are critical for myelin removal in a peripheral nerve injury model of wallerian degeneration. J. Neurosci. 2017;37:10258–10277. doi: 10.1523/JNEUROSCI.2085-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapetanovic R, Bokil NJ, Sweet MJ. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: a ticking time bomb in ageing. Ageing Res. Rev. 2015;24:40–53. doi: 10.1016/j.arr.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Wiley CD, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calfee CS, Matthay MA. Clinical immunology: culprits with evolutionary ties. Nature. 2010;464:41–42. doi: 10.1038/464041a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol. 2018;9:832. doi: 10.3389/fimmu.2018.00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo H, et al. Mitochondrial stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep. 2019;26:945–954 e944. doi: 10.1016/j.celrep.2018.12.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinti M, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014;44:1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- 50.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ongradi J, Stercz B, Kovesdi V, Vertes L. Immunosenescence and vaccination of the elderly, I. Age-related immune impairment. Acta Microbiol. Immunol. Hung. 2009;56:199–210. doi: 10.1556/AMicr.56.2009.3.1. [DOI] [PubMed] [Google Scholar]
- 52.Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J. Pathol. 2007;211:144–156. doi: 10.1002/path.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiecolt-Glaser JK, Wilson SJ, Madison A. Marriage and gut (microbiome) feelings: tracing novel dyadic pathways to accelerated aging. Psychosom. Med. 2019;81:704–710. doi: 10.1097/PSY.0000000000000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmadi S, et al. Metformin reduces aging-related leaky gut and improves cognitive function by beneficially modulating gut microbiome/goblet cell/mucin axis. J. Gerontol. A Biol. Sci. Med. Sci. 2020;75:e9–e21. doi: 10.1093/gerona/glaa056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, et al. Lipoteichoic acid from the cell wall of a heat killed Lactobacillus paracasei D3-5 ameliorates aging-related leaky gut, inflammation and improves physical and cognitive functions: from C. elegans to mice. Geroscience. 2020;42:333–352. doi: 10.1007/s11357-019-00137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toward R, Montandon S, Walton G, Gibson GR. Effect of prebiotics on the human gut microbiota of elderly persons. Gut Microbes. 2012;3:57–60. doi: 10.4161/gmic.19411. [DOI] [PubMed] [Google Scholar]
- 57.Claesson MJ, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 58.Bian G, et al. The gut microbiota of healthy aged Chinese is similar to that of the healthy young. mSphere. 2017;2:e00327–17. doi: 10.1128/mSphere.00327-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kong F, Deng F, Li Y, Zhao J. Identification of gut microbiome signatures associated with longevity provides a promising modulation target for healthy aging. Gut Microbes. 2019;10:210–215. doi: 10.1080/19490976.2018.1494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellulu MS, et al. Obesity and inflammation: the linking mechanism and the complications. Arch. Med. Sci. 2017;13:851–863. doi: 10.5114/aoms.2016.58928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee H, Lee IS, Choue R. Obesity, inflammation and diet. Pediatr. Gastroenterol. Hepatol. Nutr. 2013;16:143–152. doi: 10.5223/pghn.2013.16.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernier M, et al. Disulfiram treatment normalizes body weight in obese mice. Cell Metab. 2020;32:203–214 e204. doi: 10.1016/j.cmet.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moulton VR. Sex hormones in acquired immunity and autoimmune disease. Front. Immunol. 2018;9:2279. doi: 10.3389/fimmu.2018.02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perheentupa A, Huhtaniemi I. Aging of the human ovary and testis. Mol. Cell Endocrinol. 2009;299:2–13. doi: 10.1016/j.mce.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 65.Maggio M, et al. SHBG, sex hormones, and inflammatory markers in older women. J. Clin. Endocrinol. Metab. 2011;96:1053–1059. doi: 10.1210/jc.2010-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maggio M, et al. Association of hormonal dysregulation with metabolic syndrome in older women: data from the InCHIANTI study. Am. J. Physiol. Endocrinol. Metab. 2007;292:E353–E358. doi: 10.1152/ajpendo.00339.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumru S, Godekmerdan A, Yilmaz B. Immune effects of surgical menopause and estrogen replacement therapy in peri-menopausal women. J. Reprod. Immunol. 2004;63:31–38. doi: 10.1016/j.jri.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 68.Giglio T, et al. Immune cell circulating subsets are affected by gonadal function. Life Sci. 1994;54:1305–1312. doi: 10.1016/0024-3205(94)00508-7. [DOI] [PubMed] [Google Scholar]
- 69.Kamada M, et al. B cell subsets in postmenopausal women and the effect of hormone replacement therapy. Maturitas. 2001;37:173–179. doi: 10.1016/S0378-5122(00)00180-8. [DOI] [PubMed] [Google Scholar]
- 70.Deguchi K, et al. Postmenopausal changes in production of type 1 and type 2 cytokines and the effects of hormone replacement therapy. Menopause. 2001;8:266–273. doi: 10.1097/00042192-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 71.Rais M, Wilson RM, Urbanski HF, Messaoudi I. Androgen supplementation improves some but not all aspects of immune senescence in aged male macaques. Geroscience. 2017;39:373–384. doi: 10.1007/s11357-017-9979-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2011;10:319–329. doi: 10.1016/j.arr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010;34:J258–J265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Asthana A, et al. Effects of smoking intensity and cessation on inflammatory markers in a large cohort of active smokers. Am. Heart J. 2010;160:458–463. doi: 10.1016/j.ahj.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frodermann V, et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019;25:1761–1771. doi: 10.1038/s41591-019-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xia S, et al. An update on inflamm-aging: mechanisms, prevention, and treatment. J. Immunol. Res. 2016;2016:8426874. doi: 10.1155/2016/8426874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qian, F. & Montgomery, R. R. Quantitative imaging of lineage-specific Toll-like receptor-mediated signaling in monocytes and dendritic cells from small samples of human blood. J. Vis. Exp. 3741 (2012). [DOI] [PMC free article] [PubMed]
- 79.Esplin BL, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 2011;186:5367–5375. doi: 10.4049/jimmunol.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP) Cell Signal. 2012;24:835–845. doi: 10.1016/j.cellsig.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 82.Chien Y, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hashimoto M, et al. Loss of HuR leads to senescence-like cytokine induction in rodent fibroblasts by activating NF-kappaB. Biochim. Biophys. Acta. 2014;1840:3079–3087. doi: 10.1016/j.bbagen.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 84.Zhang G, et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497:211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adler AS, et al. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nasto LA, et al. ISSLS prize winner: inhibition of NF-kappaB activity ameliorates age-associated disc degeneration in a mouse model of accelerated aging. Spine (Philos. Pa 1976). 2012;37:1819–1825. doi: 10.1097/BRS.0b013e31824ee8f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z, et al. Cohesin-mediated NF-kappaB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J. Exp. Med. 2019;216:152–175. doi: 10.1084/jem.20181505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laberge RM, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015;17:1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herranz N, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015;17:1205–1217. doi: 10.1038/ncb3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer. 2014;14:547–558. doi: 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- 91.Narita M. Juxtacrine regulation of cellular senescence. BMB Rep. 2019;52:3–4. doi: 10.5483/BMBRep.2019.52.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009;15:3003–3026. doi: 10.2174/138161209789058110. [DOI] [PubMed] [Google Scholar]
- 93.Marchal J, Pifferi F, Aujard F. Resveratrol in mammals: effects on aging biomarkers, age-related diseases, and life span. Ann. N. Y. Acad. Sci. 2013;1290:67–73. doi: 10.1111/nyas.12214. [DOI] [PubMed] [Google Scholar]
- 94.Dludla PV, et al. Exploring the comparative efficacy of metformin and resveratrol in the management of diabetes-associated complications: a systematic review of preclinical studies. Nutrients. 2020;12:739. doi: 10.3390/nu12030739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 97.Bulavin DV, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004;36:343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 98.Vermeulen L, et al. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18:2238–2251. doi: 10.1016/j.cellsig.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 100.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Alspach E, et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014;4:716–729. doi: 10.1158/2159-8290.CD-13-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967–977. doi: 10.1016/j.cmet.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 103.Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350:568–571. doi: 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 104.Wang C, et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity. 2018;48:675–687.e677. doi: 10.1016/j.immuni.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 105.Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe. 2016;19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kerur N, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2018;24:50–61. doi: 10.1038/nm.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363:eaat8657. doi: 10.1126/science.aat8657. [DOI] [PubMed] [Google Scholar]
- 108.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018;215:1287–1299. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thomas CA, et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell. 2017;21:319–331 e318. doi: 10.1016/j.stem.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simon M, et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 2019;29:871–885.e875. doi: 10.1016/j.cmet.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saleh A, Macia A, Muotri AR. Transposable elements, inflammation, and neurological disease. Front. Neurol. 2019;10:894. doi: 10.3389/fneur.2019.00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.De Cecco M, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566:73–78. doi: 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Olivieri F, et al. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res. Rev. 2013;12:1056–1068. doi: 10.1016/j.arr.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 114.Rippo MR, et al. MitomiRs in human inflamm-aging: a hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014;56:154–163. doi: 10.1016/j.exger.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 115.Arenas-Padilla M, Mata-Haro V. Regulation of TLR signaling pathways by microRNAs: implications in inflammatory diseases. Cent. Eur. J. Immunol. 2018;43:482–489. doi: 10.5114/ceji.2018.81351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Olivieri F, et al. Toll like receptor signaling in “inflammaging”: microRNA as new players. Immun. Ageing. 2013;10:11. doi: 10.1186/1742-4933-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y, Shi X. MicroRNAs in the regulation of TLR and RIG-I pathways. Cell Mol. Immunol. 2013;10:65–71. doi: 10.1038/cmi.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Venugopal P, et al. Differential expression of microRNAs let-7a, miR-125b, miR-100, and miR-21 and interaction with NF-kB pathway genes in periodontitis pathogenesis. J. Cell Physiol. 2018;233:5877–5884. doi: 10.1002/jcp.26391. [DOI] [PubMed] [Google Scholar]
- 119.Cuervo AM, Macian F. Autophagy and the immune function in aging. Curr. Opin. Immunol. 2014;29:97–104. doi: 10.1016/j.coi.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan P, Ye Y, Mao J, He L. Autophagy and immune-related diseases. Adv. Exp. Med. Biol. 2019;1209:167–179. doi: 10.1007/978-981-15-0606-2_10. [DOI] [PubMed] [Google Scholar]
- 121.Zhong L, Simard MJ, Huot J. Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation. FASEB J. 2018;32:4070–4084. doi: 10.1096/fj.201701536R. [DOI] [PubMed] [Google Scholar]
- 122.Neves J, Sousa-Victor P, Jasper H. Rejuvenating strategies for stem cell-based therapies in aging. Cell Stem Cell. 2017;20:161–175. doi: 10.1016/j.stem.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 2011;13:506–512. doi: 10.1038/ncb0511-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Josephson AM, et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl Acad. Sci. USA. 2019;116:6995–7004. doi: 10.1073/pnas.1810692116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chambers SM, et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nishikawa K, et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Investig. 2010;120:3455–3465. doi: 10.1172/JCI42528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lepperdinger G. Inflammation and mesenchymal stem cell aging. Curr. Opin. Immunol. 2011;23:518–524. doi: 10.1016/j.coi.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tateda K, Matsumoto T, Miyazaki S, Yamaguchi K. Lipopolysaccharide-induced lethality and cytokine production in aged mice. Infect. Immun. 1996;64:769–774. doi: 10.1128/iai.64.3.769-774.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nomellini V, Gomez CR, Gamelli RL, Kovacs EJ. Aging and animal models of systemic insult: trauma, burn, and sepsis. Shock. 2009;31:11–20. doi: 10.1097/SHK.0b013e318180f508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and alzheimer’s disease: the last 22 years. J. Alzheimers Dis. 2016;54:853–857. doi: 10.3233/JAD-160488. [DOI] [PubMed] [Google Scholar]
- 131.Phani S, Loike JD, Przedborski S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012;18:S207–S209. doi: 10.1016/S1353-8020(11)70064-5. [DOI] [PubMed] [Google Scholar]
- 132.Kannel WB, Vasan RS. Is age really a non-modifiable cardiovascular risk factor? Am. J. Cardiol. 2009;104:1307–1310. doi: 10.1016/j.amjcard.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Smykiewicz P, Segiet A, Keag M, Zera T. Proinflammatory cytokines and ageing of the cardiovascular-renal system. Mech. Ageing Dev. 2018;175:35–45. doi: 10.1016/j.mad.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 134.Ramji DP, Davies TS. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26:673–685. doi: 10.1016/j.cytogfr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Moss JW, Ramji DP. Cytokines: roles in atherosclerosis disease progression and potential therapeutic targets. Future Med. Chem. 2016;8:1317–1330. doi: 10.4155/fmc-2016-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023–2038. doi: 10.1016/S0140-6736(16)30173-8. [DOI] [PubMed] [Google Scholar]
- 137.Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi: 10.1016/j.immuni.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lencel P, Magne D. Inflammaging: the driving force in osteoporosis? Med. Hypotheses. 2011;76:317–321. doi: 10.1016/j.mehy.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 139.Calle MC, Fernandez ML. Inflammation and type 2 diabetes. Diabetes Metab. 2012;38:183–191. doi: 10.1016/j.diabet.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 140.Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. 2019;133:40–50. doi: 10.1182/blood-2018-06-856500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Papadaki HA, et al. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: evidence for a tumor necrosis factor alpha-mediated effect. Blood. 2002;99:1610–1619. doi: 10.1182/blood.V99.5.1610. [DOI] [PubMed] [Google Scholar]
- 142.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130:1693–1698. doi: 10.1182/blood-2017-06-780882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lu HR, Vandeplassche G, Wouters L, Borgers M. Beta-blockade in the ischemic reperfused working rabbit heart: dissociation of beta-adrenergic blocking and protective effects. Arch. Int. Pharmacodyn. Ther. 1989;301:165–181. [PubMed] [Google Scholar]
- 144.Reynaud D, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–673. doi: 10.1016/j.ccr.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stifter G, et al. Over-expression of tumor necrosis factor-alpha in bone marrow biopsies from patients with myelodysplastic syndromes: relationship to anemia and prognosis. Eur. J. Haematol. 2005;75:485–491. doi: 10.1111/j.1600-0609.2005.00551.x. [DOI] [PubMed] [Google Scholar]
- 146.Agita A, Alsagaff MT. Inflammation, immunity, and hypertension. Acta Med. Indones. 2017;49:158–165. [PubMed] [Google Scholar]
- 147.Fulop T, Witkowski JM, Olivieri F, Larbi A. The integration of inflammaging in age-related diseases. Semin. Immunol. 2018;40:17–35. doi: 10.1016/j.smim.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 148.Fulop T, et al. Frailty, inflammation and immunosenescence. Interdiscip. Top. Gerontol. Geriatr. 2015;41:26–40. doi: 10.1159/000381134. [DOI] [PubMed] [Google Scholar]
- 149.Leonardi GC, et al. Ageing: from inflammation to cancer. Immun. Ageing. 2018;15:1. doi: 10.1186/s12979-017-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee Y, et al. Synergistic inhibition effect of TNIK inhibitor KY-05009 and receptor tyrosine kinase inhibitor dovitinib on IL-6-induced proliferation and Wnt signaling pathway in human multiple myeloma cells. Oncotarget. 2017;8:41091–41101. doi: 10.18632/oncotarget.17056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jeffery V, et al. IL-6 signaling regulates small intestinal crypt homeostasis. J. Immunol. 2017;199:304–311. doi: 10.4049/jimmunol.1600960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 153.Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 154.Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yuan A, Chen JJ, Yao PL, Yang PC. The role of interleukin-8 in cancer cells and microenvironment interaction. Front. Biosci. 2005;10:853–865. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 157.Bissell MJ, Radisky D. Putting tumours in context. Nat. Rev. Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ruscetti M, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020;181:424–441 e421. doi: 10.1016/j.cell.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 161.Bu L, et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38:4887–4901. doi: 10.1038/s41388-019-0765-y. [DOI] [PubMed] [Google Scholar]
- 162.Sahai E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020;20:174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Erez N, et al. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 164.Erez N, et al. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013;437:397–402. doi: 10.1016/j.bbrc.2013.06.089. [DOI] [PubMed] [Google Scholar]
- 165.Richards KE, et al. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770–1778. doi: 10.1038/onc.2016.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marin-Aguilar F, et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2020;19:e13050. doi: 10.1111/acel.13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Grosse L, et al. Defined p16(high) senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32:87–99 e86. doi: 10.1016/j.cmet.2020.05.002. [DOI] [PubMed] [Google Scholar]
- 169.Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ma S, et al. Caloric restriction reprograms the single-cell transcriptional landscape of rattus norvegicus aging. Cell. 2020;180:984–1001.e1022. doi: 10.1016/j.cell.2020.02.008. [DOI] [PubMed] [Google Scholar]
- 171.Zhu X, et al. Activation of Sirt1 by resveratrol inhibits TNF-alpha induced inflammation in fibroblasts. PLoS ONE. 2011;6:e27081. doi: 10.1371/journal.pone.0027081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu L, et al. Inhibition of NF-kappaB signaling pathway by resveratrol improves spinal cord injury. Front. Neurosci. 2018;12:690. doi: 10.3389/fnins.2018.00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Said RS, El-Demerdash E, Nada AS, Kamal MM. Resveratrol inhibits inflammatory signaling implicated in ionizing radiation-induced premature ovarian failure through antagonistic crosstalk between silencing information regulator 1 (SIRT1) and poly(ADP-ribose) polymerase 1 (PARP-1) Biochem. Pharmacol. 2016;103:140–150. doi: 10.1016/j.bcp.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 174.Buhrmann C, et al. Sirt1 is required for resveratrol-mediated chemopreventive effects in colorectal cancer cells. Nutrients. 2016;8:145. doi: 10.3390/nu8030145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Moiseeva O, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12:489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 176.Anisimov VN, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–2773. doi: 10.4161/cc.7.17.6625. [DOI] [PubMed] [Google Scholar]
- 177.Anisimov VN, et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging. 2011;3:148–157. doi: 10.18632/aging.100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Martin-Montalvo A, et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Espada L, et al. Loss of metabolic plasticity underlies metformin toxicity in aged Caenorhabditis elegans. Nat. Metab. 2020;2:1316–1331. doi: 10.1038/s42255-020-00307-1. [DOI] [PubMed] [Google Scholar]
- 180.Zhu X, et al. Effect of metformin on cardiac metabolism and longevity in aged female mice. Front. Cell Dev. Biol. 2020;8:626011. doi: 10.3389/fcell.2020.626011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009;27:351–357. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. doi: 10.1016/j.cell.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013;17:954–964. doi: 10.1016/j.cmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166:822–839. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ermolaeva M, Neri F, Ori A, Rudolph KL. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 2018;19:594–610. doi: 10.1038/s41580-018-0020-3. [DOI] [PubMed] [Google Scholar]
- 186.Zhang W, Qu J, Liu GH, Belmonte JCI. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020;21:137–150. doi: 10.1038/s41580-019-0204-5. [DOI] [PubMed] [Google Scholar]
- 187.Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell. 2015;14:924–932. doi: 10.1111/acel.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu XS, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247 e217. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 190.Narita M, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 191.Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169–1178. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xie W, et al. DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell. 2018;33:309–321 e305. doi: 10.1016/j.ccell.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bianchessi V, et al. Methylation profiling by bisulfite sequencing analysis of the mtDNA non-coding region in replicative and senescent endothelial cells. Mitochondrion. 2016;27:40–47. doi: 10.1016/j.mito.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 194.Shock LS, et al. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl Acad. Sci. USA. 2011;108:3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Koch CM, et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013;23:248–259. doi: 10.1101/gr.141945.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sakaki M, et al. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS ONE. 2017;12:e0171431. doi: 10.1371/journal.pone.0171431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Beerman I, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–425. doi: 10.1016/j.stem.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sun D, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hernando-Herraez I, et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun. 2019;10:4361. doi: 10.1038/s41467-019-12293-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Maegawa S, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bork S, et al. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010;9:54–63. doi: 10.1111/j.1474-9726.2009.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fernandez AF, et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res. 2015;25:27–40. doi: 10.1101/gr.169011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Benetti R, Garcia-Cao M, Blasco MA. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007;39:243–250. doi: 10.1038/ng1952. [DOI] [PubMed] [Google Scholar]
- 204.Diao, D. et al. Telomeric epigenetic response mediated by Gadd45a regulates stem cell aging and lifespan. EMBO Rep. 19, e45494 (2018). [DOI] [PMC free article] [PubMed]
- 205.Meer MV, Podolskiy DI, Tyshkovskiy A, Gladyshev VN. A whole lifespan mouse multi-tissue DNA methylation clock. Elife. 2018;7:e40675. doi: 10.7554/eLife.40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Heyn H, et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl Acad. Sci. USA. 2012;109:10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Field AE, et al. DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell. 2018;71:882–895. doi: 10.1016/j.molcel.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bell CG, et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 2019;20:249. doi: 10.1186/s13059-019-1824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018;19:371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 210.Gronniger E, et al. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rakyan VK, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zykovich A, et al. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell. 2014;13:360–366. doi: 10.1111/acel.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yuan T, et al. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 2015;11:e1004996. doi: 10.1371/journal.pgen.1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging. 2011;3:1018–1027. doi: 10.18632/aging.100395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Horvath S, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhang Q, et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019;11:54. doi: 10.1186/s13073-019-0667-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bell JT, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Marioni RE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chen BH, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging. 2016;8:1844–1865. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Perna L, et al. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenetics. 2016;8:64. doi: 10.1186/s13148-016-0228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.McCartney DL, et al. Investigating the relationship between DNA methylation age acceleration and risk factors for Alzheimer’s disease. Alzheimers Dement. 2018;10:429–437. doi: 10.1016/j.dadm.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Day JJ, et al. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013;16:1445–1452. doi: 10.1038/nn.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat. Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sung HY, et al. Amyloid protein-mediated differential DNA methylation status regulates gene expression in Alzheimer’s disease model cell line. Biochem. Biophys. Res Commun. 2011;414:700–705. doi: 10.1016/j.bbrc.2011.09.136. [DOI] [PubMed] [Google Scholar]
- 227.De Jager PL, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mastroeni D, et al. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol. Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Chouliaras L, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging. 2013;34:2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Marioni RE, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015;44:1388–1396. doi: 10.1093/ije/dyu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Horvath S, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;14:491–495. doi: 10.1111/acel.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Breitling LP, et al. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenetics. 2016;8:21. doi: 10.1186/s13148-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zheng Y, et al. Blood epigenetic age may predict cancer incidence and mortality. EBioMedicine. 2016;5:68–73. doi: 10.1016/j.ebiom.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Takahashi A, et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol. Cell. 2012;45:123–131. doi: 10.1016/j.molcel.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 235.Nelson DM, et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016;17:158. doi: 10.1186/s13059-016-1017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sanders YY, et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013;1:8–16. doi: 10.1016/j.redox.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chandra T, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Martin N, et al. Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK(4)a regulation. EMBO J. 2013;32:982–995. doi: 10.1038/emboj.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Martin N, Beach D, Gil J. Ageing as developmental decay: insights from p16(INK4a.) Trends Mol. Med. 2014;20:667–674. doi: 10.1016/j.molmed.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 240.Soriano-Canton R, et al. Regulation of the p19(Arf)/p53 pathway by histone acetylation underlies neural stem cell behavior in senescence-prone SAMP8 mice. Aging Cell. 2015;14:453–462. doi: 10.1111/acel.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhai Y, et al. Histone deacetylase inhibitor valproic acid promotes the induction of pluripotency in mouse fibroblasts by suppressing reprogramming-induced senescence stress. Exp. Cell Res. 2015;337:61–67. doi: 10.1016/j.yexcr.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 242.Sen P, et al. Histone acetyltransferase p300 induces de novo super-enhancers to drive cellular senescence. Mol. Cell. 2019;73:684–698 e688. doi: 10.1016/j.molcel.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Veitia RA, Govindaraju DR, Bottani S, Birchler JA. Aging: somatic mutations, epigenetic drift and gene dosage imbalance. Trends Cell Biol. 2017;27:299–310. doi: 10.1016/j.tcb.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 244.Carvalhal Marques F, Volovik Y, Cohen E. The roles of cellular and organismal aging in the development of late-onset maladies. Annu. Rev. Pathol. 2015;10:1–23. doi: 10.1146/annurev-pathol-012414-040508. [DOI] [PubMed] [Google Scholar]
- 245.Eisenstein M. Centenarians: great expectations. Nature. 2012;492:S6–S8. doi: 10.1038/492S6a. [DOI] [PubMed] [Google Scholar]
- 246.Nacarelli T, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019;21:397–407. doi: 10.1038/s41556-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ido Y, et al. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE. 2015;10:e0115341. doi: 10.1371/journal.pone.0115341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Han X, et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 2016;15:416–427. doi: 10.1111/acel.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Jadeja RN, et al. Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging. 2018;10:1306–1323. doi: 10.18632/aging.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Liu L, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Beerman I, Rossi DJ. Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell. 2015;16:613–625. doi: 10.1016/j.stem.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kung AL, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–277. [PMC free article] [PubMed] [Google Scholar]
- 253.Rebel VI, et al. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl Acad. Sci. USA. 2002;99:14789–14794. doi: 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Chan WI, et al. The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol. Cell Biol. 2011;31:5046–5060. doi: 10.1128/MCB.05830-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Katsumoto T, Yoshida N, Kitabayashi I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008;99:1523–1527. doi: 10.1111/j.1349-7006.2008.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Turgeon N, et al. HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE. 2013;8:e73785. doi: 10.1371/journal.pone.0073785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zimberlin CD, et al. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. 2015;29:2070–2080. doi: 10.1096/fj.14-257931. [DOI] [PubMed] [Google Scholar]
- 258.Wilting RH, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Heideman MR, et al. Sin3a-associated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. Haematologica. 2014;99:1292–1303. doi: 10.3324/haematol.2013.092643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sun G, Yu RT, Evans RM, Shi Y. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc. Natl Acad. Sci. USA. 2007;104:15282–15287. doi: 10.1073/pnas.0704089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lee S, et al. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 2009;42:711–720. doi: 10.1111/j.1365-2184.2009.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Simic P, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol. Med. 2013;5:430–440. doi: 10.1002/emmm.201201606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Rafalski VA, et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat. Cell Biol. 2013;15:614–624. doi: 10.1038/ncb2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Li W, et al. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang H, et al. SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of Wnt signaling. Cell Stem Cell. 2016;18:495–507. doi: 10.1016/j.stem.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 266.Ming M, Qiang L, Zhao B, He YY. Mammalian SIRT2 inhibits keratin 19 expression and is a tumor suppressor in skin. Exp. Dermatol. 2014;23:207–209. doi: 10.1111/exd.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ryall JG, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Rimmele P, et al. Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. 2014;3:44–59. doi: 10.1016/j.stemcr.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Matsui K, et al. NAD-dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res Commun. 2012;418:811–817. doi: 10.1016/j.bbrc.2012.01.109. [DOI] [PubMed] [Google Scholar]
- 270.Straume B, Forsdahl A. [Accessibility and waiting time in general practice. A patient study in Northern Norway in 1987] Tidsskr. Nor. Laegeforen. 1990;110:3484–3488. [PubMed] [Google Scholar]
- 271.Sharma GG, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell Biol. 2010;30:3582–3595. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Beerman I, et al. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15:37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Avgustinova A, Benitah SA. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016;17:643–658. doi: 10.1038/nrm.2016.76. [DOI] [PubMed] [Google Scholar]
- 274.Faralli H, et al. UTX demethylase activity is required for satellite cell-mediated muscle regeneration. J. Clin. Investig. 2016;126:1555–1565. doi: 10.1172/JCI83239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wight RG, Cochrane T. A comparison of the effects of xylometazoline on nasal airflow, and on blood flux as measured by laser Doppler flowmetry. Acta Otolaryngol. 1989;108:284–289. doi: 10.3109/00016488909125529. [DOI] [PubMed] [Google Scholar]
- 276.Boonsanay V, et al. Regulation of skeletal muscle stem cell quiescence by Suv4-20h1-dependent facultative heterochromatin formation. Cell Stem Cell. 2016;18:229–242. doi: 10.1016/j.stem.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 277.Criscione SW, et al. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016;2:e1500882. doi: 10.1126/sciadv.1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Camozzi D, et al. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus. 2014;5:427–440. doi: 10.4161/nucl.36289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Forsberg F, Brunet A, Ali TML, Collas P. Interplay of lamin A and lamin B LADs on the radial positioning of chromatin. Nucleus. 2019;10:7–20. doi: 10.1080/19491034.2019.1570810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Briand N, Collas P. Laminopathy-causing lamin A mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus. 2018;9:216–226. doi: 10.1080/19491034.2018.1449498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lenain C, et al. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017;27:1634–1644. doi: 10.1101/gr.225763.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 283.Ghosh S, et al. Lamin A is an endogenous sirt6 activator and promotes SIRT6-mediated DNA repair. Cell Rep. 2015;13:1396–1406. doi: 10.1016/j.celrep.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 284.Liu B, et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16:738–750. doi: 10.1016/j.cmet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 285.Mattioli E, et al. Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell. 2018;17:e12824. doi: 10.1111/acel.12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Columbaro M, et al. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol. Life Sci. 2005;62:2669–2678. doi: 10.1007/s00018-005-5318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Filesi I, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol. Genomics. 2005;23:150–158. doi: 10.1152/physiolgenomics.00060.2005. [DOI] [PubMed] [Google Scholar]
- 288.McCord RP, et al. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23:260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics. 2013;8:28–33. doi: 10.4161/epi.23366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shah PP, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Pal S, Tyler JK. Epigenetics and aging. Sci. Adv. 2016;2:e1600584. doi: 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Payer LM, Burns KH. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019;20:760–772. doi: 10.1038/s41576-019-0165-8. [DOI] [PubMed] [Google Scholar]
- 293.Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351:aac7247. doi: 10.1126/science.aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Chandra T, Kirschner K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016;40:161–167. doi: 10.1016/j.ceb.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 295.De Cecco M, et al. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging. 2013;5:867–883. doi: 10.18632/aging.100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Green CD, et al. Impact of dietary interventions on noncoding RNA networks and mRNAs encoding chromatin-related factors. Cell Rep. 2017;18:2957–2968. doi: 10.1016/j.celrep.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 297.Oberdoerffer P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Ishak CA, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell. 2016;64:1074–1087. doi: 10.1016/j.molcel.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Blaudin de The FX, et al. Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE-1 repression. EMBO J. 2018;37:e97374. doi: 10.15252/embj.201797374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Tchasovnikarova IA, et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. 2015;348:1481–1485. doi: 10.1126/science.aaa7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Liu N, et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature. 2018;553:228–232. doi: 10.1038/nature25179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Weimer S, et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 2014;5:3563. doi: 10.1038/ncomms4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Johnson LC, et al. Amino acid and lipid associated plasma metabolomic patterns are related to healthspan indicators with ageing. Clin. Sci. 2018;132:1765–1777. doi: 10.1042/CS20180409. [DOI] [PubMed] [Google Scholar]
- 305.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 306.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 307.Zhang W, et al. SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature. 2018;560:661–665. doi: 10.1038/s41586-018-0437-z. [DOI] [PubMed] [Google Scholar]
- 308.Mitchell SJ, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014;6:836–843. doi: 10.1016/j.celrep.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Mercken EM, et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell. 2014;13:787–796. doi: 10.1111/acel.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Singh SK, et al. Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. J. Exp. Med. 2013;210:987–1001. doi: 10.1084/jem.20121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Leko V, et al. SIRT1 is dispensable for function of hematopoietic stem cells in adult mice. Blood. 2012;119:1856–1860. doi: 10.1182/blood-2011-09-377077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ou X, et al. SIRT1 deficiency compromises mouse embryonic stem cell hematopoietic differentiation, and embryonic and adult hematopoiesis in the mouse. Blood. 2011;117:440–450. doi: 10.1182/blood-2010-03-273011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.North BJ, et al. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11:437–444. doi: 10.1016/S1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 314.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Brown K, et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3:319–327. doi: 10.1016/j.celrep.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Lang A, et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging. 2016;8:484–505. doi: 10.18632/aging.100905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Castex J, et al. Inactivation of Lsd1 triggers senescence in trophoblast stem cells by induction of Sirt4. Cell Death Dis. 2017;8:e2631. doi: 10.1038/cddis.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radic. Biol. Med. 2016;100:164–174. doi: 10.1016/j.freeradbiomed.2016.04.197. [DOI] [PubMed] [Google Scholar]
- 319.Yang L, et al. Sirtuin 5: a review of structure, known inhibitors and clues for developing new inhibitors. Sci. China Life Sci. 2017;60:249–256. doi: 10.1007/s11427-016-0060-7. [DOI] [PubMed] [Google Scholar]
- 320.Polletta L, et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. 2015;11:253–270. doi: 10.1080/15548627.2015.1009778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Denu RA, Hematti P. Effects of oxidative stress on mesenchymal stem cell biology. Oxid. Med Cell Longev. 2016;2016:2989076. doi: 10.1155/2016/2989076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Pan H, et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016;26:190–205. doi: 10.1038/cr.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Mohrin M, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 325.Kim JB, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 326.Chen K, et al. Heterochromatin loosening by the Oct4 linker region facilitates Klf4 binding and iPSC reprogramming. EMBO J. 2020;39:e99165. doi: 10.15252/embj.201899165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 328.Bhutani N, et al. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Mertens J, et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell. 2015;17:705–718. doi: 10.1016/j.stem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020;19:513–532. doi: 10.1038/s41573-020-0067-7. [DOI] [PubMed] [Google Scholar]
- 331.Serrano M, Barzilai N. Targeting senescence. Nat. Med. 2018;24:1092–1094. doi: 10.1038/s41591-018-0141-4. [DOI] [PubMed] [Google Scholar]
- 332.Fontana L. Interventions to promote cardiometabolic health and slow cardiovascular ageing. Nat. Rev. Cardiol. 2018;15:566–577. doi: 10.1038/s41569-018-0026-8. [DOI] [PubMed] [Google Scholar]
- 333.Wu D, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559:637–641. doi: 10.1038/s41586-018-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Pietrocola F, et al. Aspirin recapitulates features of caloric restriction. Cell Rep. 2018;22:2395–2407. doi: 10.1016/j.celrep.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Baumann K. Rejuvenating senolytics. Nat. Rev. Mol. Cell Biol. 2018;19:543. doi: 10.1038/s41580-018-0047-5. [DOI] [PubMed] [Google Scholar]
- 336.Bussian TJ, et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–582. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Shyh-Chang N, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Li S, et al. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol. Cell. 2015;60:408–421. doi: 10.1016/j.molcel.2015.09.024. [DOI] [PubMed] [Google Scholar]
- 339.Liu PS, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017;18:985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- 340.Yu L, et al. Structural insights into a novel histone demethylase PHF8. Cell Res. 2010;20:166–173. doi: 10.1038/cr.2010.8. [DOI] [PubMed] [Google Scholar]
- 341.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Cao J, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–1385. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Angermueller C, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods. 2016;13:229–232. doi: 10.1038/nmeth.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Cheung P, et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell. 2018;173:1385–1397 e1314. doi: 10.1016/j.cell.2018.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Smallwood SA, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Rotem A, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Cusanovich DA, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–914. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Nagano T, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Chen S, Lake BB, Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019;37:1452–1457. doi: 10.1038/s41587-019-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Liu L, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 2019;10:470. doi: 10.1038/s41467-018-08205-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-kappaB signalling. Mech. Ageing Dev. 2018;170:30–36. doi: 10.1016/j.mad.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Meng J, et al. The decay of redox-stress response capacity is a substantive characteristic of aging: revising the redox theory of aging. Redox Biol. 2017;11:365–374. doi: 10.1016/j.redox.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012;16:18–31. doi: 10.1016/j.cmet.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Kops GJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 355.Parkes TL, et al. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat. Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 356.Schriner SE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 357.Paul BD, Sbodio JI, Snyder SH. Cysteine metabolism in neuronal redox homeostasis. Trends Pharm. Sci. 2018;39:513–524. doi: 10.1016/j.tips.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharm. Toxicol. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- 359.Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 2015;14:1–7. doi: 10.1111/acel.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Biefer HRC, Elkhal A, Cesarovic N, Emmert MY. NAD+ the disregarded molecule in cardiac metabolism. Eur. Heart J. 2020;41:983–986. doi: 10.1093/eurheartj/ehaa044. [DOI] [PubMed] [Google Scholar]
- 361.Tarrago MG, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD(+) decline. Cell Metab. 2018;27:1081–1095 e1010. doi: 10.1016/j.cmet.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Zhu X, et al. Fine-tuning of PGC1alpha expression regulates cardiac function and longevity. Circ. Res. 2019;125:707–719. doi: 10.1161/CIRCRESAHA.119.315529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Zou Y, et al. Illuminating NAD(+) metabolism in live cells and in vivo using a genetically encoded fluorescent sensor. Dev. Cell. 2020;53:240–252.e247. doi: 10.1016/j.devcel.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Zhang H, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 366.Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016;17:679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Zhu X, et al. Nicotinamide adenine dinucleotide replenishment rescues colon degeneration in aged mice. Signal Transduct. Target Ther. 2017;2:17017. doi: 10.1038/sigtrans.2017.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Katsyuba E, et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature. 2018;563:354–359. doi: 10.1038/s41586-018-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Pirinen E, et al. Niacin cures systemic NAD(+) Deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. 2020;31:1078–1090 e1075. doi: 10.1016/j.cmet.2020.04.008. [DOI] [PubMed] [Google Scholar]
- 370.Goodman RP, et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature. 2020;583:122–126. doi: 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Patgiri A, et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat. Biotechnol. 2020;38:309–313. doi: 10.1038/s41587-019-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 373.Zhao Y, et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 2015;21:777–789. doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Zou Y, et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018;13:2362–2386. doi: 10.1038/s41596-018-0042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Horecker BL. The pentose phosphate pathway. J. Biol. Chem. 2002;277:47965–47971. doi: 10.1074/jbc.X200007200. [DOI] [PubMed] [Google Scholar]
- 376.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Bjelakovic G, et al. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 378.Hecker L, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014;6:231ra247. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Das A, et al. Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell. 2019;176:944–945. doi: 10.1016/j.cell.2019.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Khrapko K, Vijg J. Mitochondrial DNA mutations and aging: devils in the details? Trends Genet. 2009;25:91–98. doi: 10.1016/j.tig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 382.Passos JF, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Gupta A, et al. SOD2 activity is not impacted by hyperoxia in murine neonatal pulmonary artery smooth muscle cells and mice. Int. J. Mol. Sci. 2015;16:6373–6390. doi: 10.3390/ijms16036373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Yalcin S, et al. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3(-/-) mice. EMBO J. 2010;29:4118–4131. doi: 10.1038/emboj.2010.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Klotz LO, et al. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Rabinovitch RC, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21:1–9. doi: 10.1016/j.celrep.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 387.Zhao Y, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol. Cancer. 2017;16:79. doi: 10.1186/s12943-017-0648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Maryanovich M, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012;14:535–541. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 389.Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8:188–199. doi: 10.1016/j.stem.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Niedernhofer LJ, et al. Nuclear genomic instability and aging. Annu. Rev. Biochem. 2018;87:295–322. doi: 10.1146/annurev-biochem-062917-012239. [DOI] [PubMed] [Google Scholar]
- 391.Vermeij WP, et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016;537:427–431. doi: 10.1038/nature19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Puente BN, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Gong G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Sack, M. N. & Finkel, T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb. Perspect. Biol. 4, a013102 (2012). [DOI] [PMC free article] [PubMed]
- 396.Guarente L. Linking DNA damage, NAD(+)/SIRT1, and aging. Cell Metab. 2014;20:706–707. doi: 10.1016/j.cmet.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 397.Luna A, Aladjem MI, Kohn KW. SIRT1/PARP1 crosstalk: connecting DNA damage and metabolism. Genome Integr. 2013;4:6. doi: 10.1186/2041-9414-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013–1021. doi: 10.1016/j.cmet.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–1198. doi: 10.1126/science.aab3389. [DOI] [PubMed] [Google Scholar]
- 400.Xie Z, et al. Early telomerase inactivation accelerates aging independently of telomere length. Cell. 2015;160:928–939. doi: 10.1016/j.cell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Vulliamy T, et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 402.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–2170. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 403.Ju Z, et al. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat. Med. 2007;13:742–747. doi: 10.1038/nm1578. [DOI] [PubMed] [Google Scholar]
- 404.Choudhury AR, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 405.Schaetzlein S, et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell. 2007;130:863–877. doi: 10.1016/j.cell.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Chen LY, et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol. Cell. 2012;47:839–850. doi: 10.1016/j.molcel.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Amano H, et al. Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell Metab. 2019;29:1274–1290 e1279. doi: 10.1016/j.cmet.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Munoz-Lorente MA, Cano-Martin AC, Blasco MA. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019;10:4723. doi: 10.1038/s41467-019-12664-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Liu D, Xu Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011;15:1669–1678. doi: 10.1089/ars.2010.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2010;2:a001040. doi: 10.1101/cshperspect.a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Wanka C, et al. Synthesis of cytochrome C oxidase 2: a p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene. 2012;31:3764–3776. doi: 10.1038/onc.2011.530. [DOI] [PubMed] [Google Scholar]
- 413.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 414.Wang J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–1919. doi: 10.1161/CIRCULATIONAHA.115.016457. [DOI] [PubMed] [Google Scholar]
- 415.Heo JR, et al. Resveratrol induced reactive oxygen species and endoplasmic reticulum stressmediated apoptosis, and cell cycle arrest in the A375SM malignant melanoma cell line. Int. J. Mol. Med. 2018;42:1427–1435. doi: 10.3892/ijmm.2018.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Saito S, et al. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J. Biol. Chem. 2002;277:12491–12494. doi: 10.1074/jbc.C200093200. [DOI] [PubMed] [Google Scholar]
- 417.Chen Z, et al. Wip1 deficiency impairs haematopoietic stem cell function via p53 and mTORC1 pathways. Nat. Commun. 2015;6:6808. doi: 10.1038/ncomms7808. [DOI] [PubMed] [Google Scholar]
- 418.Yi W, et al. Phosphatase Wip1 controls antigen-independent B-cell development in a p53-dependent manner. Blood. 2015;126:620–628. doi: 10.1182/blood-2015-02-624114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Zhang L, et al. Inhibition of wild-type p53-induced phosphatase 1 promotes liver regeneration in mice by direct activation of mammalian target of rapamycin. Hepatology. 2015;61:2030–2041. doi: 10.1002/hep.27755. [DOI] [PubMed] [Google Scholar]
- 420.Kang C, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Schneider JG, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 422.Ching JK, et al. Impaired insulin-stimulated glucose transport in ATM-deficient mouse skeletal muscle. Appl. Physiol. Nutr. Metab. 2013;38:589–596. doi: 10.1139/apnm-2012-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Fang EF, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24:566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 425.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 426.Fang EF, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Fang EF, et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016;17:308–321. doi: 10.1038/nrm.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Someya S, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Vakhrusheva O, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 430.Herranz D, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Lee JH, et al. Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging. Nucleic Acids Res. 2019;47:8548–8562. doi: 10.1093/nar/gkz568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Vida A, Marton J, Miko E, Bai P. Metabolic roles of poly(ADP-ribose) polymerases. Semin. Cell Dev. Biol. 2017;63:135–143. doi: 10.1016/j.semcdb.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 434.Burkle A, Diefenbach J, Brabeck C, Beneke S. Ageing and PARP. Pharm. Res. 2005;52:93–99. doi: 10.1016/j.phrs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 435.Burton DGA, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018;43:17–25. doi: 10.1016/j.arr.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 436.Furman D, et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019;25:1822–1832. doi: 10.1038/s41591-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Demaria M, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356:1026–1030. doi: 10.1126/science.aam7928. [DOI] [PubMed] [Google Scholar]
- 439.Salminen A. Activation of immunosuppressive network in the aging process. Ageing Res. Rev. 2020;57:100998. doi: 10.1016/j.arr.2019.100998. [DOI] [PubMed] [Google Scholar]
- 440.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 441.Malaquin N, Carrier-Leclerc A, Dessureault M, Rodier F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015;6:94. doi: 10.3389/fgene.2015.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Bonafe M, Sabbatinelli J, Olivieri F. Exploiting the telomere machinery to put the brakes on inflamm-aging. Ageing Res. Rev. 2020;59:101027. doi: 10.1016/j.arr.2020.101027. [DOI] [PubMed] [Google Scholar]
- 443.Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat. Rev. Endocrinol. 2013;9:228–240. doi: 10.1038/nrendo.2013.29. [DOI] [PubMed] [Google Scholar]
- 444.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 445.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020;21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 446.Childs BG, et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017;16:718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Franceschi C, et al. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018;14:576–590. doi: 10.1038/s41574-018-0059-4. [DOI] [PubMed] [Google Scholar]
- 448.Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014;289:35237–35245. doi: 10.1074/jbc.R114.619304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Borras C, et al. Extracellular vesicles and redox modulation in aging. Free Radic. Biol. Med. 2020;149:44–50. doi: 10.1016/j.freeradbiomed.2019.11.032. [DOI] [PubMed] [Google Scholar]
- 450.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Yoshida M, et al. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 2019;30:329–342.e325. doi: 10.1016/j.cmet.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Goulielmaki E, et al. Tissue-infiltrating macrophages mediate an exosome-based metabolic reprogramming upon DNA damage. Nat. Commun. 2020;11:42. doi: 10.1038/s41467-019-13894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Cabral J, Ryan AE, Griffin MD, Ritter T. Extracellular vesicles as modulators of wound healing. Adv. Drug Deliv. Rev. 2018;129:394–406. doi: 10.1016/j.addr.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 454.Rani S, Ritter T. The exosome - a naturally secreted nanoparticle and its application to wound healing. Adv. Mater. 2016;28:5542–5552. doi: 10.1002/adma.201504009. [DOI] [PubMed] [Google Scholar]
- 455.Gallet R, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur. Heart J. 2017;38:201–211. doi: 10.1093/eurheartj/ehw240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev. Immunol. 2014;32:609–634. doi: 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.O’Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019;19:273–281. doi: 10.1038/s41577-019-0128-5. [DOI] [PubMed] [Google Scholar]
- 458.Wang H, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020;21:298–308. doi: 10.1038/s41590-019-0589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Mills EL, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470 e413. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Tomas L, et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur. Heart J. 2018;39:2301–2310. doi: 10.1093/eurheartj/ehy124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018;19:526–537. doi: 10.1038/s41590-018-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex-linking immunity and metabolism. Nat. Rev. Endocrinol. 2012;8:743–754. doi: 10.1038/nrendo.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Theodoropoulou S, Copland DA, Liu J, Dick AD. Role of interleukin 33/ST2 axis in the immune-mediated pathogenesis of age-related macular degeneration. Lancet. 2015;385:S97. doi: 10.1016/S0140-6736(15)60412-3. [DOI] [PubMed] [Google Scholar]
- 464.Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017;18:488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 465.Lenaers G, Bonneau D, Delneste Y, Papon N. Dysfunctional T cell mitochondria lead to premature aging. Trends Mol. Med. 2020;26:799–800. doi: 10.1016/j.molmed.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 466.van Beek AA, et al. Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol. 2019;40:113–127. doi: 10.1016/j.it.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 467.Minhas PS, et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019;20:50–63. doi: 10.1038/s41590-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Tian Q, et al. Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell. 2020;19:e13059. doi: 10.1111/acel.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Kuro-o M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat. Rev. Nephrol. 2013;9:650–660. doi: 10.1038/nrneph.2013.111. [DOI] [PubMed] [Google Scholar]
- 470.John GB, Cheng CY, Kuro-o M. Role of Klotho in aging, phosphate metabolism, and CKD. Am. J. Kidney Dis. 2011;58:127–134. doi: 10.1053/j.ajkd.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Kuro OM. Phosphate as a pathogen of arteriosclerosis and aging. J. Atheroscler. Thromb. 2021;28:203–213. doi: 10.5551/jat.RV17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Sosa P, et al. Hyperphosphatemia promotes senescence of myoblasts by impairing autophagy through ilk overexpression, a possible mechanism involved in sarcopenia. Aging Dis. 2018;9:769–784. doi: 10.14336/AD.2017.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Olmos G, et al. Hyperphosphatemia induces senescence in human endothelial cells by increasing endothelin-1 production. Aging Cell. 2017;16:1300–1312. doi: 10.1111/acel.12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Chen K, et al. Klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ. Res. 2021;128:492–507. doi: 10.1161/CIRCRESAHA.120.317348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Abbenhardt C, et al. Biomarkers of one-carbon metabolism are associated with biomarkers of inflammation in women. J. Nutr. 2014;144:714–721. doi: 10.3945/jn.113.183970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Ron-Harel N, et al. Defective respiration and one-carbon metabolism contribute to impaired naive T cell activation in aged mice. Proc. Natl Acad. Sci. USA. 2018;115:13347–13352. doi: 10.1073/pnas.1804149115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Yu W, et al. One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell. 2019;75:1147–1160 e1145. doi: 10.1016/j.molcel.2019.06.039. [DOI] [PubMed] [Google Scholar]
- 479.Chen S, et al. Serine supports IL-1beta production in macrophages through mTOR signaling. Front. Immunol. 2020;11:1866. doi: 10.3389/fimmu.2020.01866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Lind MV, et al. One-carbon metabolism markers are associated with cardiometabolic risk factors. Nutr. Metab. Cardiovasc. Dis. 2018;28:402–410. doi: 10.1016/j.numecd.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 481.Balsa E, et al. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun. 2020;11:2714. doi: 10.1038/s41467-020-16423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Menendez JA, Joven J. One-carbon metabolism: an aging-cancer crossroad for the gerosuppressant metformin. Aging. 2012;4:894–898. doi: 10.18632/aging.100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Biragyn A, Ferrucci L. Gut dysbiosis: a potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. 2018;19:e295–e304. doi: 10.1016/S1470-2045(18)30095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535:65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 485.Schirmer M, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167:1125–1136.e1128. doi: 10.1016/j.cell.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Schaupp L, et al. Microbiota-induced type I interferons instruct a poised basal state of dendritic cells. Cell. 2020;181:1080–1096.e1019. doi: 10.1016/j.cell.2020.04.022. [DOI] [PubMed] [Google Scholar]
- 487.Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity. 2017;46:549–561. doi: 10.1016/j.immuni.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 488.Ang QY, et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 Cells. Cell. 2020;181:1263–1275.e1216. doi: 10.1016/j.cell.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Blacher E, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019;572:474–480. doi: 10.1038/s41586-019-1443-5. [DOI] [PubMed] [Google Scholar]
- 490.Violi F, Nocella C, Carnevale R. Gut microbiota and myocardial infarction. Eur. Heart J. 2020;41:2221–2222. doi: 10.1093/eurheartj/ehaa222. [DOI] [PubMed] [Google Scholar]
- 491.Galkin F, et al. Human gut microbiome aging clock based on taxonomic profiling and deep learning. iScience. 2020;23:101199. doi: 10.1016/j.isci.2020.101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Kaushik S, Cuervo AM. Proteostasis and aging. Nat. Med. 2015;21:1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 493.Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Enam C, Geffen Y, Ravid T, Gardner RG. Protein quality control degradation in the nucleus. Annu. Rev. Biochem. 2018;87:725–749. doi: 10.1146/annurev-biochem-062917-012730. [DOI] [PubMed] [Google Scholar]
- 495.Steffen KK, Dillin A. A ribosomal perspective on proteostasis and aging. Cell Metab. 2016;23:1004–1012. doi: 10.1016/j.cmet.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 496.Walker CL, Pomatto LCD, Tripathi DN, Davies KJA. Redox regulation of homeostasis and proteostasis in peroxisomes. Physiol. Rev. 2018;98:89–115. doi: 10.1152/physrev.00033.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20:214–225. doi: 10.1016/j.cmet.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep. 2017;19:1083–1090. doi: 10.1016/j.celrep.2017.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Zhang Y, Manning BD. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle. 2015;14:2011–2017. doi: 10.1080/15384101.2015.1044188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Rousseau A, Bertolotti A. An evolutionarily conserved pathway controls proteasome homeostasis. Nature. 2016;536:184–189. doi: 10.1038/nature18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl Acad. Sci. USA. 2015;112:15790–15797. doi: 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Arias E, et al. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol. Cell. 2015;59:270–284. doi: 10.1016/j.molcel.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Lamming DW, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014;5:5659. doi: 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- 507.Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharm. Toxicol. 2015;55:353–371. doi: 10.1146/annurev-pharmtox-010814-124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Peng Y, et al. Improved proteostasis in the secretory pathway rescues Alzheimer’s disease in the mouse. Brain. 2016;139:937–952. doi: 10.1093/brain/awv385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Sorrentino V, et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Poewe W, et al. Parkinson disease. Nat. Rev. Dis. Prim. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 511.Soares TR, et al. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019;49:92–103. doi: 10.1016/j.arr.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Yerbury JJ, Farrawell NE, McAlary L. Proteome homeostasis dysfunction: a unifying principle in ALS pathogenesis. Trends Neurosci. 2020;43:274–284. doi: 10.1016/j.tins.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 513.Li Y, et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019;38:e98786. doi: 10.15252/embj.201798786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Ryu D, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016;22:879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 515.La Spada AR. PPARGC1A/PGC-1alpha, TFEB and enhanced proteostasis in Huntington disease: defining regulatory linkages between energy production and protein-organelle quality control. Autophagy. 2012;8:1845–1847. doi: 10.4161/auto.21862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- 517.Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:731–745. doi: 10.1038/s41580-018-0068-0. [DOI] [PubMed] [Google Scholar]
- 518.Teske BF, et al. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell. 2013;24:2477–2490. doi: 10.1091/mbc.e13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Zhao Q, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE. 2007;2:e835. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Tian Y, et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt) Cell. 2016;165:1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Merkwirth C, et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Nargund AM, et al. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Taylor RC. Aging and the UPR(ER) Brain Res. 2016;1648:588–593. doi: 10.1016/j.brainres.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 525.Frakes AE, Dillin A. The UPR(ER): sensor and coordinator of organismal homeostasis. Mol. Cell. 2017;66:761–771. doi: 10.1016/j.molcel.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 526.Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Daniele JR, et al. UPR(ER) promotes lipophagy independent of chaperones to extend life span. Sci. Adv. 2020;6:eaaz1441. doi: 10.1126/sciadv.aaz1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Schinzel RT, et al. The hyaluronidase, TMEM2, promotes ER homeostasis and longevity independent of the UPR(ER) Cell. 2019;179:1306–1318 e1318. doi: 10.1016/j.cell.2019.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Finkel T. The metabolic regulation of aging. Nat. Med. 2015;21:1416–1423. doi: 10.1038/nm.3998. [DOI] [PubMed] [Google Scholar]
- 530.Lopez-Otin C, et al. Metabolic control of longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 531.Kauppila TES, Kauppila JHK, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab. 2017;25:57–71. doi: 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 532.Jang JY, Blum A, Liu J, Finkel T. The role of mitochondria in aging. J. Clin. Investig. 2018;128:3662–3670. doi: 10.1172/JCI120842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Guo Y, et al. Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell. 2018;175:1430–1442 e1417. doi: 10.1016/j.cell.2018.09.057. [DOI] [PubMed] [Google Scholar]
- 534.Gbel J, et al. Mitochondria-endoplasmic reticulum contacts in reactive astrocytes promote vascular remodeling. Cell Metab. 2020;31:791–808 e798. doi: 10.1016/j.cmet.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Shen M, et al. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat. Neurosci. 2019;22:386–400. doi: 10.1038/s41593-019-0338-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Kim J, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Ying Z, et al. Short-term mitochondrial permeability transition pore opening modulates histone lysine methylation at the early phase of somatic cell reprogramming. Cell Metab. 2018;28:935–945 e935. doi: 10.1016/j.cmet.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 538.MacVicar T, et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature. 2019;575:361–365. doi: 10.1038/s41586-019-1738-6. [DOI] [PubMed] [Google Scholar]
- 539.Leeman DS, et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018;359:1277–1283. doi: 10.1126/science.aag3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Castellano BM, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Wyant GA, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171:642–654 e612. doi: 10.1016/j.cell.2017.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Abu-Remaileh M, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Lapierre LR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Titorenko VI, Terlecky SR. Peroxisome metabolism and cellular aging. Traffic. 2011;12:252–259. doi: 10.1111/j.1600-0854.2010.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Elsner M, Gehrmann W, Lenzen S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes. 2011;60:200–208. doi: 10.2337/db09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Dixit E, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Benjamin D, Hall MN. TSC on the peroxisome controls mTORC1. Nat. Cell Biol. 2013;15:1135–1136. doi: 10.1038/ncb2849. [DOI] [PubMed] [Google Scholar]
- 548.Zhang J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.He X, et al. Single-cell omics in ageing: a young and growing field. Nat. Metab. 2020;2:293–302. doi: 10.1038/s42255-020-0196-7. [DOI] [PubMed] [Google Scholar]
- 550.Wang S, et al. Single-cell transcriptomic atlas of primate ovarian aging. Cell. 2020;180:585–600 e519. doi: 10.1016/j.cell.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 551.Bian Z, et al. Deciphering human macrophage development at single-cell resolution. Nature. 2020;582:571–576. doi: 10.1038/s41586-020-2316-7. [DOI] [PubMed] [Google Scholar]
- 552.Pang H, Jia W, Hu Z. Emerging applications of metabolomics in clinical pharmacology. Clin. Pharm. Ther. 2019;106:544–556. doi: 10.1002/cpt.1538. [DOI] [PubMed] [Google Scholar]
- 553.Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. 2013;342:1243259. doi: 10.1126/science.1243259. [DOI] [PubMed] [Google Scholar]
- 554.Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Majidinia M, Reiter RJ, Shakouri SK, Yousefi B. The role of melatonin, a multitasking molecule, in retarding the processes of ageing. Ageing Res. Rev. 2018;47:198–213. doi: 10.1016/j.arr.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 556.Eisenberg T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016;22:1428–1438. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Harrison DE, et al. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014;13:273–282. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Sinha M, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Biesemann N, et al. Myostatin regulates energy homeostasis in the heart and prevents heart failure. Circ. Res. 2014;115:296–310. doi: 10.1161/CIRCRESAHA.115.304185. [DOI] [PubMed] [Google Scholar]
- 560.Gredilla R, Sanz A, Lopez-Torres M, Barja G. Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 2001;15:1589–1591. doi: 10.1096/fj.00-0764fje. [DOI] [PubMed] [Google Scholar]
- 561.Cabreiro F, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Robida-Stubbs S, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.McAlpine CS, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. 2019;566:383–387. doi: 10.1038/s41586-019-0948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Nagpal R, et al. Gut microbiome and aging: physiological and mechanistic insights. Nutr. Healthy Aging. 2018;4:267–285. doi: 10.3233/NHA-170030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Zhavoronkov A, Mamoshina P. Deep aging clocks: the emergence of AI-based biomarkers of aging and longevity. Trends Pharm. Sci. 2019;40:546–549. doi: 10.1016/j.tips.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 566.Zhavoronkov A, et al. Artificial intelligence for aging and longevity research: Recent advances and perspectives. Ageing Res. Rev. 2019;49:49–66. doi: 10.1016/j.arr.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 567.Jonsson BA, et al. Brain age prediction using deep learning uncovers associated sequence variants. Nat. Commun. 2019;10:5409. doi: 10.1038/s41467-019-13163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 569.Krishnamurthy J, et al. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Beausejour CM, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Chicas A, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012;23:2066–2075. doi: 10.1091/mbc.e11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003;13:1549–1556. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 574.Hewitt G, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Mah, L. J., El-Osta, A. & Karagiannis, T. C. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia24, 679–686 (2010). [DOI] [PubMed]
- 576.Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Lindner AB, et al. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl Acad. Sci. USA. 2008;105:3076–3081. doi: 10.1073/pnas.0708931105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 579.Aird KM, Zhang R. Detection of senescence-associated heterochromatin foci (SAHF) Methods Mol. Biol. 2013;965:185–196. doi: 10.1007/978-1-62703-239-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Johnson AA, et al. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15:483–494. doi: 10.1089/rej.2012.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22:42–49. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Goyarts E, Muizzuddin N, Maes D, Giacomoni PU. Morphological changes associated with aging: age spots and the microinflammatory model of skin aging. Ann. N. Y. Acad. Sci. 2007;1119:32–39. doi: 10.1196/annals.1404.006. [DOI] [PubMed] [Google Scholar]
- 583.Aon MA, et al. Untangling determinants of enhanced health and lifespan through a multi-omics approach in mice. Cell Metab. 2020;32:100–116 e104. doi: 10.1016/j.cmet.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]