

Abstract
Recently, accumulating evidence has highlighted the role of endothelial dysfunction in COVID‐19 progression. Coronary microvascular dysfunction (CMD) plays a pivotal role in cardiovascular disease (CVD) and CVD‐related risk factors (eg, age, gender, hypertension, diabetes mellitus, and obesity). Equally, these are also risk factors for COVID‐19. The purpose of this review was to explore CMD pathophysiology in COVID‐19, based on recent evidence. COVID‐19 mechanisms were reviewed in terms of imbalanced renin‐angiotensin‐aldosterone‐systems (RAAS), systemic inflammation and immune responses, endothelial dysfunction, and coagulatory disorders. Based on these mechanisms, we addressed CMD pathophysiology within the context of COVID‐19, from five perspectives. The first was the disarrangement of local RAAS and Kallikrein‐kinin‐systems attributable to SARS‐Cov‐2 entry, and the concomitant decrease in coronary microvascular endothelial angiotensin I converting enzyme 2 (ACE2) levels. The second was related to coronary microvascular obstruction, induced by COVID‐19‐associated systemic hyper‐inflammation and pro‐thrombotic state. The third was focused on how pneumonia/acute respiratory distress syndrome (ARDS)‐related systemic hypoxia elicited oxidative stress in coronary microvessels and cardiac sympathetic nerve activation. Fourthly, we discussed how autonomic nerve dysfunction mediated by COVID‐19‐associated mental, physical, or physiological factors could elicit changes in coronary blood flow, resulting in CMD in COVID‐19 patients. Finally, we analyzed reciprocity between the coronary microvascular endothelium and perivascular cellular structures due to viremia, SARS‐CoV‐2 dissemination, and systemic inflammation. These mechanisms may function either consecutively or intermittently, finally culminating in CMD‐mediated cardiovascular symptoms in COVID‐19 patients. However, the underlying molecular pathogenesis remains to be clarified.
Keywords: coronary microvascular dysfunction, COVID‐19, SARS‐CoV‐2
Abbreviations
- ACE2
Angiotensin I converting enzyme 2
- Ang II
Angiotensin II
- Ang(1–7)
Angiotension 1–7
- AT1R
Angiotensin II type 1 receptor
- BNP
Brain natriuretic peptide
- CMD
Coronary microvascular dysfunction
- COVID‐19
Coronavirus disease 2019
- CVD
Cardiovascular disease
- DM
Diabetes mellitus
- eNOS
endothelial nitric oxide synthase
- HTN
Hypertension
- ICU
Intensive care unit
- NOX‐2
NADPH oxidase 2
- RAAS
Renin‐angiotensin‐aldosterone system
- ROS
Reactive oxygen species
- VWF
von Willebrand Factor
1. INTRODUCTION
At the end of 2019, coronavirus disease 2019 (COVID‐19) was defined as a global emergency by the World Health Organization. 1 The disease went worldwide within months and was transmitted and exacerbated by international travel and human‐to‐human contact. 2 COVID‐19 has demonstrated a wide spectrum of clinical manifestations in asymptomatic to critically ill patients, ranging from severe pneumonia, respiratory failure, cardiovascular events, diffuse intravascular coagulation, multi‐organ failure, sepsis, septic shock, and death. 3 , 4 Moreover, elderly males (>65 years) with underlying CVD, for example, hypertension, myocardial ischemia, heart failure, and arrhythmias, have been confirmed as independent risk factors for disease severity and in‐hospital mortality. 5 , 6
Prior epidemiological statistics from China reported that gross case fatality percentages for different comorbidities were as follows: 10.5% for established CVD, 7.3% for diabetes mellitus (DM), 6.0% for hypertension (HTN), 6.3% for respiratory disease, and 5.6% for cancer. 7 Furthermore, a recent meta‐analysis recruited 49,076 confirmed COVID‐19 cases, indicating that patients with pre‐existing CVD risk factors (eg, DM and HTN) and those with established CVD were more vulnerable to SARS‐CoV‐2, with a higher risk of developing severe disease. 8 Additionally, autopsy findings from Germany showed that the majority (85%) of the deceased patients had established CVD, followed by lung disease (55%), kidney disease (34%), central nervous system (CNS) disease (35%), and DM (21%). 9 Coronary microvascular dysfunction (CMD) is closely related to these co‐morbidities.
COVID‐19 is caused by a novel β‐coronavirus SARS‐CoV‐2, which is a single‐stranded RNA virus of 60‐140nm in diameter. The virus shares approximately 79% sequence identity to SARS‐CoV, which was responsible for another epidemic in 2003. 10 , 11 , 12 The ACE2 receptor is well documented to facilitate SARS‐CoV‐2 entry into the human body. Emerging clinical evidence has shown that COVID‐19 is implicated in renin‐angiotensin‐aldosterone‐system (RAAS) dysregulation, hyper‐inflammation, and coagulatory dysfunction. 13 , 14 , 15 However, further evidence has also underscored the importance of microvascular endothelial dysfunction in pathophysiology. Microvascular endothelial dysfunction is an important mechanism of microvascular dysfunction. SARS‐CoV‐2 has been proposed to infect blood vessels after lung incursion, thereby inducing vascular endothelial injury, activating hemostasis and coagulation, ultimately leading to thrombotic disorders in COVID‐19 patients. 16 , 17 The surface area of the microvascular endothelium in the human circulation is greater than that of the macrovascular endothelium, and the microvascular endothelium is the primary barrier for blood‐tissue exchange. 18 , 19 In addition, any microstructural (eg, microvascular remodeling, capillary density changes) or functional alterations of microvessels (eg, in endothelium, vascular smooth muscle cells) can affect organ perfusion and metabolism, resulting in direct organ injury. 20
Like other microcirculatory‐dependent organs, heart hemodynamics and metabolic homeostasis are regulated by coronary microcirculation. 21 Moreover, CMD primarily presents as chest pain syndrome, accounting for myocardial ischemia in some cases, irrespective of the presence/absence of obstructive epicardial coronary vessels. 22 During SARS‐CoV‐2 infection, manifestations of cardiovascular dysfunction include acute coronary syndrome, myocarditis, pericarditis, heart failure, and arrhythmias, 23 with their incidences remaining obscure. Thus, the accumulating evidence suggests a dysfunctional role for the coronary microvasculature during SARS‐CoV‐2 infections. 24 , 25 , 26 , 27 In this review, we would discuss COVID‐19 mechanisms in terms of imbalanced renin‐angiotensin‐aldosterone systems (RAAS), systemic inflammation and immune responses, endothelial dysfunction, and coagulatory disorders. Based on these mechanisms, we will address the CMD pathogenesis within the context of COVID‐19, from five perspectives, according to existing evidence.
2. COVID‐19 PATHOPHYSIOLOGICAL MECHANISMS
2.1. RAAS imbalance
RAAS is an important hormonal system that modulates blood pressure and maintains fluid homeostasis. However, localized RAAS over‐activation in organs and tissues augments cell growth, facilitating the proliferation and inflammation of organs and tissues. 27 , 28 A major component of RAAS is the glycoprotein metalloprotease, angiotensin I converting enzyme 2 (ACE2), which catalyses multiple substrates, including kinins, apelin, neurotension, dynorphin, ghrelin, angiotensins, and des‐Arginin9‐bradykinin. 29 , 30 ACE2 mRNA is deemed to be expressed in all organs, whereas the ACE2 protein is abundantly expressed in the epithelium of the lungs and small intestine, endothelial cells of arteries and veins, arterial smooth muscle cells, cardiomyocytes, and adipocytes. 31 , 32
ACE2 downregulation stimulates a variety of ACE2‐associated pathways in the lungs, including the Kallikrein‐kinin system, which controls vascular permeability and vasodilatation, and assists RAAS with blood pressure regulation. Kallikrein‐kinin system is also involved in endothelial dysfunction, via the production of reactive oxygen species (ROS). 33 However, pulmonary RAAS is most frequently disturbed by ACE2 dysfunction. As ACE2 levels decrease, hyperactivity of angiotensin II (Ang II)/angiotensin II type 1 receptor (AT1R) axis occurs, whereas hypoactivity of ACE2/angiotensin 1–7 (Ang(1–7))/Mas receptor axis also causes detrimental effects (eg, vasoconstriction, hypertrophy, fibrosis, proliferation, and increased ROS). 34 , 35 , 36
Increasing Ang II levels also elevates aldosterone levels. 37 This hormone is important for fluid maintenance as it promotes water and sodium retention and potassium excretion, leading to systemic electrolyte disturbance and arrhythmias. Hyper‐aldosteronemia causes hypokalemia in COVID‐19 patients. 38 However, Ang II elevation also triggers immune responses, promoting coagulatory and thrombosis states, and induces endothelial cells expressing tissue factors and plasminogen activator inhibitor‐1 (PAI‐1) via AT1R, resulting in imbalanced PAI‐1/tissue‐plasminogen activator, facilitating the hyper‐coagulatory state. 17
2.2. Systemic inflammation and immune responses
It is well acknowledged that inflammation is closely related to immune responses. Non‐specific immune cells such as phagocytes, dendritic cells, and natural killer cells express pattern recognition receptors which identify pathogen‐associated or danger‐associated molecular patterns. The protective effects of type I interferon (interferon‐α and interferon‐β) are potentially antagonized by SARS‐CoV (or SARS‐CoV‐2) via pattern recognition receptors. This leads to the release of danger‐associated molecular patterns and cytokines from infected cells, excessive monocyte/macrophage expression and polymorphonuclear neutrophil infiltration and T‐cell apoptosis, resulting in a hyper‐inflammatory state. 17
Immune responses appear to benefit the host hyper‐inflammatory state during the early stages of SARS‐CoV‐2 infection. It has been reported that plasma IL‐6 levels are significantly increased in critically ill COVID‐19 patients, whereas CD4+ T, CD8+ T cells, and natural killer cells are decreased, indicating an immunosuppressive state (S. Wan, unpublished observations). With the exacerbation of infection, cytokine release becomes dysregulated, and the immune system becomes suppressed, generating secondary hemophagocytic lymphohistiocytosis. This state is characterized by fulminant cytokine release, primarily driven by viral infection, and reported in approximately 3.7–4.3% of sepsis cases. 39 , 40 A recent retrospective study demonstrated that levels of IL‐1β, IL‐6, IL‐8, and soluble tumor necrosis factor receptor 1 were positively correlated to COVID‐19 severity, respectively. 41 The release of IL‐6 and granulocyte‐macrophage colony‐stimulating factor by T lymphocytes and monocytes, respectively, was concluded as central to viral‐induced cytokine syndrome. 42
2.3. Endothelial dysfunction and coagulatory disorders
Autopsy and/or biopsy findings have implicated endothelial dysfunction as a key component in COVID‐19 pathogenesis. 15 , 43 , 44 It is speculated that when SARS‐CoV‐2 invades the host, immune responses are triggered, causing pro‐inflammatory cytokine release, leading to a pro‐inflammation state. This causes acute injury to both epithelial and microvascular endothelial cells. 45 Upon endothelial cell edema and apoptosis, and concomitant permeability in alveolar microvessels, SARS‐CoV‐2 enters the blood stream, causing vascular RAAS derangement, invoking viremia or sepsis, finally culminating in multi‐organ damage. SARS‐CoV‐2 entry into the blood stream is key to its dissemination throughout the host, with the heart believed to be the first destination of pulmonary circulation outflow. 46 Sepsis is defined as severe endothelial dysfunction syndrome and is generated in response to endo‐ and exo‐vascular infections. The infections may cause reversible or irreversible injury to the microcirculation, finally leading to septic shock, multiple organ failure, and even death. 47 About 20–35% of COVID‐19 patients were with septic shock. 48 Microvasculature dysfunction is an important characteristic in septic shock. Cardiogenic shock, from imbalanced oxygen supply and demands of the heart, is also reported in COVID‐19 patients. 49
Overexpressed monocytes/macrophages and polymorphonuclear neutrophils can derive tissue factors, neutrophils, and extracellular traps, respectively, triggering the endo‐ and exo‐genetic coagulation pathway and causing imbalance between inflammation and coagulation. The complement system is an essential component of the innate immune system and is activated mainly via three pathways (ie, canonical, alternative, and lectin pathways). The activation of the complement system induces a cascade of events, such as the generation of a variety of bioactive molecules, including C3a, C5a, and membrane attack complex, resulting in coagulatory dysfunction and thrombotic vasculopathy. 17 Buja and colleagues described that pulmonary microthrombi were frequently observed in deceased COVID‐19 patients at autopsy, with pulmonary thromboembolism as a common fatal complication. 50 Electron micrographs of the lung tissues from COVID‐19 patients revealed deformed and elongated capillaries, and damaged endothelial ultrastructure with the presence of intracellular SARS‐CoV‐2 viruses. 51 Diffuse alveolar damage (characterized by endothelial cell damage, capillary leakage, activation of type II pneumocytes, formation of hyaline membranes, and accumulation of pulmonary macrophages) was confirmed at autopsy. Additionally, capillary endotheliitis and microthrombi were also identified in alveolar capillaries and small pulmonary vessels, authenticating pulmonary thrombotic microangiopathy. 52 , 53 Ceceri et al. proposed that diffuse alveolar endothelial damage, leading to progressive endothelial pulmonary syndrome with microvascular thrombosis, could account for the COVID‐19 pathogenesis in patients with ARDS. 54
Coagulation dysfunction, as characterized by elevated levels of D‐dimers and fibrinogen degradation products, is associated with COVID‐19 morbidity and mortality. D‐dimer levels are independent risk factors and a prognostic marker for COVID‐19 infection. 55 Elevated D‐dimer levels, together with prolonged prothrombin times and thrombocytopenia in COVID‐19 patients, indicate the occurrence of disseminated intravascular coagulation (DIC). 14 , 56 Approximately 71.4% of DIC was reported in deceased COVID‐19 patients. 57 This condition may result from activated monocytes/macrophages and endothelial cells, due to the release of cytokines, tissue factors, and von Willebrand factor (VWF, major protein component in Weibel‐Palade bodies in the endothelium). COVID‐19 patients reportedly presented with elevated VWF suggestive of massive endothelial stimulation and damage, and platelet activation. 58 A recent cross‐sectional study revealed a significant increase in VWF antigens and soluble P‐selectin (a marker of stimulated endothelial cells and platelets). In addition, soluble thrombomodulin (a specific marker of endothelial cell activation) was also shown to be related to the in‐hospital mortality of COVID‐19 patients. These observations demonstrated endotheliopathy could be implicated in COVID‐19 patients. 59
3. THE ROLE OF COVID‐19 IN CMD
3.1. The relationship between COVID‐19 and CMD
Myocardial injury has been observed in severe COVID‐19 patients requiring intensive care. According to prior reports, the dramatic elevation of troponin (a marker of acute myocardial injury) in COVID‐19 patients may serve as an additional sensitive prognostic predictor associated with COVID‐19 severity and mortality. 60 , 61 However, Atallah et al stated that troponin levels should not be used as an isolated indicator of myocardial injury, as such elevations could be related to non‐coronary mechanisms, 62 which was supported by cumulative findings in deceased COVID‐19 patients with prior elevation of troponin levels in clinical scenario but mild pathological myocardial changes at autopsy. 50
However, Vrsalovic and co‐workers highlighted the role of coronary small‐vessel disease in troponin elevation, due to a pro‐thrombotic state and endothelial dysfunction induced by SARS‐CoV‐2. Troponin elevation was also identified in patients with previous asymptomatic CAD but subsequent type 2 myocardial infarction, as well as patients with cytokine storm‐induced myocardial injury and myocarditis attributable to SARS‐CoV‐2 virus. 63
Cardiovascular injury during a COVID‐19 infection could lead to heart failure, as characterized by elevated brain natriuretic peptide (BNP) or N‐terminal pro‐BNP (NT‐proBNP) levels. These elevations are associated with the increased in‐hospital mortality of COVID‐19 patients. 64 , 65 The ACE2 distribution would alter in the case of pathologic conditions. Guo et al. observed the ACE2 expression chiefly in the endothelial and smooth muscle cells in fibrotic lung tissues, but in type 2 alveolar epithelial cells (AT2) in normal lungs. During heart failure, ACE2 expression is detected in cardiomyocytes, rather than arterial vascular cells in the normal heart. These findings suggest that SARS‐CoV‐2 may invade the bloodstream by damaging alveolar systems and proceed into the heart. 46
SARS‐CoV‐2 virus particles are generally spherical with some pleomorphism under electron microscopy, with a varied diameter (60–140 nm) and quite distinctive spikes (about 9–12 nm), and render virions a solar corona appearance. This morphology is consistent with the Coronaviridae family. 66 Owing to the diameter of SARS‐CoV‐2 virus of approximately 60–140 nm, such viruses can presumably invade coronary microcirculation to cause CMD. Consistent with the conjecture, Fox and colleagues detected viral particles that were in accordance with the SARS‐CoV‐2 virus, in a coronary endothelial cell, by electron microscopy. 67 However, evaluation for SARS‐CoV‐2 infection by electron microscopy is often unreliable as normal cellular structures are often reported as being virus. In addition, in two recent studies, endothelial cell infection by SARS‐CoV‐2 in the heart was validated by means of in situ hybridization and in the setting of microvascular thrombosis. 68 , 69 This provides potent evidence of coronary microvascular involvement in COVID‐19, indicating direct coronary small vessel injury. Despite the lack of consensus as to the CMD definition to date, it was described as perturbation of the function of coronary small‐vessels or irregular coronary microvascular resistance on account of injured coronary perfusion and detrimental coronary blood flow. 70 Four major types of CMD have been classified, the mechanisms of which include luminal obstruction, vascular remodeling, endothelial dysfunction, smooth muscle cell dysfunction, automatic dysfunction, and extravascular compression (Table 1). 71
TABLE 1.
Classification and pathogenic mechanisms of CMD
| Type | Underlying clinical condition | Pathogenic mechanism |
|---|---|---|
| Type 1 (Primary): CMD in the absence of CAD and myocardial disease | Traditional coronary risk factors (smoking, HTN, hyperlipidemia, diabetes, and insulin‐resistant states) |
Structural Vascular remodeling Functional Endothelial dysfunction SMC dysfunction |
| Type 2: CMD in the presence of myocardial disease |
Primary cardiomyopathies Hypertrophic cardiomyopathy Dilated cardiomyopathy Secondary cardiomyopathies Cardiomyopathy secondary to Anderson‐Fabry's disease Myocardial amyloidosis Myocarditis Aortic stenotic cardiomyopathy |
Structural Vascular remodeling Luminal obstruction Functional SMC dysfunction Extravascular Extramural compression |
| Type 3: CMD in the presence of obstructive CAD |
Stable CAD Acute coronary syndromes with or without ST‐segment elevation |
Structural Luminal obstruction Functional Endothelial dysfunction SMC dysfunction |
| Type 4: Iatrogenic CMD |
Reperfusion injury (No‐reflow phenomenon/ micro‐embolization) PCI‐related micro‐embolization Coronary artery grafting Heart transplantation |
Structural Luminal obstruction Functional Autonomic dysfunction |
Abbreviations: CAD, coronary artery disease; CMD, coronary microvascular dysfunction; HTN, hypertension; PCI, percutaneous coronary intervention; SMC, smooth muscle cell.
3.2. Age, gender, CMD, and COVID‐19
Cardiovascular symptoms are frequently observed in elderly COVID‐19 patients as well as those with chronic diseases such as HTN, DM, established CVD, and chronic obstructive pulmonary disease. 72 Critically, both aging and the gender of male per se are confirmed as independent risk factors for COVID‐19 and are associated with infection‐related endothelial dysfunction. 5 , 6 , 73 With increasing age, coronary arteries decline in endothelial function and are characterized by reduced coronary flow reserves, and a diminution in vascular reactivity, however, baseline flow capacity remains intact. 74 , 75 In addition, diminished cardiomyocytic population and cardiomyocytic hypertrophy were identified in aging murine heart, with reduction of capillary density and remodeling of microvessels, in compensation for the increased metabolic demand. 76
Testosterone plays a negative role in the activation, survival, and differentiation of B cells (which is closely related to acquired immunity in humans), whereas estrogen plays a beneficial role. Elderly men were more predisposed to atherosclerosis. 77 Myocardiocytic senescence in healthy men often develops a decade earlier than in women, in contrast to the more rapid decline in vascular reactivity in postmenopausal women than their male counterparts. Estrogen receptor signaling exerts a protective role in female mice upon infection. 70 , 78 However, the majority of CMD patients are peri‐ and postmenopausal women. These populations were more vulnerable to cardiovascular risk factors when compared with younger women, with similar or higher CVD incidence in postmenopausal women versus the males in the same age group. 79 , 80 A plausible explanation would be the reduction in estrogen levels and impairment of endothelial function in peri‐menopausal women, attributable to interrupted nitric oxide generation, augmented oxidative stress, and increased pro‐inflammatory cytokines. 70
Estrogen also activates endothelial nitric oxide synthase (eNOS) via the PI3K/Akt signaling pathway, thereby producing nitric oxide and dilating coronary arteries. 81 , 82 Moreover, estrogen replacement therapy has been reported to increase coronary blood flow, and reverse detrimental changes in coronary resistance vessels. 83 In addition, estrogen could serve as an up‐regulator of angiotensinogen, while downregulating renin, angiotensin‐converting enzyme, and angiotensin II type 1 receptor. 84 These molecules are important for RAAS regulation, while RAAS per se plays a critical part in HTN, CAD, and HF progression. SARS‐CoV‐2 invades the cell via ACE2 receptor. However, the ACE2 gene is an X‐located gene, and ACE2 protein mainly modulates cardiac homeostasis (including cytobiology of cardiomyocytes, cardiac fibroblasts, and coronary endothelial cells) via RAAS. 85 , 86
With age advance, both men and women suffer from decreased immunity, which facilitates SARS‐CoV‐2 infection. Similarly, changes in coronary microvessels mediated by senescence and declining gonadal hormone levels, generate a declining health status, which may predispose to a worsened COVID‐19 prognosis in those with CVD or CVD risk factors.
3.3. Potential implications of COVID‐19 in CMD
Cardiovascular metabolic risk factors (ie, HTN, DM, obesity, and hypercholesterolemia) and CVD are all related to CMD. 79 Responses of microvascular to cardiovascular risk factors include endothelial barrier dysfunction, oxidative stress, vasomotor function impairment, microvessel density alteration, leukocyte‐endothelial adhesion, and platelet recruitment or thrombosis, as well as microvascular remodeling. 87 , 88 , 89 , 99 Chronic heart diseases can promote CMD progression. Conversely, CMD may also facilitate hypertrophy, fibrosis of cardiomyocytes as well as microvascular rarefaction in chronic heart diseases. 91 Given the prominence of HTN, DM, obesity, and CVD in COVID‐19 comorbidities, chronic CMD may play important roles during COVID‐19 progression. 92 , 93
Coronary microvessels may be directly injured by SARS‐CoV‐2 second wave infection (eg, viremia, hyper‐inflammation, ARDS‐induced hypoxia, RAAS imbalance, and automatic or sympathetic nerve activation), or indirectly by damage to perivascular cells (eg, cardiomyocytes edema and/or pericyte injury). However, the resultant or consequent CMD would be a co‐consequence of direct and indirect factors secondary to SARS‐CoV‐2 infection.
Coronary microvascular dysfunction may contribute to viremia, as part of systemic endothelial dysfunction. SARS‐CoV‐2 RNA has been confirmed to be present in coronary microvessels in a small number of studies and has also been detected in blood as well. 94 Observation of overtly elevated biomarkers such as VWF, D‐dimer, and fibrinogen degradation products suggested endothelial activation and hypercoagulation state. In general, the evidence above strongly supports the likelihood of endothelial damage, as ACE2 receptors are also expressed in endothelial cells. 58 , 94 , 95 , 96 Hence, we might postulate that subsequent to primary attacks of the pulmonary epithelial cells and damages to the perialveolar structure including the epithelial‐endothelial barrier, SARS‐CoV‐2 enters the blood circulation, including the heart and coronary microvessels. The resulting viremia induces coronary microvascular endothelial injury, including endothelial cell activation, endothelial structural, and functional changes, thereby resulting in hypercoagulation, microthrombosis, and myocardial injury. Therefore, this evidence suggests that viremia/sepsis may contribute to the COVID‐19 mechanisms in CMD.
3.3.1. Decreased coronary microvascular endothelial ACE2 levels
It has been shown that decreased local ACE2 levels in coronary microvessels led to increased AngII/Ang(1–7) ratios, resulting in a RAAS imbalance in coronary small‐vessels. As AngII/Ang(1–7) ratios increase, the Ang II‐AT1R pathway in the coronary small vasculature becomes overactive. It is accepted that AT1R activation induces vascular constriction, aldosterone production and release (followed by electrolyte disturbance, especially hyperkalemia, which is a mechanism in ventricular arrhythmia), vasopressin increase and cardiac hypertrophy, and autonomic nerve dysfunction. 97 Similarly, imbalanced Ang II/Ang(1–7) decreases eNOS levels, which function as vasodilators and tissue factor downregulators, thereby increasing NOX‐2 activity and promoting ROS production. 98 Additionally, the bradykinin system is responsible for vascular permeability and vasodilatation, and once activated, it would exacerbate endothelial dysfunction, thereby increasing ROS production.
In short, CMD is generated by the disarrangement of local RAAS and Kallikrein‐kinin‐system, resulting in coronary microvascular constriction, eNOS/NOX‐2 imbalance, increased ROS, and vascular permeability.
3.3.2. Obstruction by atherosclerotic fragments and microthrombi
Upon internalization and duplication of SARS‐CoV‐2 in epithelial cells, multiple cytokines and chemokines, for example, IL‐6, INF‐γ, and monocyte chemotactic protein 1, are released into the blood stream. As ACE2 levels decrease, vascular permeability increases, and vasodilation capacity decreases, due to the imbalanced bradykinin system. Systemic inflammation increases coronary blood flow, resulting in the activation and rupture of pre‐existing atherosclerotic plaques, causing type 1 myocardial infarction. 99 Moreover, inflammation reduces coronary flow reserves and elevates microvascular resistance indices in patients with coronary syndrome X. 100 Recently, autopsy findings frequently validate a mass of microthrombi in coronary small arteries, which is strongly indicative of coronary microvascular injury. 101 , 102 Also, Tedeschi and colleagues reported a case of acute massive coronary thrombosis in a COVID‐19 patient (aged 60 years, male) without pre‐existing coronary plaque (confirmed by coronary angiography), wherein inflammatory and pro‐thrombotic markers were normal on admission but elevated overtly, indicating the contribution of the pro‐inflammatory and pro‐thrombotic state from COVID‐19 to coronary thrombosis. 103 However, despite the cardiac procedures in the patient, the Thrombolysis in Myocardial Infarction flow and Myocardial Blush Grade were still poor (grade 1–2 and 0, respectively), and eventually the patient deceased. Therein, CMD resulting from SARS‐CoV‐2‐mediated inflammation and coagulation disorder, and no‐flow phenomenon caused by microthrombi, might play a key role in the progression of coronary artery lesions. 103
In general, CMD can be caused by atherosclerotic fragments and microthrombi generated by systemic hyper‐inflammation and pro‐thrombotic state in coronary artery.
3.3.3. Oxidative stress due to pneumonia/ARDS‐related systemic hypoxia
CMD could also result from oxidative stress in coronary microvessels and activation of cardiac sympathetic nerves by pneumonia/ARDS‐related systemic hypoxia. Pneumonia/ARDS‐associated systemic hypoxia triggers oxidative stress in endothelial cells or myocardiocytes, stimulating several pathways for myocardium‐vascular supply and demand mismatch, leading to type 2 myocardial infarction. 65 ST‐elevated myocardial infarction, without obstructive coronary artery disease, has been observed in 39.3% of COVID‐19 patients who required urgent angiography. 104
Oxidative stress also augments sympathetic tone, mediating catecholamine elevation in the blood. 105 Sympathetic activation is negatively correlated with coronary blood flow velocity (index of coronary microvascular function) in patients with hyperglycemia and atherosclerotic risk factors, indicating a causative role of sympathetic activation in CMD progression. 106 The inference is that coronary microvascular oxidative stress induced by SARS‐CoV‐2 infection increases the sympathetic tone, thereby mediating catecholamine release and leading to CMD and consequently, myocardial toxicity results, finally leading to myocardial injury and ischemia in COVID‐19 patients.
3.3.4. Autonomic nerve dysfunction
Sympathetic control of coronary vasomotor tone and coronary blood flow is functionally significant in patients with endothelial dysfunction. Increased tone in sympathetic adrenergic nerves leads to the vasoconstriction of coronary vasculature and oxygen demand of cardiomyocytes. 107 However, cardiac sympathetic nerves are not only activated by oxidative stress, but also by other factors. DM is a risk factor for COVID‐19, and chronic hyperglycemia affects cardiac micro‐environment, resulting in CMD. 108 Cardiac autonomic dysfunction is related to cardiomyopathy and DM‐induced myocardial ischemia. 106 , 109 In addition, several reports on Takotsube cardiomyopathy in SARS‐CoV‐2 positive patients have highlighted the role of autonomic dysfunction, triggered by mental (ie, fear, anxiety, grief), physical, or physiological stress in COVID‐19‐associated CMD. 24 , 25 , 26 , 110
In summary, CMD in COVID‐19 may result from autonomic nerve dysfunction mediated by COVID‐19‐related emotional, physical or physiological factors.
3.3.5. Compromise of perivascular cells in coronary microvasculature
Compromised perivascular cells (ie, myocytes, pericytes, and adipocytes) also elicit coronary microvascular endothelial dysfunction. A recent study showed that engineered human capillary organoids consisting of endothelial cells and platelet‐derived growth factor receptor β‐positive pericytes can be infected with SARS‐CoV‐2, whereas the administration of recombinant human ACE2 has been shown to be effective against the infection. 12 Pericytes or perivascular cells envelop the endothelial layers of microvessels in the body, including the heart, and maintain the tone and integrity of microvasculature, as well as promoting angiogenesis. With an endothelial cell/pericyte ratio of 2:1–3:1, and a density of approximately 3.6 × 107 pericytes/cm3, pericytes are the second most common myocardial cell type in the heart, followed by cardiomyocytes. 111
In pericyte‐deficient murine models, the production and release of VWF from microvascular endothelial cells are augmented, facilitating platelet aggregation and formation of a hypercoagulation state, resulting in thrombotic microangiopathy. He et al. observed pericyte‐specific ACE2‐expression, with absent ACE2 expression in endothelial cells, perivascular macrophages, and fibroblasts, suggesting a modulatory role for pericytes in endothelial cells responding to thrombotic risk. Accordingly, a “COVID‐19‐pericyte hypothesis” was proposed: as the microvascular endothelial barrier is damaged, SARS‐CoV‐2 leaks out and infects pericytes. This enhances VWF production in neighboring endothelial cells and enhances platelet aggregation and fibrin deposition, eventually leading to vascular symptoms in some COVID‐19 patients (L. He, unpublished observations).
In postmortem histological analysis, Varga et al. observed apoptotic cell bodies in tissue endothelium, including the lungs, heart, and small intestine. Their findings confirmed the presence of endotheliitis, which indicated direct viral injury of microvessels at different vascular beds. By electron microscopy, the authors observed viral particles in the endothelium, plus evidence of myocardial infarction rather than myocarditis. 44 However, evaluation for SARS‐CoV‐2 infection by electron microscopy is often unreliable as normal cellular structures are often misidentified as viruses. Recently, two studies have shown endothelial cell infection by SARS‐CoV‐2 in the heart by means of in situ hybridization. 68 , 69 In addition, although rarely presented, viral particles were detected in the cardiomyocytes adjacent to myofibrils in a case from Italy. 112 Several other studies also reported SARS‐CoV‐2 RNA in the heart without describing the specific location. 94 , 113 , 114 , 115
The SARS‐CoV‐2 virus is transported from the blood stream, through the endothelial barrier to perivascular tissues such as pericytes, myocytes, and epicardial adipocytes (which share the same microcirculation with myocardiocytes 116 ) due to the endothelial apoptosis and increased vascular permeability and leakage in the context of hyper‐inflammation and immune dysregulation, rendering them to viral infection, causing perivascular tissues edema, which in turn oppresses the microvessels, resulting in CMD. Epicardial adipocytes are also involved in the modulation of endothelial function, coagulation and inflammation, and secrete harmful inflammatory factors during pathologic conditions. 116
In a word, dissemination of SARS‐CoV‐2 into perivascular cells of coronary microvessels via ongoing viremic blood stream and inflammation‐induced endothelial apoptosis causes perivascular structural cell edema. These edematous cells in return oppress coronary microvasculature, causing, or intensifying CMD. These mechanisms can aggravate pre‐existing CMD, or induce new‐onset CMD, thereby worsening heart conditions, causing different cardiovascular manifestations in some COVID‐19 patients.
4. CONCLUSIONS
A priori, these CMD mechanisms in COVID‐19 can be summarized as Figure 1 shows:
Decreased coronary microvascular endothelial ACE2 levels attributed to SARS‐Cov‐2 entry causes disarrangement of local RAAS and Kallikrein‐kinin‐systems. This results in coronary microvascular constriction, eNOS/NOX‐2 imbalance, and vascular permeability increase, therefore, leading to CMD.
Atherosclerotic fragments and microthrombi induced by COVID‐19‐associated systemic hyper‐inflammation and pro‐thrombotic state, can obstruct coronary microvessels, causing CMD.
Pneumonia/ARDS‐related systemic hypoxia can elicit oxidative stress in coronary microvessels, inducing myocardial‐vascular supply and demand mismatch, activating cardiac sympathetic nerves, and contributing to CMD.
Autonomic nerve dysfunction, mediated by COVID‐19‐associated mental, physical or physiological factors, can elicit changes in coronary blood flow, resulting in CMD in COVID‐19 patients.
Dissemination of SARS‐CoV‐2 into perivascular cells of coronary microvessels via ongoing viremic blood stream and inflammation‐induced endothelial apoptosis causes perivascular structural cell edema. These edematous cells in return oppress coronary microvasculature, causing, or intensifying CMD.
FIGURE 1.
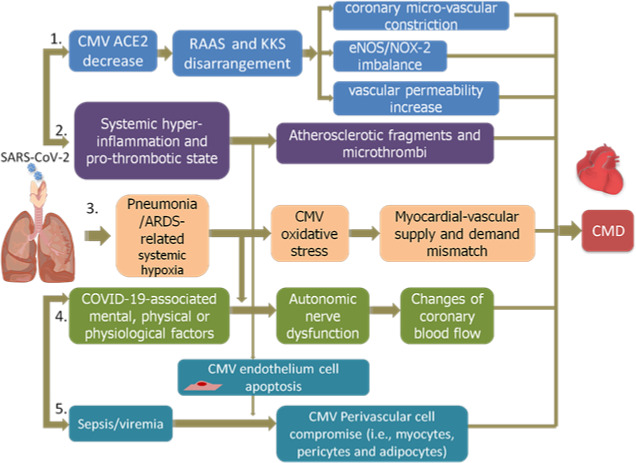
CMD mechanisms in COVID‐19. 1. Coronary microvascular endothelial ACE2 levels decrease, causing microvessels constriction, eNOS/NOX‐2 imbalance, and vascular permeability, therefore, leading to CMD; 2. Coronary microvessels obstruction caused by atherosclerotic fragments and microthrombi can also induce CMD; 3. Pneumonia/ARDS‐related systemic hypoxia elicits oxidative stress in coronary microvessels, activating cardiac sympathetic nerves, and contributing to CMD; 4. Autonomic nerve dysfunction, mediated by COVID‐19‐associated mental, physical or physiological factors, elicits changes in coronary blood flow, resulting in CMD in COVID‐19 patients; 5. SARS‐CoV‐2 disseminates into perivascular cells of coronary microvessels, causing perivascular structural cell edema, causing, or intensifying CMD. CMV, coronary micro‐vascular; RAAS, renin‐angiotensin‐aldosteron‐system; KKS, Kallikrein‐kinin‐system; eNOS, endothelial nitric oxide synthase; NOX‐2, reduced nicotinamide adenine dinucleotide phosphate oxidase 2; ARDS, acute respiratory syndrome; CMD, coronary microvascular dysfunction
These mechanisms may perform consecutively or intertwiningly, finally leading to CMD and manifesting as cardiovascular symptoms observed in COVID‐19 patients. However, the underlying molecular pathogenesis remains to be elucidated, and data collection for indices involved in coronary microvascular function in COVID‐19 patients (eg, coronary flow reserve, index of microvascular resistance, and response to intracoronary acetylcholine) is of extreme urgency.
5. PERSPECTIVES
We present a state‐of‐the‐art review of CMD pathophysiology in COVID‐19 from five aspects. As SARS‐CoV‐2‐like virus particles were detected in a coronary endothelial cells, and massive coronary small vascular microthrombi were found in deceased COVID‐19 patients, we thought our review was meaningful for further studies on SARS‐CoV‐2 induced heart injury. However, there is a need for more and larger well‐designed trials for future research.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
D.L. and T.X.: Idea for the article. J.Y., Y.L., and J.C.: Literature research. J.Y.: Manuscript drafting. All authors: Data analysis/interpretation, manuscript revision for important intellectual content, approval of final version of submitted manuscript, and agrees to ensure any questions related to the work are appropriately resolved.
ACKNOWLEDGEMENTS
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Yin J, Wang S, Liu Y, Chen J, Li D, Xu T. Coronary microvascular dysfunction pathophysiology in COVID‐19. Microcirculation. 2021;28:1–11. 10.1111/micc.12718
Yin and Wang should be considered as co‐first authors.
Contributor Information
Dongye Li, Email: dongyeli@xzhmu.edu.cn.
Tongda Xu, Email: xutongda3004@163.com.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available.
REFERENCES
- 1. Sohrabi C, Alsafi Z, O'Neill N, et al. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID‐19). Int J Surg. 2020;76:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guan W‐J, Ni Z‐Y, Hu Y, et al. in China. N Engl J Med. 2019;2020:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID‐19 and Multiorgan Response. Curr Probl Cardiol. 2020;45(8):100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN. Cardiovascular Disease, Drug Therapy, and Mortality in Covid‐19. N Engl J Med. 2020;382(25):e102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Breithaupt‐Faloppa AC, Correia CJ, Prado CM, Stilhano RS, Ureshino RP, Moreira LFP. 17β‐Estradiol, a potential ally to alleviate SARS‐CoV‐2 infection. Clinics (Sao Paulo). 2020;75:e1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID‐19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239. [DOI] [PubMed] [Google Scholar]
- 8. Krittanawong C, Virk HUH, Narasimhan B, et al. Coronavirus disease 2019 (COVID‐19) and cardiovascular risk: A meta‐analysis. Prog Cardiovasc Dis. 2020;63(4):527‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edler C, Schroder AS, Aepfelbacher M, et al. Dying with SARS‐CoV‐2 infection‐an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int J Legal Med. 2020;134:1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liya G, Yuguang W, Jian L, et al. Studies on viral pneumonia related to novel coronavirus SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV: a literature review. APMIS. 2020;128(6):423‐432. [DOI] [PubMed] [Google Scholar]
- 12. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181(4):905‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alifano M, Alifano P, Forgez P, Iannelli A. Renin‐angiotensin system at the heart of COVID‐19 pandemic. Biochimie. 2020;174:30‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castelli R, Gidaro A. Abnormal hemostatic parameters and risk of thromboembolism among patients with COVID‐19 infection. J Hematol. 2020;9:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Debuc B, Smadja DM. Is COVID‐19 a new hematologic disease? Stem Cell Rev Rep. 2021;17(1):4‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin‐angiotensin‐aldosterone system in COVID‐19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta. 2020;507:167‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13:1743‐1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adamson RH. Microvascular endothelial cell shape and size in situ. Microvasc Res. 1993;46:77‐88. [DOI] [PubMed] [Google Scholar]
- 20. Serné EH, de Jongh RT, Eringa EC, IJzerman RG, Stehouwer CDA. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension. 2007;50:204‐211. [DOI] [PubMed] [Google Scholar]
- 21. Lee J, Niederer S, Nordsletten D, et al. Coupling contraction, excitation, ventricular and coronary blood flow across scale and physics in the heart. Philos Trans A Math Phys Eng Sci. 2009;367:2311‐2331. [DOI] [PubMed] [Google Scholar]
- 22. Herrmann J, Kaski JC, Lerman A. Coronary microvascular dysfunction in the clinical setting: from mystery to reality. Eur Heart J. 2012;33:2771‐2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duan J, Wu Y, Liu C, Yang C, Yang L. Deleterious effects of viral pneumonia on cardiovascular system. Eur Heart J. 2020;41:1833‐1838. [DOI] [PubMed] [Google Scholar]
- 24. Moderato L, Monello A, Lazzeroni D, et al. Takotsubo syndrome during SARS‐CoV‐2 pneumonia: a possible cardiovascular complication. G Ital Cardiol (Rome). 2020;21:417‐420. [DOI] [PubMed] [Google Scholar]
- 25. Roca E, Lombardi C, Campana M, et al. Takotsubo Syndrome Associated with COVID‐19. Eur J Case Rep Intern Med. 2020;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen D, Nguyen T, De Bels D, Castro RJ. A case of Takotsubo cardiomyopathy with COVID 19. Eur Heart J ‐ Cardiovas Imag. 2020;21(9):1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cole‐Jeffrey CT, Liu M, Katovich MJ, Raizada MK, Shenoy V. ACE2 and microbiota: emerging targets for cardiopulmonary disease therapy. J Cardiovasc Pharmacol. 2015;66:540‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110:1103‐1107. [DOI] [PubMed] [Google Scholar]
- 29. Touyz RM, Li H, Delles C. ACE2 the Janus‐faced protein ‐ from cardiovascular protection to severe acute respiratory syndrome‐coronavirus and COVID‐19. Clin Sci (Lond). 2020;134:747‐750. [DOI] [PubMed] [Google Scholar]
- 30. Sodhi CP, Wohlford‐Lenane C, Yamaguchi Y, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des‐Arg(9) bradykinin/BKB1R axis and facilitates LPS‐induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314:L17‐L31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS‐CoV‐2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van de Veerdonk FL, Netea MG, van Deuren M, et al. Kallikrein‐kinin blockade in patients with COVID‐19 to prevent acute respiratory distress syndrome. Elife. 2020;9:e57555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1‐9. [DOI] [PubMed] [Google Scholar]
- 35. Santos RAS, Sampaio WO, Alzamora AC, et al. The ACE2/Angiotensin‐(1–7)/MAS Axis of the Renin‐Angiotensin System: Focus on Angiotensin‐(1–7). Physiol Rev. 2018;98:505‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin Converting Enzyme 2: SARS‐CoV‐2 Receptor and Regulator of the Renin‐Angiotensin System. Circ Res. 2020;126(10):1456‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoll D, Yokota R, Sanches Aragão D, Casarini DE. Both aldosterone and spironolactone can modulate the intracellular ACE/ANG II/AT1 and ACE2/ANG (1–7)/MAS receptor axes in human mesangial cells. Physiol Rep. 2019;7:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lippi G, South AM, Henry BM. Electrolyte imbalances in patients with severe coronavirus disease 2019 (COVID‐19). Ann Clin Biochem. 2020;57:262‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kyriazopoulou E, Leventogiannis K, Norrby‐Teglund A, et al. Macrophage activation‐like syndrome: an immunological entity associated with rapid progression to death in sepsis. BMC Med. 2017;15:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McElvaney OJ, McEvoy N, McElvaney OF, et al. Characterization of the Inflammatory Response to Severe COVID‐19 Illness. Am J Respir Crit Care Med. 2020;202(6):812‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou Y, Fu B, Zheng X, et al. Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. National Science Review. 2020;7(6):998–1002. 10.1093/nsr/nwaa041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carnevale S, Beretta P, Morbini P. Direct endothelial damage and vasculitis due to SARS‐CoV‐2 in small bowel submucosa of CoViD‐19 patient with diarrhea. J Med Virol. 2021;93(1):61‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395:1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guang C, Phillips RD, Jiang B, Milani F. Three key proteases–angiotensin‐I‐converting enzyme (ACE), ACE2 and renin–within and beyond the renin‐angiotensin system. Arch Cardiovasc Dis. 2012;105:373‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo J, Wei X, Li Q, et al. Single‐cell RNA analysis on ACE2 expression provides insights into SARS‐CoV‐2 potential entry into the bloodstream and heart injury. J Cell Physiol. 2020;235:9884–9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hawiger J. Heartfelt sepsis: microvascular injury due to genomic storm. Kardiol Pol. 2018;76:1203‐1216. [DOI] [PubMed] [Google Scholar]
- 48. Kasal DA, De Lorenzo A, Tibiriçá E. COVID‐19 and Microvascular Disease: Pathophysiology of SARS‐CoV‐2 Infection With Focus on the Renin‐Angiotensin System. Heart Lung Circ. 2020;29(11):1596‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chau VQ, Giustino G, Mahmood K, et al. Cardiogenic shock and hyperinflammatory syndrome in young males with COVID‐19. Circ Heart Fail. 2020;13(10):e007485. [DOI] [PubMed] [Google Scholar]
- 50. Buja LM, Wolf DA, Zhao B, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID‐19): Report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid‐19. N Engl J Med. 2020;383:120‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bösmüller H, Traxler S, Bitzer M, et al. The evolution of pulmonary pathology in fatal COVID‐19 disease: an autopsy study with clinical correlation. Virchows Arch. 2020;477(3):349‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prilutskiy A, Kritselis M, Shevtsov A, et al. SARS‐CoV‐2 Infection‐Associated Hemophagocytic Lymphohistiocytosis. Am J Clin Pathol. 2020;154(4):466‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ciceri F, Beretta L, Scandroglio AM, et al. Microvascular COVID‐19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc. 2020;22:95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colling ME, Kanthi Y. COVID‐19‐associated coagulopathy: An exploration of mechanisms. Vasc Med. 2020;25(5):471‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID‐19. Lancet Haematol. 2020;7:e438‐e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Escher R, Breakey N, Lämmle B. Severe COVID‐19 infection associated with endothelial activation. Thromb Res. 2020;190:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goshua G, Pine AB, Meizlish ML, et al. Endotheliopathy in COVID‐19‐associated coagulopathy: evidence from a single‐centre, cross‐sectional study. Lancet Haematol. 2020;7(8):e575‐e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaura A, Panoulas V, Glampson B, et al. Association of troponin level and age with mortality in 250 000 patients: cohort study across five UK acute care centres. BMJ. 2019;367:l6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Santoso A, Pranata R, Wibowo A, Al‐Farabi MJ, Huang I, Antariksa B. Cardiac injury is associated with mortality and critically ill pneumonia in COVID‐19: A meta‐analysis. Am J Emerg Med. 2020. (In press) 10.1016/j.ajem.2020.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Atallah B, Mallah SI, AbdelWareth L, AlMahmeed W, Fonarow GC. A Marker of systemic inflammation or direct cardiac injury: Should cardiac troponin levels be monitored in COVID‐19 patients? Eur Heart J Qual Care Clin Outcomes. 2020;6(3):204‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vrsalovic M, Vrsalovic PA. Cardiac troponins predict mortality in patients with COVID‐19: A meta‐analysis of adjusted risk estimates. J Infect. 2020;81(3):e99‐e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pranata R, Huang I, Lukito AA, Raharjo SB. Elevated N‐terminal pro‐brain natriuretic peptide is associated with increased mortality in patients with COVID‐19: systematic review and meta‐analysis. Postgrad Med J. 2020;96(1137):387‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Harikrishnan S, Mohanan PP, Chopra VK, et al. Cardiological society of India position statement on COVID‐19 and heart failure. Indian Heart J. 2020;72(2):75‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fox SE, Li G, Akmatbekov A, et al. Unexpected Features of Cardiac Pathology in COVID‐19 Infection. Circulation. 2020;142:1123‐1125. [DOI] [PubMed] [Google Scholar]
- 68. Bhatnagar J, Gary J, Reagan‐Steiner S, et al. Evidence of Severe Acute Respiratory Syndrome Coronavirus 2 Replication and Tropism in the Lungs, Airways, and Vascular Endothelium of Patients With Fatal Coronavirus Disease 2019: An Autopsy Case Series. J Infect Dis. 2021;223:752‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bearse M, Hung YP, Krauson AJ, et al. Factors associated with myocardial SARS‐CoV‐2 infection, myocarditis, and cardiac inflammation in patients with COVID‐19. Mod Pathol. 2021. 10.1038/s41379-021-00790-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tunc E, Eve AA, Madak‐Erdogan Z. Coronary Microvascular Dysfunction and Estrogen Receptor Signaling. Trends Endocrinol Metab. 2020;31:228‐238. [DOI] [PubMed] [Google Scholar]
- 71. Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830‐840. [DOI] [PubMed] [Google Scholar]
- 72. Shi S, Qin M, Shen B, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID‐19 in Wuhan, China. JAMA Cardiol. 2020;5:802‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Froldi G, Dorigo P. Endothelial dysfunction in Coronavirus disease 2019 (COVID‐19): Gender and age influences. Med Hypotheses. 2020;144:110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age‐related decline in women. J Am Coll Cardiol. 1994;24:471‐476. [DOI] [PubMed] [Google Scholar]
- 75. LeBlanc AJ, Hoying JB. Adaptation of the Coronary Microcirculation in Aging. Microcirculation. 2016;23:157‐167. [DOI] [PubMed] [Google Scholar]
- 76. Anversa P, Hiler B, Ricci R, Guideri G, Olivetti G. Myocyte cell loss and myocyte hypertrophy in the aging rat heart. J Am Coll Cardiol. 1986;8:1441‐1448. [DOI] [PubMed] [Google Scholar]
- 77. Sthoeger ZM, Chiorazzi N, Lahita RG. Regulation of the immune response by sex hormones. I. In vitro effects of estradiol and testosterone on pokeweed mitogen‐induced human B cell differentiation. J Immunol. 1988;141:91‐98. [PubMed] [Google Scholar]
- 78. Channappanavar R, Fett C, Mack M, Ten Eyck PP, Meyerholz DK, Perlman S. Sex‐Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J Immunol. 2017;198:4046‐4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mygind ND, Michelsen MM, Pena A, et al. Coronary Microvascular Function and Cardiovascular Risk Factors in Women With Angina Pectoris and No Obstructive Coronary Artery Disease: The iPOWER Study. J Am Heart Assoc. 2016;5:e003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Oda E, Abe M, Kato K, Watanabe K, Veeraveedu PT, Aizawa Y. Gender differences in correlations among cardiovascular risk factors. Gend Med. 2006;3:196‐205. [DOI] [PubMed] [Google Scholar]
- 81. Chen Z, Yuhanna IS, Galcheva‐Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Darkow DJ, Lu L, White RE. Estrogen relaxation of coronary artery smooth muscle is mediated by nitric oxide and cGMP. Am J Physiol. 1997;272:H2765‐2773. [DOI] [PubMed] [Google Scholar]
- 83. Mericli M, Nádasy GL, Szekeres M, et al. Estrogen replacement therapy reverses changes in intramural coronary resistance arteries caused by female sex hormone depletion. Cardiovasc Res. 2004;61:317‐324. [DOI] [PubMed] [Google Scholar]
- 84. Fischer M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;53:672‐677. [DOI] [PubMed] [Google Scholar]
- 85. Gebhard C, Regitz‐Zagrosek V, Neuhauser HK, Morgan R, Klein SL. Impact of sex and gender on COVID‐19 outcomes in Europe. Biol Sex Diff. 2020;11(1):29. 10.1186/s13293-020-00304-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem. 2000;275:33238‐33243. [DOI] [PubMed] [Google Scholar]
- 87. Granger DN, Rodrigues SF, Yildirim A, Senchenkova EY. Microvascular responses to cardiovascular risk factors. Microcirculation. 2010;17:192‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Labazi H, Trask AJ. Coronary microvascular disease as an early culprit in the pathophysiology of diabetes and metabolic syndrome. Pharmacol Res. 2017;123:114‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Strain WD, Paldánius PM. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc Diabetol. 2018;17:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Virdis A, Masi S, Colucci R, et al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. 2019;21:32. [DOI] [PubMed] [Google Scholar]
- 91. Camici PG, Tschöpe C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. 2020;116:806‐816. [DOI] [PubMed] [Google Scholar]
- 92. Bermejo‐Martin JF, Almansa R, Torres A, González‐Rivera M, Kelvin DJ. COVID‐19 as a cardiovascular disease: the potential role of chronic endothelial dysfunction. Cardiovasc Res. 2020;116(10):e132‐e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lavie CJ, Sanchis‐Gomar F, Henry BM, Lippi G. COVID‐19 and obesity: links and risks. Expert Rev Endocrinol Metabol. 15(4):215‐216. [DOI] [PubMed] [Google Scholar]
- 94. Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID‐19. Ann Intern Med. 2020;173(4):268‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhang W, Du RH, Li B, et al. Molecular and serological investigation of 2019‐nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. 2020;9:386‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang W, Xu Y, Gao R, et al. Detection of SARS‐CoV‐2 in Different Types of Clinical Specimens. JAMA. 2020;323:1843‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hu H, Garcia‐Barrio M, Jiang ZS, Chen YE, Chang L. Roles of Perivascular adipose tissue in hypertension and atherosclerosis. Antioxid Redox Signal. 2021;34(9):736‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mulvey JJ, Magro CM, Ma LX, Nuovo GJ, Baergen RN. A mechanistic analysis placental intravascular thrombus formation in COVID‐19 patients. Ann Diagn Pathol. 2020;46:151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sławiński G, Lewicka E. What should a cardiologist know about coronavirus disease 2019? Kardiol Pol. 2020;78:278‐283. [DOI] [PubMed] [Google Scholar]
- 100. Long M, Huang Z, Zhuang X, et al. Association of Inflammation and Endothelial Dysfunction with Coronary Microvascular Resistance in Patients with Cardiac Syndrome X. Arq Bras Cardiol. 2017;109:397‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Basso C, Leone O, Rizzo S, et al. Pathological features of COVID‐19‐associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J. 2020;41:3827‐3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hanley B, Naresh KN, Roufosse C, et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID‐19: a post‐mortem study. Lancet Microbe. 2020;1:e245‐e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tedeschi D, Rizzi A, Biscaglia S, Tumscitz C. Acute myocardial infarction and large coronary thrombosis in a patient with COVID‐19. Catheter Cardiovasc Interv. 2021;97(2):272‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Stefanini GG, Montorfano M, Trabattoni D, et al. ST‐Elevation Myocardial Infarction in Patients with COVID‐19: Clinical and Angiographic Outcomes. Circulation. 2020;141(25):2113‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ye S, Zhong H, Yanamadala S, Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309‐315. [DOI] [PubMed] [Google Scholar]
- 106. Takei Y, Tomiyama H, Tanaka N, Yamashina A. Close relationship between sympathetic activation and coronary microvascular dysfunction during acute hyperglycemia in subjects with atherosclerotic risk factors. Circ J. 2007;71:202‐206. [DOI] [PubMed] [Google Scholar]
- 107. Feigl EO. Neural control of coronary blood flow. J Vasc Res. 1998;35:85‐92. [DOI] [PubMed] [Google Scholar]
- 108. Kibel A, Selthofer‐Relatic K, Drenjancevic I, et al. Coronary microvascular dysfunction in diabetes mellitus. J Int Med Res. 2017;45:1901‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wackers FJ, Young LH, Inzucchi SE, et al. Detection of silent myocardial ischemia in asymptomatic diabetic subjects: the DIAD study. Diabetes Care. 2004;27:1954‐1961. [DOI] [PubMed] [Google Scholar]
- 110. Fineschi V, Michalodimitrakis M, D'Errico S, et al. Insight into stress‐induced cardiomyopathy and sudden cardiac death due to stress. A forensic cardio‐pathologist point of view. Forensic Sci Int. 2010;194:1‐8. [DOI] [PubMed] [Google Scholar]
- 111. Nees S, Weiss DR, Senftl A, et al. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: the second most frequent myocardial cell type in vitro. Am J Physiol Heart Circ Physiol. 2012;302:H69‐84. [DOI] [PubMed] [Google Scholar]
- 112. Pesaresi M, Pirani F, Tagliabracci A, et al. SARS‐CoV‐2 identification in lungs, heart and kidney specimens by transmission and scanning electron microscopy. Eur Rev Med Pharmacol Sci. 2020;24:5186‐5188. [DOI] [PubMed] [Google Scholar]
- 113. Bradley BT, Maioli H, Johnston R, et al. Histopathology and Ultrastructural Findings of Fatal COVID‐19 Infections; 2020. [DOI] [PMC free article] [PubMed]
- 114. Sekulic M, Harper H, Nezami BG, et al. Molecular Detection of SARS‐CoV‐2 Infection in FFPE Samples and Histopathologic Findings in Fatal SARS‐CoV‐2 Cases. Am J Clin Pathol. 2020;154(2):190‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tian S, Xiong Y, Liu H, et al. Pathological study of the novel coronavirus disease (COVID‐19) through postmortem core biopsies. Mod Pathol. 2019;2020:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu Y, Zhang A, Hamilton DJ, Deng T. Epicardial Fat in the Maintenance of Cardiovascular Health. Methodist Debakey Cardiovasc J. 2017;13:20‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available.