

Abstract
The molecular mechanism of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) Spike protein was characterized to identify novel therapies. The impact of tofacitinib, IL‐6R Ab, or TNFi therapy was determined on Spike protein or LPS/IFN‐γ‐induced signaling, inflammation, and metabolic reprogramming in MΦs and/or rheumatoid arthritis (RA) fibroblast‐like synoviocyte (FLS). ACE2 frequency was markedly expanded in MΦs compared to T cells and RA FLS. Tofacitinib suppresses Spike protein potentiated STAT1 signaling, whereas this function was unchanged by TNFi. Tofacitinib impairs IL‐6/IFN/LPS‐induced STAT1 and STAT3 phosphorylation in RA MΦs and FLS. Interestingly, tofacitinib had a broader inhibitory effect on the monokines, glycolytic regulators, or oxidative metabolites compared to IL‐6R Ab and TNFi in Spike‐protein‐activated MΦs. In contrast, all three therapies disrupted IFN‐α and IFN‐β secretion in response to Spike protein; nonetheless, the IFN‐γ was only curtailed by tofacitinib or IL‐6R Ab. While tofacitinib counteracted MΦ metabolic rewiring instigated by Spike protein, it was inconsequential on the glycolysis expansion mediated via HK2 and/or LDHA in the activated RA MΦ and FLS. Nevertheless, the potentiated inflammatory response and the diminished oxidative phosphorylation modulated by Spike protein and/or LPS/IFN‐γ stimulation in MΦs or RA FLS were reversed by tofacitinib. In conclusion, tofacitinib suppresses MΦ inflammation and immunometabolism triggered by Spike protein and may provide a promising strategy for COVID‐19 patients.
Keywords: Macrophages, Osteoclasts, RA FLS, Spike protein, Tofacitinib
Tofacitinib has both overlapping and distinct characteristics in cells treated with Spike protein or danger signals. The mutual effect of tofacitinib on MΦ activation syndrome (MAS) in response to Spike protein or danger signal is its ability to counteract inflammation and potentiate oxidative phosphorylation. Tofacitinib distinctly reverses Spike‐protein‐induced metabolic reprogramming by restraining glycolytic intermediates.
Introduction
The rapid worldwide spread of the novel coronavirus 2019 (COVID‐19) modulated by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has become a global health emergency. Currently, there are no vaccines or therapies for COVID‐19, and the treatment strategies are based on symptomatic care [1]. The SARS‐CoV‐2 consists of a single‐stranded viral RNA that is packaged by nucleocapsid proteins surrounded by a membrane in addition to envelope and Spike protein [1, 2]. Both SARS‐CoV RNA and envelope are indispensable for disease activity, as deletion of the envelope protein modestly negates virus growth in tissue culture and obstructs virulence in murine models [3].
SARS‐CoV‐2 infection activates excessive innate response, along with dysregulation of adaptive immune defense, resulting in tissue damage at the site and systemic level [4]. Monocytes and macrophages (MΦs) are the front‐line defense against SARS‐CoV‐2 infection. Recent studies have shown that the size and function of circulating CD14+ monocytes alter in patients with COVID‐19 compared to healthy individuals (medRxiv 2020.03.24.20042655) [5]. Interestingly, monocytes from COVID‐19+ patients express CD68+CD80+CD163+CD206+ that are markers for M1 and M2 MΦs (medRxiv 2020.03.24.20042655). Nevertheless, the dominance of the inflammatory phenotype in severe COVID‐19 patients is indicative of MΦ activation syndrome that accompanies increased serum levels of IL‐6, IL‐1β, IL‐8, GM‐CSF, CCL2, and TNF‐α, outweighing IL‐10 concentration [4]. It has been reported that GM‐CSF produced from CD14+CD16+ monocytes amplifies the inflammatory response in the lungs by promoting monocyte migration and subsequent damage (medRxiv 2020.02.12.945576) (PMCID: PMC7108005). CCL2 and CCL7 enriched in bronchoalveolar lavage fluid (BAL) from COVID‐19 patients trigger monocyte recruitment as well as M1 and M2 differentiation depending on the inflammatory or recovery status [1].
Macrophages infected with SARS‐CoV‐2 secrete elevated levels of IL‐6, which is considered to be one of the main markers of SARS‐CoV‐2 infection [6]. Moreover, in severe SARS‐CoV‐2 patients, the expansion of IL‐6 levels was accompanied by IFN‐α/β response [7] and JAK/STAT activation [4, 8]. Therefore, the impact of IL‐6R antibody (Ab) and JAK/STAT inhibitors (i) are being tested in a clinical trial for COVID‐19 therapy. Anti‐IL‐6R Ab therapy via Tocilizumab increased the serum IL‐6 levels, while the diminished C reactive protein in COVID‐19 patients resulted in improved clinical outcomes [9, 10]. Nevertheless, JAK/STATi may have a greater advantage over IL‐6 blockade as it impairs the function of multiple cytokines involved in COVID‐19 pathology including IL‐6, IL‐7, G‐CSF, GM‐CSF, and IFN‐γ [8]. Treatment with a JAK1/2i, ruxolitibm, is shown to be safe for COVID‐19 patients in phase I, then again its overall effect may depend on its suppression on IFN‐α/β function during early or late disease activity [11]. Since tofacitinib (Tofa) is utilized as a standard of care, this treatment strategy was compared in Spike‐protein‐activated macrophages with other cell types in rheumatoid arthritis (RA) patients.
In this study, we illustrate that the Spike protein stimulation of myeloid cells activates STAT1 signaling, along with potentiating the expression of M1 monokines, type I IFN response, and glycolytic intermediates in addition to diminishing the levels of oxidative mediators. We found that Tofa is superior to IL‐6R antibody (Ab) or TNF inhibitor (i) therapy in reversing the Spike‐protein‐induced inflammatory and metabolic phenotypes in MΦs. Remarkably, Tofa treatment restrained IL‐6/IFN/LPS‐induced STAT1 and STAT3 phosphorylation and inflammatory response in RA MΦs and fibroblast‐like synoviocyte (FLS), which coincided with the upregulation of peroxisome proliferator‐activated receptor γ (PPARγ). Furthermore, Tofa treatment impaired Th1 cell polarization driven by IL‐12 or Spike protein. Taken together, the efficiency of Tofa over IL‐6R Ab or TNFi therapy is manifested by STAT1 deactivation and shifting the glycolytic to oxidative metabolism in Spike‐protein‐activated MΦs, indicating that Tofa can be utilized as a promising strategy for COVID‐19 patients.
Results
Tofa therapy disrupts M1 MΦ and Th1 cell responses modulated by Spike protein
In severe COVID‐19 patients, the number of classical monocytes is increased along with an amplified inflammatory response [7]. Hence, we asked whether Spike protein hyperactivates monocyte‐derived MΦs and if this phenotype could be intercepted by RA standard of care therapies. The frequency of ACE2 was markedly higher in CD14+ monocytes (17×) compared to CD4+ T cells (Fig. 1A). In MΦs, stimulation with Spike protein triggered STAT1 phosphorylation but not STAT3 (Fig. 1B). We showed that Tofa or IL‐6R Ab treatment restrained baseline pSTAT3 signaling, while pSTAT1 was abrogated by Tofa and suppressed via IL‐6R Ab therapy. At the onset of these studies, the effective dose of Tofa was determined based on the dose–response experiments that were accomplished in MΦs and FLS (Figs. 3 and 4). We found that stimulation with Spike protein remodels naïve cells to M1 MΦs that secrete IL‐1β (4×), TNF‐α (5×), IL‐6 (84×), IL‐8 (3×), and CCL2 (2×) but not CCL5 (Fig. 1C–I). Neither Tofa, IL‐6R Ab, or TNFi treatments impacted Spike‐protein‐induced IL‐1β and IL‐8 from MΦs (Fig. 1D and G), whereas treatment with Tofa impeded TNF, IL‐6, and CCL2 production; this process was unaffected by IL‐6R Ab or TNFi therapy (IL‐6R Ab or TNFi was excluded in IL‐6 or TNF ELISA, respectively) (Fig. 1E‐F and H). We noted that type I (α and β) or II (γ) IFN responses amplified by Spike protein in MΦs (type I) or PBMCs (type II) were diminished by Tofa, IL‐6R Ab, or TNFi (expect for IFN‐γ) therapy (Fig. 1J–L). To further authenticate the specificity of Spike protein, we showed that ACE2 knockdown abrogates Spike‐protein‐induced CCL2 secretion (Supporting Information Figs. S1 and S2). Collectively, in MΦs, Spike‐protein‐driven STAT1 signaling and inflammatory phenotype are more effectively impeded by Tofa relative to IL‐6R Ab or TNFi treatment.
Figure 1.
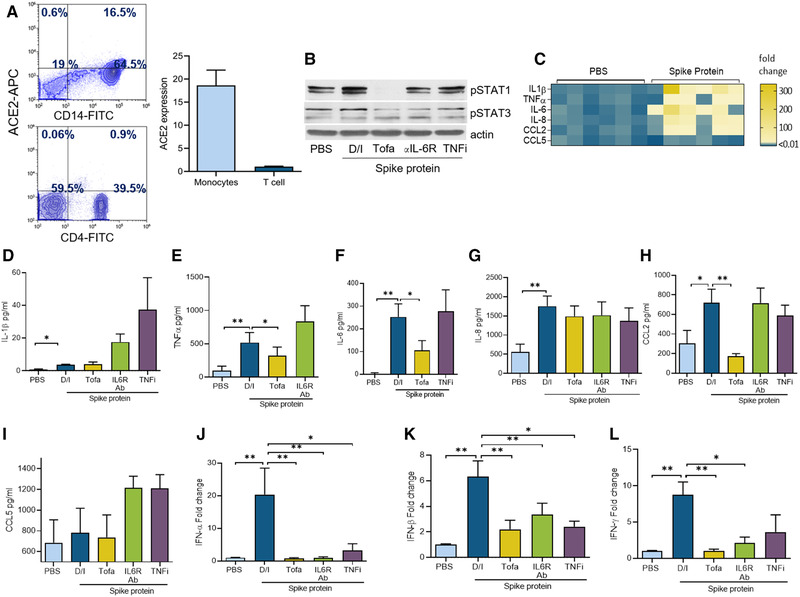
Tofacitinib restrains MΦ inflammatory phenotype as well as type I and II IFN responses activated by Spike protein. (A) ACE2 frequency was quantified in negatively selected blood monocytes (for CD14 staining) or PBMCs (for CD4 staining) using ACE2, CD14, or CD4 staining and measured by flow cytometry, n = 3 samples from two independent experiments; the gating strategy is demonstrated in Supplementary 4–6. (B) MΦs were pretreated with (DMSO/IgG control; D/I), Tofa (10 μM), anti‐IL‐6R Ab (10 μg/mL), or TNFi (10 μg/mL) o/n before PBS and Spike protein treatment (Exonbio; 30 nM) for 30 min and cell lysates were probed for pSTAT1, pSTAT3, and actin (loading control) by Western blot, n = 1 sample; data representative of three independent experiments (raw WB images are shown in Supporting Information 3). (C) MΦs were untreated (PBS) or treated with Spike protein (30 nM) for 8 h before evaluating the transcription regulation of the proinflammatory factors by real‐time RT‐PCR normalized to GAPDH, n = 3 samples from two independent experiments. MΦs (D‐K) or PBMCs (L) were pretreated with D/I, Tofa (10 μM), IL‐6R Ab (10 μg/mL), or TNFi (10 μg/mL) o/n before treating the cells with PBS or Spike protein (30 nm) for 8 h. Protein or mRNA levels of IL‐1β, TNF, IL‐6, IL‐8, CCL2, CCL5, IFN‐α, IFN‐β (D‐K) and IFN‐γ (L) was measured by ELISA or RT‐PCR, n = 3 samples from three independent experiments. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. The data were also analyzed using a two‐tailed Student's t‐test for comparisons among two groups, or one‐way ANOVA followed by Tukey's multiple comparison test among multiple groups.
Figure 3.
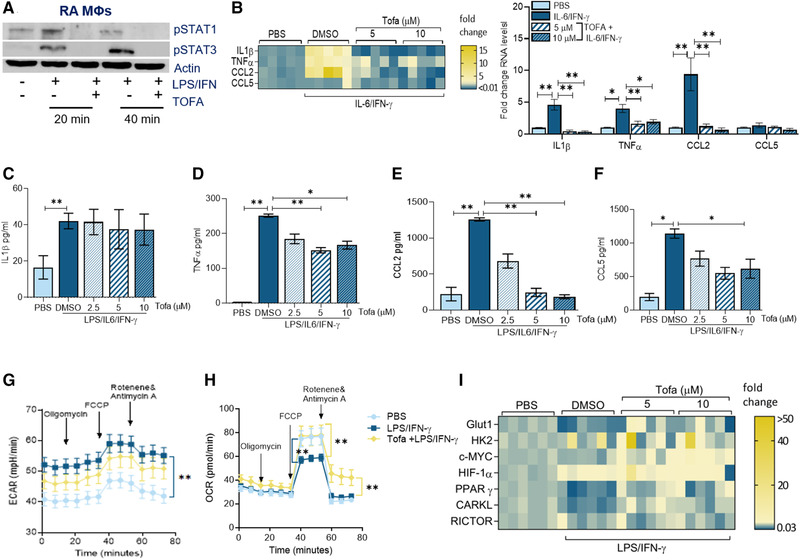
Tofacitinib impairs RA MΦ inflammatory response by expanding mitochondrial oxidative phosphorylation. (A) RA MΦs were either untreated (PBS) or treated with IL‐6/IFN‐γ (100 ng/mL each) in the presence of DMSO (‐) or Tofa (10μM; +) for 20 min or 40 min before detecting pSTAT1 and pSTAT3 or actin (loading control) by Western blotting, n = 1 sample, data representative of three independent experiments (raw WB images are shown in Supporting Information 3). (B) RA MΦs were pretreated with DMSO or Tofa (5–10 μM) o/n before PBS or IL‐6/IFN‐γ (100 ng/mL each) stimulation for 6 h followed by evaluating the transcriptional regulation of inflammatory markers including IL‐1β, TNF‐α, CCL2, and CCL5, quantified by real‐time RT‐PCR normalized to GAPDH, n = 3 samples are from two independent experiments (shown as a heatmap or bar graph). RA MΦs were pretreated with DMSO or Tofa (5–10 μM) o/n before PBS or IL‐6/IFN‐γ (100 ng/mL each) stimulation for 24 h before analyzing the protein levels of IL‐1β (C), TNF‐α (D), CCL2 (E), and CCL5 (F) by ELISA, n = 3–4 samples from two independent experiments. ECAR (G) or OCR (H) capacity was measured using Mito Stress Test Kit in RAW cells (5000/well) pretreated with PBS, LPS/IFN‐γ (100 ng/mL each) with DMSO or Tofa (10 μM) o/n before taking 12 measurements from 0 to 75 min by Seahorse Bioscience XF96 analyzer, n = 3 samples from two independent experiments. (I) RA MΦs were pretreated with DMSO, 5 μM or 10 μM of Tofa o/n before PBS or LPS/IFN‐γ (100 ng/mL each) stimulation for 6 h. Transcriptional regulation of the glycolytic (GLUT1, HK2, cMYC, and HIF1α) and the oxidative (PPARγ, CARKL, and RICTOR) intermediates were determined by real‐time RT‐PCR and normalized to actin, n = 3 samples from two independent experiments (statistical analysis is shown in Supporting Information Fig. S7). The data are shown as mean ± SEM, *p < 0.05 and **p < 0.01. One‐way ANOVA followed by Tukey's multiple comparison test among multiple groups.
Figure 4.
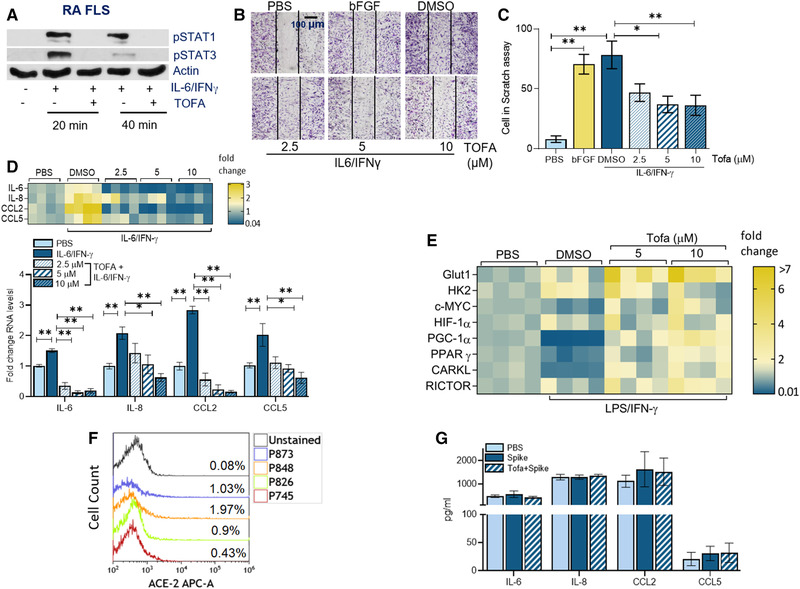
Tofacitinib dysregulates RA FLS‐induced inflammatory phenotype by potentiating mitochondrial oxidative phosphorylation. (A) RA FLS was either untreated or treated with IL‐6/IFN‐γ (100 ng/mL each) in the presence of DMSO (‐) or Tofa (10μM; +) for 20 min or 40 min before detecting pSTAT1 and pSTAT3 or actin (loading control) by Western blotting, n = 1 sample, data representative of four independent different experiments (raw WB images are shown in Supporting Information 3). (B and C) Impact of Tofa was determined on RA FLS migration using an in vitro scratch assay, counting the number of cells that re‐populated in the scratch area within 24 h. Image (B) and the number of RA FLS (C) are shown when cells were untreated (‐Ctrl) or treated with bFGF (+Ctrl) or IL‐6/IFN‐γ (100 ng/mL each) with DMSO or Tofa (2.5–10 μM), n = 1 sample from four independent experiments. (D) RA FLS was pretreated with DMSO or Tofa (2.5–10 μM) o/n before PBS or IL‐6/IFN‐γ (100 ng/mL each) stimulation for 6 h followed by evaluating the transcriptional regulation of inflammatory markers, which were normalized to GAPDH, n = 3 samples from two to three independent experiments (data are presented as a heatmap or bar graph [protein levels are shown in Supporting Information Figs. S3 and S4]). (E) RA FLS was pretreated with DMSO; 5 μM or 10 μM of Tofa o/n before PBS or LPS+IFN‐γ (100 ng/mL each) stimulation for 6 h and transcriptional regulation of the metabolic intermediates were determined by real‐time RT‐PCR, n = 4 samples are from two independent experiments (statistical analysis is shown in Supporting Information Fig. S5). (F) ACE2 frequency was quantified by cell number (cell count) compared to unstained control in RA FLS by flow cytometry, n = 1 sample, data representative of four independent experiments. (G) RA FLS was pretreated with DMSO or tofacitinib (10 μM) o/n before treating the cells with PBS or Spike protein (30 nm) for 8 h and quantifying inflammatory factors by ELISA, n = 3 samples are from two independent experiments. The data are shown as mean ± SEM, *p < 0.05 and **p < 0.01. The data were also analyzed using a two‐tailed Student's t‐test for comparisons among two groups, or one‐way ANOVA followed by Tukey's multiple comparison test among multiple groups.
Spike‐protein‐induced MΦ immunometabolism is reversed by Tofa therapy
Next, experiments were performed to elucidate whether Spike protein modulates MΦ metabolic activity and if this function is compromised by Tofa therapy. In MΦs activated by Spike protein, transcription of the master regulators of glycolysis, hexokinase (HK)2 (3x), cMYC (2x), and hypoxia‐inducible factor (HIF1α)(3x) was markedly upregulated, while glucose transporter (GLUT)1, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase (PFKFB)3 and Regulatory‐Associated Protein of TOR (RAPTOR) were not implicated in the process (Fig. 2A; Supporting Information Figs. S3–S5). Interestingly, Tofa selectively inhibited the expression of cMYC and HK2 expanded by Spike protein (Fig. 2B and C). However, TNFi therapy was inconsequential on Spike‐protein‐induced glycolytic genes, and IL‐6R Ab treatment only suppressed cMYC transcriptional regulation (Fig. 2B–D). We found that while levels of distinct oxidative metabolites, such as PPARγ and citrate, were potentiated by Tofa, the concentration of succinate was unchanged by all three biologic treatments in Spike‐protein‐activated MΦs (Fig. 2E and F, Supporting Information Fig. S8). Interestingly, similar to Tofa, IL‐6R Ab therapy reversed the transcriptional downregulation of CARKL (in the phosphate pentose pathway) modulated by Spike protein (Supporting Information Fig. S7). While ECAR was amplified by Spike protein, the OCR deregulation observed in Spike‐protein‐stimulated MΦs was counteracted by Tofa and IL‐6R Ab therapy (Fig. 2G and H). Moreover, Spike protein expansion of lactate secretion (but not pyruvate) and LDHA expression was impaired by Tofa, whereas IL‐6R Ab therapy was ineffective on this manifestation (Fig. 2I–K). Overall, Spike protein rewires naïve cells into hypermetabolic MΦs; this phenotype is inhibited by Tofa through repression of glycolysis and augmentation of oxidative phosphorylation.
Figure 2.
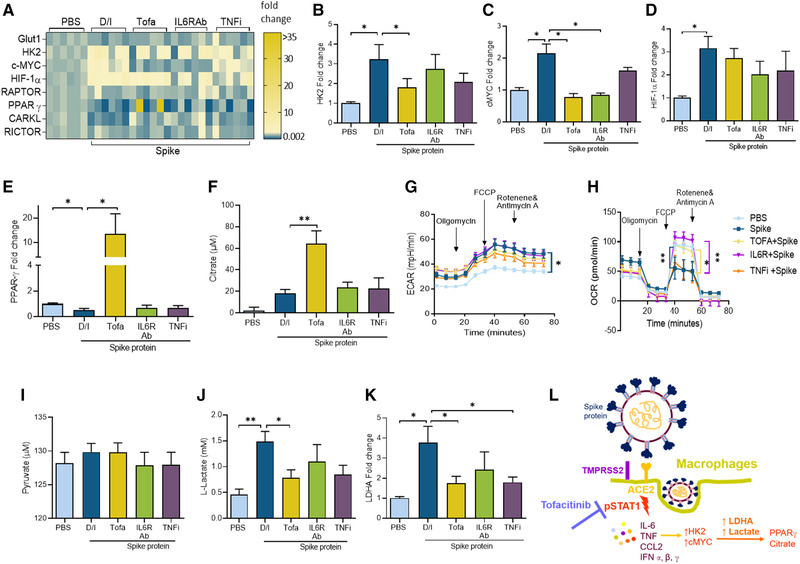
Tofacitinib reverses Spike‐protein‐induced MΦ metabolic rewiring. MΦs were pretreated with DMSO/IgG, Tofa (10 μM), 10 μg/mL of anti‐IL‐6R Ab, or TNFi o/n before PBS or Spike protein (30 nM) treatment for 8 h. The transcriptional regulation of the metabolic intermediates quantified by real‐time RT‐PCR and normalized to GAPDH is presented as a heatmap (A) or data are shown as bar graphs for HK2, cMYC, HIF1α, PPARγ, and citrate (B–F), n = 3 samples from two independent experiments. ECAR (G) and OCR (H) were measured using Mito Stress Test Kit in RAW cells (5000/well) that were untreated (PBS) or stimulated with Spike protein (300 nM plus D/I) or with Tofa (10 μM), 10 μg/mL of anti‐IL‐6R Ab or TNFi for 24 h before taking 12 measurements from 0 to 75 min by Seahorse Bioscience XF96 analyzer, n = 3 replicates from independent three experiments. Conditioned media were obtained from MΦs pretreated with D/I, Tofa (10 μM), 10 μg/mL of anti‐IL‐6R Ab, or TNFi o/n before PBS or Spike protein (30 nM) 8 h treatment before quantifying pyruvate (I) and lactate (J) by colorimetric assay or LDHA transcriptional regulation by real‐time RT‐PCR and normalized to GAPDH (K), n = 3 samples from three independent experiments. (L) Schematic figure illustrates Spike protein's mechanism of action in MΦs. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. One‐way ANOVA followed by Tukey's multiple comparison test among multiple groups.
Tofa impairs RA MΦ inflammatory phenotype by expanding mitochondrial oxidative phosphorylation
Given that treatment with Tofa restricted Spike‐protein‐induced MΦ hyperactivation, we asked whether this strategy functions comparably in response to M1 regulators in RA MΦs. We authenticated that Tofa therapy disrupts STAT1 and STAT3 phosphorylation in RA MΦs following 20 and 40 min of LPS/IFN‐γ stimulation in RA MΦs (Fig. 3A). Transcription of IL‐1β (15×), TNF‐α (2×), and CCL2 (up to14×) but not CCL5 was diminished by Tofa therapy in IL‐6/IFN‐γ‐activated RA MΦs (Fig. 3B). While IL‐1β secretion was unaffected by Tofa treatment, production of TNF‐α, CCL2, and CCL5 from RA MΦs was significantly reduced (2–7×) at 10 μM in response to LPS/IL‐6/IFN‐γ stimulation (Fig. 3C–F). Interestingly, Tofa therapy was inconsequential on LPS/IFN‐γ‐potentiated ECAR, whereas it reversed the LPS/IFN‐γ‐mediated suppression on OCR between 40 and 80 min (Fig. 3G and H). Corroborating these findings, we found that Tofa was unable to impair the glycolytic activity amplified by HK2, HIF1α, and lactate dehydrogenase (LDHA) in the activated RA MΦs (Supporting Information Figs. S1 and S2). On the contrary, Tofa could normalize the LPS/IFN‐γ dysregulated oxidative phosphorylation by expanding PPARγ transcription (2.5×) (Fig. 3I, Supporting Information Fig. S1). Collectively, these results suggest that Tofa therapy negates RA M1 MΦ rewiring by rebalancing the metabolic reprogramming toward oxidative phosphorylation.
Tofa interferes with RA FLS migration, inflammatory phenotype, and oxidative activity
Next, experiments were performed to characterize the influence of Tofa treatment on RA FLS pathogenesis. We showed that blockade of STAT1/STAT3 activation by Tofa intercepted IL‐6/IFN‐γ‐induced RA FLS migration at 5 and 10 μM (Fig. 4A–C). Similarly, Tofa therapy impaired IL‐6/IFN‐γ‐mediated IL‐6 (4–8×), IL‐8 (2–4×), CCL2 (12–18×), and CCL5 (2–3×) transcriptional upregulation (Fig. 4D). However, in RA FLS, Tofa primarily suppressed LPS/IL‐6/IFN‐γ‐induced CCL2 and CCL5 production (Supporting Information Figs. S3 and S4). It was noted that the glycolytic intermediate, HK2, enhanced by LPS/IFN in RA FLS was unchanged by Tofa therapy (Fig. 4E, Supporting Information Fig. S5). In contrast, transcriptional downregulation of PPARγ and PGC1α by LPS/IFN‐γ was overturned by Tofa treatment at 10 μM (4×, Fig. 4E, Supporting Information Fig. S5). Unlike MΦs, ACE2 was expressed on 1% of RA FLS; consequently, these cells were unresponsive to Spike protein activation (Fig. 4F and G). Taken together, Tofa can mitigate RA pannus formation by potentiating oxidative remodeling of the inflammatory RA FLS and MΦs.
Tofa alleviates RA adaptive response and bone erosion
To determine the cellular and molecular mechanisms of Tofa therapy, the impact of this treatment was examined on Th1/Th17 cell differentiation and RA bone destruction. We demonstrated that RA Th1/Th17 reprogramming was counteracted by Tofa therapy (Fig. 5A–C). Moreover, IL‐6‐mediated osteoclast formation was repressed by Tofa, in part, by inhibiting IL‐1β and CCL2 expression (Fig. 5D and E). On the contrary, RANK, RANKL, and CTSK were unaffected by Tofa therapy in IL‐6‐activated osteoclast progenitor cells (data not shown). In short, treatment with Tofa can influence RA innate and adaptive responses.
Figure 5.
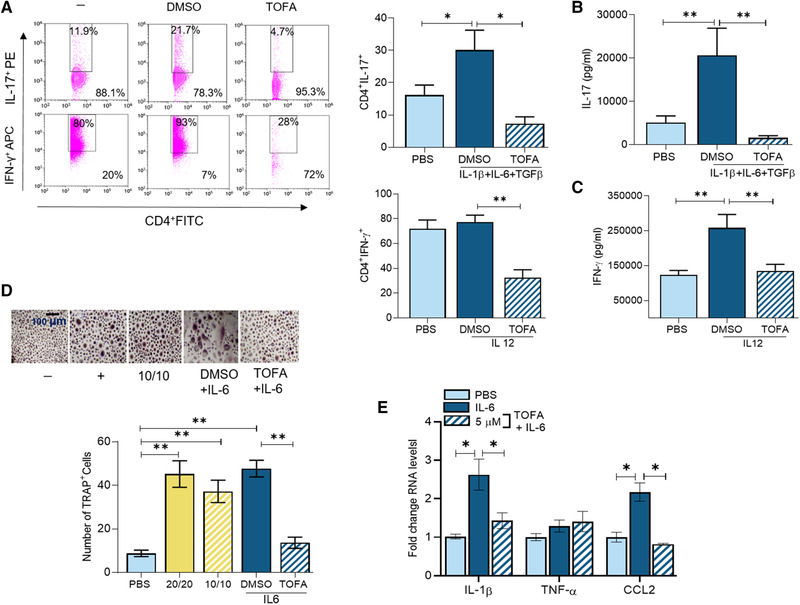
Tofacitinib disrupts Th1/Th17 cell polarization and osteoclastogenesis in RA patients. Frequency of Th1 and Th17 cells (A) measured by flow cytometry or protein levels of IL‐17 (B) and IFN‐γ (C) measure by ELISA were determined in RA PBMCs in the presence of CD3 Ab plus CD28 Ab (0.25 μg/mL each), which were either untreated (PBS) or treated with IL‐12 (10 ng/mL; Th1 +Ctl), TGF‐β (4 ng/mL) and IL‐1β + IL‐6 (20 ng/mL; Th17 +Ctl) in the presence of DMSO or Tofa (10 μM) for 4 days and cells and conditioned media were harvested for quantifying Th1 or Th17 cell frequency or IFN‐γ or IL‐17 production, n = 3 samples are from three independent experiments. (D) RA PBMCs were cultured in suboptimal levels of RANKL/M‐CSF (10 ng/mL each; 10/10) alone or with IL‐6 (200 ng/mL) in the presence of DMSO or Tofa (5 μM) for 14–21 days. The negative control was cells cultivated in 10% FBS/αMEM and the positive control was 20 ng/mL of RANKL/M‐CSF. The number of TRAP+ cells/HPF was counted on microscopic images taken at 10× magnification. (E) Osteoclast progenitor cells were either untreated (PBS) or treated with IL‐6 (100 ng/mL each) in the presence of DMSO or Tofa (5 μM) for 7 h before quantifying IL‐1β, TNF‐α, or CCL2 by real‐time RT‐PCR and normalized to GAPDH, n = 3 samples are from three independent experiments. The data are shown as mean ± SEM, * < 0.05 and **p < 0.01. The data were also analyzed using a two‐tailed Student's t‐test for comparisons among two groups, or one‐way ANOVA followed by Tukey's multiple comparison test among multiple groups.
Notably, our overall goal was to characterize the mechanism of Tofa, IL‐6R Ab, and TNFi action on Spike‐protein‐induced pathology. Moreover, the global effect of Tofa therapy was compared in Spike‐protein‐activated MΦs with RA cells that receive this therapeutic compound as a standard of care. As such, we observed that Tofa therapy had both overlapping and distinct characteristics in cells treated with Spike protein or danger signals.
The limitation of this study was our inability to access an adequate number or volume of blood samples from ongoing UIC clinical trials treating COVID‐19 patients with JAK/STAT inhibitors or IL‐6R Ab to perform well‐powered experiments. Hence, rigorous studies were conducted utilizing normal and RA human samples.
Discussion
We revealed that the immunopathology provoked by Spike protein stimulation in ACE2+ MΦs was triggered by STAT1 phosphorylation. It was noted that Tofa therapy could more effectively disrupt Spike‐protein‐driven MΦ inflammatory and metabolic phenotypes (Fig. 2L) compared to TNFi or IL‐6 Ab treatment. In contrast, type I IFN cytokines potentiated in response to Spike protein were similarly intercepted by all three biologic therapies utilized in this study. Furthermore, Tofa therapy curtailed Spike‐protein‐enhanced IFN‐γ expression in addition to the polarization of Th1/Th17 cells. While Tofa can effectively nullify glycolysis (HK2, cMYC, LDHA, and lactate) potentiated by Spike protein in myeloid cells, it is ineffective on the metabolic activity (HK2) amplified in the activated RA macrophages and FLS. In short, disruption of inflammatory response and restoration of oxidative phosphorylation are overlapping characteristics that are cultivated by Tofa therapy in macrophages and/or FLS activated by Spike protein or danger signal.
We found that the elevated responsiveness to Spike protein in MΦs (∼17–25%) relative to T cells or RA FLS (1%) was due to cell surface ACE2 expression. Recent studies demonstrate that the number of ACE2+ monocytes (∼90%) is similar in healthy and COVID‐19+ patients, however infected monocytes are morphologically bigger (medRxiv 2020.03.24.20042655). Similarly, we observed that the number of ACE2+CD11b+ myeloid cells was close to 70%, yet monocyte negative selection narrowed these cells to 20%, partially by eliminating neutrophils. Further, the necessity of hACE2 function was characterized in transgenic mice where these mice in contrast to WT animals were highly susceptible to SARS‐CoV‐2 infection [12]. hACE2 transgenic mice infected with SARS‐CoV‐2 displayed a significant increase in lung infiltration of MΦs and T cells [12]. Spike protein facilitates the entry of viral RNA by binding to its receptor‐binding domain in the S1 subunit to ACE2 on the host cell [13]. Subsequently, the viral and host membranes are fused via the Spike protein S2 subunit and TMPRSS2 [14]. Notably, the promising COVID‐19 vaccines target Spike protein and its receptor‐binding domain, which are responsible for ACE2 interaction [15, 16].
Inflammatory MΦs and T cells were absent in lung tissues of IFNAR‐/‐ compared to WT mice that were infected with SARS‐CoV2 and received AAV‐hACE2 administration, suggesting that type‐I IFN and its downstream pathways are essential for SARS‐CoV‐2 infection [17]. We showed that activation of STAT1 or STAT3 signaling was critical for IFN‐α, ‐β, and ‐γ function; while Tofa or IL‐6R Ab therapy impaired both type‐I and ‐II responses, the efficiency of TNFi was restricted to type‐I. Type‐I IFN is rapidly produced because of its antiviral activity and the ability to regulate innate immunity [18]. During acute infection, IFN type I produces CCL2 to recruit monocytes as well as to sustain antigen presentation and MHC II expression [19], whereas during chronic infection, type‐I IFN suppresses CXCL1 and CXCL2 secretion and downregulates the IFN‐γR expression on MΦs to increase the viral load and deplete CD4+T cells [18, 20]. We exhibited that Tofa therapy disrupts MΦ‐induced Th1 response (T cells express 1% ACE2) driven by Spike protein in addition to the polarization of Th1/Th17 cell cultivated by IL‐12 or IL‐6/IL‐1β. Our data indicate that Tofa therapy can be used in COVID‐19 patients to diminish the M1 MΦ‐induced Th1 cell cross‐regulation, consequently preventing T cell exhaustion. Nevertheless, the relationship between SARS‐CoV‐2 infection and type‐I IFN is complicated as two recent studies have shown contrasting data. Corroborating our findings, single‐cell transcriptome analysis of classical monocytes from severe COVID‐19+ patients revealed that type‐1 IFN was co‐expressed and capable of exacerbating the TNF/IL‐1β inflammatory phenotype [7]. On the contrary, others have shown that plasma levels of IFN‐α were low and IFN‐β was absent in severe compared to mild‐to‐moderate COVID‐19 patients [6]. Perhaps the distinct observations between the two studies were due to differences in detecting mRNA versus protein levels; as potentiated type‐I, IFN responses are short and hard to capture in plasma.
Interestingly, peripheral blood (PB) extracted from COVID‐19+ patients express CD11b, CD68, CD80, CD163, and CD206 MΦs that are markers for M1 and M2 cells (medRxiv 2020.03.24.20042655). Similar to the observations in PB, there were four different MΦ subtypes identified in COVID‐19+ BAL. In groups 1 and 2, transcription of NF‐κB, STAT1/2, and IRF1/2 was amplified and expressed s100A8, CCL2, CCL3, and CXCL10 [21]. Distinct from 1 and 2 subtypes, MΦs in groups 3 or 4 expressed oxidative mediators such as PPARα and CREB1 or PPARγ and CEBPB [21]. These results indicate that the highly plastic monocytes or MΦs in the blood or lung are capable of co‐adapting their functional diversity with the patient's immune response.
Earlier studies have postulated that IL‐6 and IFN‐mediated signaling is responsible for JAK/STAT activation in COVID‐19+ patients [22, 23]. Our results imply that Tofa intercepts Spike‐protein‐induced STAT1 phosphorylation 30 min postactivation in MΦs; rather than indirectly influencing STAT1/STAT3 activation through IL‐6 and IFN signaling. One of the many biomarkers for COVID‐19 positivity is escalated LDHA that catalyzes the conversion of pyruvate to lactate [24, 25]. Intriguingly, we showed that pyruvate levels were unchanged in Spike‐protein‐stimulated MΦs; meanwhile, lactate concentration was enhanced by Spike protein and diminished by Tofa but not IL‐6R Ab or TNFi. Surprisingly, we exhibited that LDHA transcription upregulated in Spike‐protein‐stimulated MΦs was abrogated by Tofa and TNFi therapy. Extending our findings, others have shown that glycolysis is critical for SARS‐C0V‐2 replication and monocyte inflammatory response [26].
In line with these observations, selected glycolytic modulators, cMYC, HIF1α, or HK2, were amplified in response to Spike protein or LPS/IFN‐γ; however, transcription of GLUT1, PFKFB3, and RAPTOR remained unaffected. In contrast to our findings, a recent study has shown that in SARS‐CoV‐2‐infected monocytes, transcription of GLUT1, PFKFB3, and PKM2 was upregulated [26]. Furthermore, repression of oxidative regulators such as PPARγ and citrate and induction of glycolytic intermediates including cMYC or HK2 by Spike protein were overturned by Tofa and/or IL‐6 Ab therapy but not TNFi. Macrophage hyperactivation syndrome driven by Spike protein or LPS/IFN expanded the glycolytic activity as exhibited by ECAR quantification. Still, Tofa primarily mitigates MΦ or RA FLS inflammatory phenotype via expansion of oxidative phosphorylation through PPARγ as displayed by OCR. Others have shown that the blood glucose levels were not impacted by Tofa therapy in obese mice [27], which is consistent with the lack of robust influence of Tofa on glycolysis. The potency of Tofa over IL‐6R Ab or TNFi therapy on dysregulating the Spike‐protein‐induced MΦ inflammatory or metabolic activity highlights its efficiency in COVID‐19 therapy. Our findings are limited to human cells, hence additional in vivo studies are required to compare the effectiveness of these biologics in SARS‐COV‐2 infection in the K18‐hACE2 transgenic murine model. Moreover, we were unable to obtain access to the blood samples procured from ongoing UIC clinical trials treating COVID‐19 patients with Baricitinib plus Remdesivir or Sarilumab pre‐ and posttherapy. Nevertheless, a clinical trial is in progress for evaluating the significance of Tofa therapy on SARS‐CoV2 pneumonia in Italy (ClinicalTrials.gov Identifier: NCT04332042).
We found that RA FLS migration and inflammatory response by IL‐6/IFN‐γ was intercepted via Tofa, while these cells were unresponsive to Spike protein due to negligible ACE2 expression (∼1%). Interestingly, we demonstrated that Tofa treatment in RA FLS, like MΦs, dampens the inflammatory response in part via amplifying PGC1α, PPARγ, and CARKL expression. In contrast to our findings, a recent study showed that glycolytic intermediates, HIF1α and HK2 transcription, were downregulated by Tofa therapy in RA FLS and explants [28]. Corroborating our data, these investigators exhibited that Tofa was capable of shifting the glycolytic balance into oxidative phosphorylation in RA FLS by reducing mitochondrial membrane potential and ROS [28].
In addition to Tofa's ability to negate RA inflammation, it inhibits IL‐6‐mediated osteoclastogenesis by impairing IL‐1β and CCL2 transcription in osteoclast progenitor cells. In the clinical setting, Tofa monotherapy or combinational therapy ameliorated RA bone erosion [29]. Earlier studies reported that Tofa expands osteoblast formation rather than intercepting osteoclastogenesis in KxB/N preclinical model [30]. Taken together, Tofa treatment nullifies Spike protein or M1 regulators induced metabolic activity in MΦs by rewiring glycolysis to mitochondrial oxidative phosphorylation through PPARγ and/or citrate activation.
Materials and methods
Normal and RA cells
Studies were approved by the University of Illinois at Chicago (UIC) Institutional Ethics Review Board and all donors gave informed written consent. RA patients were diagnosed according to the 1987 revised criteria from the American College of Rheumatology (ACR) [31]. Mononuclear cells were isolated by Histopaque gradient centrifugation and monocytes were isolated from normal or RA PB using a negative selection kit according to the manufacturer's instruction (StemCell Technology) [32, 33, 34, 35, 36, 37, 38, 39]. FLS from fresh RA synovial tissue (ST) were isolated by mincing and digestion in a solution of dispase, collagenase, and DNase. Cells were used between passages 3 and 9 [40, 41, 42, 43].
Seahorse assay
Glycolytic capacity (ECAR) and oxygen consumption (OCR) was tested in RAW 264.7 cells (5 × 103 cells/well) treated with PBS, Spike Protein (300 nM), or LPS/IFN‐γ (1000/500 ng/mL, respectively) with or without Tofa (10 μM) and/or 10 μg/mL of anti‐IL‐6R Ab (Tocilizumab) or TNFi (Etanercept) using the Cell Energy Phenotype Test kit (103325‐100; Agilent Technologies) as per manufacturer's instructions. Cells were preconditioned with the stimuli in 0% FBS/DMEM for 24 h before ECAR and OCR evaluation.
Real‐time RT‐PCR or ELISA
Cells or conditioned media were utilized from MΦs untreated or treated with Spike protein (Exonbio; 30 nM) for 8 h in the presence of DMSO and IgG control (D/I), Tofa (10 μM), 10 μg/mL of anti‐IL‐6R Ab, or TNFi o/n pretreatment (Tocilizumab or Etanercept). Macrophages or RA FLS were pretreated with DMSO or Tofa (2.5–10 μg/mL) o/n. Thereafter, cells were treated with PBS, IL‐6/IFN‐γ (100 ng/mL each, for inflammatory genes), or LPS/IFN (100 ng/mL each, for metabolic genes) for 6 h before real‐time RT‐PCR. Next, in vitro differentiated MΦs or RA FLS was pretreated with DMSO or Tofa (2.5–10 μM) o/n. Following therapy, cells were treated with PBS and/or LPS/IL‐6/IFN‐γ (50/100/50 ng/mL, respectively) for 24 h and conditioned media were harvested for ELISA.
Western blot analysis
Cell lysates from in vitro differentiated MΦs and RA FLS were probed for pSTAT1, pSTAT3 (1:1000; Cell Signaling), and actin (1:3000; Santa Cruz). All cytokines used in this study were purchased from R&D Systems. Additional details are provided in the Supporting Information Materials.
Scratch assay
Confluent RA FLS cultures were scratched, while cultured in 5% FBS/RPMI. Subsequently, RA FLS cultures were treated with PBS or IL‐6/IFN‐γ (200 ng/mL and 50 ng/mL, respectively) with DMSO or Tofa (2.5–10 μM). PBS or bFGF (100 ng/mL) was considered as – or + control. After 24 h, RA FLS were fixed with 10% formalin (1 h) and stained with 0.05% crystal violet (1 h). Microscopic images were used to quantify the number of RA FLS migrated into the cell‐free scratch area [43, 44].
Osteoclastogenesis
RA PBMCs were cultured in suboptimal levels of RANKL/M‐CSF (10 ng/mL each; 10/10) alone or with IL‐6 (200 ng/mL) in the presence of DMSO or Tofa (10 μM) for 14–21 days. The negative control was cells cultivated in 10% FBS/αMEM and the positive control was 20 ng/mL of RANKL/M‐CSF and osteoclasts were stained using TRAP Kit (387A‐1KT; Sigma‐Aldrich). The number of TRAP+ cells per HPF was counted on microscopic images taken at 10× magnification [35, 44].
Flow cytometry
RA PBMCs were cultured in the presence of CD3 Ab plus CD28 Ab (0.25 μg/mL each), which were untreated (PBS) or treated with IL‐12 (10 ng/mL; Th1 +Ctl) and TGF‐β (4 ng/mL) + IL‐1β + IL‐6 (20 ng/mL; Th17 + Ctl) in the presence of DMSO or Tofa (10μM) for 4 days; cells and supernatants were harvested for quantifying Th1 or Th17 cell frequency or protein levels of IFN‐γ or IL‐17 secretion. Additionally, normal negatively selected monocytes (used for CD14 staining) or PBMCs (used for CD4 staining) and RA FLS were blocked with 5% BSA for 1 h before APC‐ACE2 (R&D Systems) and FITC‐CD14 or FITC‐CD4 (eBioscience) staining for 1 h. DAPI was used to exclude dead cells.
ACE2 knockdown
Macrophages were transfected with ACE2 specific and nonspecific control siRNA (Santa Cruz Biotechnologies) at a final concentration of 100 nM using Lipofectamine 3000 (Thermo Fisher) complying with the manufacturer's instruction. The transfected cell culture media was replaced after 6 h and the MΦs were harvested after 48 h of transfection. Cell lysates were utilized to assess ACE2 expression by Western blot analysis.
Statistical analysis
For comparison among multiple groups, one‐way ANOVA followed by Tukey's multiple comparison test was employed, using Graph Pad Prism8 software. The data were also analyzed using a two‐tailed Student's t‐test for paired or unpaired comparisons between two groups. Values of p < 0.05 were considered significant.
Conflict of interest
The authors declare no financial or commercial conflict of interest
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202049159
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Shahrara had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: KP, SU, and SS. Acquisition of data: SU, KP, BR, RKZ, and SS. Analysis and interpretation of data: KP, SU, and SS.
Abbreviations
- COVID‐19
coronavirus 2019
- FLS
fibroblast‐like synoviocyte
- PPARγ
peroxisome proliferator‐activated receptor γ
- RA
rheumatoid arthritis
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
Supporting information
Supporting information
Acknowledgements
This work was supported in part by awards from the Department of Veteran's Affairs MERIT Award BX002286, the National Institutes of Health NIH AI147697, AR056099, and AR065778, Pfizer Investigator‐Initiated award, the National Psoriasis Foundation (NPF), and Chicago Biomedical Consortium (CBC) Accelerator Award. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Data Availability Statement
All findings are exhibited in the paper and the material and data are available for transparency.
References
- 1. Merad, M. and Martin, J. C. , Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020. 20: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vardhana, S. A. and Wolchok, J. D. , The many faces of the anti‐COVID immune response. J. Exp. Med. 2020. 217: e20200678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Netland, J. , DeDiego, M. L. , Zhao, J. , Fett, C. , Alvarez, E. , Nieto‐Torres, J. L. , Enjuanes, L. et al., Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010. 399: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Catanzaro, M. , Fagiani, F. , Racchi, M. , Corsini, E. , Govoni, S. and Lanni, C. , Immune response in COVID‐19: addressing a pharmacological challenge by targeting pathways triggered by SARS‐CoV‐2. Signal Transduct Target Ther 2020. 5: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wen, W. , Su, W. , Tang, H. , Le, W. , Zhang, X. , Zheng, Y. , Liu, X. et al., Immune cell profiling of COVID‐19 patients in the recovery stage by single‐cell sequencing. Cell Discov 2020. 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hadjadj, J. , Yatim, N. , Barnabei, L. , Corneau, A. , Boussier, J. , Smith, N. , Pere, H. et al., Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 2020. 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee, J. S. , Park, S. , Jeong, H. W. , Ahn, J. Y. , Choi, S. J. , Lee, H. , Choi, B. et al., Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol 2020. 5: eabd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goker Bagca, B. and Biray Avci, C. , The potential of JAK/STAT pathway inhibition by ruxolitinib in the treatment of COVID‐19. Cytokine Growth Factor Rev. 2020. 54: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Antwi‐Amoabeng, D. , Kanji, Z. , Ford, B. , Beutler, B. D. , Riddle, M. S. and Siddiqui, F. , Clinical outcomes in COVID‐19 patients treated with tocilizumab: an individual patient data systematic review. J. Med. Virol. 2020. 92: 2516–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu, X. , Han, M. , Li, T. , Sun, W. , Wang, D. , Fu, B. , Zhou, Y. et al., Effective treatment of severe COVID‐19 patients with tocilizumab. Proc. Natl. Acad. Sci. U. S. A. 2020. 117: 10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. La Rosee, F. , Bremer, H. C. , Gehrke, I. , Kehr, A. , Hochhaus, A. , Birndt, S. , Fellhauer, M. et al., The Janus kinase 1/2 inhibitor ruxolitinib in COVID‐19 with severe systemic hyperinflammation. Leukemia 2020. 34: 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bao, L. , Deng, W. , Huang, B. , Gao, H. , Liu, J. , Ren, L. , Wei, Q. et al., The pathogenicity of SARS‐CoV‐2 in hACE2 transgenic mice. Nature 2020. 583: 830–833. [DOI] [PubMed] [Google Scholar]
- 13. Barile, E. , Baggio, C. , Gambini, L. , Shiryaev, S. A. , Strongin, A. Y. and Pellecchia, M. , Potential therapeutic targeting of coronavirus spike glycoprotein priming. Molecules 2020. 25: 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoffmann, M. , Kleine‐Weber, H. and Pohlmann, S. , A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol. Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amanat, F. and Krammer, F. , SARS‐CoV‐2 vaccines: status report. Immunity 2020. 52: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Callaghan, K. P. , Blatz, A. M. and Offit, P. A. , Developing a SARS‐CoV‐2 vaccine at Warp speed. JAMA 2020. 324: 437–438. [DOI] [PubMed] [Google Scholar]
- 17. Israelow, B. , Song, E. , Mao, T. , Lu, P. , Meir, A. , Liu, F. , Alfajaro, M. M. et al., Mouse model of SARS‐CoV‐2 reveals inflammatory role of type I interferon signaling. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee, A. J. and Ashkar, A. A. , The dual nature of type I and type II interferons. Front. Immunol. 2018. 9: 2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simmons, D. P. , Wearsch, P. A. , Canaday, D. H. , Meyerson, H. J. , Liu, Y. C. , Wang, Y. , Boom, W. H. et al., Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J. Immunol. 2012. 188: 3116–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eshleman, E. M. , Delgado, C. , Kearney, S. J. , Friedman, R. S. and Lenz, L. L. , Down regulation of macrophage IFNGR1 exacerbates systemic L. monocytogenes infection. PLoS Pathog. 2017. 13: e1006388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao, M. , Liu, Y. , Yuan, J. , Wen, Y. , Xu, G. , Zhao, J. , Cheng, L. et al., Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat. Med. 2020. 26: 842–844. [DOI] [PubMed] [Google Scholar]
- 22. Qin, C. , Zhou, L. , Hu, Z. , Zhang, S. , Yang, S. , Tao, Y. , Xie, C. et al., Dysregulation of immune response in patients with coronavirus 2019 (COVID‐19) in Wuhan, China. Clin. Infect. Dis. 2020. 71: 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mehta, P. , McAuley, D. F. , Brown, M. , Sanchez, E. , Tattersall, R. S. , Manson, J. J. and Hlh Across Speciality Collaboration, U. K. , COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020. 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferrari, D. , Motta, A. , Strollo, M. , Banfi, G. and Locatelli, M. , Routine blood tests as a potential diagnostic tool for COVID‐19. Clin. Chem. Lab. Med. 2020. 58: 1095–1099. [DOI] [PubMed] [Google Scholar]
- 25. Xiong, Y. , Sun, D. , Liu, Y. , Fan, Y. , Zhao, L. , Li, X. and Zhu, W. , Clinical and high‐resolution CT features of the COVID‐19 infection: comparison of the initial and follow‐up changes. Invest. Radiol. 2020. 55: 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Codo, A. C. , Davanzo, G. G. , Monteiro, L. B. , de Souza, G. F. , Muraro, S. P. , Virgilio‐da‐Silva, J. V. , Prodonoff, J. S. et al., Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1alpha/glycolysis‐dependent axis. Cell Metab. 2020. 32: 498–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chikugo, M. , Sebe, M. , Tsutsumi, R. , Iuchi, M. , Klshi, J. , Kuroda, M. , Harada, N. et al., Effect of Janus kinase inhibition by tofacitinib on body composition and glucose metabolism. J. Med. Invest. 2018. 65: 166–170. [DOI] [PubMed] [Google Scholar]
- 28. McGarry, T. , Orr, C. , Wade, S. , Biniecka, M. , Wade, S. , Gallagher, L. , Low, C. et al., JAK/STAT blockade alters synovial bioenergetics, mitochondrial function, and proinflammatory mediators in rheumatoid arthritis. Arthritis Rheumatol. 2018. 70: 1959–1970. [DOI] [PubMed] [Google Scholar]
- 29. Conaghan, P. G. , Ostergaard, M. , Bowes, M. A. , Wu, C. , Fuerst, T. , van der Heijde, D. , Irazoque‐Palazuelos, F. et al., Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate‐naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann. Rheum. Dis. 2016. 75: 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adam, S. , Simon, N. , Steffen, U. , Andes, F. T. , Scholtysek, C. , Muller, D. I. H. , Weidner, D. et al., JAK inhibition increases bone mass in steady‐state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci. Transl. Med. 2020. 12: eaay4447. [DOI] [PubMed] [Google Scholar]
- 31. Arnett, F. C. , Edworthy, S. M. , Bloch, D. A. , McShane, D. J. , Fries, J. F. , Cooper, N. S. , Healey, L. A. et al., The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988. 31: 315–324. [DOI] [PubMed] [Google Scholar]
- 32. Chamberlain, N. D. , Kim, S. J. , Vila, O. M. , Volin, M. V. , Volkov, S. , Pope, R. M. , Arami, S. et al., Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFalpha in monocytes. Ann. Rheum. Dis. 2013. 72: 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen, Z. , Kim, S. J. , Chamberlain, N. D. , Pickens, S. R. , Volin, M. V. , Volkov, S. , Arami, S. et al., The novel role of IL‐7 ligation to IL‐7 receptor in myeloid cells of rheumatoid arthritis and collagen‐induced arthritis. J. Immunol. 2013. 190: 5256–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen, Z. , Kim, S. J. , Essani, A. B. , Volin, M. V. , Vila, O. M. , Swedler, W. , Arami, S. et al., Characterising the expression and function of CCL28 and its corresponding receptor, CCR10, in RA pathogenesis. Ann. Rheum. Dis. 2015. 74: 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim, S. J. , Chen, Z. , Chamberlain, N. D. , Essani, A. B. , Volin, M. V. , Amin, M. A. , Volkov, S. et al., Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J. Immunol. 2014. 193: 3902–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim, S. J. , Chen, Z. , Essani, A. B. , Elshabrawy, H. A. , Volin, M. V. , Volkov, S. , Swedler, W. et al., Identification of a novel Toll‐like receptor 7 endogenous ligand in rheumatoid arthritis synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2016. 68: 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim, S. J. , Chen, Z. , Essani, A. B. , Elshabrawy, H. A. , Volin, M. V. , Fantuzzi, G. , McInnes, I. B. et al., Differential impact of obesity on the pathogenesis of RA or preclinical models is contingent on the disease status. Ann. Rheum. Dis. 2017. 76: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim, S. J. , Chang, H. J. , Volin, M. V. , Umar, S. , Van Raemdonck, K. , Chevalier, A. , Palasiewicz, K. et al., Macrophages are the primary effector cells in IL‐7‐induced arthritis. Cell. Mol. Immunol. 2019. 17: 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Raemdonck, K. , Umar, S. , Palasiewicz, K. , Volkov, S. , Volin, M. V. , Arami, S. , Chang, H. J. et al., CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17‐mediated osteoclast formation in rheumatoid arthritis. Cell. Mol. Life Sci. 2020. 77: 1387–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pickens, S. R. , Chamberlain, N. D. , Volin, M. V. , Pope, R. M. , Mandelin, A. M., 2nd and Shahrara, S. , Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheumatol. 2011. 63: 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pickens, S. R. , Chamberlain, N. D. , Volin, M. V. , Pope, R. M. , Talarico, N. E. , Mandelin, A. M., 2nd and Shahrara, S. , Characterization of interleukin‐7 and interleukin‐7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheumatol. 2011. 63: 2884–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chamberlain, N. D. , Vila, O. M. , Volin, M. V. , Volkov, S. , Pope, R. M. , Swedler, W. , Mandelin, A. M., 2nd et al., TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF‐alpha levels. J. Immunol. 2012. 189: 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elshabrawy, H. A. , Volin, M. V. , Essani, A. B. , Chen, Z. , McInnes, I. B. , Van Raemdonck, K. , Palasiewicz, K. et al., IL‐11 facilitates a novel connection between RA joint fibroblasts and endothelial cells. Angiogenesis 2018. 21: 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Umar, S. , Palasiewicz, K. , Van Raemdonck, K. , Volin, M. V. , Romay, B. , Amin, M. A. , Zomorrodi, R. K. et al., IRAK4 inhibition: a promising strategy for treating RA joint inflammation and bone erosion. Cell. Mol. Immunol. 2020. 10.1038/s41423-020-0433-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
All findings are exhibited in the paper and the material and data are available for transparency.