

Abstract
Intragravidic and perinatal infections, acting through either direct viral effect or immune‐mediated responses, are recognized causes of liability for neurodevelopmental disorders in the progeny. The large amounts of epidemiological data and the wealth of information deriving from animal models of gestational infections have contributed to delineate, in the last years, possible underpinning mechanisms for this phenomenon, including defects in neuronal migration, impaired spine and synaptic development, and altered activation of microglia. Recently, dysfunctions of the neurovascular unit and anomalies of the brain vasculature have unexpectedly emerged as potential causes at the origin of behavioral abnormalities and psychiatric disorders consequent to prenatal and perinatal infections. This review aims to discuss the up‐to‐date literature evidence pointing to the neurovascular unit and brain vasculature damages as the etiological mechanisms in neurodevelopmental syndromes. We focus on the inflammatory events consequent to intragravidic viral infections as well as on the direct viral effects as the potential primary triggers. These authors hope that a timely review of the literature will help to envision promising research directions, also relevant for the present and future COVID‐19 longitudinal studies.
Keywords: blood–brain barrier, COVID‐19, cytokines, hemorrhage, inflammation, long‐COVID, maternal immune activation, neurovascular unit, prenatal viral infection, synapse
Intragravidic and perinatal infections are recognized causes of liability for neurodevelopmental disorders in the progeny. This review aims to discuss the up‐to‐date evidence pointing to the neurovascular unit and brain vasculature damages as the etiological mechanisms. We focus on the inflammatory events consequent to intragravidic viral infections as well as on the direct viral effects as the potential primary triggers.

Abbreviations
- ASD
autism spectrum disorder
- BBB
blood–brain barrier
- BCAAs
branched‐chain amino acids
- BCKDK
branched chain keto‐acid dehydrogenase
- CMV
cytomegalovirus
- COX
cyclooxygenase
- CSDS
chronic social defeat stress
- ECs
endothelial cells
- eNOS
endothelial nitric oxide synthase
- GH‐IVH
intraventricular hemorrhage of the germinal matrix
- HIV
human immunodeficiency virus
- HSV
herpes simplex virus
- IL
Interleukin
- LPA
lysophosphatidic acid
- LPS
lipopolysaccharide
- MIA
maternal immune activation
- NAc
nucleus accumbens
- NVU
neurovascular unit
- P
postnatal day
- PFC
prefrontal cortex
- P‐gp
P‐glycoprotein
- PolyI:C
polyinosinic:polycytidylic acid
- PPI
prepulse inhibition
- SARS
severe acute respiratory syndrome
- TNF
tumor necrosis factor
Introduction
Gestational stress can be produced by social and psychological adversities as well as by environmental factors, which primarily include prenatal infections. While the latter can directly cause a congenital fetal infection, all the above conditions pose a threat to the health of both the mother and the fetus through the action of immune factors. Long‐term consequences of gestational stress on offspring's mental health have been studied for almost 80 years now. The hongerwinter, the Hollands famine winter that took place right after WWII, represents the first documented testimony of how maternal prenatal stress has an impact on the occurrence of psychiatric disorders in the progeny [1, 2]. Moreover, in the following years, several authors noted a link between the increased risk of neuropsychiatric disorders and winter‐born babies, thus raising the attention on intragravidic infections as the underlying cause [3, 4, 5]. Nowadays, maternal influenza infection is one of the most well‐replicated risk factors for schizophrenia—increasing the odds ratio by 3 [5]—and substantial epidemiological evidence exists to sustain a causal link for prenatal and perinatal infections to increase, later in life in the progeny, the susceptibility to neurodevelopmental disorders such as autism spectrum disorder (ASD) [6, 7, 8] and bipolar disorder [9, 10], in addition to schizophrenia [11, 12, 13].
Recent studies have highlighted the activation of the immune system as a risk‐shared mechanism in gestational stress‐induced long‐term newborn outcomes. Evidence span from findings of increased C‐reactive protein, interleukin (IL)‐6 plasma levels [14], and enhanced expression of various cytokines at the umbilical cord in the offspring of psychologically and socially stressed mothers [15] to the observations collected in translational animal models, collectively called maternal immune activation (MIA) models [16]. Together, these findings indicate that the physiological role of cytokines and immune factors in the government of brain development is likely to be disrupted upon stress and maternal immune responses. MIA models, mostly represented by rodents exposed to Toll‐like receptors’ agonists (mainly lipopolysaccharide—LPS and polyinosinic:polycytidylic acid—PolyI:C) during the gestational periods to mimic an inflammatory response to a pathological agent, are crucial to shed light on the underpinning molecular mechanisms which link the maternal–prenatal stress with the increased risk of psychiatric disorders in the progeny [17, 18]. Of note, MIA has proved the non‐necessity of congenital infections, of which there is limited evidence for influenza virus infections [19]. The latter are mostly due to the so‐called ‘TORCH’ pathogens, a cluster of microbes that include Toxoplasma gondii, rubella virus, cytomegalovirus (CMV), and herpes simplex virus (HSV), known for their different ability to directly infect the fetus during gestation [20, 21].
Potentially dangerous viruses have a distinctive spot in the world of microbes. They are high in number [22] and, differently from bacteria, have been associated with a twofold increased risk of adult diagnosis for nonaffective psychosis and schizophrenia [23]. In recent years, several viral outbreaks have affected large areas of the world. In < 20 years we have faced the coronavirus SARS‐CoV (2002/2003)—named after the severe acute respiratory syndrome (SARS) that it causes, the ‘swine flu’ caused by the influenza virus H1N1 (2009), the MERS‐CoV (2012)—another coronavirus that prevalently spread in the Middle East area, the mosquito‐borne Zika virus (ZIKV) (2015) and since December 2019, the worst global pandemic since 1918 ‘Spanish’ influenza, caused again by another coronavirus: the SARS‐coronavirus‐2 (CoV‐2).
Till now, 88 880 pregnant women have been found positive to COVID‐19 just in the United States (CDC's data). At present, data are mostly available from third‐trimester infections and there is a growing list of associated neonatal complications [24, 25]. Major aspects, including the timing of the infection and the presence/absence of symptoms in the mothers‐to‐be, are being collected and will be evaluated. Still, marginal attention has been paid to the fetus as a patient during the pandemic and is too soon to understand the long‐term effects of SARS‐CoV‐2 infection on progeny's mental and neurological disability. The average onset for schizophrenia coincides with adolescence‐early twenties [26, 27] while the diagnosis among children with ASD comes around 3.1 years of age [28]. In the context of gestational COVID‐19 infections, if and how the virus can influence progeny neural system development will emerge in the next years.
Presently, we possess an extensive quantity of data on viral intragravidic infections and numerous MIA models. These data are being exploited to unveil the mechanisms which link the activation of the maternal immune system with the behavioral abnormalities in the offspring. These aspects, which are still not clarified, are of crucial relevance also for predicting the possible impact of the current pandemics in the neurodevelopmental trajectories of the next generation. Different processes have been brought into play to explain the effect of MIA on the offspring neurodevelopment, including (but not limited to) the altered expression of genes involved in neuronal migration [29] and the generation of an abnormal cortical phenotype [30], changes in the intrinsic excitability of neurons [18, 31], impaired spine and synaptic development [32, 33, 34], and altered activation of microglia [35, 36]. Notably, disturbances of the neurovascular unit (NVU) physiology and brain vasculature morphology have been identified as one of the leading causes of several neurological diseases, both in developing and aged brain. Moreover, multiple evidences indicate a key involvement of the vasculature in SARS‐CoV‐2 pathological manifestations [37]. The spike protein has recently been proved to alter the blood–brain barrier (BBB) function and integrity in an in vitro model with human brain microvascular endothelial cells (ECs) derived from fetal brains [38]. This review seeks to discuss state‐of‐art evidence indicating that NVU and brain vasculature damages may represent the etiological mechanism in neurodevelopmental syndromes. We specifically focus on inflammation, deriving from intragravidic viral infections, as well as on direct viral effects on the embryo, as potential primary triggers (Fig. 1). These authors hope that a timely review of the literature will help to envision research promising directions, also relevant for the present and future COVID‐19 longitudinal studies.
Fig. 1.
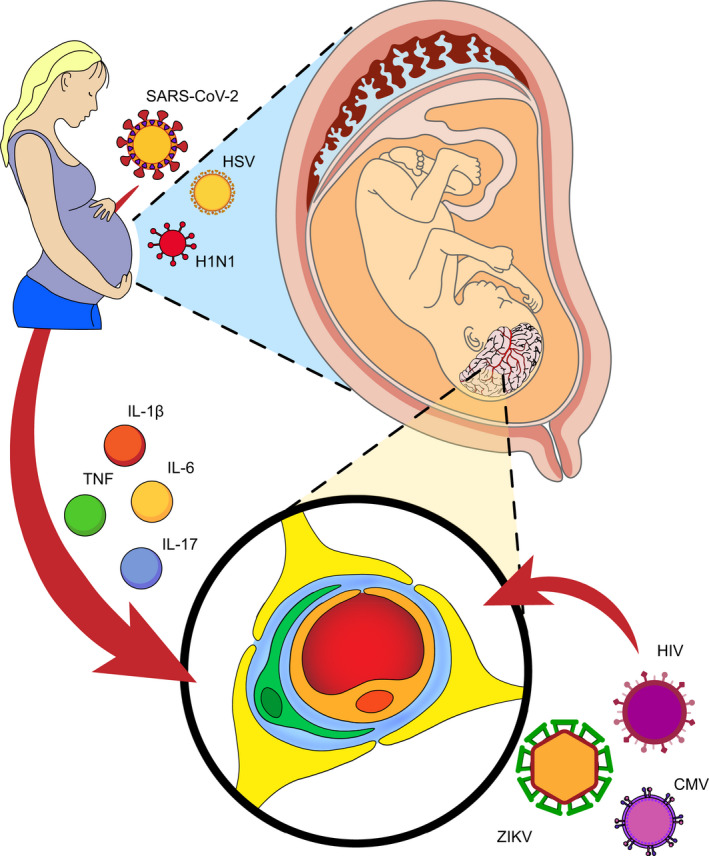
Schematic representation of how viral infections affect the fetal NVU and the brain vasculature during pregnancy. Viruses can influence vessel development by both direct (i.e., congenital infection) and immune‐mediated (i.e., inflammatory state) processes.
Neurovascular unit dysfunctions result in pathological behavioral phenotypes
The NVU is a relatively recent concept that encloses highly dynamic and interactive cell types at the interface of brain parenchyma with the peripheral blood. Including both cellular and acellular components wrapped around the ECs that form the vasculature of the brain [39], the NVU controls the trafficking of blood‐borne molecules and cells for homeostasis conservancy. The NVU starts to be formed early during brain development. Vascularization is then rapidly followed by the expression of tight junction proteins and transporters [40], allowing the brain protection from vasogenic edema, the passage of toxic substances, and microbe's invasion [41, 42]. NVU development is a step‐by‐step process in which temporal and regional differences play a critical role in the functioning of the mature adult brain [43].
Many reviews have been published about NVU impairments and neurovascular anomalies in association with neurodevelopmental disorders [44, 45, 46, 47]. BBB breakdown is considered an early marker for later cognitive dysfunctions [48]; further, neurovascular problems due to perinatal brain injuries—including inflammation, hypoxic‐ischemia, or hemorrhagic events—are recognized as one of the leading causes of long‐term mental disabilities [49, 50, 51]. Blood oxygen level–dependent fMRI in ASD patients shows a reduced signal that correlates with symptoms severity, possibly indicating a faulty neurovascular coupling process [52]. Consistently, persistent angiogenesis (i.e., the growth of new vessels from the preexisting vasculature) has been recently observed in the brain of postmortem ASD patients [53], while several neurodevelopmental disorders, including autism [54], schizophrenia, depression [55], cerebral palsy [56], and bipolar disorder [57], have been epidemiologically associated with prenatal cerebral bleeding.
Here, we will analyze the direct impact of brain vasculature and NVU dysfunctions on behavioral disorders phenocopying, focusing on the lines of evidence that directly imply NVU/vasculature early damages as etiopathological in autism, schizophrenia, and depression (Fig. 2 and Table 1).
Fig. 2.
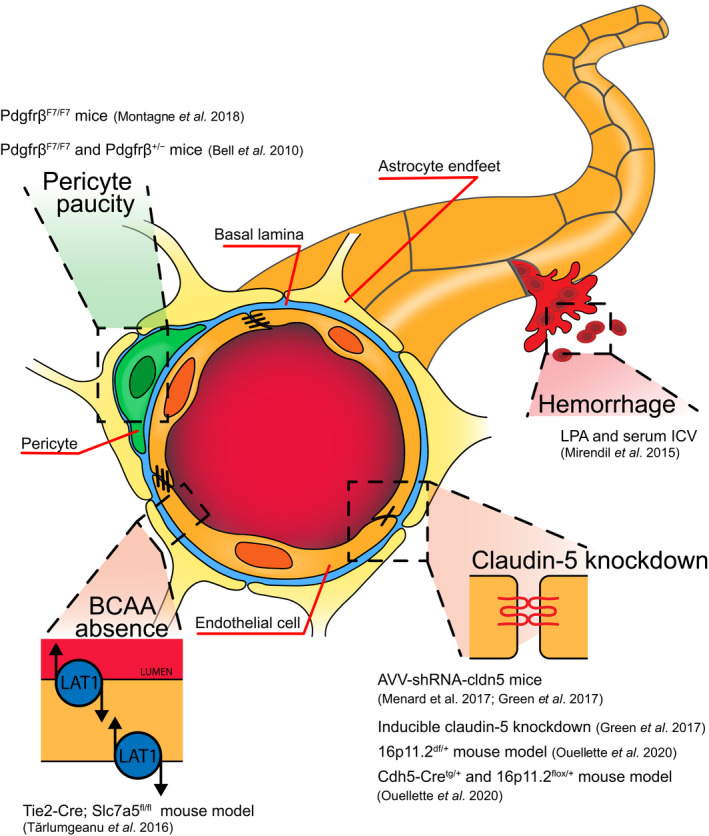
Research tools for the study of behavioral phenotypes consequent to the NVU dysfunctions. Pericyte paucity and reduced vascular coverage, hemorrhages, downregulation of the tight junction protein claudin‐5, and lack of BCAAs into the brain parenchyma result in ASD‐ and schizophrenia‐like behaviors, as well as depression and dementia.
Table 1.
Animal models of NVU dysfunctions, resulting in behavioral phenotypes. A, anxiety‐related; D, depression‐related; LC, locomotor competence; LM, learning and memory; NS, not specified; S, schizophrenia‐related; SL, sensory/locomotor.
| Mouse model | Target | Age of treatment | Vascular phenotype | Age of assessment | Analyzed behavioral category | Behavioral profile | Sex | Reference |
|---|---|---|---|---|---|---|---|---|
| 16p11.2df/+ | All cells | Constitutive | Neurovascular uncoupling | P14, P50 | A, SL, LM, LC | ASD | M | [83] |
| Cdh5‐Cretg/+; 16p11.2flox/+ | ECs | constitutive | ↓cortex vascular density ↓cortex vascular branching | P0, P50 | A, SL, LM, LC | ASD | Both | [83] |
| Tie2‐Cre; Slc7a5fl/fl | ECs | Constitutive | BCAA absence | E14.5, P2‐65 | A, LM, LC | ASD | both | [88] |
| LPA and serum ICV | none | E13.5 | Prenatal cerebral hemorrhage | P70 | S, A, LM, LC | Schizophrenia‐like | F | [93] |
| inducible claudin‐5 knockdown | ECs | P56‐84 | ↓claudin‐5 | P56‐84 | S, A, LM, LC, D | Schizophrenia‐like | NS | [97] |
| AVV‐shRNA‐cldn5 | HP, PFC | P56‐84 | ↓claudin‐5 | P56‐84 | S, A, LM, LC, D | Schizophrenia‐like/Stress resistance | NS | [97] |
| AVV‐shRNA‐cldn5 | HP, NAc | P56‐70 | ↓claudin‐5 | P56‐70 | A, D, LC | Depression | M | [103] |
| PdgfrβF7/F7 | Pericyte | Constitutive | Pericyte paucity and low vessel coverage | P14‐336 | A, SL, LM, LC | Dementia | Both | [74] |
| PdgfrβF7/F7 and Pdgfrβ+/− | Pericyte | Constitutive | Pericyte paucity and low vessel coverage | P7‐112 | LM | Dementia | NS | [75] |
The case of pericytes
Pericytes, differently from astrocytes [58], are recruited by the ECs of the brain early during development, specifically by E10 in mice [59, 60, 61] and by the 8–10th week of gestation in humans [62, 63, 64]. Pericytes promote vascular structural stability and protect the brain from blood‐borne elements and bleeding phenomena [59]. Anomalies involving pericytes biology have been proven to play key roles in the etiopathogenesis of different genetic diseases characterized by psychiatric comorbidities, including the Hereditary Hemorrhagic Telangiectasia [65, 66], a vascular dysplasia condition affecting one in 5000 people worldwide, and the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy [67, 68], the most commonly inherited small‐vessel disease. Furthermore, pericytes paucity has been recently found associated with intraventricular hemorrhage of the germinal matrix (GH‐IVH), both in premature humans and in a rabbit premature pup model [69]. GH‐IVH affects 12 000 premature babies every year just in the United States [70] and nearly 45% of all extremely premature infants [71], leading to seizures, mental retardation, cerebral palsy, and death [70, 72]. Early treatments with angiogenesis inhibitors have been found to ameliorate pericytes coverage [69] and decrease bleeding incidence in a premature animal model [73].
Among pericytes paucity models, the PdgfrβF7/F7 which bear homozygous F7 hypomorphic mutations of Pdgfrβ, and the Pdgfrβ+/−, heterozygous null Pdgfrβ mice, are well‐characterized models which show a leaky NVU [59, 74, 75]. In details, the PdgfrβF7/F7 mice display a progressive functional deficit of the white matter, manifesting as impaired spatial working memory and reduced maximum velocity on the complex running test [74]. Only later in life (i.e., 36–48 weeks), PdgfrβF7/F7 mice display hippocampus‐dependent behavioral defects [74]. On the other hand, the Pdgfrβ+/− mice show a 20% loss in pericyte coverage by 1 month of age, which is sufficient to initiate progressive vascular damage [75]. By the age of 6–8 months, also Pdgfrβ +/− mice show hippocampal‐dependent impairments in learning and spatial memory, although age‐matched PdgfrβF7/F7 mutants have worse performance [75]. The PdgfrβF7/F7 and the Pdgfrβ+/− mice are considered good models of vascular age‐dependent degenerative diseases resulting in neurodegeneration, memory, and learning impairments as Alzheimer's disease [76, 77, 78] and mild dementia [79]. Both Pdgfrβ+/− and PdgfrβF7/F7 mice show a significant reduction in pericytes (i.e., 35–75%) in the neural tube already at E14.5 [80]. At present, to the best of our knowledge, no literature data describe the behavioral phenotype resulting from a pericyte loss restricted to the embryonic time window. Therefore, whether pericyte loss during fetal development contributes to the observed behavioral consequences is presently unknown. Recently, Cuervo et al. [81] developed a new tamoxifen‐inducible Cre‐model targeting pericyte, called PDGFRβ‐P2A‐CreERT2, which will likely offer the opportunity for more detailed studies on this matter in the future. Of note, animal models of GH‐IVH, as the collagenase‐induced hemorrhage neonatal rat model, display a delay in motor abilities development and cognitive deficits [82].
Autism spectrum disorder
Recent studies have investigated the vascular contribution to ASD using autism mouse models (Fig. 2 and Table 1). This is the case for the 16p11.2 microdeletion [83], which approximately involves 2% of all ASD diagnosed cases [84] leading prevalently to large head size, intellectual disabilities, and language‐related problems in humans [85]. The 16p11.2df/+ mouse model shows alterations of the basal ganglia and synaptic defects beyond behavioral abnormalities that include stereotypic movements and hyperactivity [86]. Ouellette et al. [83] demonstrated that 16p11.2df/+ adult males are affected by a form of endothelium‐dependent neurovascular uncoupling. In particular, they evidenced in postnatal day 50 (P50) females and males, the occurrence of a reduced vessel relaxation in response to acetylcholine or adenosine, and a poor vessel constriction in response to nitric endothelial oxide synthase inhibition. Furthermore, isolated 16p11.2df/+ ECs harvested at P14—but not at P50—exhibited a scant capacity of network formation compared with WT. This phenomenon was recapitulated by ECs derived from human induced pluripotent stem cells obtained from donors carrying the 16p11.2 deletion. The authors excluded a contribution of smooth muscle cells by testing vasorelaxation in response to sodium nitroprusside and phenylephrine and by replicating most of their observations using an endothelial‐specific 16p11.2 (7qF3) deletion mouse model [83]. The paper establishes a solid and direct link between neurovascular abnormalities and autistic phenotypes. Of note, many of the observed defects were sex‐ and age‐specific. Another mutation, accountable for patients affected by ASD, but also found in cases of intellectual disability or epilepsy, is located in the gene branched‐chain keto‐acid dehydrogenase (BCKDK) [87]. The primary consequence of the mutation is the lack of branched‐chain amino acids (BCAAs), for which the brain depends on supply from the periphery. Tărlungeanu et al. [88] studied a mouse model that mimics BCKDK absence by the deletion of Slc7a5, a gene responsible for the encoding of the large amino acid transporter 1, at its physiological site of expression: the BBB. The deletion of Slc7a5 from ECs, using a Tie2Cre line, led to neuronal activity imbalance and behavioral abnormalities in the mice, mostly revertible in adults by a 3‐week long intracerebroventricular BCAAs administration. The observed behavior phenotypes spanned from motor coordination and locomotion difficulties to more typical autistic tracts, including reduced explorative behavior, limited social interactions, decreased play activity, and reduced amount of isolation‐induced ultrasonic vocalization [88]. Interestingly, BCAAs seem to be transported into the developing brain at higher levels than in the adult one [89, 90]. Tărlungeanu et al. demonstrated that brain BCAA homeostatic concentrations—which are regulated at the BBB level—are critical for brain functioning and need to be taken into account in the context of ADS pathology.
Schizophrenia
Perinatal brain hemorrhages are a well‐recognized risk factor for schizophrenia [91, 92]. Still, very few authors have directly addressed the behavioral tracts resulting from developmental intracranial hemorrhages or increased permeability of serum elements into the brain (Fig. 2 and Table 1). Mirendil et al. [93] developed an animal model of prenatal hemorrhages by intraventricular injections of the bioactive phospholipid lysophosphatidic acid (LPA) in the fetus. LPA in vivo administration is known to induce bleedings and inflammation [94, 95]. Fetuses intraventricularly treated with LPA or serum at E13.5 displayed schizophrenic‐like behaviors in adulthood; in particular, the authors found a female‐specific prepulse inhibition (PPI) deficit and anxiety‐related exploratory patterns [93]. Prenatal cerebral bleeding or diffusion of blood derivates (i.e., serum or LPA) are sufficient to induce schizophrenia symptoms in mice; if the observed sex‐specific effect was due to the developmental stage chosen for the hemorrhagic hit, remains to be addressed.
Paracellular diffusion of serum proteins at the BBB is physiologically restrained by the means of occluding junctions, which seal the adjoining ECs. Claudin‐5, encoded by the chromosomal region 22q11.21 is one of the most enriched tight junction proteins at the BBB and has been implicated in many neurological disorders, including depression and schizophrenia [44]. Deletions of the chromosomal region 22q11 confer the carrying individuals (i.e., ~ 1 in 4000) with a 30‐fold increase risk for schizophrenia and other neuropsychiatric conditions [96]. Green et al. [97] generated an inducible ECs‐specific claudin‐5 knockdown mouse demonstrating that the prolonged suppression of claudin‐5 at the BBB produces psychosis‐like behaviors. Claudin‐5 knocked‐down animals showed a schizophrenia‐like behavioral phenotype, consisting of altered spontaneous alternated Y‐maze and T‐maze performances, and reduced acoustic PPI response. The paper suggests the direct implication of claudin‐5 anomalies in psychiatric disorder symptomatology, a concept strengthened by the fact that antipsychotic treatments (i.e., lithium, haloperidol, and chlorpromazine), among others, induce an increased expression of claudin‐5 protein both in vivo and on EC cultures [97].
Depression
About 50 years are passed since the first vascular hypothesis on the origin of depression was formulated [98]. Today, multiple pieces of evidence indicate increased NVU permeability in both depressed patients [45, 99] and animal models of depression [100, 101, 102, 103] (Fig. 2 and Table 1). Menard et al. [103] recently proved that the sole downregulation of claudin‐5 in the nucleus accumbens (NAc), which allows the passage of molecules till 800 Da through the BBB [104], is sufficient to induce depression‐like behaviors in mice. The authors employed an inducible AVV‐shRNA against claudin‐5 to assess the behavioral phenotype resulting from the region‐specific downregulation of the gene. Intra‐NAc injections followed by doxycycline treatment effectively inhibited claudin‐5 expression and resulted in stress‐triggered depression‐like behaviors. Specifically, mice subjected to a subthreshold micro‐defeat routine showed anhedonia and significantly reduced their performances in the forced swim test [103]. Furthermore, mice displayed social avoidance, compared with unstressed control and unstressed AAV‐shRNA mice. Differently, mice subjected to the virus‐mediated conditional knockdown of claudin‐5 in the hippocampus—another brain region that showed claudin‐5 downregulation following depression induced by chronic social defeat stress (CSDS)—still spent more time immobile in the forced swim test, but displayed no anhedonia [103]. These results suggest a region‐specific effect of claudin‐5 expression levels on behavior (Table 1). This concept was strengthened by the fact that CSDS‐induced depression does not lead to a ubiquitous claudin‐5 downregulation throughout the brain. In particular, the mice prefrontal cortex (PFC) does not show variations of claudin‐5 expression upon CSDS [103]; on the same line, the AVV‐shRNA mediated downregulation of claudin‐5 in the PFC leads to impairments of recognition memory and working spatial memory but results in enhanced performances in the forced swim test [97]. These data indicate that the PFC is resilient to stress‐induced NVU breakdown and that the experimental downregulation of claudin‐5 in this region results in memory deficits accompanied by stress resistance. As a proof of concept for the relevance of claudin‐5 homeostasis in brain physiology [44], only a chronic treatment with the antidepressant imipramine, known to reverse CSDS‐induced depression‐like behaviors [105], was able to normalize claudin‐5 mRNA levels and behavioral phenotypes in the NAs‐AVV‐shRNA‐cldn5 mice, while an acute administration of the same drug did not affect either [103].
Do viral infections cause neurovascular unit dysfunctions linked to neurodevelopmental diseases?
The above data support the concept that alterations of the NVU result in pathological behavioral phenotypes. Consistently, NVU defects are detectable in several neurodevelopmental diseases. In the next section, we will analyze the results of studies investigating the direct effects of the mother‐to‐embryo vertical viral infection and the indirect effect caused by inflammation consequent to prenatal infections.
Direct effects of congenital infections on the NVU and the brain vasculature
During pregnancy, viruses can reach and infect the fetus mainly through intrapartum or in utero (i.e., ascending or transplacental) pathways [21]. Both routes result in the vertical transmission of the infection from the mother to the child, which poses an immediate threat to the newborn's immature immune system [106]. Of particular concern are in utero infections, which can occur at any time during the pregnancy and therefore interfere with the specific time schedule of development. Different viruses and TORCH infectious agents are known to target the fetal nervous system; some possess tropism for NVU components [107, 108] and can induce prenatal brain vasculopathies and hemorrhages (Table 2) [109, 110, 111]. These complications constitute a common event in premature babies which can evolve in cerebral palsy, epilepsy, cognitive deficits, and/or behavioral difficulties in the adult [112, 113, 114]. Here, we will summarize recent findings of direct‐viral‐effects producing brain vascular anomalies and faulty NVU establishment, in the context of intragravidic infections.
Table 2.
Brain vascular outcomes in vertically transmitted infections.
| Virus | Species | Vascular phenotype | References |
|---|---|---|---|
| CMV | Humans | Cerebral hemorrhagic infarction | [115] |
| Humans | Brain bleeding | [118] | |
| Humans | Fetus vasculitis | [111, 119, 120] | |
| HIV | Humans | Infant hemorrhagic Moyamoya Syndrome | [110] |
| Zika virus | Humans | Ischemic infarcts | [124] |
| Humans | Encephalomalacia | [125] | |
| Humans | Brain calcification | [126, 127] | |
| Monkey | Hemorrhage and vasculitis | [128] | |
| Monkey | Intracranial calcification | [129] | |
| Monkey | Increased vascular density and increased permeability | [130] | |
| Mice (type 2 interferon‐deficient) | Delayed vascular development | [131] |
Of interest is the case of a patient infected by CMV during the first 16 weeks of gestation, an important time window for brain vasculature development, who gave birth (at 21 weeks) to a child affected by cerebral hemorrhagic infarction [115]. This report is suggestive of a direct effect of CMV on the developing vasculature; in fact, even if the most frequent outcomes of CMV congenital infections are polymicrogyria, cortical dysplasia, and neuronal migration disorders [116, 117], the occurrence of neonatal brain bleedings has been reported [118]. CMV can infect ECs of the brain [108] and induce in the fetus vasculitis that can evolve and manifest as thrombosis, coagulopathy, or hemorrhage even in the absence of thrombocytopenia [111, 119, 120]. Vasculopathies are also a common complication associated with human immunodeficiency virus (HIV) infections [121], for which the NVU is believed to constitute the main brain route access. HIV, through a Tat‐mediated downregulation of the BBB tight junctions, has been demonstrated to induce a loose NVU and an increase in vitro paracellular permeability [122, 123]. Of particular interest, is the case report of hemorrhagic Moyamoya syndrome in a child with congenital HIV‐1 infection [110], an uncommon manifestation in pediatric age, probably due to the HIV‐mediated vascular damage. A higher amount of data is available on ZIKV congenital infections, which is known to have a huge impact on the cerebrovascular structure and functional development. Zika intragravidic infections can produce ischemic infarcts in human fetuses [124], encephalomalacia due to chronic infarction [125], and brain calcification in neonates [126, 127]. All these outcomes are reproduced in the offspring of ZIKV‐infected pregnant monkeys, adding to frequently occurring fetal loss, small brain size, hemorrhage, necrosis, vasculitis, and apoptosis of neuro‐progenitor cells [128]. Moreover, offspring display more severe cerebrovascular signs if born from female monkeys challenged with viral particles at early (i.e., 6–7 weeks) relative to late gestation (i.e., 12–14 weeks) [128]. Similar anomalies are also detectable in the offspring of murine models of congenital Zika infection. For example, intra‐amniotic administration of Zika particles at E15.5 leads to intracranial calcifications in the adult progeny [129], while the direct intracerebral inoculation of the virus at E14.5 induces enlargement of the brain's vessels accompanied by higher vascular density and increased permeability [130]. Furthermore, ZIKV has been shown to infect fetal brain ECs (IB4‐positive vessels) in the study of Garcez et al., where an E12.5‐infected type 2 interferon‐deficient pregnant mice model was used. In detail, at E15.5 the progeny of the infected mice showed a reduction in the vasculature percentage area and vessel branching, while by birth (i.e., at P2), pups showed only a significant decrease in branches length [131], a phenomenon possibly due to a virus‐induced delayed vascular development.
Different from the above cases, we are still experiencing a lag in information about SARS‐CoV‐2. The huge global effort of clinicians and researchers in the last year provided a clear description of COVID‐19 acute manifestations and cerebrovascular comorbidities, but our understanding of the long‐COVID neurological implications is still in its infancy. Even more complex will be the analysis of possible neurological and psychiatric sequelae due to intragravidic SARS‐CoV‐2 infections in the progeny. Gestational COVID‐19 infections could impact the developing brain, with possible consequences for behavioral impairment later in life, through several mechanisms. For example, IL‐6, the major cytokine elevating during SARS‐CoV‐2 infections [132], has been shown to boost, during intragravidic infections, the excitatory synaptogenesis in the offspring, with consequences on brain connectivity [133]. Maternal T helper 17 cells have been implicated in cortical and behavioral abnormalities in MIA offspring [30]. Also, maternal infections have been found to reduce brain offspring's potassium‐chloride co‐transporter 2 expression, thus delaying the excitatory‐to‐inhibitory GABA switch [17, 18]. Nevertheless, the NVU could directly play a role: We know that SARS‐CoV‐2 can directly affect the brain microvascular endothelium and thus force its way to the brain parenchyma [37]. MRI and autoptic studies showed endothelial activation, loss of cerebral vascular integrity, and multifocal hemorrhage in adult COVID‐19‐infected patients [134], while in children cases of Kawasaki‐like disease vasculitis have been reported [135]. Furthermore, in vitro studies recently proved that the spike protein, and even just subunits of it, is sufficient to alter the BBB integrity [38]. Current data indicate that SARS‐CoV‐2 vertical transmission is relatively uncommon, occurring in about 3.2% of pregnant women who contracted COVID‐19, while it is over 10% for ZIKV and roughly 30% for CMV [24, 25, 136]. Although it is too early to draw final conclusions, current reports on newborn outcomes show no increased risk of congenital anomalies [137]; also, cases of hypoxic‐ischemic encephalopathy [138] and birth asphyxia [139] seem extremely rare. Nonetheless, the background knowledge about TORCH and other respiratory viruses suggests that SARS‐CoV‐2 might induce permanent neurological sequelae through damage of the NVU, so further studies are needed. Besides the possible direct effect of the virus on the NVU development and structural integrity, other elements need to be considered, including the proinflammatory state induced by the infection and the high rate of premature delivery (i.e., 12.9%) correlated with COVID‐19 [137] which, by themselves, are known to be risk factors for NVU later malfunctions [49, 140].
Inflammatory‐mediated effects of gestational infections on the NVU and the brain vasculature
Tightly regulated changes in immune cell composition and cytokine expression at the maternal–fetal interface sustain and drive pregnancy. Longitudinal analysis of peripheral blood, decidua, and amniotic fluid demonstrated that the first trimester is characterized by the establishment of a mild proinflammatory environment and accumulation of immune cells at the decidua which allow trophoblast invasion, implantation, and placenta formation [141]. On the opposite, the second and third trimesters present a shift toward an anti‐inflammatory phenotype [142, 143, 144, 145, 146] which facilitates fetal growth and immune tolerance [141]. Lastly, parturition display a proinflammatory bias: IL‐6, tumor necrosis factor‐alpha (TNF‐α), and IL‐1 increase at term, suggesting a role for these cytokines in normal labor [147]. Despite their relevant role in physiological pregnancy, inflammatory states can also be dangerous for the developing fetus as demonstrated by the harmful effects of late‐pregnant serum on brain tissue, which promotes neuronal hyperexcitability and seizures, activating microglia cells, and promoting TNF‐α production [148, 149]. NVU formation and maturation during in utero development act therefore as a key brain shield to avoid these dangerous effects [150].
Upon maternal immune response to infection, acute or chronic disturbances can undermine these highly regulated immune adaptations and disrupt placental and NVU barriers ultimately affecting fetal development, pregnancy outcomes, and long‐term progeny neurodevelopment (Table 3). Adinolfi was the first to propose that MIA during the course of infection may be harmful to the developing brain of the unborn infant [151]. Leviton further extended this hypothesis, suggesting that cytokines, such as TNF‐α, may contribute to both preterm birth and periventricular white matter damage [152]. Nowadays, several maternal‐derived cytokines have been identified as critical mediators of MIA on disease‐related phenotypes in the offspring, including TNF‐α, IL‐1β, IL‐6, and IL‐17a [153]. Proinflammatory cytokines and their respective receptors are expressed in the brain and on the luminal side of cerebral ECs or NVU‐associated cells, making the NVU a sensory organ for neuro‐immune crosstalk [154, 155, 156].
Table 3.
Brain vascular outcomes in MIA models. A, anxiety‐related; E, embryonic day; GD, gestational day; LC, locomotor competence; LM, learning and memory; NA, not assessed; S, schizophrenia‐related.
| Inflammatory Agent | Dose | Target (species) | Age of treatment | Systemic features | Age of assessment | Vascular phenotype | Behavioral profile | References |
|---|---|---|---|---|---|---|---|---|
| PolyI:C | 5 or 10 mg·kg−1 | Pregnant (mice) | GD14.5 ± 1 | ↑ IL‐6 concentration in the maternal plasma after 4 h | +4/24 h | Accumulation of P‐gp substrates | NA | [169] |
| LPS | 0.1 µg·kg−1 | Fetus (sheep) | GD133±1 | – | +24 h | ↑ albumin permeability | NA | [170] |
| 30–60 µg | Uterine artery (sheep) | GD134‐136 | – | +72 h | ↑ albumin permeability | LM | [171] | |
| 100 ng·kg−1 over 24 h, followed by 250 ng·kg−1/24 h for 96 h plus boluses of 1 μg LPS at 48, 72, and 96 h | Fetus (sheep) | GD103/104 | – | +10 days | ↓vessel density, ↓ pericyte and astrocyte microvascular coverage | NA | [172] | |
| 0.2–10 mg·kg−1 | Postnatal (rats and opossum) | P0, P2, P4, P6, P8 and adult rats; P15,P20, P35, P50, P60 and adult opossum | ↑ IL‐1b and TNF‐α plasma levels | P20 and adults | ↑ permeability adulthood, preceded by claudin‐5 altered distribution | S, A | [173, 174, 175] | |
| 0.25 mg·kg−1 | pregnant (rat) | GD15 | ↑ cytokines and reactive oxygen species in the fetal brain | +6 h, 12 h and 24 h | ↓ BBB and placental barrier integrities | LM, S, LC | [177] | |
| IL‐1β | 0, 0.1, 0.5, 1 µg | Pregnant (mice) | E14‐E17 | – | +6 h | ↓ placental ECs, red blood cells clumping | NA | [176] |
Cytokines may have access to the fetal brain either by crossing the intact NVU [157, 158, 159] or by mediating NVU alteration and breakdown [160, 161, 162]. Activation of maternal immunity also presents a risk of developing autoantibodies, which have been implicated mainly in autism (i.e., maternal autoantibody‐related autism) [163], and of local immune response activations in the fetal brain, which may lead to secondary cytokines production by microglia and astrocytes and, in turn, have cell damaging properties [154, 164, 165, 166, 167, 168].
In the case of intragravidic infections, although the viruses are usually not transmitted to the fetus during pregnancy thanks to effective multiple defense mechanisms specialized to protect the mother and the fetus [42, 44], inflammatory responses in the placenta or infection‐induced systemic changes in the pregnant mother might result in more severe or prolonged disease for the mother and long‐term consequences for her newborn [43]. Viral infections modeled by the usage of PolyI:C (5 or 10 mg·kg−1) intraperitoneal injections in pregnant mice (GD15.5) acutely elevate IL‐6 concentration in the maternal plasma, concomitantly increasing fetal brain accumulation of P‐glycoprotein (P‐gp) substrates, an efflux transporter that protects the brain by limiting the transfer of substrates across the BBB [169]. The resulting BBB alteration and P‐gp decreased activity may expose the developing brain to xenobiotics and environmental toxins present in the maternal circulation.
Yan et al. [170] demonstrated an increased permeability of the NVU to albumin just 24 h after LPS 0.1 µg·kg−1 intravenous injection in 133‐day gestation fetal sheep, suggesting that this is the result of prostaglandin production as indicated by cyclooxygenase (COX‐2) expression in the fetal brain small vessels. Similarly, injections of LPS into the uterine artery of pregnant ewes resulted in extravasation of plasma albumin into the cerebellar parenchyma of the fetus [171], suggesting that both direct and maternal exposure to endotoxin inflammatory insults can compromise the fetal NVU tightness [170, 171]. Dysregulation of NVU components was also demonstrated in a model of prolonged in utero inflammation, exposing 103/104‐day gestation fetal sheep (approximately similar to the preterm human brain between 28 and 32 weeks) to a low‐dose LPS regimen [172]. This paradigm results in a significant decreased cortical and white matter microvascular density (measured by collagen type IV morphometric analysis) and reduced pericyte microvascular coverage (detected as desmin area overlapping with the vessel basal lamina) of the ovine fetal brain. Furthermore, the authors report increased astrogliosis and decreased astrocytes' end‐foot coverage of the microvasculature in the white matter, suggesting an impaired gliovascular coupling. Prolonged in utero inflammation adversely affected multiple components of cortical and white matter cerebral NVU in the preterm ovine fetus, potentially predisposing to impaired brain development.
Stolp et al. were the first to demonstrate that changes in blood vessel tight junction distribution, caused by systemic inflammatory insults during early life, precede long‐term alterations in both NVU permeability and behaviors. In particular, they conducted longitudinal studies in neonatal, adolescent, and adult rats exposed to intraperitoneal injections of 0.2 mg·kg−1 LPS during the early postnatal period [i.e., on postnatal day (P) 0, P2, P4, P6, and P8]. The systemic inflammatory response to LPS treatment—confirmed by increased IL‐1β and TNF‐α plasma levels—resulted in increased NVU permeability only in adulthood, preceded by claudin‐5 altered distribution at an earlier time point [173, 174]. Stolp et al. [173, 175] demonstrated that NVU alterations due to inflammation during brain development can occur in several phases, each one leading to different behavioral modifications: at short‐term, juvenile animals showed alteration in the PPI paradigm, while in the long‐term, changes in NVU permeability (as shown by the sucrose permeability test) were correlated with anxiety‐related behaviors (as shown by altered responses to the dark/light test).
Maternal immune activation can alter the permeability of the NVU and placental barriers [176] and so increase the circulation of inflammatory mediators to the fetal brain, such as cytokines and reactive oxygen species, ultimately leading to behavioral changes in offspring's adult life [177]. Altogether, animal experimental studies using models of MIA (Table 3), especially LPS and PolyI:C, suggest that maternal inflammatory response induced during the early postnatal period—hallmarked by excessive cytokine secretion and signal transduction, cellular immune activation, and recruitment—has been associated with altered NVU permeability, acting as mediator of abnormal brain development, leading to long‐term behavioral as well as neurochemical changes in adult offspring [178]. Brain injuries in the setting of systemic inflammatory processes can act as pathogenic mediator, not as a single hit in time [179], but holding for months or even for years and finally contributing to postinsult neuronal deficits as long‐term ongoing process and lifelong disability, including cerebral palsy, seizure disorders, sensory impairment, and cognitive limitations.
Conclusions
Neurovascular interrelationship is essential for the proper functioning and development of the brain. Many animal models of NVU dysfunctions have been shown to be lethal, as for both the PDGFR‐β−/− and the PDGF‐B−/− mice, for which the loss of pericytes results in perinatal lethality caused by microaneurysms, mostly in the brain and kidneys [80, 180] and the claudin‐5‐deficient mice, which exhibit a selective increased permeability for molecules under 800 Da [104]. Moreover, the cerebrovascular system serves as structural track and guide for the migration of various cell types. This is the case for oligodendrocyte precursors—which use vessels to migrate and finally engage neuronal axons to be enveloped with myelin sheaths [181]—and neurons—which rely on EC cues in order to find their final position [182]. Not only does the brain vasculature influence neuronal activity, but also the opposite occurs. Neural activity can indeed promote the formation of new vessels and ultimately modify vessel density and branching [183]. Furthermore, detrimental NVU events are consistently accompanied by microglia recruitment and enhanced release of cytokines, which in turn directly affect synapse function and cognitive processes [18, 168, 184]. For all these reasons, modifications of the NVU may have long‐lasting effects on behavior, even resulting in psychiatric conditions. The undergoing SARS‐CoV‐2 pandemic stresses the need to build a solid net of clinical data and preclinical research evidence to understand not only the impact of intragravidic infections on the offspring's mental health, but also the underpinning mechanisms. Various registries, as the Pregnancy CoRonavIrus Outcomes RegIsTrY—a US study led by the University of California—or the International Registry of Coronavirus Exposure in Pregnancy—an international cohort—collect data on pregnancies and longitudinal birth outcomes, representing a valuable opportunity to collect information on possible NVU dysfunctions in the context of intragravidic infections. Therapies aimed at lowering intraventricular pressure, distortion, free iron concentration, and cytokine levels in cases of posthemorrhagic brains have been already exploited. Recently, a 10‐year follow‐up study using the Drainage, Irrigation, and Fibrinolytic Therapy therapeutic approach on preterm babies presenting GH‐IVH has shown improvements in child developmental profiles, cognitive ability, and global cerebral function, reducing the overall neurodevelopmental disability [185, 186]. The future challenge will be to better understand the mechanisms through which the development of NVU and brain vasculature can shape our behavior, particularly when under the influence of inflammatory and viral hits.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MR, EL, FM, and MM wrote and edited the paper. FM, MR, and EL prepared the figures and tables.
Acknowledgements
We thank Dr Davide Pozzi (Humanitas University) and Dr. Maria Luisa Malosio (IN‐CNR) for discussion. Work in our laboratory is supported by PRIN (Ministero dell'Istruzione, dell'Università e della Ricerca, 2017A9MK4R); Ministry of Health (Ministero della Salute RF‐201602361571); FISM (2019/R‐Single/032), Ferring COVID‐19 Investigational Grant to M.M. and by Ministero della Salute (Grant No. GR‐2018‐12367117 and GR‐2019‐12370776) to EL.
References
- 1. Brown AS & Susser ES (2008) Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr Bull 34, 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stein AD & Lumey LH (2000) The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch Famine Birth Cohort Study. Hum Biol 72, 641–654. [PubMed] [Google Scholar]
- 3. Kępińska AP, Iyegbe CO, Vernon AC, Yolken R, Murray RM & Pollak TA (2020) Schizophrenia and influenza at the centenary of the 1918–1919 Spanish influenza pandemic: mechanisms of psychosis risk. Front psychiatry 11, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torrey EF, Miller J, Rawlings R & Yolken RH (1997) Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res 28, 1–38. [DOI] [PubMed] [Google Scholar]
- 5. Brown AS & Derkits EJ (2010) Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 167, 261–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, Toovey S & Prinssen EP (2014) Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10, 643–660. [DOI] [PubMed] [Google Scholar]
- 7. Brown AS (2012) Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol 72, 1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parker SE, Lijewski VA, Janulewicz PA, Collett BR, Speltz ML & Werler MM (2016) Upper respiratory infection during pregnancy and neurodevelopmental outcomes among offspring. Neurotoxicol Teratol 57, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Al‐Haddad BJS, Jacobsson B, Chabra S, Modzelewska D, Olson EM, Bernier R, Enquobahrie DA, Hagberg H, Östling S, Rajagopal L et al. (2019) Long‐term risk of neuropsychiatric disease after exposure to infection in utero . JAMA psychiatry 76, 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown AS & Meyer U (2018) Maternal immune activation and neuropsychiatric illness: a translational research perspective. Am J Psychiatry 175, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sørensen HJ, Mortensen EL, Reinisch JM & Mednick SA (2009) Association between prenatal exposure to bacterial infection and risk of Schizophrenia. Schizophr Bull 35, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fortier ME, Luheshi GN & Boksa P (2007) Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res 181, 270–277. [DOI] [PubMed] [Google Scholar]
- 13. Lydholm CN, Köhler‐Forsberg O, Nordentoft M, Yolken RH, Mortensen PB, Petersen L & Benros ME (2019) Parental infections before, during, and after pregnancy as risk factors for mental disorders in childhood and adolescence: a nationwide Danish study. Biol Psychiatry 85, 317–325. [DOI] [PubMed] [Google Scholar]
- 14. Pedersen JM, Mortensen EL, Christensen DS, Rozing M, Brunsgaard H, Meincke RH, Petersen GL & Lund R (2018) Prenatal and early postnatal stress and later life inflammation. Psychoneuroendocrinology 88, 158–166. [DOI] [PubMed] [Google Scholar]
- 15. Andersson NW, Li Q, Mills CW, Ly J, Nomura Y & Chen J (2016) Influence of prenatal maternal stress on umbilical cord blood cytokine levels. Arch Womens Ment Health 19, 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patterson PH (2009) Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav Brain Res 204, 313–321. [DOI] [PubMed] [Google Scholar]
- 17. Pozzi D, Rasile M, Corradini I & Matteoli M (2020) Environmental regulation of the chloride transporter KCC2: switching inflammation off to switch the GABA on? Transl Psychiatry 10. 10.1038/s41398-020-01027-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corradini I, Focchi E, Rasile M, Morini R, Desiato G, Tomasoni R, Lizier M, Ghirardini E, Fesce R, Morone D et al. (2017) Maternal immune activation delays excitatory‐to‐inhibitory gamma‐aminobutyric acid switch in offspring. Biol Psychiatry 83, 680–691. [DOI] [PubMed] [Google Scholar]
- 19. Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K & Karlsson H (2002) Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 virus during pregnancy. J Neurovirol 8, 353–357. [DOI] [PubMed] [Google Scholar]
- 20. Yockey LJ, Lucas C & Iwasaki A (2020) Contributions of maternal and fetal antiviral immunity in congenital disease. Science (80‐ ) 368, 608–612. [DOI] [PubMed] [Google Scholar]
- 21. Neu N, Duchon J & Zachariah P (2015) TORCH infections. Clin Perinatol 42, 77–103, viii. [DOI] [PubMed] [Google Scholar]
- 22. Dennehy JJ (2017) Evolutionary ecology of virus emergence. Ann N Y Acad Sci 1389, 124–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khandaker GM, Zimbron J, Dalman C, Lewis G & Jones PB (2012) Childhood infection and adult schizophrenia: a meta‐analysis of population‐based studies. Schizophr Res 139, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Mascio D, Khalil A, Saccone G, Rizzo G, Buca D, Liberati M, Vecchiet J, Nappi L, Scambia G, Berghella V et al. (2020) Outcome of coronavirus spectrum infections (SARS, MERS, COVID‐19) during pregnancy: a systematic review and meta‐analysis. Am J Obstet Gynecol MFM 2, 100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC et al. (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gogtay N, Vyas NS, Testa R, Wood SJ & Pantelis C (2011) Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr Bull 37, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez‐Castroman J, Leiva‐Murillo JM, Cegla‐Schvartzman F, Blasco‐Fontecilla H, Garcia‐Nieto R, Artes‐Rodriguez A, Morant‐Ginestar C, Courtet P, Blanco C, Aroca F et al. (2019) Onset of schizophrenia diagnoses in a large clinical cohort. Sci Rep 9, 9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mandell DS, Novak MM & Zubritsky CD (2005) Factors associated with age of diagnosis among children with autism spectrum disorders. Pediatrics 116, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM & Herkenham M (2012) Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun 26, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR & Huh JR (2016) The maternal interleukin‐17a pathway in mice promotes autism‐like phenotypes in offspring. Science (80‐ ) 351 , 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patrich E, Piontkewitz Y, Peretz A, Weiner I & Attali B (2016) Maternal immune activation produces neonatal excitability defects in offspring hippocampal neurons from pregnant rats treated with poly I:C. Sci Rep 6, 19106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coiro P, Padmashri R, Suresh A, Spartz E, Pendyala G, Chou S, Jung Y, Meays B, Roy S, Gautam N et al. (2015) Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav Immun 50, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elmer BM, Estes ML, Barrow SL & McAllister AK (2013) MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J Neurosci 33, 13791–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weir RK, Forghany R, Smith SEP, Patterson PH, McAllister AK, Schumann CM & Bauman MD (2015) Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun 48, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, Shibushita M, Tatematsu T, Haruwaka K, Moorhouse AJ et al. (2020) Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep 10, 21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smolders S, Notter T, Smolders SMT, Rigo J‐M & Brône B (2018) Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun 73, 51–65. [DOI] [PubMed] [Google Scholar]
- 37. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F & Moch H (2020) Endothelial cell infection and endotheliitis in COVID‐19. Lancet 395, 1417–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buzhdygan TP, DeOre BJ, Baldwin‐Leclair A, McGary H, Razmpour R, Galie PA, Potula R, Andrews AM & Ramirez SH (2020) The SARS‐CoV‐2 spike protein alters barrier function in 2D static and 3D microfluidic in vitro models of the human blood–brain barrier. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Villabona‐Rueda A, Erice C, Pardo CA & Stins MF (2019) The evolving concept of the blood brain barrier (BBB): from a single static barrier to a heterogeneous and dynamic relay center. Front Cell Neurosci 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Virgintino D, Errede M, Robertson D, Capobianco C, Girolamo F, Vimercati A, Bertossi M & Roncali L (2004) Immunolocalization of tight junction proteins in the adult and developing human brain. Histochem Cell Biol 122, 51–59. [DOI] [PubMed] [Google Scholar]
- 41. Goasdoué K, Miller SM, Colditz PB & Björkman ST (2017) Review: The blood‐brain barrier; protecting the developing fetal brain. Placenta 54, 111–116. [DOI] [PubMed] [Google Scholar]
- 42. Saunders N, Liddelow S & Dziegielewska K (2012) Barrier mechanisms in the developing brain. Front Pharmacol 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H & Gu C (2014) Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greene C, Hanley N & Campbell M (2019) Claudin‐5: gatekeeper of neurological function. Fluids Barriers CNS 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Najjar S, Pearlman DM, Devinsky O, Najjar A & Zagzag D (2013) Neurovascular unit dysfunction with blood‐brain barrier hyperpermeability contributes to major depressive disorder: a review of clinical and experimental evidence. J Neuroinflammation 10, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Katsel P, Roussos P, Pletnikov M & Haroutunian V (2017) Microvascular anomaly conditions in psychiatric disease. Schizophrenia ‐ angiogenesis connection. Neurosci Biobehav Rev 77, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sweeney MD, Zhao Z, Montagne A, Nelson AR & Zlokovic BV (2019) Blood‐brain barrier: from physiology to disease and back. Physiol Rev 99, 21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG et al. (2019) Blood‐brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 25, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bell AH, Miller SL, Castillo‐Melendez M & Malhotra A (2019) The neurovascular unit: effects of brain insults during the perinatal period. Front Neurosci 13, 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi‐Egea J‐F, Damante G, Fleiss B, Titomanlio L & Gressens P (2015) Blood‐brain barrier dysfunction in disorders of the developing brain. Front Neurosci 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mallard C, Ek CJ & Vexler ZS (2018) The myth of the immature barrier systems in the developing brain: role in perinatal brain injury. J Physiol 596, 5655–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reynell C & Harris JJ (2013) The BOLD signal and neurovascular coupling in autism. Dev Cogn Neurosci 6, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Azmitia EC, Saccomano ZT, Alzoobaee MF, Boldrini M & Whitaker‐Azmitia PM (2016) Persistent angiogenesis in the autism brain: an immunocytochemical study of postmortem cortex, brainstem and cerebellum. J Autism Dev Disord 46, 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gardener H, Spiegelman D & Buka SL (2011) Perinatal and neonatal risk factors for autism: a comprehensive meta‐analysis. Pediatrics 128, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Preti A, Cardascia L, Zen T, Pellizzari P, Marchetti M, Favaretto G & Miotto P (2000) Obstetric complications in patients with depression–a population‐based case‐control study. J Affect Disord 61, 101–106. [DOI] [PubMed] [Google Scholar]
- 56. Gardner MR (2005) Outcomes in children experiencing neurologic insults as preterm neonates. Pediatr Nurs 31, 448, 451–456. [PubMed] [Google Scholar]
- 57. Kinney DK, Yurgelun‐Todd DA, Tohen M & Tramer S (1998) Pre‐ and perinatal complications and risk for bipolar disorder: a retrospective study. J Affect Disord 50, 117–124. [DOI] [PubMed] [Google Scholar]
- 58. Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA & Temple S (2000) Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28, 69–80. [DOI] [PubMed] [Google Scholar]
- 59. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K et al. (2010) Pericytes regulate the blood‐brain barrier. Nature 468, 557–561. [DOI] [PubMed] [Google Scholar]
- 60. Daneman R, Zhou L, Kebede AA & Barres BA (2010) Pericytes are required for blood‐brain barrier integrity during embryogenesis. Nature 468, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bauer HC, Bauer H, Lametschwandtner A, Amberger A, Ruiz P & Steiner M (1993) Neovascularization and the appearance of morphological characteristics of the blood‐brain barrier in the embryonic mouse central nervous system. Brain Res Dev Brain Res 75, 269–278. [DOI] [PubMed] [Google Scholar]
- 62. Papageorghiou AT, Kennedy SH, Salomon LJ, Ohuma EO, Cheikh Ismail L, Barros FC, Lambert A, Carvalho M, Jaffer YA, Bertino E et al. (2014) International standards for early fetal size and pregnancy dating based on ultrasound measurement of crown‐rump length in the first trimester of pregnancy. Ultrasound Obstet Gynecol 44, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Allsopp G & Gamble HJ (1979) An electron microscopic study of the pericytes of the developing capillaries in human fetal brain and muscle. J Anat 128, 155–168. [PMC free article] [PubMed] [Google Scholar]
- 64. Maxwell M, Galanopoulos T, Neville‐Golden J, Hedley‐Whyte ET & Antoniades HN (1998) Cellular localization of PDGF mRNAs in developing human forebrain. Neuropathol Appl Neurobiol 24, 337–345. [DOI] [PubMed] [Google Scholar]
- 65. Galaris G, Thalgott JH & Lebrin FPG (2019) Pericytes in hereditary hemorrhagic telangiectasia. Adv Exp Med Biol 1147, 215–246. [DOI] [PubMed] [Google Scholar]
- 66. Thalgott J, Dos‐Santos‐Luis D & Lebrin F (2015) Pericytes as targets in hereditary hemorrhagic telangiectasia. Front Genet 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U & Plesnila N (2015) Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol 78, 887–900. [DOI] [PubMed] [Google Scholar]
- 68. Dziewulska D & Lewandowska E (2012) Pericytes as a new target for pathological processes in CADASIL. Neuropathology 32, 515–521. [DOI] [PubMed] [Google Scholar]
- 69. Braun A, Xu H, Hu F, Kocherlakota P, Siegel D, Chander P, Ungvari Z, Csiszar A, Nedergaard M & Ballabh P (2007) Paucity of pericytes in germinal matrix vasculature of premature infants. J Neurosci 27, 12012–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ballabh P (2010) Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res 67, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wilson‐Costello D, Friedman H, Minich N, Fanaroff AA & Hack M (2005) Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics 115, 997–1003. [DOI] [PubMed] [Google Scholar]
- 72. du Plessis AJ & Volpe JJ (2002) Perinatal brain injury in the preterm and term newborn. Curr Opin Neurol 15, 151–157. [DOI] [PubMed] [Google Scholar]
- 73. Ballabh P, Xu H, Hu F, Braun A, Smith K, Rivera A, Lou N, Ungvari Z, Goldman SA, Csiszar A et al. (2007) Angiogenic inhibition reduces germinal matrix hemorrhage. Nat Med 13, 477–485. [DOI] [PubMed] [Google Scholar]
- 74. Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, Barnes SR, Daianu M, Ramanathan A, Go A et al. (2018) Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med 24, 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R & Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A & Zlokovic BV (2013) Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun 4, 2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M & Zlokovic BV (2013) Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer’s disease. Brain Pathol 23, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA & Zlokovic BV (2016) Accelerated pericyte degeneration and blood‐brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L et al. (2015) Blood‐brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tallquist MD, French WJ & Soriano P (2003) Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol 1, E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cuervo H, Pereira B, Nadeem T, Lin M, Lee F, Kitajewski J & Lin C‐S (2017) PDGFRβ‐P2A‐CreER(T2) mice: a genetic tool to target pericytes in angiogenesis. Angiogenesis 20, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lekic T, Manaenko A, Rolland W, Krafft PR, Peters R, Hartman RE, Altay O, Tang J & Zhang JH (2012) Rodent neonatal germinal matrix hemorrhage mimics the human brain injury, neurological consequences, and post‐hemorrhagic hydrocephalus. Exp Neurol 236, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ouellette J, Toussay X, Comin CH, Costa LF, Ho M, Lacalle‐Aurioles M, Freitas‐Andrade M, Liu QY, Leclerc S, Pan Y et al. (2020) Vascular contributions to 16p11.2 deletion autism syndrome modeled in mice. Nat Neurosci 23, 1090–1101. [DOI] [PubMed] [Google Scholar]
- 84. Spiro JE & Chung WK (2012) Simons variation in individuals project (Simons VIP): a genetics‐first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron 73, 1063–1067. [DOI] [PubMed] [Google Scholar]
- 85. Blackmon K, Thesen T, Green S, Ben‐Avi E, Wang X, Fuchs B, Kuzniecky R & Devinsky O (2018) Focal cortical anomalies and language impairment in 16p11.2 deletion and duplication syndrome. Cereb Cortex 28, 2422–2430. [DOI] [PubMed] [Google Scholar]
- 86. Horev G, Ellegood J, Lerch JP, Son Y‐EE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M et al. (2011) Dosage‐dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci USA 108, 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Novarino G, El‐Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, Silhavy JL, Kara M, Khalil RO, Ben‐Omran T et al. (2012) Mutations in BCKD‐kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tărlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, Galluccio M, Tesulov M, Morelli E, Sonmez FM et al. (2016) Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lefauconnier J‐M & Trouvé R (1983) Developmental changes in the pattern of amino acid transport at the blood‐brain barrier in rats. Dev Brain Res 6, 175–182. [DOI] [PubMed] [Google Scholar]
- 90. Braun LD, Cornford EM & Oldendorf WH (1980) Newborn rabbit blood‐brain barrier is selectively permeable and differs substantially from the adult. J Neurochem 34, 147–152. [DOI] [PubMed] [Google Scholar]
- 91. Rosanoff AJ, Handy LM, Plesset IR & Brush S (1934) the etiology of so‐called schizophrenic psychoses. Am J Psychiatry 91, 247–286. [Google Scholar]
- 92. Torrey EF, Hersh SP & McCabe KD (1975) Early childhood psychosis and bleeding during pregnancy. A prospective study of gravid women and their offspring. J Autism Child Schizophr 5, 287–297. [DOI] [PubMed] [Google Scholar]
- 93. Mirendil H, Thomas EA, De Loera C, Okada K, Inomata Y & Chun J (2015) LPA signaling initiates schizophrenia‐like brain and behavioral changes in a mouse model of prenatal brain hemorrhage. Transl Psychiatry 5, e541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hao F, Tan M, Wu DD, Xu X & Cui M‐Z (2010) LPA induces IL‐6 secretion from aortic smooth muscle cells via an LPA1‐regulated, PKC‐dependent, and p38alpha‐mediated pathway. Am J Physiol Heart Circ Physiol 298, H974–H983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li Z‐G, Yu Z‐C, Yu Y‐P, Ju W‐P, Wang D‐Z, Zhan X, Wu X‐J & Zhou L (2010) Lysophosphatidic acid level and the incidence of silent brain infarction in patients with nonvalvular atrial fibrillation. Int J Mol Sci 11, 3988–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Williams NM (2011) Molecular mechanisms in 22q11 deletion syndrome. Schizophr Bull 37, 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Greene C, Kealy J, Humphries MM, Gong Y, Hou J, Hudson N, Cassidy LM, Martiniano R, Shashi V, Hooper SR et al. (2017) Dose‐dependent expression of claudin‐5 is a modifying factor in schizophrenia. Mol Psychiatry 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Coppen AJ (1960) Abnormality of the blood‐cerebrospinal fluid barrier of patients suffering from a depressive illness. J Neurol Neurosurg Psychiatry 23, 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shalev H, Serlin Y & Friedman A (2009) Breaching the blood‐brain barrier as a gate to psychiatric disorder. Cardiovasc Psychiatry Neurol 2009, 278531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cheng Y, Desse S, Martinez A, Worthen RJ, Jope RS & Beurel E (2018) TNFα disrupts blood brain barrier integrity to maintain prolonged depressive‐like behavior in mice. Brain Behav Immun 69, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sántha P, Veszelka S, Hoyk Z, Mészáros M, Walter FR, Tóth AE, Kiss L, Kincses A, Oláh Z, Seprényi G et al. (2016) Restraint stress‐induced morphological changes at the blood‐brain barrier in adult rats. Front Mol Neurosci 8, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sharma HS, Westman J, Cervós Navarro J, Dey PK & Nyberg F (1995) Probable involvement of serotonin in the increased permeability of the blood—brain barrier by forced swimming. An experimental study using Evans blue and 131I‐sodium tracers in the rat. Behav Brain Res 72, 189–196. [DOI] [PubMed] [Google Scholar]
- 103. Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, Takahashi A, Flanigan ME, Aleyasin H, LeClair KB et al. (2017) Social stress induces neurovascular pathology promoting depression. Nat Neurosci 20, 1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M & Tsukita S (2003) Size‐selective loosening of the blood‐brain barrier in claudin‐5‐deficient mice. J Cell Biol 161, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Berton O & Nestler EJ (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 7, 137–151. [DOI] [PubMed] [Google Scholar]
- 106. Al‐Haddad BJS, Oler E, Armistead B, Elsayed NA, Weinberger DR, Bernier R, Burd I, Kapur R, Jacobsson B, Wang C et al. (2019) The fetal origins of mental illness. Am J Obstet Gynecol 221, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim J, Alejandro B, Hetman M, Hattab EM, Joiner J, Schroten H, Ishikawa H & Chung D‐H (2020) Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood‐cerebrospinal fluid barrier. PLOS Pathog 16, e1008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jarvis MA & Nelson JA (2007) Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J Virol 81, 2095–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Marcos AC, Siqueira M, Alvarez‐Rosa L, Cascabulho CM, Waghabi MC, Barbosa HS, Adesse D & Stipursky J (2020) Toxoplasma gondii infection impairs radial glia differentiation and its potential to modulate brain microvascular endothelial cell function in the cerebral cortex. Microvasc Res 131, 104024. [DOI] [PubMed] [Google Scholar]
- 110. Yamanaka J, Nozaki I, Tanaka M, Uryuu H, Sato N, Matsushita T & Shichino H (2018) Moyamoya syndrome in a pediatric patient with congenital human immunodeficiency virus type 1 infection resulting in intracranial hemorrhage. J Infect Chemother 24, 220–223. [DOI] [PubMed] [Google Scholar]
- 111. Golden MP, Hammer SM, Wanke CA & Albrecht MA (1994) Cytomegalovirus vasculitis: case reports and review of the literature. Medicine (Baltimore) 73, 246–255. [PubMed] [Google Scholar]
- 112. Lekic T, Klebe D, Poblete R, Krafft PR, Rolland WB, Tang J & Zhang JH (2015) Neonatal brain hemorrhage (NBH) of prematurity: translational mechanisms of the vascular‐neural network. Curr Med Chem 22, 1214–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dunbar M & Kirton A (2018) Perinatal stroke: mechanisms, management, and outcomes of early cerebrovascular brain injury. Lancet Child Adolesc Heal 2, 666–676. [DOI] [PubMed] [Google Scholar]
- 114. Carletti A, Colleoni GG, Perolo A, Simonazzi G, Ghi T, Rizzo N & Pilu G (2009) Prenatal diagnosis of cerebral lesions acquired in utero and with a late appearance. Prenat Diagn 29, 389–395. [DOI] [PubMed] [Google Scholar]
- 115. Nigro G, La Torre R, Sali E, Auteri M, Mazzocco M, Maranghi L & Cosmi E (2002) Intraventricular haemorrhage in a fetus with cerebral cytomegalovirus infection. Prenat Diagn 22, 558–561. [DOI] [PubMed] [Google Scholar]
- 116. Shinmura Y, Kosugi I, Aiba‐Masago S, Baba S, Yong LR & Tsutsui Y (1997) Disordered migration and loss of virus‐infected neuronal cells in developing mouse brains infected with murine cytomegalovirus. Acta Neuropathol 93, 551–557. [DOI] [PubMed] [Google Scholar]
- 117. Tsutsui Y, Kosugi I, Kawasaki H, Arai Y, Han G‐P, Li L & Kaneta M (2008) Roles of neural stem progenitor cells in cytomegalovirus infection of the brain in mouse models. Pathol Int 58, 257–267. [DOI] [PubMed] [Google Scholar]
- 118. McDonald JM, Raghuveer TS & D'Alessandro MP (2001) Can congenital CMV infection lead to intracranial hemorrhage? J Perinatol 21, 402–404. [DOI] [PubMed] [Google Scholar]
- 119. Dallar Y, Tiras U, Catakli T, Gulal G, Sayar Y, Selvar B & Alioglu B (2011) Life‐threatening intracranial bleeding in a newborn with congenital cytomegalovirus infection: late‐onset neonatal hemorrhagic disease. Pediatr Hematol Oncol 28, 78–82. [DOI] [PubMed] [Google Scholar]
- 120. Suksumek N, Scott JN, Chadha R & Yusuf K (2013) Intraventricular hemorrhage and multiple intracranial cysts associated with congenital cytomegalovirus infection. J Clin Microbiol 51, 2466–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pillay B, Ramdial PK & Naidoo DP (2015) HIV‐associated large‐vessel vasculopathy: a review of the current and emerging clinicopathological spectrum in vascular surgical practice. Cardiovasc J Afr 26, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liao K, Niu F, Hu G, Guo M‐L, Sil S & Buch S (2020) HIV Tat‐mediated induction of autophagy regulates the disruption of ZO‐1 in brain endothelial cells. Tissue Barriers 8, 1748983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. András IE, Pu H, Deli MA, Nath A, Hennig B & Toborek M (2003) HIV‐1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 74, 255–265. [DOI] [PubMed] [Google Scholar]
- 124. Mulkey SB, Vezina G, Bulas DI, Khademian Z, Blask A, Kousa Y, Cristante C, Pesacreta L, du Plessis AJ & DeBiasi RL (2018) Neuroimaging findings in normocephalic newborns with intrauterine zika virus exposure. Pediatr Neurol 78, 75–78. [DOI] [PubMed] [Google Scholar]
- 125. Mulkey SB, Bulas DI, Vezina G, Fourzali Y, Morales A, Arroyave‐Wessel M, Swisher CB, Cristante C, Russo SM, Encinales L et al. (2019) Sequential neuroimaging of the fetus and newborn with in utero zika virus exposure. JAMA Pediatr 173, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Driggers RW, Ho C‐Y, Korhonen EM, Kuivanen S, Jääskeläinen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G et al. (2016) Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374, 2142–2151. [DOI] [PubMed] [Google Scholar]
- 127. Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S & Bispo de Filippis AM (2016) Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol 47, 6–7. [DOI] [PubMed] [Google Scholar]
- 128. Martinot AJ, Abbink P, Afacan O, Prohl AK, Bronson R, Hecht JL, Borducchi EN, Larocca RA, Peterson RL, Rinaldi W et al. (2018) Fetal neuropathology in zika virus‐infected pregnant female rhesus monkeys. Cell 173, 1111–1122.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cui L, Zou P, Chen E, Yao H, Zheng H, Wang Q, Zhu J‐N, Jiang S, Lu L & Zhang J (2017) Visual and motor deficits in grown‐up mice with congenital zika virus infection. EBioMedicine 20, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shao Q, Herrlinger S, Yang S‐L, Lai F, Moore JM, Brindley MA & Chen J‐F (2016) Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 143, 4127–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Garcez PP, Stolp HB, Sravanam S, Christoff RR, Ferreira JCCG, Dias AA, Pezzuto P, Higa LM, Barbeito‐Andrés J, Ferreira RO et al. (2018) Zika virus impairs the development of blood vessels in a mouse model of congenital infection. Sci Rep 8, 12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X et al. (2020) Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mirabella F, Desiato G, Mancinelli S, Fossati G, Rasile M, Morini R, Markicevic M, Grimm C, Amegandjin C, Termanini A et al. (2020) Transient Maternal IL‐6 boosts glutamatergic synapses and disrupts hippocampal connectivity in the offspring. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S & Griffith B (2020) COVID‐19‐associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology 296, E119–E120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Viner RM & Whittaker E (2020) Comment Kawasaki‐like disease : emerging complication during the COVID‐19 pandemic. Lancet 6736, 19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kotlyar AM, Grechukhina O, Chen A, Popkhadze S, Grimshaw A, Tal O, Taylor HS & Tal R (2021) Vertical transmission of coronavirus disease 2019: a systematic review and meta‐analysis. Am J Obstet Gynecol 224, 35–53.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Woodworth KR, Olsen EO, Neelam V, Lewis EL, Galang RR, Oduyebo T, Aveni K, Yazdy MM, Harvey E, Longcore ND et al. (2020) Birth and infant outcomes following laboratory‐confirmed SARS‐CoV‐2 infection in pregnancy — SET‐NET, 16 jurisdictions, March 29–October 14, 2020. MMWR Morb Mortal Wkly Rep 69, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jain P, Thakur A, Kler N & Garg P (2020) Manifestations in Neonates Born to COVID‐19 positive mothers. Indian J Pediatr 87, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zeng L, Xia S, Yuan W, Yan K, Xiao F, Shao J & Zhou W (2020) Neonatal early‐onset infection with SARS‐CoV‐2 in 33 neonates born to mothers With COVID‐19 in Wuhan, China. JAMA Pediatr 174, 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Dammann O & Leviton A (1997) Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42, 1–8. [DOI] [PubMed] [Google Scholar]
- 141. Mor G, Aldo P & Alvero AB (2017) The unique immunological and microbial aspects of pregnancy. Nat Rev Immunol 17, 469–482. [DOI] [PubMed] [Google Scholar]
- 142. Kraus TA, Sperling RS, Engel SM, Lo Y, Kellerman L, Singh T, Loubeau M, Ge Y, Garrido JL, Rodríguez‐García M et al. (2010) Peripheral blood cytokine profiling during pregnancy and post‐partum periods. Am J Reprod Immunol 64, 411–426. [DOI] [PubMed] [Google Scholar]
- 143. Förger F & Villiger PM (2020) Immunological adaptations in pregnancy that modulate rheumatoid arthritis disease activity. Nat Rev Rheumatol 16, 113–122. [DOI] [PubMed] [Google Scholar]
- 144. Kraus TA, Engel SM, Sperling RS, Kellerman L, Lo Y, Wallenstein S, Escribese MM, Garrido JL, Singh T, Loubeau M et al. (2012) Characterizing the pregnancy immune phenotype: results of the viral immunity and pregnancy (VIP) study. J Clin Immunol 32, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Graham C, Chooniedass R, Stefura WP, Becker AB, Sears MR, Turvey SE, Mandhane PJ, Subbarao P & HayGlass KT (2017) In vivo immune signatures of healthy human pregnancy: Inherently inflammatory or anti‐inflammatory? PLoS One 12, e0177813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gomez‐Lopez N, Romero R, Xu Y, Miller D, Leng Y, Panaitescu B, Silva P, Faro J, Alhousseini A, Gill N et al. (2018) The immunophenotype of amniotic fluid leukocytes in normal and complicated pregnancies. Am J Reprod Immunol 79, e12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Opsjłn SL, Wathen NC, Tingulstad S, Wiedswang G, Sundan A, Waage A & Austgulen R (1993) Tumor necrosis factor, interleukin‐1, and interleukin‐6 in normal human pregnancy. Am J Obstet Gynecol 169, 397–404. [DOI] [PubMed] [Google Scholar]
- 148. Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA & Pittman QJ (2008) Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA 105, 17151–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Cipolla MJ, Pusic AD, Grinberg YY, Chapman AC, Poynter ME & Kraig RP (2012) Pregnant serum induces neuroinflammation and seizure activity via TNFα. Exp Neurol 234, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Cipolla MJ (2013) The adaptation of the cerebral circulation to pregnancy: mechanisms and consequences. J Cereb Blood Flow Metab 33, 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Adlinolfi M (1993) Infectious diseases in pregnancy, cytokines and neurological impairment: an hypothesis. Dev Med Child Neurol 35, 549–553. [PubMed] [Google Scholar]
- 152. Leviton A (1993) Preterm birth and cerebral palsy: is tumor necrosis factor the missing link? Dev Med Child Neurol 35, 553–558. [DOI] [PubMed] [Google Scholar]
- 153. Jiang NM, Cowan M, Moonah SN & Petri WA Jr (2018) The impact of systemic inflammation on neurodevelopment. Trends Mol Med 24, 794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hopkins SJ & Rothwell NJ (1995) Cytokines and the nervous system. I: expression and recognition. Trends Neurosci 18, 83–88. [PubMed] [Google Scholar]
- 155. Engblom D, Ek M, Saha S, Ericsson‐Dahlstrand A, Jakobsson P‐J & Blomqvist A (2002) Prostaglandins as inflammatory messengers across the blood‐brain barrier. J Mol Med 80, 5–15. [DOI] [PubMed] [Google Scholar]
- 156. Lauranzano E, Campo E, Rasile M, Molteni R, Pizzocri M, Passoni L, Bello L, Pozzi D, Pardi R, Matteoli M et al. (2019) A microfluidic human model of blood‐brain barrier employing primary human astrocytes. Adv Biosyst 1800335, 1800335. [DOI] [PubMed] [Google Scholar]
- 157. Banks WA, Ortiz L, Plotkin SR & Kastin AJ (1991) Human interleukin (IL) 1 alpha, murine IL‐1 alpha and murine IL‐1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther 259, 988–996. [PubMed] [Google Scholar]
- 158. Banks WA, Kastin AJ & Gutierrez EG (1993) Interleukin‐1 alpha in blood has direct access to cortical brain cells. Neurosci Lett 163, 41–44. [DOI] [PubMed] [Google Scholar]
- 159. Banks WA, Kastin AJ & Gutierrez EG (1994) Penetration of interleukin‐6 across the murine blood‐brain barrier. Neurosci Lett 179, 53–56. [DOI] [PubMed] [Google Scholar]
- 160. Quagliarello VJ, Wispelwey B, Long WJJ & Scheld WM (1991) Recombinant human interleukin‐1 induces meningitis and blood‐brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J Clin Invest 87, 1360–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wright JL & Merchant RE (1994) Blood‐brain barrier changes following intracerebral injection of human recombinant tumor necrosis factor‐alpha in the rat. J Neurooncol 20, 17–25. [DOI] [PubMed] [Google Scholar]
- 162. Saija A, Princi P, Lanza M, Scalese M, Aramnejad E & De Sarro A (1995) Systemic cytokine administration can affect blood‐brain barrier permeability in the rat. Life Sci 56, 775–784. [DOI] [PubMed] [Google Scholar]
- 163. Ramirez‐Celis A, Becker M, Nuño M, Schauer J, Aghaeepour N & Van de Water J (2021) Risk assessment analysis for maternal autoantibody‐related autism (MAR‐ASD): a subtype of autism. Mol Psychiatry. 10.1038/s41380-020-00998-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ & Peterson PK (1992) Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis 166, 847–853. [DOI] [PubMed] [Google Scholar]
- 165. Lee SC, Liu W, Dickson DW, Brosnan CF & Berman JW (1993) Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL‐1 beta. J Immunol 150, 2659–2667. [PubMed] [Google Scholar]
- 166. Merrill JE (1992) Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: normal and pathological. Dev Neurosci 14, 1–10. [DOI] [PubMed] [Google Scholar]
- 167. Chao CC, Hu S, Sheng WS & Peterson PK (1995) Tumor necrosis factor‐alpha production by human fetal microglial cells: regulation by other cytokines. Dev Neurosci 17, 97–105. [DOI] [PubMed] [Google Scholar]
- 168. Pozzi D, Menna E, Canzi A, Desiato G, Mantovani C & Matteoli M (2018) The communication between the immune and nervous systems: the role of IL‐1β in synaptopathies. Front Mol Neurosci 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bloise E, Petropoulos S, Iqbal M, Kostaki A, Ortiga‐Carvalho TM, Gibb W & Matthews SG (2017) Acute effects of viral exposure on P‐glycoprotein function in the mouse fetal blood‐brain barrier. Cell Physiol Biochem 41, 1044–1050. [DOI] [PubMed] [Google Scholar]
- 170. Yan E, Castillo‐Meléndez M, Nicholls T, Hirst J & Walker D (2004) Cerebrovascular responses in the fetal sheep brain to low‐dose endotoxin. Pediatr Res 55, 855–863. [DOI] [PubMed] [Google Scholar]
- 171. Hutton LC, Castillo‐Melendez M & Walker DW (2007) Uteroplacental inflammation results in blood brain barrier breakdown, increased activated caspase 3 and lipid peroxidation in the late gestation ovine fetal cerebellum. Dev Neurosci 29, 341–354. [DOI] [PubMed] [Google Scholar]
- 172. Disdier C, Awa F, Chen X, Dhillon SK, Galinsky R, Davidson JO, Lear CA, Bennet L, Gunn AJ & Stonestreet BS (2020) Lipopolysaccharide‐induced changes in the neurovascular unit in the preterm fetal sheep brain. J Neuroinflammation 17, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Stolp HB, Dziegielewska KM, Ek CJ, Habgood MD, Lane MA, Potter AM & Saunders NR (2005) Breakdown of the blood‐brain barrier to proteins in white matter of the developing brain following systemic inflammation. Cell Tissue Res 320, 369–378. [DOI] [PubMed] [Google Scholar]
- 174. Stolp HB, Dziegielewska KM, Ek CJ, Potter AM & Saunders NR (2005) Long‐term changes in blood–brain barrier permeability and white matter following prolonged systemic inflammation in early development in the rat. Eur J Neurosci 22, 2805–2816. [DOI] [PubMed] [Google Scholar]
- 175. Stolp HB, Johansson PA, Habgood MD, Dziegielewska KM, Saunders NR & Ek CJ (2011) Effects of neonatal systemic inflammation on blood‐brain barrier permeability and behaviour in juvenile and adult rats. Cardiovasc Psychiatry Neurol 2011, 469046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Chudnovets A, Lei J, Na Q, Dong J, Narasimhan H, Klein SL & Burd I (2020) Dose‐dependent structural and immunological changes in the placenta and fetal brain in response to systemic inflammation during pregnancy. Am J Reprod Immunol 84, e13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Simões LR, Sangiogo G, Tashiro MH, Generoso JS, Faller CJ, Dominguini D, Mastella GA, Scaini G, Giridharan VV, Michels M et al. (2018) Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J Psychiatr Res 100, 71–83. [DOI] [PubMed] [Google Scholar]
- 178. Monji A, Kato T & Kanba S (2009) Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci 63, 257–265. [DOI] [PubMed] [Google Scholar]
- 179. Malaeb S & Dammann O (2009) Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol 24, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Lindahl P, Johansson BR, Levéen P & Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF‐B‐deficient mice. Science 277, 242–245. [DOI] [PubMed] [Google Scholar]
- 181. Tsai H‐H, Niu J, Munji R, Davalos D, Chang J, Zhang H, Tien A‐C, Kuo CJ, Chan JR, Daneman R et al. (2016) Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science (80‐ ) 351, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Segarra M, Aburto MR, Cop F, Llaó‐Cid C, Härtl R, Damm M, Bethani I, Parrilla M, Husainie D, Schänzer A et al. (2018) Endothelial Dab1 signaling orchestrates neuro‐glia‐vessel communication in the central nervous system. Science 361, eaao2861. [DOI] [PubMed] [Google Scholar]
- 183. Lacoste B, Comin CH, Ben‐Zvi A, Kaeser PS, Xu X, Costa L F & Gu C (2014) Sensory‐related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 83, 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Tomasoni R, Morini R, Lopez‐Atalaya JPJP, Corradini I, Canzi A, Rasile M, Mantovani C, Pozzi D, Garlanda C, Mantovani A et al. (2017) Lack of IL‐1R8 in neurons causes hyperactivation of IL‐1 receptor pathway and induces MECP2‐dependent synaptic defects. Elife 6, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Whitelaw A, Jary S, Kmita G, Wroblewska J, Musialik‐Swietlinska E, Mandera M, Hunt L, Carter M & Pople I (2010) Randomized trial of drainage, irrigation and fibrinolytic therapy for premature infants with posthemorrhagic ventricular dilatation: developmental outcome at 2 years. Pediatrics 125, e852–e858. [DOI] [PubMed] [Google Scholar]
- 186. Luyt K, Jary SL, Lea CL, Young GJ, Odd DE, Miller HE, Kmita G, Williams C, Blair PS, Hollingworth W et al. (2020) Drainage, irrigation and fibrinolytic therapy (DRIFT) for posthaemorrhagic ventricular dilatation: 10‐year follow‐up of a randomised controlled trial. Arch Dis Child ‐ Fetal Neonatal Ed 105 , 466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]