

Abstract
Inborn errors of immunity (IEIs) are a group of genetically defined disorders leading to defective immunity. Some IEIs have been linked to mutations of immune receptors or signaling molecules, resulting in defective signaling of respective cascades essential for combating specific pathogens. However, it remains incompletely understood why in selected IEIs, such as X‐linked lymphoproliferative syndrome type 2 (XLP‐2), hypo‐immune response to specific pathogens results in persistent inflammation. Moreover, mechanisms underlying the generation of anticytokine autoantibodies are mostly unknown. Recently, IEIs have been associated with coronavirus disease 2019 (COVID‐19), with a small proportion of patients that contract severe COVID‐19 displaying loss‐of‐function mutations in genes associated with type I interferons (IFNs). Moreover, approximately 10% of patients with severe COVID‐19 possess anti‐type I IFN‐neutralizing autoantibodies. Apart from IEIs that impair immune responses to severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), SARS‐CoV‐2 encodes several proteins that suppress early type I IFN production. One primary consequence of the lack of type I IFNs during early SARS‐CoV‐2 infection is the increased inflammation associated with COVID‐19. In XLP‐2, resolution of inflammation rescued experimental subjects from infection‐induced mortality. Recent studies also indicate that targeting inflammation could alleviate COVID‐19. In this review, we discuss infection‐induced inflammation in IEIs, using XLP‐2 and COVID‐19 as examples. We suggest that resolving inflammation may represent an effective therapeutic approach to these diseases.
Keywords: anticytokine autoantibodies, COVID‐19, inborn errors of immunity, SARS‐CoV‐2, XLP‐2
Inborn error of immunity (IEI) like XLP‐2 is susceptible to infection‐induced inflammation. Autoantibodies to interferons (IFNs), the late onset of immunodeficiency, impair responses to microbes. Immunodeficiency nature of severe coronavirus disease 2019 (COVID‐19) is comprised of IEIs in IFN‐I pathway, autoantibodies to IFN‐I, and suppression of IFN‐I by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). The lack of IFN‐I responses leads to uncontrolled SARS‐CoV‐2 replication, excess infection‐induced inflammation that is exemplified by severe COVID‐19 syndromes.
Abbreviations
- APS‐I
autoimmune polyendocrine syndrome I
- ARDS
acute respiratory distress syndrome
- COVID‐19
coronavirus disease 2019
- DAMP
damage‐associated molecular pattern
- EBV
Epstein–Barr virus
- HLH
hemophagocytic lymphohistiocytosis
- IEIs
inborn errors of immunity
- IFN
interferon
- IFNAR1/2
IFN‐α receptor ½
- IL
interleukin
- IRF
IFN regulatory factor
- ISG
interferon‐stimulated gene
- M protein
membrane protein
- MERS‐CoV
Middle East respiratory syndrome coronavirus
- MMR
measles, mumps, and rubella
- MSMD
Mendelian susceptibility to mycobacterial disease
- N protein
nucleocapsid protein
- NLRP3
NOD‐, LRR‐, and pyrin domain‐containing protein 3
- NOD2
nucleotide‐binding oligomerization domain‐containing protein 2
- NSP
nonstructural protein
- PAMP
pathogen‐associated molecular pattern
- PRR
pattern recognition receptor, S protein, spike protein
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- SOCS1
suppressor of cytokine signaling 1
- STAT1
signal transducer and activator of transcription 1
- TLR
Toll‐like receptor
- Treg cell
regulatory T cell
- XIAP
X‐linked inhibitor of apoptosis protein
- XLP‐2
X‐linked lymphoproliferative syndrome type 2
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is a novel coronavirus that is causing the human coronavirus disease 2019 (COVID‐19) pandemic. SARS‐CoV‐2 was first identified in December 2019, and as of April 2, 2021, the COVID‐19 pandemic has led to over 130 million infections and more than 2.8 million deaths worldwide. Like other coronaviruses (CoVs), including SARS‐CoV and Middle East respiratory syndrome coronavirus (MERS‐CoV), SARS‐CoV‐2 is a positive‐sense single‐stranded RNA virus [1]. The 29.9‐kilobase SARS‐CoV‐2 genome encodes the viral spike (S), envelope (E), membrane (M), and nucleocapsid (N) structural proteins, as well as 16 nonstructural proteins (nsp1‐nsp16) [1, 2]. The S, E, and membrane protein (M proteins) form the viral envelope that encapsulates the viral nucleocapsid consisting of nucleocapsid protein (N protein) and genomic RNA. S protein binds to angiotensin‐converting enzyme 2 (ACE2), its receptor on host cells, to trigger fusion of the viral particle with the host cell membrane, enabling delivery of the viral RNA genome into the cytoplasm of the host cell. SARS‐CoV‐2 infection can be asymptomatic or symptomatic, with ~ 20% of symptomatic patients developing severe pneumonia and dyspnea, which can be accompanied by numerous complications and even death. Despite intensive global efforts to characterize SARS‐CoV‐2 infection, the pathogenic mechanism underlying COVID‐19 remains incompletely understood [3, 4, 5].
Inborn errors of immunity (IEIs), also known as primary immunodeficiencies, are a group of genetically defined disorders leading to defective innate or adaptive immunity. Which immune responses are impaired depends on the associated genetic mutation [6]. For example, mutation of genes involved in the recombination of immunoglobulins and T‐cell receptors, such as RAG, DNA‐PK, or DNA ligase IV, induces severe combined immune deficiency (SCID) because adaptive immunity is abrogated. Other IEIs include those leading to antibody deficiency, immune dysregulation, congenital defects in phagocytes, autoinflammatory disorders, complement deficiencies, or autoantibody generation [6]. Some IEIs confer susceptibility to infection by specific pathogens, such as Epstein–Barr virus (EBV), cytomegalovirus (CMV), herpes simplex virus (HSV), Candida, or mycobacteria [7]. The pathogenic mechanisms of some IEIs can be directly inferred from gene impairment; for example, the inability to tackle any infection in the case of SCID is due to the absence of B cells and T cells, while deficiency in interleukin (IL)‐6R signaling molecules fails to induce T helper 17 (Th17) and to control pyrogenic bacterial infection and fungal infection [6]. For a few IEIs, such as X‐linked lymphoproliferative syndrome type 2 (XLP‐2), despite the identity of a mutated immune receptor or signaling molecule being known, it remains incompletely understood how an initial hyporesponse to the specific pathogen exacerbates inflammation.
With regard to COVID‐19, ongoing efforts to characterize in detail immune responses to SARS‐CoV‐2 have revealed early ineffective innate immune responses and an eventual inflammatory storm in patients with severe COVID‐19. Moreover, type I interferon (IFN) autoantibodies and mutation of several type I IFN‐associated genes have been identified in a subpopulation of such patients [8, 9], revealing a pivotal role of type I IFN to control SARS‐CoV‐2 and of host immune defect in the pathogenesis of severe COVID‐19 infection. Furthermore, SARS‐CoV‐2 suppresses the expression and signaling of type I IFNs, leading to excess inflammation. In this review, we discuss recent progress in studying anti‐IFN antibodies and IEIs involving XLP‐2 and IFN, as well as their relevance to infection‐induced inflammation in COVID‐19 [10].
Inborn errors of immunity
Up to 430 IEIs have been documented, and they have been classified into 10 categories by the International Union of Immunological Societies Expert Committee [6]. IEIs are often associated with inflammation. Certain IEIs involve gain‐of‐function mutations in genes encoding, for example, inflammasome components such as NOD‐, LRR‐, and pyrin domain‐containing protein 3 (NLRP3), pyrin, NLRP1, or NLRC4, yielding constitutively active inflammasomes that induce auto‐inflammation [11]. Mutations that generate deficiency of negative signaling molecules, such as Foxp3 or IL‐10R, also lead to uncontrollable inflammation. For other IEIs, inflammation is attributable to infection [12]. Notably, the sensitivity to pathogens cannot be directly inferred from the function of the mutated gene, with varying degrees of redundancy in pathogen sensitivity [13]. For instance, despite that IRAK4 and MyD88 are the mediators of several Toll‐like receptor (TLR) signaling, both deficiencies are indispensable for controlling pyrogenic bacteria but not for most other pathogens [14, 15]. Likewise, TLR3 deficiency selectively impairs immunity to some viral infections, such as herpes simplex virus encephalitis [16]. Autosomal recessive mutation of UNC93B, encoding a protein essential for the production of type I/III IFNs, impairs TLR3 response and induces development of herpes simplex virus 1 (HSV‐1) encephalitis (HSE), but patients respond normally to other TLRs and viruses [17]. In addition to this unpredictable sensitivity to selective pathogens, an unresolved question is how disruption of a pro‐inflammatory signal leads to the inflammatory disorder associated with infection‐linked IEIs. For example, XLP‐2 represents a specific type of IEI in which an initial hyporesponse to EBV infection results in persistent inflammation characterized by hemophagocytic lymphohistiocytosis (HLH). XLP‐2 is selected for discussion in this review because of the similarity with severe COVID‐19 in which defective early immunity leads to uncontrollable inflammation. Additionally, the generation of anti‐IFN‐γ autoantibodies, with unknown mechanism, shows a phenocopy of inborn error of IFN‐γ pathway deficiencies, which confers susceptibility to specific infection [18]. An analog was found in a fraction of severe COVID‐19 patients carry anti‐type I IFN antibodies [8]. In this review, focusing on the relevance to COVID‐19, we limit our discussion on IEIs of XLP‐2 and IFNs, and autoantibodies to IFNs.
Infection‐induced inflammation in XLP‐2
Signaling defects associated with XLP‐2
XLP‐2 is an IEI caused by a deficiency in X‐linked inhibitor of apoptosis protein (XIAP) [19, 20, 21]. XLP‐2 is often associated with infection from EBV, resulting in excess cytokine production and HLH due to hyperactivation of lymphocytes and macrophages [22]. Recent studies have revealed that XIAP mutation also contributes to pediatric‐onset Crohn's disease in some patients [23, 24, 25]. Even though XIAP is an anti‐apoptotic protein that specifically inactivates caspase‐3, caspase‐7, and caspase‐9 [26], it also participates in several critical immune signaling cascades independently of its anti‐apoptotic function.
Figure 1 summarizes the known signing processes mediated by XIAP. The best‐characterized signaling defect in XIAP‐deficient cells relates to the nucleotide‐binding oligomerization domain‐containing protein 2 (NOD2) pathway (Fig. 1A). XIAP interacts with receptor‐interacting protein kinase 2 (RIPK2), the scaffold protein that mediates NOD2 activation, by introducing K63 poly‐ubiquitination on RIPK2 for signalosome formation, leading to MAPK and NF‐κB activation and the expression of pro‐inflammatory cytokines [27, 28, 29, 30]. NOD2 is a cytoplasmic pattern recognition receptor (PRR) that senses bacterial peptidoglycan fragments containing muramyl dipeptide (MDP). Consequently, XIAP deficiency impairs NOD2 signaling, accounting for the failure of Xiap − /− mice to handle Listeria monocytogenes infection [31].
Fig. 1.
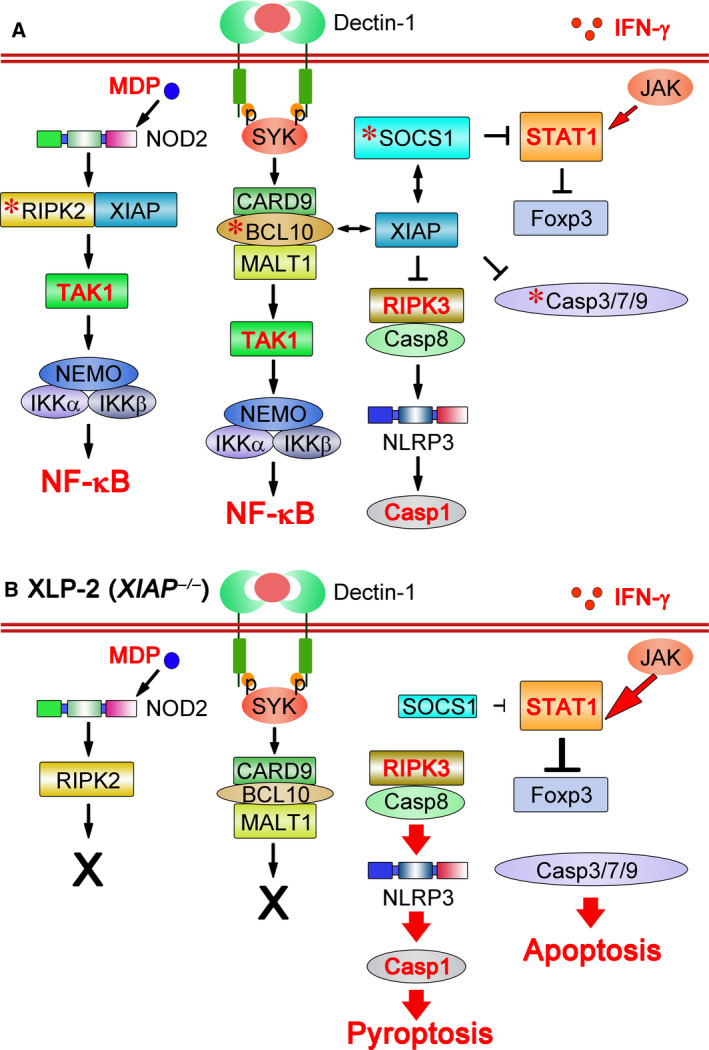
Signaling defects in XIAP‐deficient individuals. (A) XIAP regulates NOD2, dectin‐1, and Treg cells by binding to RIPK2, BCL10, and SOCS1. Binding of XIAP to RIPK2 polyubiquitinates RIPK2 and activates NOD2 signaling [27, 28, 29, 30]. XIAP also binds and stabilizes BCL10 to enable dectin‐1 signaling [33]. Interaction of XIAP with SOCS1 stabilizes this latter, helping to maintain regulatory T‐cell populations by suppressing IFN‐γ‐STAT1 signaling [34]. XIAP also inhibits RIPK3 and caspase‐8‐mediated NLRP3 inflammasome activation [41, 42]. Moreover, it binds and inactivates caspase‐3, caspase‐7, and caspase‐9. *, XIAP‐interacting protein. That most TLR activation is XIAP‐independent is not shown in the figure. (B) Infection by EBV, Listeria monocytogenes or Candida albicans induces signaling defects in XLP‐2 (XIAP‐deficient) cells. As a consequence of XIAP loss, the NOD2 signaling cascade is blocked and dectin‐1‐initiated signal transduction is diminished. In addition, reduced SOCS1 levels fail to antagonize STAT1‐directed Foxp3 destabilization, leading to attenuated Treg function. The absence of XIAP also facilitates caspase‐8‐dependent NLRP3 inflammasome activation, bypassing the dual signal requirement, resulting in caspase‐1 activation and pyroptosis. XIAP deficiency increases the activity of caspase‐3, caspase‐7, and caspase‐9 for apoptosis induction. Most TLR signaling activity remains intact, leading to TNF expression, which primes necroptosis.
Canonical NF‐κB activation and the TNF/IL‐6 production induced by various TLRs are intact in Xiap −/− cells [28, 31, 32, 33], and XIAP deficiency impairs the dectin‐1‐induced NF‐κB activation mediated by BCL10 [33] (Fig. 1). XIAP promotes BCL10 K63 ubiquitination and stabilizes BCL10. In the absence of XIAP, BCL10 levels are downregulated, resulting in diminished dectin‐1‐initiated NF‐κB activation and TNF/IL‐6 production [33]. Defective dectin‐1 signaling results in Xiap −/− mice being incapable of controlling Candida albicans infection [33].
XIAP deficiency also leads to defects in regulatory T (Treg) cells [34]. Tregs are critical components for maintaining peripheral immune tolerance [35]. XIAP‐deficient Tregs appear normal, but inflammatory cues can reprogram them into IFN‐γ‐secreting cells [34]. Foxp3 expression is reduced in Xiap −/− regulatory T cell (Treg cells), and they lost suppressive functions. These Xiap −/− Treg cell defects have been linked to downregulated expression of suppressor of cytokine signaling 1 (SOCS1; Fig. 1), a negative regulator of cytokine signaling that stabilizes Foxp3 expression by suppressing signal transducer and activator of transcription 1 and 3 (STAT1‐ and STAT3)‐directed IFN‐γ and IL‐17 production [36]. XIAP binds SOCS1 and promotes its stabilization [34].
XIAP knockout does not affect the sensitivity of thymocytes, conventional T cells, and embryonic fibroblasts to cell death [37]. However, XIAP deficiency confers susceptibility to apoptosis on invariant natural killer (iNKT) cells and mucosal‐associated invariant T (MAIT) cells, with enhanced expression of caspase‐3, caspase‐7, and BH3‐interacting domain death agonist (BID) in iNKT cells [38]. In contrast, XIAP‐deficient myeloid cells are prone to inflammasome activation. Inflammasome activation requires two signals: one from innate receptors to activate the expression of NLRP3 and pro‐IL‐1β, and a second danger signal to promote inflammasome assembly [39, 40]. In the absence of XIAP, TLR signal treatment alone drives RIPK3–caspase‐8‐mediated NLRP3 inflammasome activation, circumventing the requirement for a second signal [41, 42]. The susceptibility of Xiap −/− myeloid cells to inflammasome activation implies that the inflammatory cues generated by infection in XIAP‐null individuals arise from inflammasomes. Apart from Tregs, it is interesting to note that XIAP deficiency primarily affects signaling in myeloid cells.
Infection‐induced inflammation in XLP‐2
Epstein–Barr virus is the primary pathogen triggering XLP‐2 syndromes in humans. The predominance of EBV infection associated with XLP‐2 is likely linked to the prevalence (90%) of EBV in adults, meaning that XLP‐2 infants are exposed early to EBV. The exact mechanism of how EBV induces XLP‐2 syndrome remains unclear [43]. In healthy individuals, control of EBV infection is highly dependent on CD8+ T cells [44, 45]. Notably, EBV‐specific CD8+ T cells from XLP‐2 patients exhibit effector‐memory phenotypes and elicit strong IFN‐γ production against EBV extracts [46], indicating an intact efficacy of EBV‐specific CD8+ T cells. In contrast, impaired NOD2 signaling in XLP‐2 patients may explain their susceptibility to Crohn's disease [47]. NOD2 binds MDP derived from bacterial peptidoglycans, and mutation of NOD2 is linked to a higher risk of Crohn's disease (for review, see [48]). It is generally believed that by sensing MDP from microbiota, NOD2 confers protective immunity against harmful intestinal bacterial. Defective NOD2 signaling due to XIAP deficiency thereby increases the risk of Crohn's disease.
Xiap −/− mice can be used to understand how infection triggers inflammation in XLP‐2 patients. XIAP knockout mice are normal when housed in specific pathogen‐free (SPF) environments [37, 49, 50]. However, they become vulnerable upon infection by selected pathogens, including L. monocytogenes, Chlamydophila pneumoniae, C. albicans, or murine γ‐herpesvirus 68, displaying XLP‐2‐like syndromes and high mortality [31, 33, 41, 51]. By using a low‐dose C. albicans treatment survived by all wild‐type (WT) mice, the susceptibility of XIAP‐deficient mice to pathogen was shown to be a consequence of infection‐induced inflammation [33]. Consistent with defective dectin‐1 signaling, serum levels of TNF, IL‐6, and MCP‐1 were significantly lower in Xiap −/− mice than WT mice 4 h after C. albicans infection treatment. In contrast, high inflammatory cytokine levels were detected in Xiap −/− mouse after 1 week, correlating with the high fungal loads, indicating that inflammation is a consequence of pathogen persistence [33]. Almost all Xiap −/− mice succumbed to C. albicans within 15 days of infection.
The mortality associated with infection in XIAP‐deficient mice may be rescued by targeting inflammation. During inflammation, prostaglandins (eicosanoids) activate the generation of pro‐resolving lipid mediators—including resolvins, protectins, and lipoxins. These eicosanoids act in various ways to resolve inflammation [52, 53]. Administration of resolvin D1 effectively suppresses C. albicans infection‐induced inflammatory cytokine generation and prevents infection‐triggered mortality in Xiap −/− mice [33]. Transfer of XIAP‐sufficient Tregs after C. albicans infection also rescued Xiap −/− mice from infection‐induced death [34], accompanied by nearly complete abrogation of inflammatory cytokine. While infection in many IEIs needs antimicrobial treatment, these results illustrate an extreme case in IEI that infection‐induced inflammation is a dominant cause of pathogenesis and lethality and that resolution of inflammation is sufficient to control infection in the absence of antimicrobial reagents.
Figure 2 is a summary on how XIAP deficiency confers infection‐induced uncontrollable inflammation in XLP‐2. Upon infection by the pathogens that XIAP‐null myeloid cells cannot handle (such as EBV, L. monocytogenes, or C. albicans), the pathogens continuously multiply, leading to excess tissue damage and cell death. That cell death plays a critical role in the transition from hypo‐immune responses to inflammation. XIAP‐deficient cells are more vulnerable than their normal counterparts to induction of pyroptosis, apoptosis, and necroptosis. The low threshold of inflammasome activation in XIAP‐null cells elevates caspase‐1 production and enhances pyroptotic death [41, 42] (Fig. 1B). Loss of XIAP removes suppression of caspase‐3, caspase‐7, and caspase‐9 and enhances apoptotic death (Fig. 1B), as observed in iNKT and MAIT cells [38]. TLRs and TNFR signaling mostly remain intact in XIAP‐deficient myeloid cells [28, 31, 32, 33], allowing TNF generation and TNF‐directed necroptosis and apoptosis [54, 55]. Excessive pathogen‐associated molecular patterns (PAMPs) as a result of prolonged pathogen presence, together with the generation and release of damage‐associated molecular patterns (DAMPs) in the death process, act as persistent stimuli to activate innate immune receptors. The intact TLR and other innate immune receptor signaling cascades in XIAP‐deficient myeloid cells respond to emerging DAMPs, facilitating low‐responding XIAP‐null myeloid cells to convert to high inflammatory cytokine‐producing cells. Compounding the extent of inflammation, CD8+ T cells are fully reactive to EBV in XLP‐2 patients, fueled by inflammatory antigen‐presenting cells (e.g., dendritic cells), which generate the large amounts of IFN‐γ that contribute to HLH. Anti‐IFN‐γ‐neutralizing antibodies have been approved for use in pediatric primary HLH patients, even though not directly on XLP‐2 patients, before standard hematopoietic stem cell transplantation [56, 57], indicating that blockade of IFN‐γ‐mediated inflammation alleviates HLH symptoms. It is interesting to note that anti‐IFN‐γ plus anti‐TNF treatment effectively prevents experimental HLH induced by poly I:C plus LPS [58]. Therefore, pathogen‐induced cell death, DAMP/PAMP‐mediated activation of innate signaling, and inflammatory cell death together control the switch from an initial low‐level response to the persistent inflammation seen in IEIs like XLP‐2.
Fig. 2.
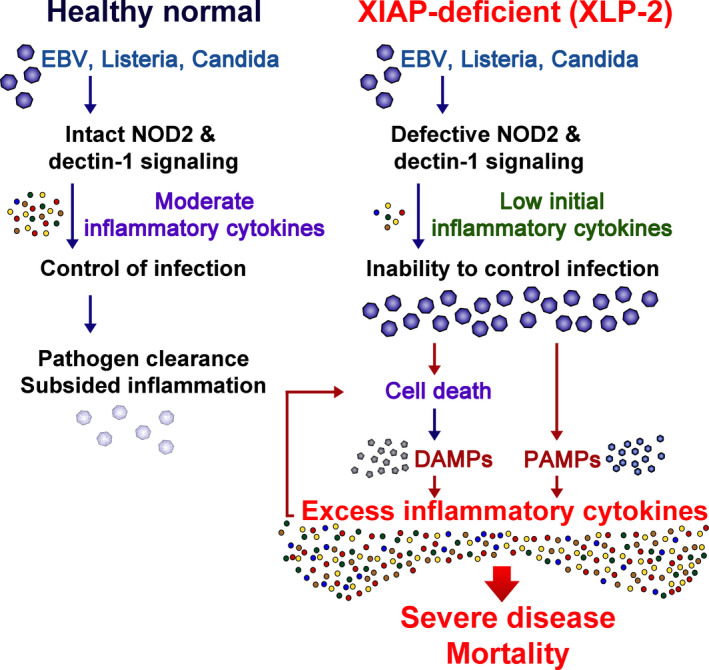
How does infection trigger persistent inflammation in XLP‐2? During infection by EBV, Listeria monocytogenes, or Candida albicans in normal healthy individuals (left), intact innate immunity generates moderate amounts of inflammatory cytokines such as TNF, IL‐6, and IFN‐γ that help control pathogen replication. Consequently, the pathogens are cleared and inflammatory cytokines subside. In contrast, XIAP‐deficient individuals cannot handle the same infections due to defective NOD2 and dectin‐1 signaling pathways (right). Accordingly, inflammatory cytokine levels are low upon immediate infection. The inability to control the pathogen results in increased pathogen load and PAMPs. Moreover, XIAP‐deficient cells are more susceptible to apoptosis, pyroptosis, and necroptosis, leading to the release of DAMPs. This buildup of PAMPs and DAMPs results in persistent release of inflammatory cytokines that further induce inflammatory cell death. The excess in inflammatory cytokines leads to severe disease and mortality.
IEIs associated with lack of interferons
Human immunodeficiency of IFN‐γ
Interferons, first identified in 1957, are a group of cytokines that play critical roles in endowing immunity to infectious diseases [59]. Based on their cognate receptors, IFNs can be classified into three different groups, that is, types I, II, and III. Below, we discuss the role of these IFNs in human immunity, with a particular focus on type I and type II IFNs. Figure 3 is a summary of the role of IFN‐γ—the sole type II IFN, in host immunity to microbes. IFN‐γ is exclusively produced by T cells and natural killer (NK) cells—forms a homodimer and binds to heterodimeric IFN‐γ receptor comprising IFN‐γR1 and IFN‐γR2 (IFNGR1, IFNGR2) subunits. The resultingly activated IFN‐γ receptor induces STAT1 phosphorylation via receptor‐associated JAK1 and JAK2 proteins. Phosphorylated STAT1 forms a homodimer, enabling it to bind to conserved IFN‐γ activation sites (GAS) in DNA elements to drive the transcription of interferon‐stimulated genes (ISGs) [60, 61]. IFN‐γ receptor, like IFN‐α receptor (IFNAR), is expressed in most cell types, but IFN‐γ is well‐known as a key factor in macrophage activation, and macrophages are the major target for IFN‐γ‐mediated activity [62].
Fig. 3.
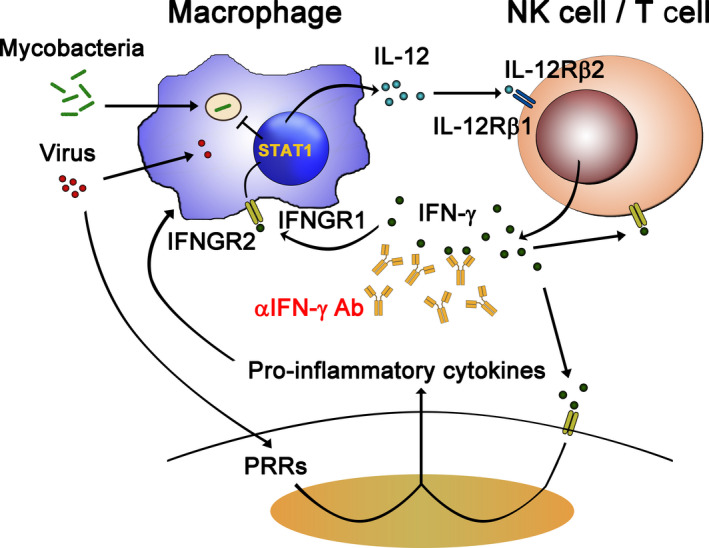
Role of IFN‐γ in host immunity to microbes. Intramacrophagic pathogens (such as mycobacteria) infect macrophages and strongly induce production of IL‐12, which binds IL‐12 receptors (IL‐12Rβ1 and IL‐12Rβ2 heterodimer) and activates T/NK cells to produce IFN‐γ. IFN‐γ then binds IFN‐γ receptors (IFNGR1 and IFNGR2 heterodimer), enabling STAT1 activation and expression of ISGs to eliminate the engulfed microbes. IFN‐γ also stimulates other cell types to induce ISG expression in a similar way. Anti‐IFN‐γ autoantibodies have been identified in adult patients with disseminated mycobacterial infections [18]. The direct role of IFN‐γ in antiviral responses has not been established. During viral infection, cells recognize viral invasion through PRRs and they produce pro‐inflammatory cytokines. The pro‐inflammatory cytokines further enhance IFN‐γ production. The synergistic effects of IFN‐γ and pro‐inflammatory cytokines may orchestrate the host immune system to tackle the viral invasion. However, hyperactivity of this pro‐inflammatory response may also cause cytokine release syndrome and cause host tissue damage. Thus, blocking these over‐reacting cytokine responses by means of anticytokine antibodies, such as anti‐IFN‐γ antibodies, might have a clinical benefit.
In humans, how IFNs function in immunity against infectious diseases has been further illustrated by studying IFN‐related inborn genetic disorders. Mendelian susceptibility to mycobacterial disease (MSMD) features a selective and inherited predisposition to nonvirulent mycobacteria and other intramacrophagic pathogens. To date, 17 gene disorders have been found in patients with MSMD [63, 64]. Even though it can arise via mutation of various genes, MSMD presents physiological homogeneity in that all contributory genetic causes to MSMD affect IFN‐γ‐dependent pathways. The affected genes encode proteins involved in inducing either IFN‐γ production [IL‐12B, IFNG, IL12RB1, IL12RB2, ILR23R, ISG15, TYK2, IKBKG, IFN regulatory factor 8 (IRF8), RORC, SPPL2A, and T‐BET] or control IFN‐γ function (IFNGR1, IFNGR2, JAK1, STAT1, and CYBB). However, viral infection is less commonly observed in IFN‐γ‐deficient MSMD patients [65].
Anti‐IFN‐γ autoantibodies
Anticytokine autoantibodies represent an emerging etiology causing immune dysregulation in human diseases [18]. By binding to cognate cytokines, autoantibodies interrupt signaling transduction and impair the biological functions of cognate cytokines, leading to physiological dysregulation in patients. Therefore, anticytokine autoantibodies are deemed a late‐onset immunodeficiency [6, 66]. Anti‐granulocyte–macrophage colony‐stimulating factor (GM‐CSF) autoantibodies were the first anticytokine autoantibodies to be comprehensively characterized as causing human disease [67]. The presence of these autoantibodies halts maturation of alveolar macrophages by blocking the function of GM‐CSF and leading to pulmonary alveolar proteinosis, which is a pathological indication of surfactant accumulation in lung tissue [68]. The disease relevance of anticytokine autoantibodies is reinforced by the fact that patients harboring autoantibodies to various cytokines are being increasingly identified, including against IFNs γ and α, as well as IL‐12, IL‐6, and IL‐17 [18, 69, 70, 71, 72, 73, 74, 75, 76]. Among them, IFNs (including IFN‐γ and some type I IFNs) are the most common targets of the autoantibodies reported in patients with various clinical manifestations. Notably, the genetic basis of anticytokine autoantibodies remains mostly unknown, as they are catalogued under ‘Phenocopies of IEI’ defined by IUIS Expert Committee [6].
Anti‐IFN‐γ autoantibodies were first reported in a small group of adult patients displaying disseminated mycobacterial infections. Similar to MSMD patients, the autoantibodies block signaling of endogenous IFN‐γ via IFN‐γ receptors [71, 72]; therefore, autoantibodies to IFN‐γ phenocopy MSMD in adults [77]. Initially, this trait was considered a very rare illness, but patients with these autoantibodies are being increasingly reported over the last decade and primarily from South‐East Asia, South China, and Japan [18, 78, 79]. Interestingly, anti‐IFN‐γ autoantibody production is strongly linked to specific HLA class II alleles (DRB1*16:02/DQB1*05:02 in patients from South China and Taiwan, or DRB1*15:02/DQB1*05:01 in those from South‐East Asia) [80]. These two alleles are almost exclusively found in those regions, providing further genetic evidence of the restricted geography of this autoantibody‐related disease. Recently, anti‐IFN‐γ autoantibodies were reported as the major etiology of Talaromyces marneffei infection—a dimorphic fungal pathogen only distributed in South China and South‐East Asia—in nonhuman immunodeficiency virus‐infected patients from South China [81, 82]. Thus, through both inborn genetics and autoantibodies, IFN‐γ has been demonstrated to play critical roles in controlling mycobacterial and intramacrophagic pathogens, such as bacteria and fungi (Fig. 3).
Viral infection and IFN‐γ
Similar to other human viral infections, SARS‐CoV‐2 infection results in a broad spectrum of clinical manifestations, from asymptomatic cases to fatalities. Activation of the immune system and production of inflammatory cytokines are essential processes in natural antiviral immune responses. However, hyperactivation of the immune system results in an acute increase in pro‐inflammatory cytokines, which can cause cytokine release syndrome (CRS) [83]. High levels of serum inflammatory cytokines are observed in patients with severe COVID‐19 [58, 84, 85, 86, 87, 88, 89]. In severe cases of SARS‐CoV‐2 infection, patients suffer systemic symptoms of varying severity, including acute respiratory distress syndrome (ARDS) and systemic inflammatory response syndrome (SIRS), both of which are associated with aggressive inflammatory responses and elevated levels of pro‐inflammatory cytokines. CRS is associated with lung injury, multiple organ failure, and mortality [90, 91].
It is generally recognized that type II IFN is responsible for antimycobacterial responses. What role IFN‐γ plays in controlling SARS‐CoV‐2 infection remains largely unknown. Relative to type I IFNs, its role in viral infections is not prominent. However, IFN‐γ treatment does induce strong STAT1 activation and protection from viral invasion in in vitro cell models. IFN‐γ treatment also inhibited SARS‐CoV‐2 replication in vitro [92]. The antiviral potential of IFN‐γ is further supported by the fact that varicella zoster virus (VZV) reactivation is observed in approximately one‐third of patients harboring anti‐IFN‐γ autoantibodies, and some sporadic cases of CMV, HSV, or VZV infection have been reported in patients with IFN‐γ receptor deficiency [93]. Moreover, IFN‐γ plays multiple roles in myeloid cell activation and can influence maturation of adaptive immunity, which may affect the development of adequate innate and adaptive immunity to SARS‐CoV‐2 infection. Elevated IFN‐γ levels have been reported in some cases of COVID‐19‐associated severe pneumonia and other CRS [94, 95]. Indeed, among the elevated cytokines induced by SARS‐CoV‐2 infection, IFN‐γ and TNF are the two major ones that together cause massive cell death [58]. Notably, an IEI patient with STAT1 gain‐of‐function was asymptomatic after SARS‐CoV‐2 infection [10]. Nevertheless, to our knowledge, no IFN‐γ‐deficient patient, with either IEI in IFN‐γ pathway or anti‐IFN‐γ autoantibodies, with SARS‐CoV‐2 infection, has been reported.
Immunodeficiency in type I IFN with viral infection
At least 17 type I IFN functional genes have been reported in humans (including 12 subtypes of IFN‐α, IFN‐β, IFN‐ω, IFN‐ε, and IFN‐κ) [96]. Figure 4 summarizes IFN‐I and TLR3 responses to viral infection. Type I IFNs bind to heterodimeric IFNARs and induce dimerization of receptor subunit IFNARs 1 and 2 (IFNAR1, IFNAR2). Activation of these IFNARs further induces phosphorylation of STAT1 and STAT2 via Janus kinase 1 (JAK1) and tyrosine kinases (such as TYK2). Phosphorylated STAT1/STAT2 interacts with IRF9 to form the ISGF complex, which binds to the IFN‐sensitive response element (ISRE) on chromosomes to enhance transcription of hundreds of IFN‐stimulated genes (ISGs) [61, 97, 98].
Fig. 4.
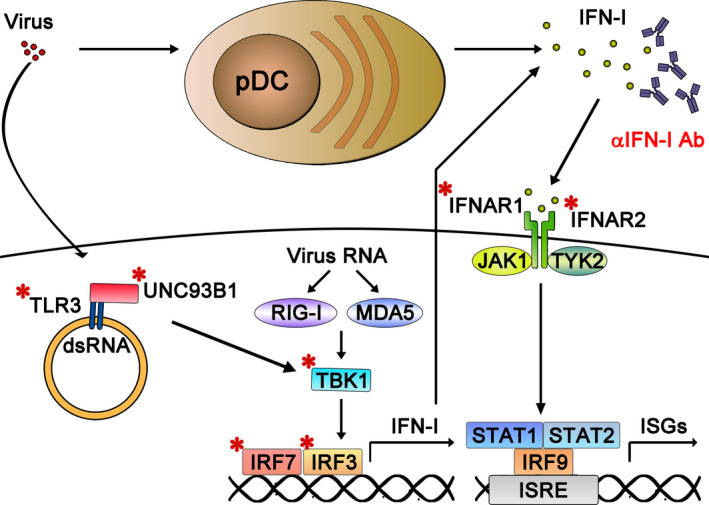
Virus‐induced type I IFN and TLR3 responses and their dysregulation in severe COVID‐19 pneumonia. Nonhematopoietic cells sense viral invasion through a panel of PPRs (such as TLR3, RIG‐I, MDA‐5) and produce type I IFNs by activating IRF3/7. Moreover, plasmacytoid dendritic cells (pDCs) have an extraordinary ability to detect virus and to secrete type I IFNs, particularly IFN‐α. These type I IFNs act via autocrine or paracrine modes to induce antiviral immunity in pDCs and surrounding cells through IFNARs (IFNAR1 and INFAR2 heterodimer). Recently, IEI in the genes encoding TLR3, Unc93B1, TBK1, IRF3, IRF7, IFNAR1, and IFNAR2 (marked with *) have been shown to impair induction of type I/III IFN responses to viral infection, and this scenario has been linked to severe COVID‐19 disease [9]. Moreover, the presence of neutralizing autoantibodies against IFN‐α and IFN‐ω, which can block the function of these crucial antiviral cytokines, can also cause severe COVID‐19‐related pneumonia [8].
Type I IFNs are well characterized for their potent antiviral effects in vitro and in vivo. The role of type I IFN‐mediated antiviral immunity has recently re‐emerged due to the identification of inborn errors in type I IFN‐ and IFNAR‐deficient patients. STAT2 is involved in type I and type III IFN signaling pathways, and STAT2 deficiency has been found in patients suffering from severe viral infections upon receipt of live‐attenuated vaccine against measles, mumps, and rubella (MMR) [99]. A patient displaying MMR vaccine‐related disease and life‐threatening influenza possessed a mutant version of IRF9, a component of the transcriptional activator complex ISGF3 together with STAT1 and STAT2 [100]. Genuine human type I IFN deficiency was illustrated by the identification of the first person with an IFNAR2 deficiency that abolished type I IFN signaling, which was described in a child that suffered fatal encephalitis after being inoculated with MMR vaccine [101]. A severe adverse reaction to MMR vaccine has also been reported in a family displaying IFNAR1 deficiency. Similarly, viscerotropic disease induced by yellow fever vaccination was also reported for a 14‐year‐old girl with compound heterozygous IFNAR1 deficiency [47]. These human IFN deficiencies demonstrate that type I IFNs are crucial for protective immunity against live‐attenuated vaccines, at least for MMR and yellow fever. Consistent with those results, a partial gain‐of‐function mutation in IFN‐induced helicase C‐domain‐containing protein 1 (IFIH1; A946T), critical for responses to RNA virus, confers host resistance to lethal encephalomyocarditis virus infection [102].
For anti‐IFN‐I autoantibodies, anti‐IFN‐α and anti‐IFN‐ω autoantibodies are common in patients with autoimmune polyendocrine syndrome I (APS‐I), an inherited autoimmune disease caused by mutation in the gene Autoimmune regulator (AIRE) responsible for thymic function in developing immunotolerance [103]. Notably, anticytokine autoantibodies are generated precede the onset of APS‐I syndromes [104]. Apart from displaying autoimmunity, APS‐I patients also suffer various fungal infections, most frequently by chronic mucocutaneous candidiasis. The identification in such patients with neutralizing autoantibodies to IL17A/F and IL22, which are key cytokines in Th17 immunity, explains their susceptibility to fungal infection [76]. Anti‐IFN‐α and other type I IFN autoantibodies have been found in some patients with other diseases, such as systemic lupus erythematosus [105], Sjögren's syndrome [106], and thymoma [75], and even in healthy individuals [107]. The association between anti‐IFN‐I autoantibodies and viral disease has been illustrated by the link of anti‐IFN‐α autoantibodies to severe VZV infection [108]. More recently, anti‐IFN‐α autoantibodies are also identified in patients with life‐threatening yellow fever vaccine‐associated diseases [109].
Type I IFN immunity in COVID‐19
Genetic defects of type I IFN immunity in severe COVID‐19
Given that severe COVID‐19 disease is less frequent in individuals younger than 50 years old, it is possible that the severe disease in these patients is due to an undiagnosed immunodeficiency, particularly in the type I IFN pathway [110]. This notion has been supported by Zhang et al. [9], who searched for loss‐of‐function (LOF) variants at the 13 gene loci known to be involved in type I IFN immunity. They identified LOF variants in 23 out of 659 (3.5%) COVID‐19 patients with life‐threatening pneumonia. In addition to the inborn monogenetic errors in TLR3, IRF7, and IRF9 that have been reported as underlying life‐threatening influenza pneumonia, the same study identified LOF variants at another five genes involved in TLR3‐ and TLR7‐dependent type I immunity. LOF variants were found in genes involved in TLR signaling to elicit type I IFN production (TLR3, UNC93B1, TCAM1, TBK1, and IRF3) and in type IFN‐I signal amplification (IRF7, IFNAR1, and IFNAR2; Fig. 4). Notably, these 23 patients had never been hospitalized for other life‐threatening viral diseases. These data demonstrate not only the essential role of type I IFNs to control SARS‐CoV‐2 infection but also that inborn genetic disorders in type I IFNs are significant factors predisposing to severe illness. Moreover, X‐linked LOF variants have also been identified in the TLR7 gene of young male patients with fatal SAR2‐CoV‐2 infection [111]. A recent genetic study of COVID‐19 patients by GWAS approach also revealed low expression of IFNAR2 associated with severe COVID‐19 disease and identified IFN‐inducible Oligoadenylate synthetase (OAS) as another COVID‐19‐linked gene [112]. Some of the IFN‐I‐ and TLR3‐associated genetic defects identified in COVID‐19 patients with critical pneumonia are marked in Fig. 4.
Autoantibodies to type I IFNs impede the type I IFN response in COVID‐19
The first indication that autoantibodies to type I IFNs might be a contributory factor to severe COVID‐19 pneumonia was the observation of severe illness in APS‐I patients, who are well known to carry autoantibodies to type I IFNs [113]. By screening for autoantibodies to type I IFNs in 987 patients with life‐threatening COVID‐19 pneumonia, Bastard et al. [8] observed that 101 (10.2%) of them had neutralizing anti‐IFN‐α and/or anti‐IFN‐ω autoantibodies. These autoantibodies were not observed in asymptomatic or mild SARS‐CoV‐2‐infected individuals and were only found in 0.33% of healthy individuals. Typically, such autoantibodies are detected within 1–2 weeks after SARS‐CoV‐2 infection. However, identification of such autoantibodies in two patients before infection, together with nondetectable type I IFNs in the severe COVID‐19 cases, suggests that these autoantibodies pre‐existed in these patients and are a cause of the severe COVID‐19 pneumonia rather than a consequence of SARS‐CoV‐2 infection. These autoantibodies impair the ability of IFN‐α2 to block SARS‐CoV‐2 infection in human cells in vitro, potentially explaining why SARS‐CoV‐2 infection is so severe in autoantibody‐producing patients. These observations imply that IFN‐α and IFN‐ω are specifically critical in controlling the pathogenesis of COVID‐19‐linked pneumonia. These autoantibodies to type I IFNs have only recently been identified and represent highly relevant factors predisposing individuals to severe SARS‐CoV‐2 infection. Understanding in detail the functions of type I IFNs in patients with these autoantibodies might provide important insights into the natural course of severe COVID‐19 infection.
SARS‐CoV‐2 inhibits type I IFN production and responses
In addition to inborn mutation or autoantibodies, several studies have identified an attenuated expression of type I IFNs in COVID‐19 patients. Dendritic cells taken from acutely infected patients present diminished production of IFN‐α and IFN‐β [114]. A systems biology analysis of severe COVID‐19 patients showed reduced expression of IFN‐I in their myeloid cells [115], which was accompanied by increased plasma levels of inflammatory molecules. Gene expression profiles of monocytes from COVID‐19 patients with ARDS revealed deficient responses to IFN [116]. Notably, influenza patients do not show the attenuated type I/III IFN responses observed in COVID‐19 patients [87, 117]. Even though IL‐6 plasma levels are elevated to similar levels in COVID‐19 and influenza A virus‐infected patients, SARS‐CoV‐2 infection induces IFN signaling (STAT1 and IRF3) in dendritic cells and T cells that is distinct from the signals (STAT3 and NF‐κB) activated by influenza A virus infection [117]. In a similar study, profoundly impaired production of IFN‐λ and type I IFNs was found in COVID‐19 patients, accompanied by highly elevated expression of pro‐inflammatory cytokines, in contrast to uniformly produced type I and type III IFNs in patients with influenza [87]. These results suggest that SARS‐CoV‐2 selectively suppresses type I IFNs immediately upon infection [87, 117, 118, 119, 120, 121].
Other CoVs, including SARS‐CoV and MERS, are known to antagonize expression of type I/III IFNs and to attenuate their downstream signaling [111, 122]. SARS‐CoV delays early type I IFN expression in a mouse model of the virus, enabling higher viral titers, an accumulation of inflammatory monocyte–macrophages, vascular leakage, increased levels of lung cytokines, and attenuated virus‐specific T‐cell responses [123]. Depletion of inflammatory monocyte–macrophages rescued the mice from SARS‐CoV‐induced lethality, and early administration of IFN‐β also prevented severe SARS‐CoV‐mediated disease. Type I IFNs also display a similar protective role in MERS [124].
SARS‐CoV‐2 inhibits production of type I IFNs and suppresses their signaling activity in vitro. It has been shown that NSP1, NSP3, NSP12, NSP13, NSP14, ORF3, ORF6, and M proteins from SARS‐CoV‐2 inhibit IFN‐β promoter activation, and ORF6 also inhibits ISRE and ISG56 promoter activation and suppresses STAT1 nuclear translocation [119]. In another study, ORF3b was shown to be a potent suppressor of type I IFN induction [125]. Similarly, ORF6, ORF8, and N proteins from SARS‐CoV‐2 suppress activation of the IFN‐β and κB promoters, with ORF6 and ORF8 further inhibiting the ISRE promoter [126]. Additionally, NSP13, NSP14, NSP15, and ORF6 attenuate production and signaling of type I IFNs [127]. SARS‐CoV‐2 suppresses type I IFN expression by its NSP16 protein‐inhibiting mRNA splicing, its NSP1 protein‐restricting mRNA translation, and NSP8/9 interfering with protein trafficking to the cell surface [128]. SARS‐CoV‐2 NSP3 protein (a papain‐like PLpro protease) binds ISG15 and cleaves it from IRF3 to attenuate type I IFN responses [129]. NSP3 also directly cleaves IRF3 to suppress type I IFN signaling [130]. In addition, SARS‐CoV‐2 N protein binds retinoic acid‐inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA‐5), molecular sensors of cytosolic double‐stranded RNA, and the immediate signal protein mitochondrial antiviral signal (MAVS) and it inhibits the RIG‐I/MDA‐5‐MAVS signal cascade [131, 132]. Together, these results show that SARS‐CoV‐2 encodes several proteins inhibiting type I and type III IFNs, both at the expression and signaling level.
Link between early type I IFN deficiency and inflammation in severe COVID‐19 cases
The afore‐described results indicate that some severe COVID‐19 patients are unable to mount an effective innate anti‐SARS‐CoV‐2 response due to genetic defects in type I IFNs or they possess autoantibodies against type I IFN, whereas other patients with severe COVID‐19 suffer from defective type I IFNs responses because they are suppressed by SARS‐CoV‐2. Figure 5 summarizes how deficiency in type I IFNs leads to excess inflammation in COVID‐19. Type I IFNs attenuate inflammation during early viral infection, acting before inflammatory cytokines to minimize damage from inflammation [133]. In severe cases of COVID‐19, expression of type I IFNs is attenuated and delayed, yet inflammatory cytokine and chemokine production is enhanced [87, 118, 121]. For patients with no IFN‐I‐associated IEIs or anti‐IFN‐I antibodies, early administration of type I IFNs alleviates severe COVID‐19 disease [120, 134, 135, 136], resulting in reduced viral load and decreased serum levels of IL‐6 and C‐reactive protein (CRP) [134, 135]. These results indicate that the diminished early type I IFN responses contribute to subsequent hyperinflammation syndromes in patients with severe COVID‐19 disease. The inability to control SARS‐CoV‐2 due to type I IFN insufficiency results in unrestricted viral replication, causing extensive tissue damage and cell death [89]. Uncontrolled viral replication leads to excess PAMPs for further inflammatory cytokine stimulation (Fig. 5). Among inflammatory cytokines, for example, high serum IL‐6 levels predict poor COVID‐19 prognosis [85, 86]. The role of IL‐6/STAT3 in severe COVID‐19 is also implicated by that loss‐of‐function STAT3 mutant protected against SARS‐CoV‐2 infection [10].
Fig. 5.
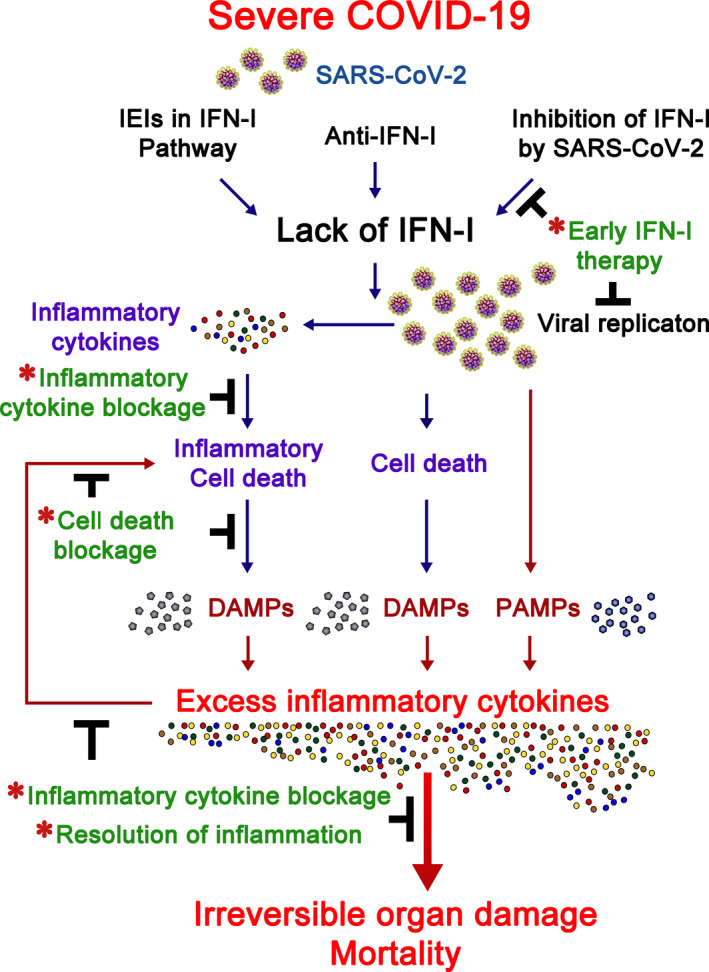
Infection‐induced inflammation in COVID‐19. Type I IFNs are critical to early control of SARS‐CoV‐2 infection. A small proportion of patients with severe COVID‐19 bear IEI of type I IFN pathway genes. Other such patients carry anti‐IFN‐I antibodies, and SARS‐CoV‐2 suppresses the expression of type I IFNs and their downstream signaling molecules in other severe COVID‐19 cases. Deficiency in type I IFN cascades leads to unrestricted viral replication and enhanced inflammatory cytokine production. PAMPs arising from SARS‐CoV‐2 infection, DAMPs from cell death induced by viral replication, and DAMPs from inflammatory cell death activated by inflammatory cytokines trigger excess inflammatory cytokine production that, in turn, induce more inflammatory cell death. This scenario may lead to irreversible organ damage and patient mortality. Infection‐induced inflammation in COVID‐19 may be alleviated (marked by *) by early type I IFN administration, inflammatory cytokine blockage, inflammatory cell death blockage, or resolution of inflammation. For details of each therapeutic approach, refer to the text.
Similar to XLP‐2, cell death likely plays a pivotal role in the transition to irreversible inflammation in severe COVID‐19 [137, 138]. Based on homology of SARS‐CoV‐2 ORF3a with that of SARS‐CoV, the SARS‐CoV ORF3a being a K+ channel, and ORF3‐mediated K+ efflux enhancing NLRP3 inflammasome activation, SARS‐CoV‐2 is likely to activate inflammasomes [139]. This scenario was validated by active NLRP3 inflammasomes being found in the peripheral blood mononuclear cells of COVID‐19 patients and in lung histology of COVID patients postmortem [140], supporting that SARS‐CoV‐2 infection induces pyroptosis [137, 138]. Pyroptotic death has also been detected in the pneumocytes and endothelial cells of lung biopsies from COVID patients postmortem [89]. Importantly, high levels of TNF are always recorded in patients with severe COVID‐19 [84, 85, 87, 89], and TNF primes for necroptosis and apoptosis [137, 138]. SARS‐CoV‐2 infection triggers caspase‐8 activation, leading to apoptosis and necroptosis in a lung epithelial cell line [141]. Importantly, both monocytes and T lymphocytes from COVID‐19 patients exhibit increased apoptotic signals [117], and pronounced apoptosis and necroptosis have been found in lung tissue from COVID‐19 patients postmortem [89, 141]. In addition, apoptotic gene signatures in plasmacytoid dendritic cells correlate with COVID‐19 disease severity [142]. Moreover, inflammatory cell death arising from pyroptosis, apoptosis, and necroptosis (termed PANoptosis) can be induced by a combination of TNF and IFN‐γ [58], and this is likely key to the pathogenesis of COVID‐19. Therefore, in the absence of early type I IFNs to repress SARS‐CoV‐2 replication, SARS‐CoV‐2 triggers extensive cell death. SARS‐CoV‐2‐mediated cell death and inflammatory cell death together enhance the release of DAMPs that further stimulate innate receptors for more inflammatory cytokine production, in turn inducing even more inflammatory cell death (Fig. 5). The extensive inflammation and cell death associated with SARS‐CoV‐2 may lead to the irreversible organ damage and mortality linked to COVID‐19. Thus, infection‐induced inflammation and cell death play dominant roles in the pathogenesis of COVID‐19, resulting in irreversible organ damage and even mortality.
Therapeutic implications for COVID‐19
Figure 5 also marks the potential therapeutic approaches to COVID‐19 intervention. First, it is necessary to screen for IFN‐I‐associated IEIs and the presence of anti‐IFN‐I antibodies in severe COVID‐19 patients. Such screening will enable provision of specialized clinical care to respective COVID‐19 patients, potentially slowing disease progression. Despite the narrow sensitivity to viral infection for each monogenic IEI defect, extension of screening to genes encoding proteins required for responses to other viruses, such as SH2D1A, XIAP, ITK, NOS2, and DBR1, could also be helpful in COVID‐19 patient assessments [7].
For patients lacking IFN‐I‐associated IEIs or anti‐IFN‐I autoantibodies, SARS‐CoV‐2 suppresses the early production of type I IFNs that leads to pathogenesis. The low levels of such IFNs in patients with severe COVID‐19 and consequently elevated inflammation imply that type I IFN administration is a potential treatment for COVID‐19. Initial clinical studies support the efficacy of such treatment. Early IFN‐α or IFN‐β administration improves clinical outcomes, increases viral clearance, decreases in‐hospital COVID‐19 deaths, and enables more rapid recovery [120, 134, 135, 136]. Notably, IFN‐I treatment should be beneficial for the COVID‐19‐infected IEI patients who cannot mount IFN‐I production. Indeed, the IFN‐α2b treatment showed a clinical benefit for two COVID‐19 patients with TLR3 or IRF3 mutation [143]. Importantly, even in COVID‐19 patient with autoantibodies against IFN‐α and IFN‐ω treatment with IFN‐β is shown to resolve syndromes [144], supporting the therapeutic application of IFN‐β in patients bearing anti‐IFN‐α/IFN‐ω autoantibodies. However, the timing of early type I IFN administration is critical in treating COVID‐19. In mouse infection models of SARS‐CoV and MERS, delayed IFN‐β treatment exacerbated disease, resulting in increased infiltration and activation of myeloid cells in lung tissues and elevated inflammatory cytokines, leading to fatal pneumonia [123, 124]. Similar outcomes have been reported for COVID‐19 cases, in which late IFN‐α2b treatment delayed recovery and increased mortality [120].
High levels of inflammatory cytokines have been implicated in contributing to severe cases of COVID‐19, so various anticytokine monoclonal antibodies have been subjected to clinical trials to determine their suitability for treating severe COVID‐19‐linked pneumonia. Even though there is discrepancy on the efficacy of IL‐6 neutralization [145], recent clinical trials found that anti‐IL‐6R improves clinical outcomes and survival in both moderately and critically ill COVID‐19 patients [146, 147]. Preliminary results also suggest that other inflammatory cytokines may represent promising targets. Monoclonal antibodies to GM‐CSF receptor showed a promising therapeutic effect for COVID‐19 pneumonia in clinical trial [148], reflecting the likely role of macrophage overactivation in severe COVID‐19 infection. A beneficial effect of anti‐TNF in COVID‐19 therapy has been suggested [149], which warrants further characterization. Tests on anti‐IFN‐γ (NCT04324021) monoclonal antibodies are ongoing [150]. In addition, monitoring the natural course of SARS‐CoV‐2 infection in patients with natural autoantibodies to IFN‐γ and GM‐CSF might provide important data informing the design of such treatments.
Persistent infection‐induced inflammation is a shared feature of COVID‐19 and XLP‐2. Though caused by different viruses (EBV versus SARS‐CoV‐2) and the clinical syndromes of COVID‐19 being distinct from the HLH of XLP‐2 [151, 152], inflammatory cell death likely represents a turning point from infection to irreversible inflammation in both COVID‐19 and XLP‐2. Inhibition of inflammatory cell death triggered by a combination of anti‐TNF and anti‐IFN‐γ significantly rescued SARS‐CoV‐2‐induced mortality in a COVID‐19 mouse model [58], suggesting a potential application in the treatment of COVID‐19 patients.
Alternatively, tackling inflammation may help control infection‐induced pathologies in IEIs, and it is likely to be effective in COVID‐19 therapy. One well‐known example of a respective therapy is dexamethasone, which acts by reducing inflammation, and it has been approved for use to improve disease status and to reduce the mortality associated with moderately and severely ill COVID‐19 patients [153, 154]. Recent study of a rhesus macaque COVID‐19 model has also shown that treatment with baricitinib, a JAK1/JAK2 inhibitor, resolves lung inflammation by suppressing the production of inflammatory cytokines by macrophages and by inhibiting neutrophil recruitment, without affecting type I IFN signaling [155]. Early clinical trial results of baricitinib, by itself or in combination with remdesivir, on hospitalized COVID‐19 patients, showed reduced inflammation and favorable clinical outcomes [156, 157], supporting the efficacy of JAK1/JAK2 inhibitors in resolving SARS‐CoV‐2 infection‐induced inflammation. From observations of IEIs in a XLP‐2 mouse model, infection‐induced inflammation and mortality can be prevented by using resolvin D or ex vivo‐expanded Tregs without the need for antimicrobial agents [33, 34], suggesting that similar approaches could be tested for their efficacy in controlling the persistent inflammation associated with COVID‐19. Mesenchymal stem cells, characterized by their anti‐inflammatory activities and Treg‐inducing capacity, have also been used in a small number of COVID‐19 patients, and respective clinical trials are ongoing [158].
Altogether, a variety of therapeutic approaches targeted to several steps contributing to the persistent inflammation of COVID‐19 are currently being investigated. These pathways are distinct from the processes exerted by antiviral reagents, such as blocking viral entry by anti‐SARS‐CoV‐2 antibodies or inhibiting viral replication via small‐molecule antiviral drugs, so the inflammation‐targeting therapies are completely independent of antiviral treatments. Based on the prominent role of persistent inflammation in COVID‐19 pathogenesis, resolving inflammation is likely to be as effective as antiviral therapy at treating COVID‐19 patients.
Conclusion
In this review, we have explored the pathogenic mechanisms underlying specific IEIs and COVID‐19. Anti‐IFN‐I antibodies and IFN‐I‐associated IEIs are found in patients with severe COVID‐19. Anticytokine autoantibodies were first identified many years ago, and they have been found to cause various diseases, including autoimmune and infectious diseases. However, the importance of late‐onset immunodeficiency remains underestimated, in particular for infectious diseases. The COVID‐19 pandemic has had an unparalleled impact, but it is also presenting an opportunity to better understand the roles of anticytokine autoantibodies in infectious diseases. Thus, investigations of anticytokine autoantibodies against type I IFNs and even other cytokines are warranted in patients with SARS‐CoV‐2 infection, and indeed other infections, to establish which host factors predispose to infectious diseases. Careful dissection of clinical manifestations and physiological responses during infectious, autoimmune, and other diseases, including COVID‐19, in patients having anticytokine autoantibodies, will help develop new diagnoses and treatments.
For COVID‐19 patients harboring inborn mutations in type I IFN signaling pathways or with autoantibodies against type I IFNs, they represent the latest examples that genetic mutation or phenocopies of IEI exacerbate infection‐induced ARDS. For other patients without apparent genetic deficiency, COVID‐19 may be reminiscent of immunodeficiency diseases in which early type I IFN responses are suppressed, leading to an eventual breakout of inflammation. This feature of persistent infection‐induced inflammation is shared by IEIs such as XLP‐2. Cell death most likely largely accounts for the transition from persistent viral infection to inflammation in both IEIs and COVID‐19. Inhibition of cell death and resolving persistent inflammation thus represent potential approaches for tackling IEIs and COVID‐19. Several agents based on such mechanisms and undergoing clinical trials are highly promising COVID‐19 therapies, including early administration of type I IFNs, use of anti‐TNF and/or anti‐IFN‐γ antibodies, or application of JAK1 or STAT1 inhibitors. Importantly, alleviation of infection‐induced inflammation should be conducted synergistically with antiviral treatments to achieve optimal therapeutic effects in COVID‐19 patients. Overall, SARS‐CoV‐2 infection‐induced inflammation is a primary cause of COVID‐19 pathogenesis and so it should be a primary target for COVID‐19 therapies.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
C‐LK and M‐ZL conducted a literature search and wrote the manuscript; I‐TC designed and prepared the figures.
Acknowledgements
We thank Dr. John O'Brien for editing the manuscript. This work was supported by grants CMRPD1I0152 from Chang Gung Memorial Hospital and MOST 109‐2628‐B‐182‐014 (CLK) and MOST 109‐2326‐B‐001‐008 (MZL) from the Ministry of Science and Education, Taiwan.
Contributor Information
Cheng‐Lung Ku, Email: clku@cgu.edu.tw.
Ming‐Zong Lai, Email: mblai@gate.sinica.edu.tw.
References
- 1. V'Kovski P, Kratzel A, Steiner S, Stalder H & Thiel V (2021) Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol 19, 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Finkel Y, Mizrahi O, Nachshon A, Weingarten‐Gabbay S, Morgenstern D, Yahalom‐Ronen Y, Tamir H, Achdout H, Stein D, Israeli O et al. (2020) The coding capacity of SARS‐CoV‐2. Nature 589, 125–130. [DOI] [PubMed] [Google Scholar]
- 3. Morens DM & Fauci AS (2020) Emerging pandemic diseases: how we got to COVID‐19. Cell 182, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, Ellingson MK, Mao T, Oh JE, Israelow B et al. (2020) Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature 584, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schultze JL & Aschenbrenner AC (2021) COVID‐19 and the human innate immune system. Cell 184, 1671–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tangye SG, Al‐Herz W, Bousfiha A, Chatila T, Cunningham‐Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T et al. (2020) Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol 40, 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Casanova JL, Su HC & Effort CHG (2020) A global effort to define the human genetics of protective immunity to SARS‐CoV‐2 infection. Cell 181, 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, Dorgham K, Philippot Q, Rosain J, Beziat V et al. (2020) Auto‐antibodies against type I IFNs in patients with life‐threatening COVID‐19. Science 370, eabd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada‐Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C et al. (2020) Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science 370, eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyts I, Bucciol G, Quinti I, Neven B, Fischer A, Seoane E, Lopez‐Granados E, Gianelli C, Robles‐Marhuenda A, Jeandel PY et al. (2021) Coronavirus disease 2019 in patients with inborn errors of immunity: an international study. J Allergy Clin Immunol 147, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Jesus AA, Canna SW, Liu Y & Goldbach‐Mansky R (2015) Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 33, 823–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischer A & Rausell A (2016) Primary immunodeficiencies suggest redundancy within the human immune system. Sci Immunol 1, eaah5861. [DOI] [PubMed] [Google Scholar]
- 13. Casanova JL & Abel L (2018) Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin Immunol 36, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C et al. (2003) Pyogenic bacterial infections in humans with IRAK‐4 deficiency. Science 299, 2076–2079. [DOI] [PubMed] [Google Scholar]
- 15. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y et al. (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho‐Shimizu V, Lorenzo L, Puel A et al. (2007) TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527. [DOI] [PubMed] [Google Scholar]
- 17. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N et al. (2006) Herpes simplex virus encephalitis in human UNC‐93B deficiency. Science 314, 308–312. [DOI] [PubMed] [Google Scholar]
- 18. Ku CL, Chi CY, von Bernuth H & Doffinger R (2020) Autoantibodies against cytokines: phenocopies of primary immunodeficiencies? Hum Genet 139, 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux‐Laucat F, Revy P et al. (2006) XIAP deficiency in humans causes an X‐linked lymphoproliferative syndrome. Nature 444, 110–114. [DOI] [PubMed] [Google Scholar]
- 20. Latour S & Aguilar C (2015) XIAP deficiency syndrome in humans. Semin Cell Dev Biol 39, 115–123. [DOI] [PubMed] [Google Scholar]
- 21. Picard C, Al‐Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham‐Rundles C, Etzioni A, Holland SM, Klein C et al. (2015) Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol 35, 696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Filipovich AH, Zhang K, Snow AL & Marsh RA (2010) X‐linked lymphoproliferative syndromes: brothers or distant cousins? Blood 116, 3398–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeissig Y, Petersen BS, Milutinovic S, Bosse E, Mayr G, Peuker K, Hartwig J, Keller A, Kohl M, Laass MW et al. (2015) XIAP variants in male Crohn's disease. Gut 64, 66–76. [DOI] [PubMed] [Google Scholar]
- 24. Amininejad L, Charloteaux B, Theatre E, Liefferinckx C, Dmitrieva J, Hayard P, Muls V, Maisin JM, Schapira M, Ghislain JM et al. (2018) Analysis of genes associated with monogenic primary immunodeficiency identifies rare variants in XIAP in patients With Crohn's disease. Gastroenterology 154, 2165–2177. [DOI] [PubMed] [Google Scholar]
- 25. Aguilar C, Lenoir C, Lambert N, Begue B, Brousse N, Canioni D, Berrebi D, Roy M, Gerart S, Chapel H et al. (2014) Characterization of Crohn disease in X‐linked inhibitor of apoptosis‐deficient male patients and female symptomatic carriers. J Allergy Clin Immunol 134, 1131–1141.e9. [DOI] [PubMed] [Google Scholar]
- 26. Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M & Salvesen GS (2005) XIAP inhibits caspase‐3 and ‐7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J 24, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, Knoefel WT & Reed JC (2009) XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 106, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, Rieser E, Rickard JA, Bankovacki A, Peschel C et al. (2012) The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 46, 746–758. [DOI] [PubMed] [Google Scholar]
- 29. Goncharov T, Hedayati S, Mulvihill MM, Izrael‐Tomasevic A, Zobel K, Jeet S, Fedorova AV, Eidenschenk C, deVoss J, Yu K et al. (2018) Disruption of XIAP‐RIP2 association blocks nod2‐mediated inflammatory signaling. Mol Cell 69, 551–565.e7. [DOI] [PubMed] [Google Scholar]
- 30. Hrdinka M, Schlicher L, Dai B, Pinkas DM, Bufton JC, Picaud S, Ward JA, Rogers C, Suebsuwong C, Nikhar S et al. (2018) Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J 37, e99372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bauler LD, Duckett CS & O'Riordan MX (2008) XIAP regulates cytosol‐specific innate immunity to Listeria infection. PLoS Pathog 4, e1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ & Vucic D (2008) c‐IAP1 and c‐IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)‐induced NF‐kappaB activation. J Biol Chem 283, 24295–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsieh WC, Chuang YT, Chiang IH, Hsu SC, Miaw SC & Lai MZ (2014) Inability to resolve specific infection generates innate immunodeficiency syndrome in Xiap‐/‐ mice. Blood 124, 2847–2857. [DOI] [PubMed] [Google Scholar]
- 34. Hsieh WC, Hsu TS, Chang YJ & Lai MZ (2018) IL‐6 receptor blockade corrects defects of XIAP‐deficient regulatory T cells. Nat Commun 9, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K & Ohkura N (2020) Regulatory T cells and human disease. Ann Rev Immunol 38, 541–566. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, Morita R, Asakawa M, Chinen T & Yoshimura A (2011) SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN‐{gamma} and IL‐17A production. J Exp Med 208, 2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harlin H, Reffey SB, Duckett CS, Lindsten T & Thompson CB (2001) Characterization of XIAP‐deficient mice. Mol Cell Biol 21, 3604–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gerart S, Siberil S, Martin E, Lenoir C, Aguilar C, Picard C, Lantz O, Fischer A & Latour S (2013) Human iNKT and MAIT cells exhibit a PLZF‐dependent proapoptotic propensity that is counterbalanced by XIAP. Blood 121, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evavold CL & Kagan JC (2019) Inflammasomes: threat‐assessment organelles of the innate immune system. Immunity 51, 609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Swanson KV, Deng M & Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwalder M et al. (2014) XIAP restricts TNF‐ and RIP3‐dependent cell death and inflammasome activation. Cell Rep 7, 1796–1808. [DOI] [PubMed] [Google Scholar]
- 42. Lawlor KE, Feltham R, Yabal M, Conos SA, Chen KW, Ziehe S, Grass C, Zhan Y, Nguyen TA, Hall C et al. (2017) XIAP loss triggers RIPK3‐ and caspase‐8‐driven IL‐1beta activation and cell death as a consequence of TLR‐MyD88‐induced cIAP1‐TRAF2 degradation. Cell Rep 20, 668–682. [DOI] [PubMed] [Google Scholar]
- 43. Aguilar C & Latour S (2015) X‐linked inhibitor of apoptosis protein deficiency: more than an X‐linked lymphoproliferative syndrome. J Clin Immunol 35, 331–338. [DOI] [PubMed] [Google Scholar]
- 44. Taylor GS, Long HM, Brooks JM, Rickinson AB & Hislop AD (2015) The immunology of Epstein‐Barr virus‐induced disease. Annu Rev Immunol 33, 787–821. [DOI] [PubMed] [Google Scholar]
- 45. Latour S & Fischer A (2019) Signaling pathways involved in the T‐cell‐mediated immunity against Epstein‐Barr virus: Lessons from genetic diseases. Immunol Rev 291, 174–189. [DOI] [PubMed] [Google Scholar]
- 46. Lopez‐Granados E, Stacey M, Kienzler AK, Sierro S, Willberg CB, Fox CP, Rigaud S, Long HM, Hislop AD, Rickinson AB et al. (2014) A mutation in X‐linked inhibitor of apoptosis (G466X) leads to memory inflation of Epstein‐Barr virus‐specific T cells. Clin Exp Immunol 178, 470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, Shirkani A, Changi‐Ashtiani M, Rokni‐Zadeh H, Sayar EH et al. (2019) Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med 216, 2057–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Al Nabhani Z, Dietrich G, Hugot JP & Barreau F (2017) Nod2: The intestinal gate keeper. PLoS Pathog 13, e1006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Olayioye MA, Kaufmann H, Pakusch M, Vaux DL, Lindeman GJ & Visvader JE (2005) XIAP‐deficiency leads to delayed lobuloalveolar development in the mammary gland. Cell Death Differ 12, 87–90. [DOI] [PubMed] [Google Scholar]
- 50. Rumble JM, Oetjen KA, Stein PL, Schwartzberg PL, Moore BB & Duckett CS (2009) Phenotypic differences between mice deficient in XIAP and SAP, two factors targeted in X‐linked lymphoproliferative syndrome (XLP). Cell Immunol 259, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prakash H, Albrecht M, Becker D, Kuhlmann T & Rudel T (2010) Deficiency of XIAP leads to sensitization for Chlamydophila pneumoniae pulmonary infection and dysregulation of innate immune response in mice. J Biol Chem 285, 20291–20302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Basil MC & Levy BD (2016) Specialized pro‐resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 16, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Serhan CN & Levy BD (2018) Resolvins in inflammation: emergence of the pro‐resolving superfamily of mediators. J Clin Invest 128, 2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan FK, Luz NF & Moriwaki K (2015) Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 33, 79–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wallach D, Kang TB, Dillon CP & Green DR (2016) Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352, aaf2154. [DOI] [PubMed] [Google Scholar]
- 56. Vallurupalli M & Berliner N (2019) Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood 134, 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, Degar B, Garrington TP, Sevilla J, Putti MC et al. (2020) Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N Engl J Med 382, 1811–1822. [DOI] [PubMed] [Google Scholar]
- 58. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS et al. (2020) Synergism of TNF‐alpha and IFN‐gamma triggers inflammatory cell death, tissue damage, and mortality in SARS‐CoV‐2 infection and cytokine shock syndromes. Cell 184, 149–168.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Isaacs A, Lindenmann J & Andrewes CH (1957) Virus interference. I. The interferon. Proc R Soc London B Biol Sci 147, 258–267.13465720 [Google Scholar]
- 60. Platanias LC (2005) Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 5, 375–386. [DOI] [PubMed] [Google Scholar]
- 61. Schneider WM, Chevillotte MD & Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hu X & Ivashkiv LB (2009) Cross‐regulation of signaling pathways by interferon‐gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bustamante J (2020) Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet 139, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kerner G, Rosain J, Guerin A, Al‐Khabaz A, Oleaga‐Quintas C, Rapaport F, Massaad MJ, Ding JY, Khan T, Ali FA et al. (2020) Inherited human IFN‐γ deficiency underlies mycobacterial disease. J Clin Invest 130, 3158–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bustamante J, Boisson‐Dupuis S, Abel L & Casanova JL (2014) Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN‐γ immunity. Semin Immunol 26, 454–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Browne SK (2014) Anticytokine autoantibody‐associated immunodeficiency. Annu Rev Immunol 32, 635–657. [DOI] [PubMed] [Google Scholar]
- 67. Kitamura T, Tanaka N, Watanabe J, Uchida K, Kanegasaki S, Yamada Y & Nakata K (1999) Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony‐stimulating factor. J Exp Med 190, 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Trapnell BC, Carey BC, Uchida K & Suzuki T (2009) Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM‐CSF stimulation of macrophages. Curr Opin Immunol 21, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, Mamani‐Matsuda M, Jouanguy E, Gendrel D & Casanova JL (2008) Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL‐6. J Immunol 180, 647–654. [DOI] [PubMed] [Google Scholar]
- 70. Patel SY, Ding L, Brown MR, Lantz L, Gay T, Cohen S, Martyak LA, Kubak B & Holland SM (2005) Anti‐IFN‐γ autoantibodies in disseminated nontuberculous mycobacterial infections. J Immunol 175, 4769–4776. [DOI] [PubMed] [Google Scholar]
- 71. Kampmann B, Hemingway C, Stephens A, Davidson R, Goodsall A, Anderson S, Nicol M, Scholvinck E, Relman D, Waddell S et al. (2005) Acquired predisposition to mycobacterial disease due to autoantibodies to IFN‐gamma. J Clin Invest 115, 2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Doffinger R, Helbert MR, Barcenas‐Morales G, Yang K, Dupuis S, Ceron‐Gutierrez L, Espitia‐Pinzon C, Barnes N, Bothamley G, Casanova JL et al. (2004) Autoantibodies to interferon‐γ in a patient with selective susceptibility to mycobacterial infection and organ‐specific autoimmunity. Clin Infect Dis 38, e10–e14. [DOI] [PubMed] [Google Scholar]
- 73. Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, Eskelin P, Perheentupa J, Husebye E, Kadota Y et al. (2006) Anti‐interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med 3, e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sim BT, Browne SK, Vigliani M, Zachary D, Rosen L, Holland SM & Opal SM (2013) Recurrent Burkholderia gladioli suppurative lymphadenitis associated with neutralizing anti‐IL‐12p70 autoantibodies. J Clin Immunol 33, 1057–1061. [DOI] [PubMed] [Google Scholar]
- 75. Meager A, Wadhwa M, Dilger P, Bird C, Thorpe R, Newsom‐Davis J & Willcox N (2003) Anti‐cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon‐alpha, interferon‐omega and interleukin‐12 in patients with thymoma and/or myasthenia gravis. Clin Exp Immunol 132, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas‐Morales G, Picard C, Cobat A, Ouachee‐Chardin M, Toulon A, Bustamante J et al. (2010) Autoantibodies against IL‐17A, IL‐17F, and IL‐22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Puel A & Casanova JL (2013) Autoantibodies against cytokines: back to human genetics. Blood 121, 1246–1247. [DOI] [PubMed] [Google Scholar]
- 78. Browne SK, Burbelo PD, Chetchotisakd P, Suputtamongkol Y, Kiertiburanakul S, Shaw PA, Kirk JL, Jutivorakool K, Zaman R, Ding L et al. (2012) Adult‐onset immunodeficiency in Thailand and Taiwan. N Engl J Med 367, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chi CY, Chu CC, Liu JP, Lin CH, Ho MW, Lo WJ, Lin PC, Chen HJ, Chou CH, Feng JY et al. (2013) Anti‐IFN‐γ autoantibodies in adults with disseminated nontuberculous mycobacterial infections are associated with HLA‐DRB1*16:02 and HLA‐DQB1*05:02 and the reactivation of latent varicella‐zoster virus infection. Blood 121, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 80. Ku CL, Lin CH, Chang SW, Chu CC, Chan JF, Kong XF, Lee CH, Rosen EA, Ding JY, Lee WI et al. (2016) Anti‐IFN‐γ autoantibodies are strongly associated with HLA‐DR*15:02/16:02 and HLA‐DQ*05:01/05:02 across Southeast Asia. J Allergy Clin Immunol 137, 945–948.e8. [DOI] [PubMed] [Google Scholar]
- 81. Guo J, Ning XQ, Ding JY, Zheng YQ, Shi NN, Wu FY, Lin YK, Shih HP, Ting HT, Liang G et al. (2020) Anti‐IFN‐gamma autoantibodies underlie disseminated Talaromyces marneffei infections. J Exp Med 217, e20190502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tang BS, Chan JF, Chen M, Tsang OT, Mok MY, Lai RW, Lee R, Que TL, Tse H, Li IW et al. (2010) Disseminated penicilliosis, recurrent bacteremic nontyphoidal salmonellosis, and burkholderiosis associated with acquired immunodeficiency due to autoantibody against gamma interferon. Clin Vaccine Immunol 17, 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR & Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76, 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Del Valle DM, Kim‐Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A et al. (2020) An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med 26, 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, Munoz‐Ruiz M, McKenzie DR, Hayday TS, Francos‐Quijorna I et al. (2020) A dynamic COVID‐19 immune signature includes associations with poor prognosis. Nat Med 26, 1623–1635. [DOI] [PubMed] [Google Scholar]
- 87. Galani IE, Rovina N, Lampropoulou V, Triantafyllia V, Manioudaki M, Pavlos E, Koukaki E, Fragkou PC, Panou V, Rapti V et al. (2021) Untuned antiviral immunity in COVID‐19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol 22, 32–40. [DOI] [PubMed] [Google Scholar]
- 88. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, Damoraki G, Gkavogianni T, Adami ME, Katsaounou P et al. (2020) Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe 27, 992–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li S, Jiang L, Li X, Lin F, Wang Y, Li B, Jiang T, An W, Liu S, Liu H et al. (2020) Clinical and pathological investigation of patients with severe COVID‐19. JCI Insight 5, e138070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang Z, Pan H & Jiang B (2020) Type I IFN deficiency: an immunological characteristic of severe COVID‐19 patients. Signal Transduct Target Ther 5, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS & Manson JJ (2020) COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Busnadiego I, Fernbach S, Pohl MO, Karakus U, Huber M, Trkola A, Stertz S & Hale BG (2020) Antiviral activity of type I, II, and III interferons counterbalances ACE2 inducibility and restricts SARS‐CoV‐2. MBio 11, e01928‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Novelli F & Casanova J‐L (2004) The role of IL‐12, IL‐23 and IFN‐γ in immunity to viruses. Cytokine Growth Factor Rev 15, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, Li W, Tong Q, Yi J, Zhao L et al. (2020) Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS‐CoV‐2 infected patients. EBioMedicine 55, 102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA & Mackall CL (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pestka S, Krause CD & Walter MR (2004) Interferons, interferon‐like cytokines, and their receptors. Immunol Rev 202, 8–32. [DOI] [PubMed] [Google Scholar]
- 97. McNab F, Mayer‐Barber K, Sher A, Wack A & O'Garra A (2015) Type I interferons in infectious disease. Nat Rev Immunol 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lazear HM, Schoggins JW & Diamond MS (2019) Shared and distinct functions of type I and type III Interferons. Immunity 50, 907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, McGovern N, Cant AJ, Hackett SJ, Ghazal P et al. (2013) STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA 110, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, Kula T, Drutman S, Belkaya S, Rattina V et al. (2018) Life‐threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med 215, 2567–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Duncan CJ, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC, Butler KM, Morfopoulou S, Brown JR, Hubank M et al. (2015) Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med 7, 307ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, Arkatkar T, Clough C, Dai X, Khim S et al. (2017) The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol 18, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE et al. (1997) Positional cloning of the APECED gene. Nat Genet 17, 393–398. [DOI] [PubMed] [Google Scholar]
- 104. Wolff AS, Sarkadi AK, Marodi L, Karner J, Orlova E, Oftedal BE, Kisand K, Olah E, Meloni A, Myhre AG et al. (2013) Anti‐cytokine autoantibodies preceding onset of autoimmune polyendocrine syndrome type I features in early childhood. J Clin Immunol 33, 1341–1348. [DOI] [PubMed] [Google Scholar]
- 105. Suit BE, Axelrod D, Moutsopoulos HM, Decker JL & Hooks JJ (1983) Detection of anti‐interferon antibodies in systemic lupus erythematosus. Clin Exp Rheumatol 1, 133–135. [PubMed] [Google Scholar]
- 106. Burbelo PD, Browne S, Holland SM, Iadarola MJ & Alevizos I (2019) Clinical features of Sjogren's syndrome patients with autoantibodies against interferons. Clin Transl Med 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ross C, Hansen MB, Schyberg T & Berg K (1990) Autoantibodies to crude human leucocyte interferon (IFN), native human IFN, recombinant human IFN‐alpha 2b and human IFN‐gamma in healthy blood donors. Clin Exp Immunol 82, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ansari R, Rosen LB, Lisco A, Gilden D, Holland SM, Zerbe CS, Bonomo RA & Cohen JI (2020) Primary and acquired immunodeficiencies associated with severe varicella‐zoster infections. Clin Infect Dis ciaa1274. 10.1093/cid/ciaa1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bastard P, Michailidis E, Hoffmann HH, Chbihi M, Le Voyer T, Rosain J, Philippot Q, Seeleuthner Y, Gervais A, Materna M et al. (2021) Auto‐antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J Exp Med 218, e20202486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang SY, Zhang Q, Casanova JL, Su HC; COVID Team (2020) Severe COVID‐19 in the young and healthy: monogenic inborn errors of immunity? Nat Rev Immunol 20, 455–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. van der Made CI, Simons A, Schuurs‐Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, van Deuren RC, Steehouwer M, van Reijmersdal SV, Jaeger M et al. (2020) Presence of genetic variants among young men with severe COVID‐19. JAMA 324, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pairo‐Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD et al.(2021) Genetic mechanisms of critical illness in Covid‐19. Nature 591, 92–98. [DOI] [PubMed] [Google Scholar]
- 113. Beccuti G, Ghizzoni L, Cambria V, Codullo V, Sacchi P, Lovati E, Mongodi S, Iotti GA & Mojoli F (2020) A COVID‐19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in Lombardy, Italy: letter to the editor. J Endocrinol Invest 43, 1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhou R, To KK, Wong YC, Liu L, Zhou B, Li X, Huang H, Mo Y, Luk TY, Lau TT et al. (2020) Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity 53, 864–877.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Arunachalam PS, Wimmers F, Mok CKP, Perera R, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak‐Yin Tsang O et al. (2020) Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science 369, 1210–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yao C, Bora SA, Parimon T, Zaman T, Friedman OA, Palatinus JA, Surapaneni NS, Matusov YP, Cerro Chiang G, Kassar AG et al. (2021) Cell‐type‐specific immune dysregulation in severely Ill COVID‐19 patients. Cell Rep 34, 108590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhu L, Yang P, Zhao Y, Zhuang Z, Wang Z, Song R, Zhang J, Liu C, Gao Q, Xu Q et al. (2020) Single‐cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID‐19 and influenza patients. Immunity 53, 685–696.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D et al. (2020) Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 181, 1036 – 1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L et al. (2020) Activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun 11, 3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, Qiu F, Wang X, Zou X, Wan D et al. (2020) Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID‐19 patients. Cell Host Microbe 28, 455–464.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier‐Gobeaux C et al. (2020) Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 369, 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Page C, Goicochea L, Matthews K, Zhang Y, Klover P, Holtzman MJ, Hennighausen L & Frieman M (2012) Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J Virol 86, 13334–13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK & Perlman S (2016) Dysregulated type I interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe 19, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Channappanavar R, Fehr AR, Zheng J, Wohlford‐Lenane C, Abrahante JE, Mack M, Sompallae R, McCray PB Jr, Meyerholz DK & Perlman S (2019) IFN‐I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest 129, 3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, Sauter D, Gifford RJ, USFQ‐COVID19 Consortium , Nakagawa S et al. (2020) SARS‐CoV‐2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep 32, 108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y & Ge XY (2020) The ORF6, ORF8 and nucleocapsid proteins of SARS‐CoV‐2 inhibit type I interferon signaling pathway. Virus Res 286, 198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, Cai JP, Jin DY, To KK, Chan JF et al. (2020) SARS‐CoV‐2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect 9, 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, Bhat P, Ollikainen N, Quinodoz SA, Loney C et al. (2020) SARS‐CoV‐2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G et al. (2020) Papain‐like protease regulates SARS‐CoV‐2 viral spread and innate immunity. Nature 87, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Moustaqil M, Ollivier E, Chiu HP, Van Tol S, Rudolffi‐Soto P, Stevens C, Bhumkar A, Hunter DJB, Freiberg AN, Jacques D et al. (2021) SARS‐CoV‐2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect 10, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, Kang D, Liu X, Gao C & Wang PH (2020) Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. Signal Transduct Target Ther 5, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, Pan P, Wu K & Wu J (2020) SARS‐CoV‐2 nucleocapsid protein interacts with RIG‐I and represses RIG‐mediated IFN‐beta production. Viruses 13, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hoagland DA, Moller R, Uhl SA, Oishi K, Frere J, Golynker I, Horiuchi S, Panis M, Blanco‐Melo D, Sachs D et al. (2021) Leveraging the antiviral type I interferon system as a first line of defense against SARS‐CoV‐2 pathogenicity. Immunity 54, 557–570.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hung IF‐N, Lung K‐C, Tso EY‐K, Liu R, Chung TW‐H, Chu M‐Y, Ng Y‐Y, Lo J, Chan J, Tam AR et al. (2020) Triple combination of interferon beta‐1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID‐19: an open‐label, randomised, phase 2 trial. Lancet 395, 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, Wang ZH, Tebbutt SJ, Kollmann TR & Fish EN (2020) Interferon‐alpha2b treatment for COVID‐19. Front Immunol 11, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Monk PD, Marsden RJ, Tear VJ, Brookes J, Batten TN, Mankowski M, Gabbay FJ, Davies DE, Holgate ST, Ho L‐P et al. (2021) Safety and efficacy of inhaled nebulised interferon beta‐1a (SNG001) for treatment of SARS‐CoV‐2 infection: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Respir Med. 9, 196–206. 10.1016/S2213-2600(20)30511-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lee S, Channappanavar R & Kanneganti TD (2020) Coronaviruses: innate immunity, inflammasome activation, inflammatory cell death, and cytokines. Trends Immunol 41, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ivanisenko NV, Seyrek K, Kolchanov NA, Ivanisenko VA & Lavrik IN (2020) The role of death domain proteins in host response upon SARS‐CoV‐2 infection: modulation of programmed cell death and translational applications. Cell Death Discov 6, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yap JKY, Moriyama M & Iwasaki A (2020) Inflammasomes and pyroptosis as therapeutic targets for COVID‐19. J Immunol 205, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, Goncalves AV, Perucello DB, Andrade WA, Castro R et al. (2021) Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J Exp Med 218, e20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, Shen J, Zhou Y, Shi ZL, Zhou P et al. (2020) SARS‐CoV‐2 triggers inflammatory responses and cell death through caspase‐8 activation. Signal Transduct Target Ther 5, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Liu C, Martins AJ, Lau WW, Rachmaninoff N, Chen J, Imberti L, Mostaghimi D, Fink DL, Burbelo PD, Dobbs K et al. (2021) Time‐resolved systems immunology reveals a late juncture linked to fatal COVID‐19. Cell 184, 1836–1857.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lévy R, Bastard P, Lanternier F, Lecuit M, Zhang SY & Casanova JL (2021) IFN‐α2a therapy in two patients with inborn errors of TLR3 and IRF3 infected with SARS‐CoV‐2. J Clin Immunol 41, 26–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bastard P, Lévy R, Henriquez S, Bodemer C, Szwebel TA & Casanova JL (2021) Interferon‐β therapy in a patient with incontinentia pigmenti and autoantibodies against type I IFNs infected with SARS‐CoV‐2. J Clin Immunol. [Epub a head of print]. 10.1007/s10875-021-01023-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rubin EJ, Longo DL & Baden LR (2021) Interleukin‐6 receptor inhibition in Covid‐19 ‐ cooling the inflammatory soup. N Engl J Med 384, 1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Investigators R‐C, Gordon AC, Mouncey PR, Al‐Beidh F, Rowan KM, Nichol AD, Arabi YM, Annane D, Beane A, van Bentum‐Puijk W et al. (2021) Interleukin‐6 receptor antagonists in critically Ill patients with Covid‐19. N Engl J Med 384, 1491–1502. 10.1056/NEJMoa2100433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Horby PW, Pessoa‐Amorim G, Peto L, Brightling CE, Sarkar R, Thomas K, Jeebun V, Ashish A, Tully R, Chadwick D et al. (2021). Tocilizumab in patients admitted to hospital with 3 COVID‐19 (RECOVERY): preliminary results of a 4 randomised, controlled, open‐label, platform trial. medRxiv [PREPRINT]. [Google Scholar]
- 148. Cremer PC, Abbate A, Hudock K, McWilliams C, Mehta J, Chang SY, Sheng CC, Van Tassell B, Bonaventura A, Vecchié A et al. (2021) Mavrilimumab in patients with severe COVID‐19 pneumonia and systemic hyperinflammation (MASH‐COVID): an investigator initiated, multicentre, double‐blind, randomised, placebo‐controlled trial. Lancet Rheumatol. [Epub a head of print]. 10.1016/S2665-9913(21)00070-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Taxonera C, Sagastagoitia I, Alba C, Manas N, Olivares D & Rey E (2020) 2019 novel coronavirus disease (COVID‐19) in patients with inflammatory bowel diseases. Aliment Pharmacol Ther 52, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bonaventura A, Vecchié A, Wang TS, Lee E, Cremer PC, Carey B, Rajendram P, Hudock KM, Korbee L, Van Tassell BW et al. (2020) Targeting GM‐CSF in COVID‐19 pneumonia: rationale and strategies. Front Immunol 11, 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Clark KEN, Nevin WD, Mahungu T, Lachmann H & Singh A (2020) Assessment of the haemophagocytic lymphohistiocytosis HScore in patients with COVID‐19. Clin Infect Dis. [Epub a head of print]. 10.1093/cid/ciaa1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lorenz G, Moog P, Bachmann Q, La Rosee P, Schneider H, Schlegl M, Spinner C, Heemann U, Schmid RM, Algul H et al. (2020) Title: cytokine release syndrome is not usually caused by secondary hemophagocytic lymphohistiocytosis in a cohort of 19 critically ill COVID‐19 patients. Sci Rep 10, 18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Tomazini BM, Maia IS, Cavalcanti AB, Berwanger O, Rosa RG, Veiga VC, Avezum A, Lopes RD, Bueno FR, Silva M et al. (2020) Effect of dexamethasone on days alive and ventilator‐free in patients with moderate or severe acute respiratory distress syndrome and COVID‐19: the CoDEX Randomized clinical trial. JAMA 324, 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. WHO Rapid Evidence Appraisal for COVID‐19 Therapies (REACT) Working Group , Sterne JAC, Murthy S, Diaz JV, Slutsky AS, Villar J, Angus DC, Annane D, Azevedo LCP, Berwanger O et al. (2020) Association between administration of systemic corticosteroids and mortality among critically Ill PATIENTS With COVID‐19: a meta‐analysis. JAMA 324, 1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hoang TN, Pino M, Boddapati AK, Viox EG, Starke CE, Upadhyay AA, Gumber S, Nekorchuk M, Busman‐Sahay K, Strongin Z et al. (2021) Baricitinib treatment resolves lower‐airway macrophage inflammation and neutrophil recruitment in SARS‐CoV‐2‐infected rhesus macaques. Cell 184, 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, Marconi VC, Ruiz‐Palacios GM, Hsieh L, Kline S et al. (2021) Baricitinib plus remdesivir for hospitalized adults with Covid‐19. N Engl J Med 384, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Bronte V, Ugel S, Tinazzi E, Vella A, De Sanctis F, Cane S, Batani V, Trovato R, Fiore A, Petrova V et al. (2020) Baricitinib restrains the immune dysregulation in patients with severe COVID‐19. J Clin Invest 130, 6409–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Barros I, Silva A, de Almeida LP & Miranda CO (2021) Mesenchymal stromal cells to fight SARS‐CoV‐2: taking advantage of a pleiotropic therapy. Cytokine Growth Factor Rev 58, 114–133. [DOI] [PMC free article] [PubMed] [Google Scholar]