

Abstract
Background and Aims
Thrombocytopenia has been described in most patients with acute and chronic liver failure. Decreased platelet production and decreased half‐life of platelets might be a consequence of low levels of thrombopoietin (TPO) in these patients. Platelet production is tightly regulated to avoid bleeding complications after vessel injury and can be enhanced under elevated platelet destruction as observed in liver disease. Thrombopoietin (TPO) is the primary regulator of platelet biogenesis and supports proliferation and differentiation of megakaryocytes.
Approach and Results
Recent work provided evidence for the control of TPO mRNA expression in liver and bone marrow (BM) by scanning circulating platelets. The Ashwell‐Morell receptor (AMR) was identified to bind desialylated platelets to regulate hepatic thrombopoietin (TPO) production by Janus kinase (JAK2)/signal transducer and activator of transcription (STAT3) activation. Two‐thirds partial hepatectomy (PHx) was performed in mice. Platelet activation and clearance by AMR/JAK2/STAT3 signaling and TPO production were analyzed at different time points after PHx. Here, we demonstrate that PHx in mice led to thrombocytopenia and platelet activation defects leading to bleeding complications, but unaltered arterial thrombosis, in these mice. Platelet counts were rapidly restored by up‐regulation and crosstalk of the AMR and the IL‐6 receptor (IL‐6R) to induce JAK2‐STAT3‐TPO activation in the liver, accompanied by an increased number of megakaryocytes in spleen and BM before liver was completely regenerated.
Conclusions
The AMR/IL‐6R‐STAT3‐TPO signaling pathway is an acute‐phase response to liver injury to reconstitute hemostasis. Bleeding complications were attributable to thrombocytopenia and platelet defects induced by elevated PGI2, NO, and bile acid plasma levels early after PHx that might also be causative for the high mortality in patients with liver disease.
Abbreviations
- AMR
Ashwell‐Morell receptor
- Asgr1
asialoglycoprotein receptor 2
- Asgr2
asialoglycoprotein receptor 2
- BDL
bile‐duct–ligated
- BM
bone marrow
- CRP
collagen‐related peptide
- Gplb
glycoprotein Ib
- hIL‐6
hyper‐IL6
- IL‐6R
IL‐6 receptor
- JAK2
Janus kinase 2
- MKs
megakaryocytes
- NO
nitric oxide
- PAR4
protease‐activated receptor 4
- PGI2
prostaglandin I2, prostacyclin
- PHx
partial hepatectomy
- PS
phosphatidylserine
- RCA‐1
Ricinus communis agglutinin
- Ser157
serine 157
- Ser239
serine 239
- SNP
sodium nitroprusside
- STAT3
signal transducer and activator of transcription 3
- STAT5
signal transducer and activator of transcription 5
- TC
taurocholate
- TCDC
taurochenodeoxycholate
- TDC
taurodeoxycholate
- TLC
taurolithocholate
- TPO
thrombopoietin
- U46619
thromboxane A2 analog
- VASP
vasodilator‐stimulated phosphoprotein
- vWF
von Willebrand factor
- WT
wild type
An adequate supply of fully functional platelets is necessary to achieve hemostasis after vascular damage. Platelet production is tightly regulated to avoid either bleeding or arterial thrombosis according to low or high platelet counts. In humans, ~1011 platelets are daily produced and removed. However, platelet production can be enhanced under conditions of platelet destruction (e.g., in response to acute infection). The primary regulator of platelet production is thrombopoietin (TPO) that supports the survival, proliferation, and differentiation of megakaryocytes (MKs), the platelet precursors.( 1 ) In general, the plasma concentration of TPO is inversely related to the mass of platelets and MKs. However, the mechanisms how circulating TPO levels are controlled have been discussed for decades. Whereas some researchers believe that TPO levels in plasma are maintained by its uptake and metabolism by myeloproliferative leukemia virus oncogene receptors on platelets and MKs,( 2 , 3 ) others provided evidence for sensing circulating platelet levels to control TPO mRNA expression in liver and bone marrow (BM).( 4 , 5 ) Recently, Grozovsky et al. have highlighted the role of sialic acid loss and platelet removal by the hepatic Ashwell‐Morell receptor (AMR). They found platelet production to be regulated by the AMR that controls hepatic TPO production by the Janus kinase 2 (JAK2)/signal transducer and activator of transcription (STAT3) signaling pathway.( 6 ) Loss of sialic acid determines the life span of platelets that triggers platelet removal by hepatic AMR. Desialylated platelets bind to the AMR to induce hepatic expression of TPO mRNA and protein to regulate platelet production.
Thrombocytopenia has been described in most patients with acute and chronic liver failure that are causative for bleeding problems in these patients.( 7 ) Decreased platelet production and decreased half‐life of platelets might be a consequence of low levels of TPO in these patients.( 8 , 9 , 10 , 11 ) Besides, platelet activation defects have been reported in experimental mice( 12 ) and patients with cirrhosis.( 13 , 14 , 15 , 16 ) In bile duct ligated (BDL) mice, elevated nitric oxide (NO) and prostacyclin levels induced phosphorylation of the platelet inhibitor, vasodilator‐stimulated phosphoprotein (VASP), leading to prolonged platelet activation defects and defective hemostasis in BDL mice.( 17 , 18 )
However, platelet production following activation of the AMR and JAK2/STAT3 signaling was reported in healthy persons. The consequences of cytokine signaling and reduced platelet life span upon liver injury are open for study. Moreover, regulation of plasma TPO levels by hepatic AMR is also open for study in mice with reduced liver tissue (e.g., after partial hepatectomy [PHx]).
In this study, we provide evidence that enhanced platelet turnover and cytokine signaling synergistically control TPO homeostasis in the remaining liver tissue by activation of the AMR and IL‐6R to restore platelet counts in the early phase after PHx in mice. Platelet activation defects were moderate and occurred early after PHx, resulting in bleeding complications at days 1 and 3 after PHx, whereas arterial thrombosis was not prevented.
Materials and Methods
Animals
Specific pathogen‐free C57BL/6J mice were obtained from Janvier Labs. IL‐6r−/− and Asgr2−/− mice were obtained from The Jackson Laboratory (Bar Harbor, Maine) and the Animal Facility of the Heinrich‐Heine‐University of Düsseldorf, Germany. Experiments were performed with male mice at the age of 8‐16 weeks. All animal experiments were conducted according to the Declaration of Helsinki and the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals. The protocol was approved by the Heinrich‐Heine‐University Animal Care Committee and by the State Agency for Nature, Environment and Consumer Protection of North Rhine‐Westphalia (LANUV, NRW; Permit Nos. 84‐02.04.2015.A462, O 86/12, and 84‐02.04.2016.A493).
All animals for the experiments underwent 2/3 partial hepatectomy, while sham or native (untreated) animals were used as control groups. Therefore a modification of the method of Greene and Puder with two separate ligatures and removal of the gallbladder was used ( 49 ). General physiological parameters such as total blood cell count and specific organ weight were determined at different time points, accompanied by analysis of liver tissue and platelets isolated from the peripheral blood of the mice. In addition, hisotological analysis of the bone marrow and spleen were performed. All analyses and their standard procedures, as well as the antibodies or peptides used for the experiments, are listed in detail in the supporting information.
Statistical Analysis
Data are provided as arithmetic means ± SEM, statistical analysis was made by one‐way or two‐way ANOVA and student’s t‐test, where applicable using GraphPad Prism, Version 7.02. If not stated different, significant differences are indicated in the graphs, while * specifies the difference between PHx versus sham and wildtype versus knock‐out, respectively, and # specifies the difference between experimental time points of one group.
Results
PHx in Mice is Associated With Thrombocytopenia and Anemia at Early Time Points
PHx was performed, and two thirds of liver in wild‐type (WT) mice was removed. Sham‐operated control mice underwent the same surgical procedure without ligation and removal of liver tissue. Blood cell counts and liver weight were analyzed in both groups. Platelet counts were significantly reduced by 16.41% 1 day after surgery and fully restored 7 days after PHx (Fig. 1A) whereas platelet size was enhanced at 1 and 3 days after surgery (Fig. 1D) as compared to platelets from sham controls. Likewise, red blood cell (RBC) count was significantly reduced by 8.75% 1 day after PHx and restored 7 days after PHx (Fig. 1B) whereas no major alterations in white blood cell (WBC) counts were detected (Fig. 1C). Liver weight ratio to body weight was significantly reduced 1, 3, and 7 days after PHx. In line with recent data, liver was fully regenerated 14 days after PHx (Fig. 1E).( 19 )
FIG. 1.
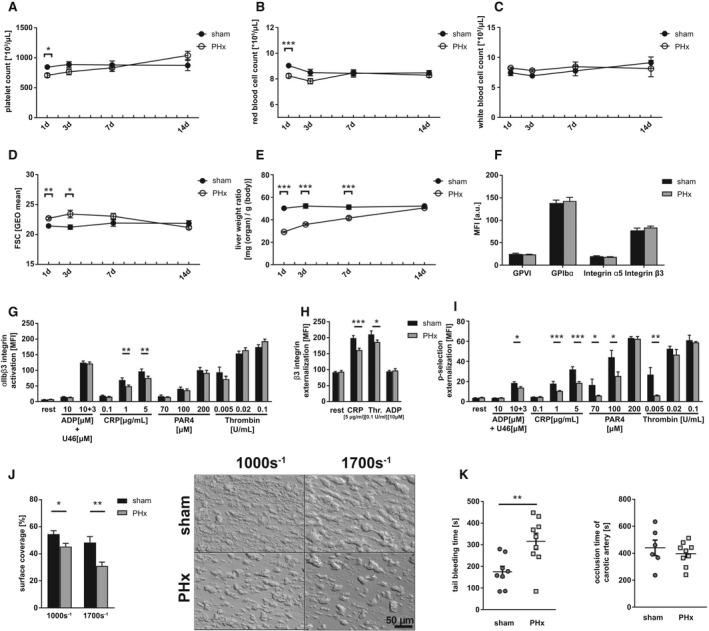
Reduced total blood cell count and platelet activation defects in thrombosis and hemostasis 1 day after PHx. (A) Platelet count, (B) RBC count, and (C) WBC count of C57Bl6/J mice undergoing PHx. Sham operation served as control (n = 1 day: 22 sham, 30 PHx; 3 days: 22 sham, 26 PHx; 7 days: 16 sham, 20 PHx; 14 days: 8 sham, 12 PHx). (D) Platelet size of PHx and sham animals measured by flow cytometry using the geometrical mean of GPIb‐positive platelets. (n = 1 day: 10 sham, 14 PHx; 3/7/14 days: 6 sham, 8 PHx). (E) Calculated liver weight ratio (n = 5 sham, 8 PHx). (F) Expression of indicated glycoproteins on the surface of platelets measured by mean fluorescence intensity (MFI) in flow cytometric analysis (n = 11 sham + 18 PHx). (G) Activation of ⍺IIbβ3 integrin on the platelet surface with indicated agonists (n = 6 sham, 9 PHx). (H) Externalization of the β3 integrin subunit upon platelet stimulation (n = 6 sham, 8 PHx). (I) Externalization of P‐selectin on the platelet surface with indicated agonists (n = 6 sham, 9 PHx). (J) Thrombus formation on a collagen matrix under arterial shear rates with representative pictures (1.000 s−1: n = 6 sham, 5 PHx; 1.700 s−1: n = 5 sham, 9 PHx). (K) In vivo analysis of hemostatic dysfunction by tail bleeding time (n = 8 sham, 9 PHx) and the Fe3Cl‐induced injury of the carotid artery (n = 6 sham, 9 PHx). Depicted are mean values + SEM; *P < 0.05; **P < 0.01; ***P < 0.001, using a paired Student t test. Abbreviations: FSC, forward scatter; GEO, geometric; GPVI, glycoprotein VI.
PHx Provokes Platelet Activation Defects and Reduced Thrombus Formation Resulting in Impaired Hemostasis at Early Phase After PHx
A detailed analysis of platelets 1 day after PHx revealed unaltered glycoprotein exposure at the surface of platelets isolated from PHx mice (Fig. 1F). Platelet activation was analyzed by antibody binding to activated integrin αIIbβ3 (fibrinogen receptor) and determination of P‐selectin exposure as a marker for degranulation using flow cytometry (Fig. 1G‐I). Reduced integrin activation restricted to stimulation of platelets with collagen‐related peptide (CRP) that activates the major collagen receptor, glycoprotein VI, was detected (Fig. 1G) whereas the number of integrin αIIbβ3 at the platelet surface was reduced in response to CRP and thrombin (Fig. 1H). Degranulation of platelets in hepatectomized mice was significantly reduced in response to the second‐wave mediators, ADP + U46619 (thromboxane A2 analog), intermediate and high concentrations of CRP, low and intermediate concentrations of the thrombin receptor activator, protease‐activated receptor 4 (PAR4), peptide, and low dose of thrombin (Fig. 1I). To analyze platelet activation under more physiological conditions, flow chamber experiments were performed. Platelet adhesion and three‐dimensional thrombus formation was analyzed at arterial shear rates (1.000 sec−1, 1.700 sec−1) using collagen coated coverslips (Fig. 1J). Thrombus formation was significantly reduced when we perfused whole blood isolated from hepatectomized mice through the chamber compared to whole blood from sham controls.
Next, we investigated whether platelet activation defects are relevant in vivo. To this end, bleeding time experiments were conducted and time was recorded when bleeding stopped after cutting the tail tip of mice. Bleeding time was significantly prolonged 1 day after PHx compared to sham controls (Fig. 1K). In addition, we examined arterial thrombus formation in mice 1 day after PHx. Fe3Cl‐induced injury of the carotid artery led to occlusion of the vessel after 439 ± 57 seconds in hepatectomized mice and 409 ± 29 seconds in sham controls (Fig. 1K), suggesting no major alterations in arterial thrombosis after PHx in mice despite defective hemostasis.
Further analysis of platelet activation and thrombus formation 3 days after PHx revealed significantly enhanced the exposure of glycoprotein Ib (GPIb)α and integrin β3 at the platelet surface of hepatectomized mice in contrast to 1 day after PHx (Fig. 2A), whereas integrin αIIbβ3 activation was still significantly reduced in response to intermediate and high concentrations of CRP, but unaltered in response to G‐protein‐coupled receptor activation such as thrombin and ADP/U46619 (Fig. 2B). In contrast to day 1 after PHx, the exposure of integrin β3 was significantly enhanced in resting and ADP‐stimulated platelets from PHx mice compared to sham controls (Fig. 2C). P‐selectin exposure as a marker for degranulation was only moderately, but still significantly, reduced at the surface of platelets from PHx mice when they were stimulated with CRP or thrombin (Fig. 2D). Moderate platelet activation defects translated into impaired thrombus formation under flow ex vivo using a shear rate of 1.000 sec−1, but not at a shear rate of 1.700 sec−1 (Fig. 2E). Moreover, bleeding time was still significantly reduced, but to a lesser extent, compared to 1 day after PHx (Fig. 2F). However, no differences in platelet activation and thrombus formation were detected 7 (Supporting Fig. S1A‐E) and 14 days (Supporting Fig. S2A‐E) after PHx.
FIG. 2.
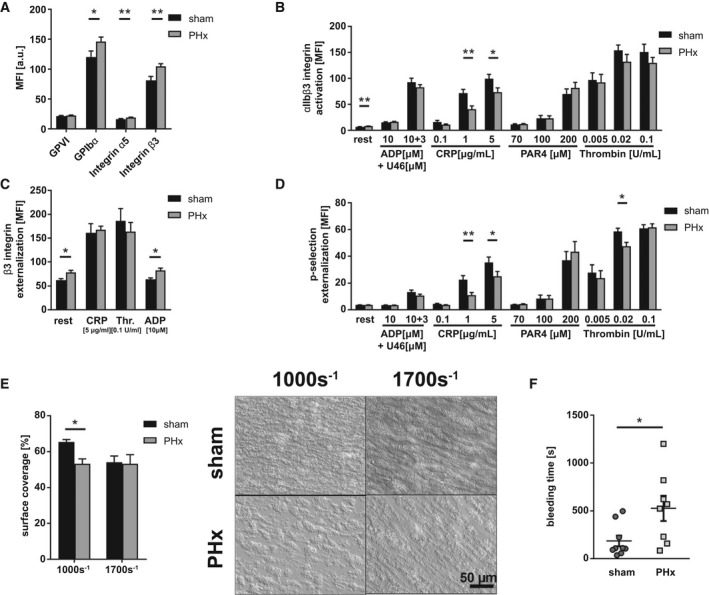
Platelet activation defects and reduced thrombus formation lead to bleeding complications 3 days after PHx. (A) Expression of indicated glycoproteins on the surface of platelets measured by MFI in flow cytometric analysis (n = 13 sham, 18 PHx). (B) Externalization of P‐selectin on the platelet surface with indicated agonists (n = 9 sham, 12 PHx). (C) Externalization of the β3 integrin subunit upon platelet stimulation (n = 8 sham, 9 PHx). (D) Activation of ⍺IIbβ3 integrin on the platelet surface with indicated agonists (n = 9 sham, 12 PHx). (E) Thrombus formation on a collagen matrix under arterial shear rates with representative pictures (1.000 and 1.700 s−1; n = 3 sham, 4 PHx). (F) In vivo analysis of hemostatic dysfunction by tail bleeding time (n = 9 sham, 8 PHx). Depicted are mean values + SEM; *P < 0.05; **P < 0.01, using paired Student t test. Abbreviations: a.u., arbitray units; MFI, mean fluorescence intensity; Thr., thrombin.
Defective Platelet Activation After PHx is Caused by Elevated Nitric Oxide, Prostacyclin, and Bile Acid Plasma Levels
To investigate the mechanisms responsible for reduced platelet activation and thrombus formation 1 and 3 days after PHx, we analyzed the phosphorylation of VASP, a negative regulator of platelet activation. VASP can be phosphorylated at serine 157 (Ser157) by protein kinase A, which is activated by prostaglandin I2 (prostacyclin, PGI2), and at serine 239 (Ser239) by protein kinase G, which is activated by nitric oxide (NO).( 18 , 20 , 21 ) We started to measure the plasma concentration of PGI2 by enzyme‐linked immunosorbent assay (ELISA). At day 1 after PHx, PGI2 plasma levels were significantly enhanced and returned to normal levels at day 3 after PHx compared to sham controls (Fig. 3A). Furthermore, we investigated the stable NO metabolites, nitrate and nitrite, by ELISA. At 1 day after surgery, levels of NO metabolites were enhanced whereas no major alterations were observed 3 days after PHx (Fig. 3B). According to enhanced PGI2 plasma levels, we detected enhanced phosphorylation of VASP at Ser157 in platelets of hepatectomized mice compared to controls using western blotting analysis. However, no major alterations in the phosphorylation of VASP at Ser157 were detected 3 days after PHx (Fig. 3C,D and Supporting Fig. S3A,B). In line with enhanced NO metabolites, enhanced VASP phosphorylation at Ser239 was detected in platelets from hepatectomized mice stimulated with the corresponding NO analog, sodium nitroprusside (SNP). However, no alterations were detected 3 days after PHx (Fig. 3E,F and Supporting Fig. S3A,B).
FIG. 3.
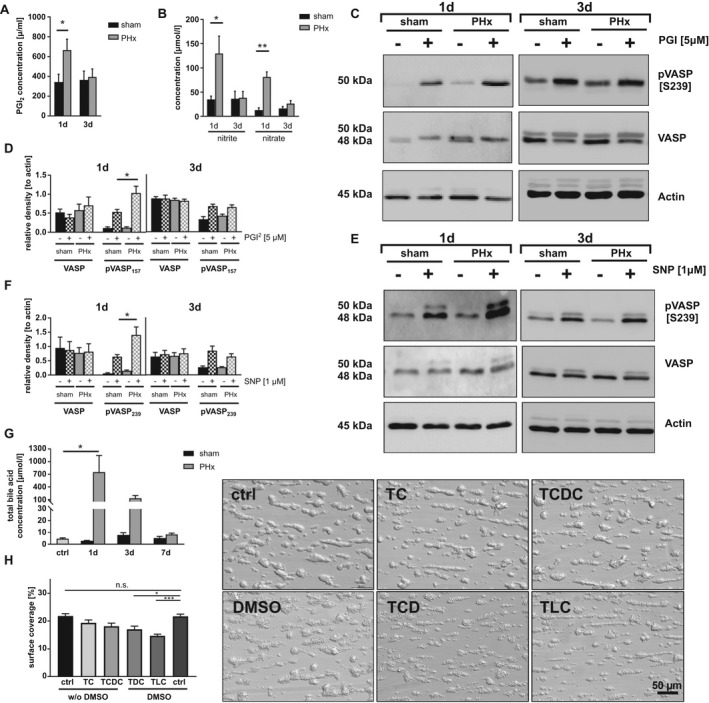
Platelet activation defects are provoked by enhanced plasma levels of NO, prostacyclin, and bile acids. (A) PGI2 plasma levels (n = 1 day: 6 sham, 6 PHx; 3 days: 3 sham, 4 PHx) and (B) indirect NO measurement by the plasma metabolites nitrite and nitrate 1 and 3 days after PHx (n = 1 day: 4 sham, 4 PHx; 3 days: 3 sham, 6 PHx). (C) Representative phosphorylation of VASP at Ser157 in platelets stimulated with 5 µM of PGI2 (D) and densitometric analysis (n = 4 sham, 5 PHx). (E) Representative phosphorylation of VASP at Ser239 and (F) densitometric analysis (n = 5 sham + 4 PHx). (G) Total bile acid content in serum of murine platelets (n = 5 control animals [ctrl]; 1 day: 3 sham, 6 PHx; 3 day: 3 sham, 4 PHx; 7 days: 3 sham, 3 PHx). (H) Thrombus formation under arterial shear rate (1.700 s−1). Whole‐blood samples were pretreated with different bile acids (concentration = 50 µM) for 30 minutes. Indicated bile acids are dissolved in 0.01% DMSO. Representative images are shown for each treatment (n = 6 per group). Depicted are mean values + SEM; *P < 0.05; **P < 0.01, using paired Student t test. Abbreviations: n.s., not significant; pVASP, phosphorylated VASP.
Previously, we have shown that enhanced plasma levels of bile acids account for platelet activation defects in BDL mice.( 17 ) Therefore, we analyzed total plasma levels of bile acids in hepatectomized mice. Enhanced total bile acid plasma levels were detected in mice at days 1 and 3 after PHx compared to sham controls, consistent with published data (Fig. 3G).( 22 ) To provide evidence that bile acids induce platelet activation defects, we incubated whole blood with different tauro‐conjugated endogenous bile acids given that especially taurine‐conjugated bile acids are present in plasma of hepatectomized mice (Supporting Fig. S4A) and analyzed platelet adhesion and thrombus formation at arterial shear rates ex vivo. Incubation of whole blood with the hydrophilic bile acids, taurocholate (TC) or taurochenodeoxycholate (TCDC), did not alter thrombus formation under flow whereas the addition of hydrophobic taurodeoxycholate (TDC) or taurolithocholate (TLC) to whole blood led to significantly reduced thrombus formation under flow compared to controls (Fig. 3H). However, no alterations were detected when platelets were incubated with 50 µM of TC, TCDC, TDC, and TLC, respectively, and platelet aggregation in response to PAR4 peptide and CRP was analyzed under static conditions (Supporting Fig. S4B‐E). These results suggest that hydrophobic bile acids affect platelet activation under dynamic conditions besides high‐plasma concentrations of PGI2 and NO.
Efficiently Restored TPO Production is Mediated by Desialylated Platelet Uptake Into the Diseased Liver by AMR and JAK2‐STAT3 Signaling
TPO, the major regulator of platelet production, is primarily produced in the liver.( 23 ) The paradox, early restoration of platelet counts 3 days after PHx, a time point where liver tissue is still strongly reduced in hepatectomized mice, prompted us to analyze TPO production in the remaining liver tissue in further detail. Senescent desialylated platelets stimulate TPO production.( 6 ) Therefore, we first characterized circulating platelets for phosphatidylserine (PS) exposure at the platelet surface as a marker for procoagulant activity, which might be also important for recognition and subsequent removal of senescent platelets.( 24 ) At early time points after surgery with enhanced levels of PGI2, NO, and bile acids, no major alterations in PS were detected between platelets from hepatectomized mice and sham controls (Fig. 4A). However, 7 days after PHx, decreased PS exposure on the surface of PHx platelets was detected following platelet activation with CRP. Reduced PS returned to basal levels 14 days after surgery when liver tissue regeneration was completed. Aging platelets lose sialic acid, the terminal carbohydrate moiety that covers the underlying galactose residues, a ligand for the hepatic AMR. Desialylated aged platelets are removed from the circulation by hepatic AMR to regulate TPO production by JAK2‐STAT3.( 25 ) We speculated that platelets following PHx may be cleared in the liver by glycan‐dependent mechanisms. Thus, we next investigated the sialic acid content of circulating platelets post PHx. The fraction of desialylated platelets was examined by platelet lectin binding, such as Ricinus communis agglutinin (RCA‐1), a lectin that recognizes terminal galactose residues (Fig. 4B). Reduced lectin binding was detected 1 day after PHx, reflecting a reduced percentage of desialylated platelets. This suggests that desialylated platelet clearance is accelerated at early time points after PHx (Fig. 4B). In contrast, we detected enhanced lectin binding of platelets from hepatectomized mice at day 7 after surgery. Thiazole orange staining revealed an elevated number of newly produced platelets 7 days after PHx, but no alterations at any other time point (1, 3, and 14 days; Fig. 4C). These results suggest a reciprocal relationship between two clearance mechanisms at early and late time points after PHx: (1) reduced desialylated platelet clearance at early time points (day 1) and (2) increased clearance of PS‐positive platelets at late time points (day 7), a notion that is supported by galactose exposure at day 1 and reduced PS exposure with increased RCA‐1 binding of PHx platelets at day 7. Enhanced numbers of RNA‐rich, newly produced platelets 7 days after PHx (Fig. 4A‐C) further support the notion. Platelet clearance can also be induced by vWF binding to GPIb, leading to the release of neuraminidase and subsequent platelet desialylation.( 26 ) Analysis of neuraminidase 1 (Neu‐1) expression after PHx revealed that platelets of PHx mice have a higher Neu‐1 expression on their surface compared to sham controls (Supporting Fig. S5A). In vitro, von Willebrand factor (vWF) binding to GPIb induced by the snake venom botrocetin led to increased Neu‐1 expression in sham operated mice whereas up‐regulation of Neu‐1 was almost inhibited in PHx mice (Fig. 4D). At the same time, no difference in plasma activity of Neu‐1 was detected (Supporting Fig. S5B). This effect of vWF‐GPIb–mediated increase of Neu‐1 expression was confirmed by blocking vWF binding to GPIb (Fig. 4D). In addition, Neu‐1 expression was also reduced when platelets from PHx mice were stimulated with 1 µg/mL of CRP. However, Neu‐1 activity was unaltered whereas vWF plasma levels were enhanced in mice 24 hours after PHx compared to sham controls (Supporting Fig. S5A‐C). Thus, reduced platelet activation early after PHx probably leads to reduced neuraminidase exposure that might account for the reduced number of desialylated platelets in the circulation of PHx mice, suggesting that shear‐stress–induced vWF binding, at least partially, contributes to the reduced number of lectin‐positive platelets that were detected after PHx.
FIG. 4.
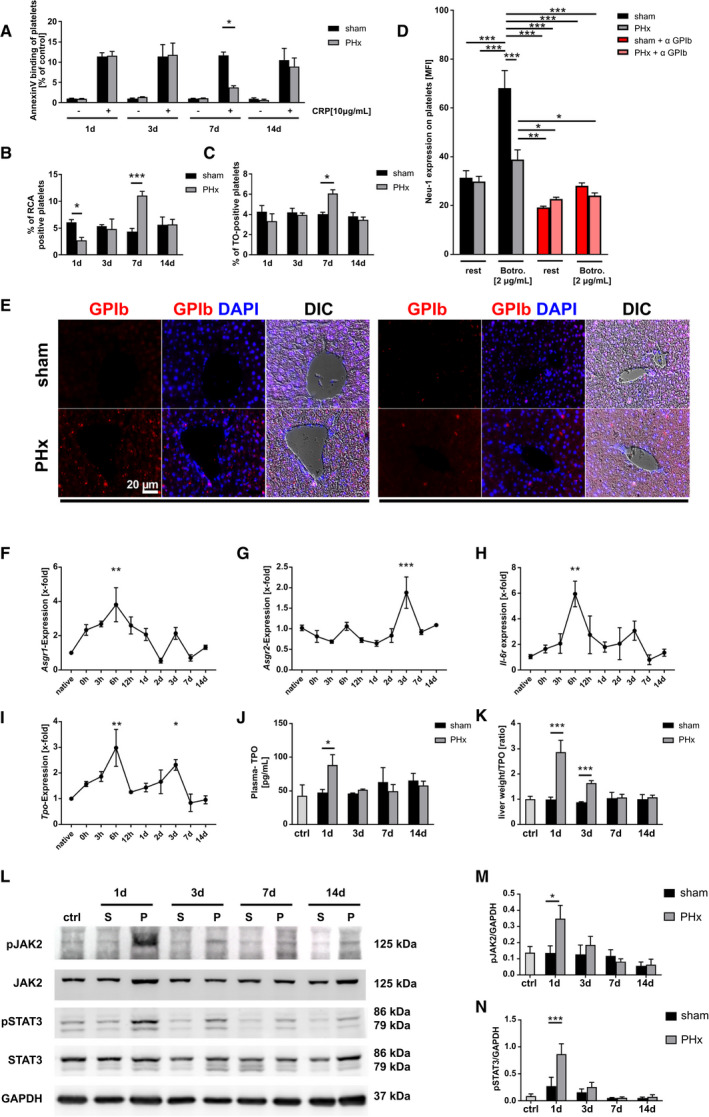
Efficiently restored TPO production is mediated by desialylated platelet uptake into diseased liver by the AMR and JAK2‐STAT3 signaling. (A) Annexin V binding of GPIb‐positive platelets stimulated with CRP in regard to control group of each time point (n = 1 + 3 days: 8 sham + 9 PHx; 7 + 14 days: 4 sham, 4 PHx). (B) Analysis of platelets positive for binding of the RCA‐1 lectin at indicated time points (n = 4 sham, 5 PHx). (C) Thiazol orange staining for RNA‐rich, young GPIb‐positive platelets measured by flow cytometry (n = 8 sham + PHx for 1 + 3 days; 4 sham, 5 PHx for 7 + 14 days). (D) Neu‐1 expression at the platelet surface of sham and PHx‐operated mice was determined after Botrocetin (Botro.) and anti‐GPIb⍺ treatment of platelets (n = 6). (E) Representative images showing staining of paraffin‐embedded liver tissue sections for GPIb‐positive platelets at indicated time points. Nucleus staining performed with DAPI. Merge with differential interference contrast (DIC) is shown (n = 4). (F‐I) Gene expression of different target genes was analyzed using the 2−ΔΔCt method (n = native animals 3; 0 hours: 4 animals; 3 hours to 14 days: 5‐6 animals). (J) TPO level measured in plasma of mice with (K) calculated plasma TPO levels in regard to measured liver weight of the same animals (n = control 3; 1 day = 6 sham, 7 PHx; 3/7/14 days = 4 mice per group). (L) Representative immunoblottings of total liver cell lysates using anti‐pJAK2 and anti‐JAK2 antibodies and anti‐pSTAT3 and anti‐STAT3 monoclonal antibodies. GAPDH was used as a loading control. (M,N) Densitometric analysis of pJAK2 and pSTAT3 with regard to loading control (n = 4). Depicted are mean values + SEM; *P < 0.05; **P < 0.01; ***P < 0.001, using ordinary two‐way ANOVA with Bonferroni’s post hoc test, (D) was analyzed using three‐way ANOVA with Bonferroni’s post hoc test; (F) to (I) were analyzed using ordinary one‐way ANOVA with Dunnett’s post hoc test. Abbreviations: ANOVA, analysis of variance; ctrl, control; DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; MFI, mean fluorescence intensity; pJAK2, phosphorylated JAK2; pSTAT3, phosphorylated STAT3; TO, thiazol orange.
Concomitant with reduced lectin binding of circulating platelets, we identified platelets invading into the liver at day 1 and to a lesser extent at day 3 (Fig. 4E). This might be attributable to the binding of desialylated platelets to the AMR inducing hepatic TPO gene transcription and translation to regulate platelet production( 6 ) in the remaining liver tissue early after PHx. Therefore, gene expression of both subunits of the AMR, asialoglycoprotein receptor 1 (Asgr1) and asialoglycoprotein receptor 2 (Asgr2), were examined in the remaining liver tissue at different time points after surgery. Expression of the Asgr1 was enhanced with a peak at 6 hours and a smaller peak at 3 days whereas Asgr2 expression was enhanced only at 3 days after PHx (Fig. 4F,G). Different reports demonstrated that IL‐6 could stimulate thrombopoiesis through TPO in an inflammatory environment.( 27 , 28 ) Here, the expression of the IL‐6R in the remaining liver tissue was measured. In line with increased Asgr1/2 expression in the remaining liver, expression of Il‐6r was elevated in liver of hepatectomized mice with a similar expression pattern observed in Asgr1 expression (Fig. 4H). Enhanced expression of Asgr1/2 and Il‐6r was accompanied by enhanced Tpo expression 6 hours and 3 days after PHx (Fig. 4I), despite a significant reduction in liver weight to body weight observed at 1‐3 days after PHx (Fig. 1E). In plasma, enhanced TPO levels were detected at day 1 after PHx compared to sham controls (Fig. 4J). A significant elevation of synthesized and released TPO was also evident at day 3, when relative TPO levels were measured in comparison to total liver mass (Fig. 4K). AMR signals through JAK2 and the acute‐phase response STAT3.( 6 ) Thus, phosphorylation of JAK2 and STAT3 at different time points after PHx was examined (Fig. 4L). In line with enhanced plasma TPO levels, phosphorylation of JAK2 and STAT3 was enhanced 1 day after PHx using western blottings (Fig. 4L‐N and Supporting Fig. S6A,B). Hence, the data show that glycan‐dependent clearance of platelets is occurring in the liver followed by up‐regulation of AMR‐dependent JAK2‐STAT3 regulated TPO production.
Enhanced AMR‐JAK2‐TPO Signaling After PHx Provokes Elevated Megakaryopoiesis in Spleen and BM
Megakaryopoiesis is mainly controlled by TPO. To investigate whether increased AMR‐JAK2‐TPO signaling affects megakaryopoiesis in spleen and BM to restore platelet counts, spleen weight to body weight ratio was measured at different time points after PHx. The ration was significantly increased 3 and 7 days after PHx compared to sham controls (Fig. 5A,B). Accordingly, the number of MKs in spleen and BM was enhanced 3 and 7 days after PHx (Fig. 5E‐H). In line with splenomegaly, clearance of platelets was observed by an increased fraction of platelets in spleen of mice 7 days after PHx compared to sham controls, suggesting clearance of PS‐positive platelets at this time point by splenic macrophage scavenger receptors (Fig. 5C,D). This accelerated platelet sequestration into spleen may explain the increase in spleen size at later time points. Taken together, the increase in MKs in spleen and BM supports the increased number of produced platelets. The data further suggest that enhanced clearance of PS‐positive platelets in the spleen either allows platelets to circulate with enhanced RCA binding to platelets at day 7 after PHx or young platelet express more terminal galactose that is not recognized by galactose receptors such as the AMR (Fig. 4B,C).
FIG. 5.
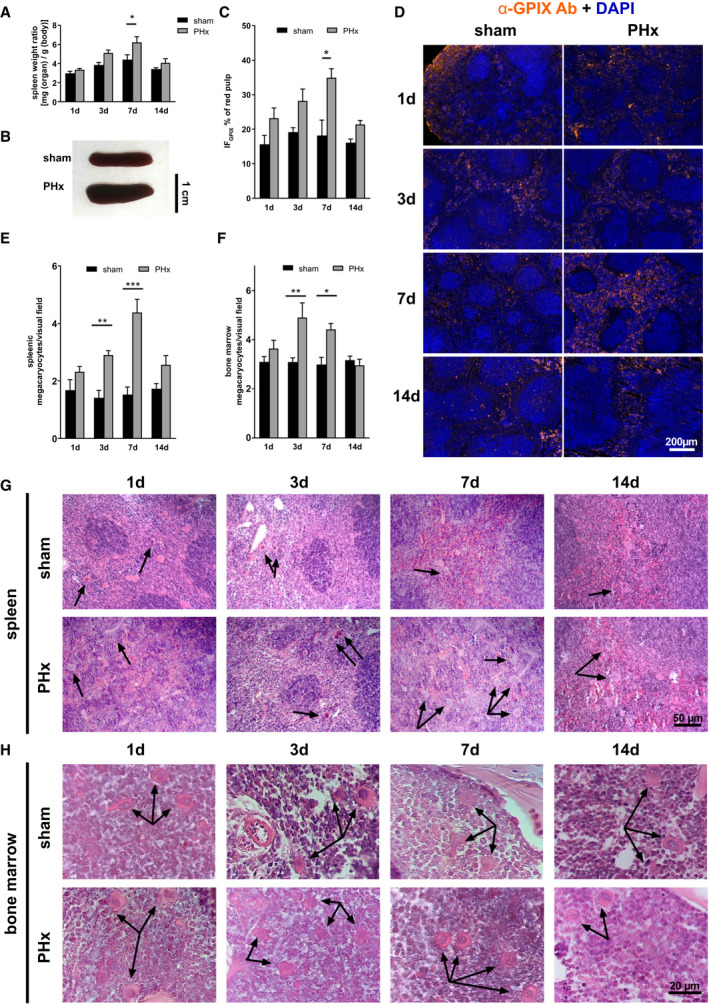
Elevated megakaryopoiesis is responsible for moderate splenomegaly after PHx. (A) Calculated spleen weight ratio (n = 1 day: 10 sham, 14 PHx; 3 days: 9 sham, 9 PHx; 7 days: 8 sham, 10 PHx, 14 days: 5 sham, 5 PHx) with (B) representative images of spleen tissue 7 days after operation. (C) Platelet sequestration in spleen sections of sham and PHx‐operated mice. A platelet‐specific marker (GPIX) was combined with DAPI staining to distinguish between red and white pulp of the spleen. Total GPIX‐positive fluorescence area [IFGPIX/µm2] of the red pulp of spleen sections was determined with (D) representative IF staining of splenic sequestration (n = 4). (E) Counted megakaryocytes in paraffin‐embedded spleen tissue (n = 5 sham, 5 PHx) and (F) BM of the femur (n = 4 sham, 4 PHx). (G) Representative images from spleen tissue and (H) BM tissue of HE staining. Arrowheads indicate MKs. Depicted are mean values + SEM; *P < 0.05; **P < 0.01, using ordinary two‐way ANOVA with Bonferroni’s post hoc test. Abbreviations: Ab, antibody; ANOVA, analysis of variance; DAPI, 4′,6‐diamidino‐2‐phenylindole; GPIX, glycoprotein IX; IF, immunofluorescence.
Cytokine Signaling by the IL‐6R and STAT Signaling Plays a Crucial Role in TPO Homeostasis After PHx
IL‐6R expression is significantly increased 6 hours after PHx (Fig. 4G). To investigate, in further detail, how IL‐6R signaling is involved in TPO homeostasis and restoration of platelet counts after PHx, we analyzed IL‐6r−/− mice at steady state (native) and after PHx (up to 24 hours after surgery because of high mortality of IL‐6r−/− mice). IL‐6r−/− mice showed significantly enhanced platelet counts under native conditions compared to wild‐type (WT) mice. However, platelet counts were strongly reduced in IL‐6r−/− mice as well as in WT mice 1 day after PHx (Fig. 6A). Platelets from IL‐6R‐deficient mice showed enhanced size under native conditions and after PHx compared to WT controls, as measured by flow cytometry (Fig. 6B). Thiazole orange staining revealed an elevated number of young platelets in IL‐6R‐deficient mice after PHx compared to native conditions as well as to WT mice after PHx (Fig. 6C). A significantly reduced number of desialylated platelets in IL‐6R‐deficient mice under native conditions provided evidence for an enhanced platelet clearance of desialylated platelets (Fig. 6D). Expression of the AMR subunits, Asgr1 and Asgr2, was significantly up‐regulated in IL‐6R‐deficient mice under native conditions. Early after PHx, expression of the subunit, Asgr1, was enhanced as well, supporting the notion of increased desialylated platelet uptake by the AMR (Fig. 6E). No difference was detected in expression of Asgr2 at any time point after PHx (Fig. 6F). Interestingly, Il‐6r was likewise up‐regulated in liver of native Asgr2−/− mice (Supporting Fig. S7F). Accordingly, Tpo expression in liver of native mice and in the remaining tissue after PHx was significantly enhanced in IL‐6R‐deficient mice (Fig. 6G). We next analyzed the downstream signaling pathway following AMR activation in the liver. Phosphorylation of JAK2 and signal transducer and activator of transcription 5 (STAT5) was indistinguishable between WT and IL‐6R‐deficient mice under native conditions whereas phosphorylation of STAT3 was significantly reduced in IL‐6R‐deficient mice (Fig. 6H,L and Supporting Fig. S7A). However, 1 day after PHx, phosphorylation of STAT3 was up‐regulated in both groups whereas STAT5 seemed not to be regulated after PHx. However, phosphorylation of JAK2 was significantly enhanced in liver of WT, but only by trend, in IL‐6R‐deficient mice (Fig. 6H,I and Supporting Fig. S7B). In line with enhanced Tpo expression in liver of IL‐6R knockout mice, enhanced TPO protein levels were observed in livers of WT and IL‐6R‐deficient mice isolated 1 day after PHx in (Fig. 6J and Supporting Fig. S7B). Accordingly, TPO plasma levels were enhanced in IL‐6R knockout mice under native conditions and 1 day after PHx. However, plasma levels of TPO were significantly lower 12 hours after PHx compared to WT controls, suggesting attenuated release of TPO from the liver into the circulation (Fig. 6K).
FIG. 6.
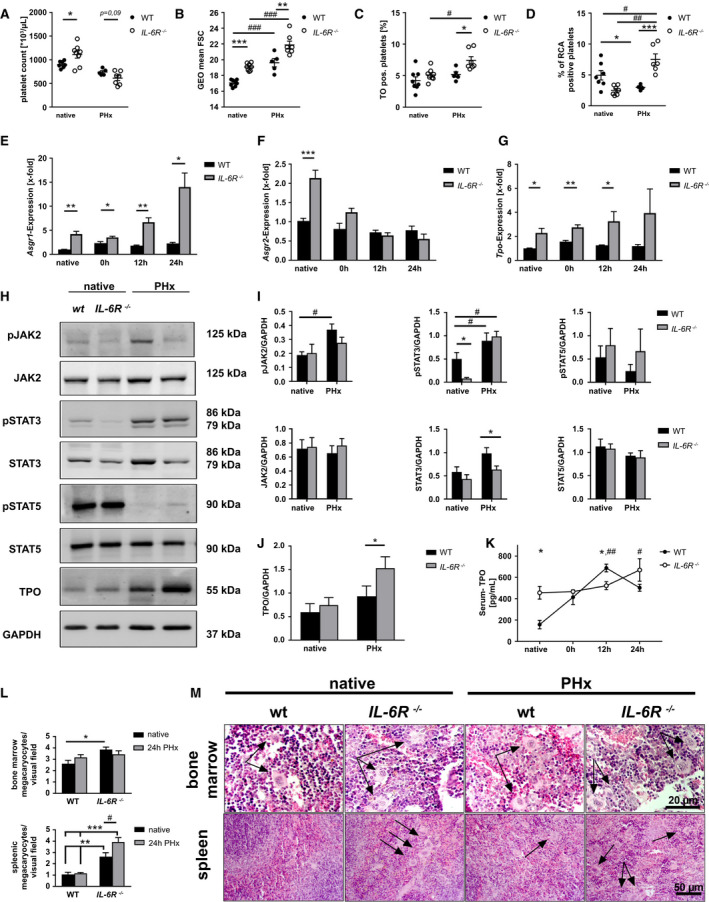
Elevated cytokine signaling controls TPO homeostasis in the remaining liver tissue thyrough IL‐6R and STAT signaling in vivo. (A) Platelet count and (B) platelet size measured by geometrical mean and (C) thiazol orange staining of GPIb‐positive platelets of WT and IL‐6R−/− mice before and 24 hours after PHx (n = native: 8 WT, 8 IL‐6R−/− ; PHx: 5 WT, 7 IL‐6R−/− ). (D) Analysis of platelets positive for binding of RCA‐1 lectin (n = native: 8 WT, 8 IL‐6R−/− ; PHx: 5 WT, 7 IL‐6R−/− ). (E‐G) Gene expression was analyzed using the 2−ΔΔCt method (n = 4‐5 WT per time point + 5 IL‐6R−/− per time point). (H) Representative immunoblottings of total liver cell lysates using anti‐pJAK2 + anti‐JAK2 antibodies, anti‐pSTAT5 + STAT5, and anti‐pSTAT3 + anti‐STAT3 monoclonal antibodies. TPO polyclonal antibody was used with GAPDH as a loading control (n = 5 for each group). (I) Densitometric analysis of immunoblottings for JAK2, STAT5, and STAT3 and their phospho‐variant calculated with GAPDH as a loading control. (J) Densitometric analysis of immunoblottings for TPO. (K) Serum concentration of TPO in WT and IL‐6R −/− mice (n = 4 WT + 5 IL‐6R−/− ). (L) Counted MKs in paraffin‐embedded BM of femur tissue (n = 4 WT, 4 IL‐6R−/− ) and spleen tissue (n = 5 WT, 5 IL‐6R−/− ). (M) Representative images from BM and spleen tissue of HE staining. Arrows indicate MKs. Depicted are mean values + SEM; special characters indicate for difference between time points (#) or WT and IL‐6R−/− (*); #,*P < 0.05; ##,**P < 0.01; ###,***P < 0.001, using paired Student t test; multiple t tests with the Holm‐Sidak method (E‐G) or two‐way ANOVA with Sidak’s post hoc (K,L). Abbreviations: ANOVA, analysis of variance; FSC, forward scatter; GEO, geometric; pJAK2, phosphorylated JAK2; pSTAT3, phosphorylated STAT3; pSTAT5, phosphorylated STAT5; TO, thiazol orange.
Subsequently, we analyzed whether accelerated desialylated platelet clearance affects the number of MKs in the BM and spleen of IL‐6R‐deficient mice 1 day after PHx, as suggested by decreased number of RCA‐1 binding platelets at early time points after PHx (Fig. 4C). An increased number of MKs was observed in BM and spleen of IL‐6R‐deficient mice under native conditions compared to WT controls (Fig. 6L,M). After PHx, the number of MKs was significantly enhanced in spleen, but not in BM, of IL‐6R‐deficient mice (Fig. 6L,M). In line with unaltered counts of MKs in BM and spleen between sham controls and PHx mice 1 day after PHx (Fig. 5C‐F), no differences were observed in native WT mice compared to 1 day after PHx. Increase in BM MKs in response to TPO stimulation was only overserved at 3‐7 days after PHx. Spleen weight was comparable between WT and IL‐6R knockout mice under native conditions and after PHx (Supporting Fig. S7E). However, platelet sequestration into spleen of Il‐6r−/− mice was enhanced under native conditions and 1 day after PHx compared to WT controls (Supporting Fig. S7C,D). The unaltered spleen weight in IL‐6R‐deficient mice points to enhanced platelet destruction in IL‐6R‐deficient mice, as suggested by a significantly reduced number of desialylated platelets in IL‐6R‐deficient mice under native conditions (Fig. 6D).
To analyze whether recombinant IL‐6 is able to stimulate thrombopoiesis after PHx, we treated mice with recombinant hyper‐IL6 (hIL‐6) because hIL‐6 has a 10 times higher bioactivity than IL‐6 alone and a longer bioavailability in vivo.( 29 ) Although no differences in platelet counts could be observed after hIL6 treatment, enhanced platelet size was measured in hIL‐6‐treated mice compared to controls (Supporting Fig. S8A,B). Additionally, hIL‐6 application induced enhanced TPO plasma levels, which was accompanied by an enhanced number of MKs in spleen, but not in BM, of hIL‐6‐treated mice 24 hours after the operation (Supporting Fig. S8C‐G). These results give an indication that treatment with recombinant IL‐6 induces thrombopoiesis after PHx. However, further experiments in the near future are needed to clarify the role of IL‐6 signaling for thrombopoiesis.
Discussion
Here, we report that platelet counts were rapidly restored after PHx by the AMR/IL‐6R/JAK2‐STAT signaling pathway, leading to elevated TPO expression and release from the remaining liver tissue to reconstitute hemostasis at early time points when liver tissue had not been fully regenerated. This strongly suggests that enhanced platelet turnover and cytokine signaling synergistically control TPO homeostasis in the remaining liver tissue. In addition, we provide evidence that platelet activation defects were moderate because of elevated plasma levels of NO, PGI2, and bile acids early after PHx, leading to defective hemostasis at days 1 and 3 after surgery.
This report shows how TPO homeostasis and restoration of platelet counts is regulated in mice with significantly reduced liver tissue. Thrombocytopenia in liver disease develops as a consequence of decreased platelet production, increased destruction/platelet turnover, and increased consumption or splenic sequestration,( 7 , 30 ) all markers that were also found in mice after PHx. Depressed TPO levels in chronic liver diseases result in a reduced rate of platelet production given that TPO regulates both platelet production and maturation, which is impaired in chronic liver disease.( 30 ) Thus, regulation of TPO is a long‐known factor in liver disease. Given that the liver is the major site of TPO production,( 31 ) it is reasonable to expect decreased plasma levels in patients with liver disease. Indeed, TPO mRNA levels in liver were slightly decreased in cirrhosis( 32 ) and/or chronic hepatitis( 33 , 34 , 35 ) and normalized after liver transplantation.( 36 , 37 ) However, increased serum TPO levels in hepatitis C and patients with cirrhosis have also been published.( 11 , 38 ) This might be attributable to the time point of the TPO measurements given that we observed a biphasic up‐regulation of TPO expression at early time points after PHx with increased cytokine signaling that might be different (e.g., in chronic liver failure with chronic inflammation).
In liver disease, thrombocytopenia is an indicator of advanced disease and poor prognosis. However, thrombocytopenia is often accompanied by platelet activation defects, and this all together might contribute to bleeding diathesis in liver failure.( 30 , 39 ) Enhanced bleeding risk is described in patients with liver transplantation.( 39 ) However, high platelet counts are beneficial for outcome after liver resection and liver transplantation not only with regard to bleeding complications, but also because of their ability to promote liver regeneration.( 40 ) Here, we report that bleeding complications in mice are related to decreased platelet counts and platelet activation defects at days 1 and 3 after PHx, but probably also because of a reduction of coagulation factors that are produced in the liver. However, the remaining liver tissue can rescue platelet counts by up‐regulation of the AMR/IL‐6R/JAK2‐STAT signaling pathway already 6 hours after surgical treatment, at least in mice (Fig. 4E‐H). Given that PGI2, NO, and bile acid levels normalize already at 3 (NO and PGI2; Fig. 3A,B) and 7 days (bile acids; Fig. 3G) after PHx, platelet counts and platelet activation are already rehabilitated 7 days after PHx when liver regeneration is not fully completed (Fig. 1E). This might be different in chronic liver disease in patients or experimental mice (e.g., in BDL mice) where bile acids circulate in plasma for weeks, and platelet activation defects, including defective hemostasis, were detected until 21 days after bile duct ligation.( 17 ) Interestingly, arterial thrombosis was not prevented by concomitant low platelet counts and platelet defects 1 day after PHx (Fig. 1K). However, thrombosis has been also described in patients with liver failure despite reduced synthesis of procoagulant proteins, platelet defects, and bleeding complications.( 39 , 41 ) There are reports about platelet defects in knockout mice, which do protect these mice against thrombosis but do not alter hemostasis, for example, in phospholipase D1–deficient mice,( 42 ) suggesting that there might be coexisting processes of hemostasis that may not cause thrombosis and vessel occlusion and the other way around.
To date, recognition of circulating desialylated platelets by hepatic AMR and regulation of TPO mRNA expression and secretion by JAK2‐STAT3 signaling were shown under steady‐state conditions.( 6 ) Recently, Kupffer cells were identified as the main trigger of platelet clearance. The researchers described a mechanism that includes the collaboration of the macrophage galactose lectin with the AMR on Kupffer cells to remove desialylated platelets from the circulation in native mice.( 43 ) Regulatory feedback mechanisms in liver disease with enhanced cytokine signaling are still open for study. Thus, the underlying mechanisms are crucial for the understanding of enhanced platelet turnover/destruction and reduced platelet counts in disease. However, our data do not clearly show whether hepatocytes and/or Kupffer cells are responsible for platelet clearance and TPO production after PHx. Thus, further experiments are needed in the near future to identify the cell population in the liver that can efficiently restore platelet counts after PHx.
Enhanced destruction of platelets has been described in liver disease and therefore is a common feature in chronic and acute liver failure.( 30 , 39 ) To date, how the AMR‐JAK2‐STAT3 signaling pathway is regulated in liver disease remains open for study. Our study shows that IL‐6R signaling in the liver is involved in TPO hemostasis and megakaryopoiesis upon elevated cytokine signaling as it occurs after PHx. Thus, elevated cytokine plasma levels account for the regulation of platelet counts upon accelerated platelet clearance. Different reports suggested that IL‐6 is able to stimulate thrombopoiesis through TPO in an inflammatory environment.( 27 , 28 ) This, together with the fact that the AMR signaling pathway shares similarities with that of the IL‐6R, prompted us to investigate the role of IL‐6R in TPO homeostasis in more detail. First, we detected elevated IL‐6R mRNA in the liver after PHx as already observed for the AMR (Fig. 4G). IL‐6R knockout mice exhibited reduced platelet counts after PHx like WT mice, but their platelets were increased in size and they had significantly more young platelets in the circulation, suggesting an enhanced platelet turnover compared to WT mice. Loss of the IL‐6R in liver tissue was compensated by an increase of AMR subunits Asgr1 and Asgr2 mRNA, the latter only under native conditions (Fig. 6E,F). This compensation led to excessive TPO mRNA levels in liver and plasma under native conditions and at different time points after PHx (Fig. 6G). However, the increase in TPO was not able to restore platelet counts 1 day after PHx compared to WT controls (Fig. 6A). This might be attributable to the dysregulation of platelet counts and enhanced platelet turnover under native conditions, reflected by an enhanced number of MKs in BM and spleen (Fig. 6L,M), enhanced platelet size, and reduced desialylation of platelets leading to enhanced expression of AMR and TPO (Fig. 6B‐D). Taken together, this provides evidence for an involvement of IL‐6R signaling in TPO homeostasis and platelet count regulation in mice with reduced liver tissue under inflammatory conditions. The fact that loss of IL‐6R is compensated by enhanced expression of Asgr1/2 provides evidence for a crosstalk between the AMR and IL‐6R to regulate TPO levels and megakaryopoiesis in BM and spleen. This conclusion is strengthened by enhanced IL‐6R mRNA levels in liver of ASGR2 knockout mice (Supporting Fig. S5D). IL‐6 is critically involved in hepatocyte proliferation and liver regeneration after PHx.( 44 , 45 ) To investigate whether IL‐6 is able to stimulate thrombopoiesis, we treated mice with recombinant hIL‐6. Elevated TPO plasma levels and an enhanced number of MKs in spleen of hIL‐6‐treated mice 24 hours after PHx gave a first indication that treatment with recombinant IL‐6 induces thrombopoiesis after PHx. Given that recent studies revealed that IL‐6 trans‐signaling through the soluble IL‐6/IL‐6R complex controls liver regeneration besides the classical IL‐6 signaling,( 46 ) we used the recombinant fusion protein, hIL‐6, which consists of soluble IL‐6R and IL‐6, which mimics IL‐6 trans‐signaling.( 47 ) In the study of Behnke et al., it was already shown that administration of hIL‐6 results in improved liver regeneration by Ki67 and phospho‐H3 staining in liver tissue after PHx.( 48 ) Taken together, it will be of great interest to investigate the effects of recombinant IL‐6 on the restoration of platelet counts and megakaryopoiesis in further detail. Therefore, the analysis of different models of IL‐6 signaling, such as IL‐6R knockout, recombinant hIL‐6, or glycoprotein 130 transgenic mice, could be helpful to shed light on the impact of IL‐6 (trans‐) signaling on thrombopoiesis after PHx or other inflammatory diseases that are accompanied by thrombocytopenia.
Of note, we here used IL‐6R‐deficient mice until 24 hours after PHx as the high mortality rate in these mice because of the important role of the IL‐6 signaling pathway after PHx.
In conclusion, our study identifies the AMR/IL‐6R/STAT3/TPO signaling pathway as a highly efficient and acute‐phase response to liver injury, cytokine signaling, and loss of liver tissue to restore platelet count and function. TPO and platelet activation defects induced by enhanced PGI2, NO, and bile acid plasma levels are responsible for bleeding complications early after PHx that might also be causative for bleeding problems and high mortality in patients with liver disease.
Author Contributions
J.S., K.M.H., D.H., and M.E. conceived the project, designed the experiments, analyzed data, and wrote the manuscript. F.R., K.B., N.F.M., M.B., L.W., M.M.L., and D.H. performed experiments and analyzed data. I.K. conducted animal experiments.
Supporting information
Supplementary Material
Acknowledgment
We thank Martina Spelleken, Vanessa Herbetz, and Nicole Eichhorst for excellent technical assistance.
Supported by a grant from the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 974, Düsseldorf.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest 2005;115:3339‐3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver‐Moore K, et al. Regulation of thrombopoietin levels by c‐mpl‐mediated binding to platelets. Blood 1996;87:2154‐2161. [PubMed] [Google Scholar]
- 3. Kuter DJ, Rosenberg RD. The reciprocal relationship of thrombopoietin (c‐Mpl ligand) to changes in the platelet mass during busulfan‐induced thrombocytopenia in the rabbit. Blood 1995;85:2720‐2730. [PubMed] [Google Scholar]
- 4. McCarty JM, Sprugel KH, Fox NE, Sabath DE, Kaushansky K. Murine thrombopoietin mRNA levels are modulated by platelet count. Blood 1995;86:3668‐3675. [PubMed] [Google Scholar]
- 5. McIntosh B, Kaushansky K. Transcriptional regulation of bone marrow thrombopoietin by platelet proteins. Exp Hematol 2008;36:799‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The Ashwell‐Morell receptor regulates hepatic thrombopoietin production via JAK2‐STAT3 signaling. Nat Med 2015;21:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lisman T, Luyendyk JP. Platelets as modulators of liver diseases. Semin Thromb Hemost 2018;44:114‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aoki Y, Hirai K, Tanikawa K. Mechanism of thrombocytopenia in liver cirrhosis: kinetics of indium‐111 tropolone labelled platelets. Eur J Nucl Med 1993;20:123‐129. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt KG, Rasmussen JW, Bekker C, Madsen PE. Kinetics and in vivo distribution of 111‐In‐labelled autologous platelets in chronic hepatic disease: mechanisms of thrombocytopenia. Scand J Haematol 1985;34:39‐46. [DOI] [PubMed] [Google Scholar]
- 10. Stein SF, Harker LA. Kinetic and functional studies of platelets, fibrinogen, and plasminogen in patients with hepatic cirrhosis. J Lab Clin Med 1982;99:217‐230. [PubMed] [Google Scholar]
- 11. Witters P, Freson K, Verslype C, Peerlinck K, Hoylaerts M, Nevens F, et al. Review article: blood platelet number and function in chronic liver disease and cirrhosis. Aliment Pharmacol Ther 2008;27:1017‐1029. [DOI] [PubMed] [Google Scholar]
- 12. Laffi G, Marra F, Failli P, Ruggiero M, Cecchi E, Carloni V, et al. Defective signal transduction in platelets from cirrhotics is associated with increased cyclic nucleotides. Gastroenterology 1993;105:148‐156. [DOI] [PubMed] [Google Scholar]
- 13. Laffi G, Cominelli F, Ruggiero M, Fedi S, Chiarugi VP, La Villa G, et al. Altered platelet function in cirrhosis of the liver: impairment of inositol lipid and arachidonic acid metabolism in response to agonists. Hepatology 1988;8:1620‐1626. [DOI] [PubMed] [Google Scholar]
- 14. Laffi G, Marra F, Gresele P, Romagnoli P, Palermo A, Bartolini O, et al. Evidence for a storage pool defect in platelets from cirrhotic patients with defective aggregation. Gastroenterology 1992;103:641‐646. [DOI] [PubMed] [Google Scholar]
- 15. Ordinas A, Escolar G, Cirera I, Vinas M, Cobo F, Bosch J, et al. Existence of a platelet‐adhesion defect in patients with cirrhosis independent of hematocrit: studies under flow conditions. Hepatology 1996;24:1137‐1142. [DOI] [PubMed] [Google Scholar]
- 16. Owen JS, Hutton RA, Day RC, Bruckdorfer KR, McIntyre N. Platelet lipid composition and platelet aggregation in human liver disease. J Lipid Res 1981;22:423‐430. [PubMed] [Google Scholar]
- 17. Gowert N, Klier M, Reich M, Reusswig F, Donner L, Keitel V, et al. Defective platelet activation and bleeding complications upon cholestasis in mice. Cell Physiol Biochem 2017;41:2133‐2149. [DOI] [PubMed] [Google Scholar]
- 18. Walter U, Eigenthaler M, Geiger J, Reinhard M. Role of cyclic nucleotide‐dependent protein kinases and their common substrate VASP in the regulation of human platelets. Adv Exp Med Biol 1993;344:237‐249. [DOI] [PubMed] [Google Scholar]
- 19. Michalopoulos GK, Grompe M, Theise ND. Assessing the potential of induced liver regeneration. Nat Med 2013;19:1096‐1067. [DOI] [PubMed] [Google Scholar]
- 20. Horstrup K, Jablonka B, Honig‐Liedl P, Just M, Kochsiek K, Walter U, et al. Phosphorylation of focal adhesion vasodilator‐stimulated phosphoprotein at Ser157 in intact human platelets correlates with fibrinogen receptor inhibition. Eur J Biochem 1994;225:21‐27. [DOI] [PubMed] [Google Scholar]
- 21. Nolte C, Eigenthaler M, Horstrup K, Hönig‐Liedl P, Walter U. Synergistic phosphorylation of the focal adhesion‐associated vasodilator‐stimulated phosphoprotein in intact human platelets in response to cGMP‐ and cAMP‐elevating platelet inhibitors. Biochem Pharmacol 1994;48:1569‐1575. [DOI] [PubMed] [Google Scholar]
- 22. Csanaky IL, Aleksunes LM, Tanaka Y, Klaassen CD. Role of hepatic transporters in prevention of bile acid toxicity after partial hepatectomy in mice. Am J Physiol Gastrointest Liver Physiol 2009;297:G419‐G433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nomura S, Ogami K, Kawamura K, Tsukamoto I, Kudo Y, Kanakura Y, et al. Cellular localization of thrombopoietin mRNA in the liver by in situ hybridization. Exp Hematol 1997;25:565‐572. [PubMed] [Google Scholar]
- 24. Pereira J, Palomo I, Ocqueteau M, Soto M, Aranda E, Mezzano D. Platelet aging in vivo is associated with loss of membrane phospholipid asymmetry. Thromb Haemost 1999;82:1318‐1321. [PubMed] [Google Scholar]
- 25. Sorensen AL, Rumjantseva V, Nayeb‐Hashemi S, Clausen H, Hartwig JH, Wandall HH, et al. Role of sialic acid for platelet life span: exposure of beta‐galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor‐expressing liver macrophages and hepatocytes. Blood 2009;114:1645‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riswari SF, Tunjungputri RN, Kullaya V, Garishah FM, Utari GSR, Farhanah N, et al. Desialylation of platelets induced by Von Willebrand factor is a novel mechanism of platelet clearance in dengue. PLoS Pathogens 2019;15:e1007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burmester H, Wolber EM, Freitag P, Fandrey J, Jelkmann W. Thrombopoietin production in wild‐type and interleukin‐6 knockout mice with acute inflammation. J Interferon Cytokine Res 2005;25:407‐413. [DOI] [PubMed] [Google Scholar]
- 28. Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin‐6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood 2001;98:2720‐2725. [DOI] [PubMed] [Google Scholar]
- 29. Peters M, Blinn G, Solem F, Fischer M, Meyer zum Büschenfelde KH, Rose‐John S. In vivo and in vitro activities of the gp130‐stimulating designer cytokine hyper‐IL‐6. J Immunol 1998;161:3575‐3581. [PubMed] [Google Scholar]
- 30. Mitchell O, Feldman DM, Diakow M, Sigal SH. The pathophysiology of thrombocytopenia in chronic liver disease. Hepat Med 2016;8:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sungaran R, Markovic B, Chong BH. Localization and regulation of thrombopoietin mRNa expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood 1997;89:101‐107. [PubMed] [Google Scholar]
- 32. Martin TG III, Somberg KA, Meng YG, Cohen RL, Heid CA, Sauvage FJ, Shuman MA. Thrombopoietin levels in patients with cirrhosis before and after orthotopic liver transplantation. Ann Intern Med 1997;127:285‐288. [DOI] [PubMed] [Google Scholar]
- 33. Koruk M, Derya Onuk M, Akçay F, Cemil Savas M. Serum thrombopoietin levels in patients with chronic hepatitis and liver cirrhosis, and its relationship with circulating thrombocyte counts. Hepatogastroenterology 2002;49:1645‐1648. [PubMed] [Google Scholar]
- 34. Panasiuk A, Prokopowicz D, Zak J, Panasiuk B. Reticulated platelets as a marker of megakaryopoiesis in liver cirrhosis; relation to thrombopoietin and hepatocyte growth factor serum concentration. Hepatogastroenterology 2004;51:1124‐1128. [PubMed] [Google Scholar]
- 35. Panasiuk A, Prokopowicz D, Zak J, Panasiuk B, Wysocka J. Inhibition of activated blood platelets by interferon alpha 2b in chronic hepatitis C. Hepatogastroenterology 2004;51:1417‐1421. [PubMed] [Google Scholar]
- 36. Peck‐Radosavljevic M, Wichlas M, Zacherl J, Stiegler G, Stohlawetz P, Fuchsjäger M, et al. Thrombopoietin induces rapid resolution of thrombocytopenia after orthotopic liver transplantation through increased platelet production. Blood 2000;95:795‐801. [PubMed] [Google Scholar]
- 37. Sezai S, Kamisaka K, Ikegami F, Usuki K, Urabe A, Tahara T, et al. Regulation of hepatic thrombopoietin production by portal hemodynamics in liver cirrhosis. Am J Gastroenterol 1998;93:80‐82. [DOI] [PubMed] [Google Scholar]
- 38. Aref S, Mabed M, Selim T, Goda T, Khafagy N. Thrombopoietin (TPO) levels in hepatic patients with thrombocytopenia. Hematology 2004;9:351‐356. [DOI] [PubMed] [Google Scholar]
- 39. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol 2002;37:280‐287. [DOI] [PubMed] [Google Scholar]
- 40. Takahashi K, Liang C, Oda T, Ohkohchi N. Platelet and liver regeneration after liver surgery. Surg Today 2020;50:974‐983. [DOI] [PubMed] [Google Scholar]
- 41. Intagliata NM, Davis JPE, Caldwell SH. Coagulation pathways, hemostasis, and thrombosis in liver failure. Semin Respir Crit Care Med 2018;39:598‐608. [DOI] [PubMed] [Google Scholar]
- 42. Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers MEJ, Boesl M, et al. Impaired alpha(IIb)beta(3) integrin activation and shear‐dependent thrombus formation in mice lacking phospholipase D1. Sci Signal 2010;3:ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med 2020;217:e20190723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blindenbacher A, Wang X, Langer I, Savino R, Terracciano L, Heim MH. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology 2003;38:674‐682. [DOI] [PubMed] [Google Scholar]
- 45. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin‐6‐deficient mice. Science 1996;274:1379‐1383. [DOI] [PubMed] [Google Scholar]
- 46. Fazel Modares N, Polz R, Haghighi F, Lamertz L, Behnke K, Zhuang Y, et al. IL‐6 trans‐signaling controls liver regeneration after partial hepatectomy. Hepatology 2019;70:2075‐2091. [DOI] [PubMed] [Google Scholar]
- 47. Fischer M, Goldschmitt J, Peschel C, Brakenhoff JPG, Kallen KJ, Wollmer A, et al. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol 1997;15:142‐145. [DOI] [PubMed] [Google Scholar]
- 48. Behnke K, Zhuang Y, Xu HC, Sundaram B, Reich M, Shinde PV, et al. B cell‐mediated maintenance of cluster of differentiation 169‐positive cells is critical for liver regeneration. Hepatology 2018;68:2348‐2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greene, AK , Puder, M . Partial hepatectomy in the mouse: technique and perioperative management. J Invest Surg, 2003;16:99–102. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material