

Abstract
Background and aims
Polygenic risk scores (PRS) may soon be used to predict inflammatory bowel disease (IBD) risk in prevention efforts. We leveraged exome-sequence and SNP array data from 29,358 individuals in the multi-ethnic, randomly-ascertained health system-based BioMe biobank to define effects of common and rare IBD variants on disease prediction and pathophysiology.
Methods
PRS were calculated from European, African-American, and Ashkenazi Jewish (AJ) reference case-control studies, and a meta-GWAS run using all three association datasets. PRS were then combined using regression to assess which combination of scores best predicted IBD status in European, AJ, Hispanic, and African American cohorts in BioMe. Additionally, rare variants were assessed in genes associated with very early onset IBD (VEO-IBD), by estimating genetic penetrance in each BioMe population.
Results
Combining risk scores based on association data from distinct ancestral populations improved IBD prediction for every population in BioMe and significantly improved prediction among European ancestry UK Biobank individuals. Lower predictive power for non-Europeans was observed, reflecting in part substantially lower African IBD case-control reference sizes. We replicated associations for two VEO-IBD genes, ADAM17 and LRBA, with high dominant model penetrance in BioMe. Autosomal recessive LRBA risk alleles are associated with severe, early-onset autoimmunity; we show that heterozygous carriage of an African-predominant LRBA protein-altering allele is associated with significantly decreased LRBA and CTLA-4 expression with T cell activation.
Conclusions
Greater genetic diversity in African populations improves prediction across populations, and generalizes some VEO-IBD genes. Increasing African-American IBD case-collections should be prioritized to reduce health disparities and enhance pathophysiologic insight.
Keywords: IBD, PRS, VEO-IBD
Lay Summary:
IBD prediction was improved when using association data from multiple ancestry groups relative to single population data. We also identified rare population-specific variants which may help lead to targeted treatment.
Background and Aims
Inflammatory bowel diseases (IBD) include both Crohn’s disease (CD) and ulcerative colitis (UC), and are complex diseases for which genome-wide association studies (GWAS) have identified over 200 significant loci1–3. Substantial population-based differences in the variance accounted for by major IBD effect loci have been shown, even for relatively common variants shared across populations. NOD2 risk alleles in European populations that are not present in in African4,5 (with the exception of European admixture) or Asian6(p2) populations; conversely, common alleles in the TNFSF15 locus confer substantially greater effects in Far East Asian compared to European ancestry cohorts2. Thus far, IBD genetics studies have been disproportionately focused on individuals of European descent.
In addition to GWAS, a variety of approaches to predict traits using genome-wide SNP data have been reported. A prominent emerging finding is that prediction models developed in one population have substantially reduced performance when applied to different populations7. In IBD, early detection and treatment can significantly improve treatment outcomes8,9. Therefore, the ability to accurately predict genetic disease risk in individuals across ancestries is a critical avenue that may positively affect patient outcomes, as early interventions and even preventive measures are being considered and developed.
Genetic variants exert their effects through selected cell subsets, and comparisons of credible single nucleotide polymorphism (SNP) set lists with transcriptionally active regions have implicated T cells generally in IBD, with particular enrichments in Th17 cells10,11. Rare, typically autosomal recessive genes with higher effect sizes than NOD2 typically present relatively early in life (very early-onset, VEO-IBD genes), and provide insight into mechanisms whereby single genes may drive IBD susceptibility. However, a recent report demonstrates that even patients carrying rare, high impact alleles have higher background polygenic risk scores (PRS)12, highlighting that the complex polygenic nature of IBD encompasses both common and rare genetic variation. By definition, rare variants are more likely to be specific to selected populations, with African ancestry populations being the most diverse.
In this study, we utilized the diverse ancestry included within the BioMe Biobank, which recruited from a primary care-predominant clinic in New York City, to estimate the ability of common variant PRS to predict IBD status across populations and to explore the genetic penetrance of rare variants in VEO-IBD genes. Analyses include comparison of different models for SNP selection and the integration of IBD association summary statistics collected from single and multiple ancestral populations, including European, Ashkenazi Jewish (AJ), and African American GWAS case-control cohorts. We replicate features of our PRS models in the UK Biobank and, using a replication African-American case-control cohort, implicate a role for rare, African-American predominant variants in the VEO-IBD genes ADAM17 and LRBA.
Methods
Phenotype validation in BioMe
The Mount Sinai BioMe Biobank contains genetic data on 32,595 patients who were seen at high-throughput Mount Sinai primary care clinics. BioMe recruitment does not select for specific traits or diseases and be should be representative of the general population in and around NYC. After rigorous phenotype validation 19,541 individuals were retained, of which 339 were IBD cases (273 CD, 28 UC, and 37 individuals who were classified as both) and 19,202 were controls. IBD cases were defined according to the presence of 2+ ICD codes for IBD (555.*,556.*,K50.*, and K51.*) as well as one or more IBD-related medications. To qualify as a healthy control for this study individuals could not have been diagnosed with inflammatory conditions or have been treated with any IBD-related medications.
BioMe genotype data quality control (QC)
GSA array data for 32,595 participants and 635,623 variants was stratified by race, then individuals with a heterozygosity rate that surpassed +/− 6 standard deviations of the mean, call rate <95%, sex discrepancies, or indeterminate sex were removed. Duplicates were determined using KING13, and one of each pair of 28 duplicates was excluded based on missingness rate. Sites were removed with call rate <95%, HWE p-value threshold of p < 1e-5 (African or European Americans) or p < 1e-13 in Hispanic Americans. This resulted in the retention of 31,911 individuals and 604,869 sites, which was imputed against 1000 Genomes using the Michigan Imputation Server14.
BioMe exome sequence data quality
Whole exome sequencing data were processed with Kappa library prep reagents, restricted to the exome using the IDT xGen capture platform, and sequenced on Illumina v4 HiSeq 2500 systems (8,761,478 total sites; 31,250 samples). QC included removal of contaminated, low coverage, genotype-exome discordant, irreconcilable gender discordant, and duplicate pairs of uncertain status. The sample with greater missingness from duplicate pairs of clear status were removed. Sites with missingness >0.02, or allelic balance (AB) <0.30 or >0.80 were removed. The final data set contained 30,813 samples and 3,257,000 sites.
Association case-control cohorts used for risk score calculation
Association summary statistics from European, AJ, and African American IBD GWAS studies were used, which totaled 12,882 IBD cases and 21,770 controls for the European study2, 2,066 CD and 559 UC cases and 3,633 controls for the AJ study15, and 2,229 IBD cases (1,646 of which were CD) and 5,002 controls for the African American study16. Additionally, a meta GWAS analysis was performed using the program METAL17 which included summary statistics from the European, African American, and AJ studies2,15,16.
PRS calculation and selection
SNPs to be included in risk score calculation were selected using 6 different association p-value cutoffs (1, 0.5, 0.05, 5x10−4, 5x10−6, 5x10−8) and 4 r2 value cutoffs (0.2, 0.4, 0.6, and 0.8)18. Scores were calculated using imputed SNPs with MAF > 0.001 in the relevant population for each individual Clumping information files with the maximally predictive sets of SNPs for each population in which pruning and thresholding performed the best are available in Supplementary Tables S1 (European BioMe prediction) and S2 (AJ BioMe prediction).
Scores were evaluated for predictive accuracy using adjusted partial R2 values calculated using the package rsq19 in R20 by comparing logistic regression models with disease state as the dependent variable and risk score, age, sex, and smoking status as independent variables to models with risk scores excluded. To ensure that results were robust 100 iterations of the evaluation were also run for each risk score, using a random selection of half of the cases and half of the controls for each model.
Multiple-ancestry risk scores
The most predictive scores originating from each association dataset were then included together as variables in regression models to predict IBD status, similar to the study of diabetes genetic risk by Márquez-Luna et al.21. These models predicted IBD status based on risk score combinations, gender, age, smoking status, and minor allele counts for top IBD-associated SNPs (Supplementary Table S5). These top causal SNPs were included as covariates in order to assess the impact of applying additional weight to each when predicting IBD, in addition to their contribution to overall risk scores. To identify the best set of predictors the area under the receiver operating characteristic (ROC) curve (AUC) for each model was calculated using the R package pROC22. The package pROC was also used to assess the significance associated with AUC improvements when comparing use of single association datasets and combined models.
Multi-racial risk scores cross validation
Cross validation was performed using the cv.lm command in R, based on 100 iterations with different seed values for each combination of risk scores and 3 folds per run. The average mean squared error was calculated across the 100 iterations and used to evaluate the accuracy of each model and to be certain overfitting was not occurring.
UK Biobank replication
A total of 430,470 individuals (424,835 controls and 5,635 IBD cases – 2,172 CD and 3,982 UC) of European ancestry from the UK Biobank cohort were defined as being IBD cases or controls based on the same ICD10 code definitions used for the BioMe cohort, then PRS were calculated for each individual using PRSice2 based on each association dataset (European, AJ, AA, and METAL). Prediction of IBD status was compared using the models developed for BioMe data.
Model calibration comparing BioMe and UK Biobank European ancestry populations
Individuals of European ancestry were separated into 10% bins based on PRS values, then the percentage of IBD cases within each bin was divided by the percentage of IBD cases within the full European-ancestry cohort.
VEO-IBD SNP penetrance
Penetrance values were calculated from exome data for the 61 genes23–27 included on a VEO-IBD risk testing panel using a custom R script that calculated the proportion of carriers of each SNP that had IBD (heterozygotes and homozygotes were considered together to model dominant effects, while only homozygotes were considered when modeling recessive effects). The significance of each penetrance value was calculated using Fisher’s exact test to compare the proportion of carriers with IBD to the proportion of total carriers of each SNP. SeattleSeq28 was used for annotation. All penetrance analyses were performed separated by ancestry. Penetrance values were also replicated using case-control whole genome sequence data from European and African American individuals (1,332 CD cases, 404 UC cases, and 1,644 controls).
European WGS case-control
IBD samples and matched population controls were sequenced at the Sanger Institute. DNA from whole blood underwent short-read paired-end sequencing using Illumina HiSeq X Ten machines (coverage target was 15x - median empirical coverage was 18.5x). Low-quality samples (low depth, high cross-sample contamination, high chimeric read count, outliers) were removed. Association tests included unrelated samples of European genetic ancestry (6,404 CD and 11,761 controls) from the INTERVAL blood donor cohort. Replication used Firth’s logistic regression with 10 principal components as covariates.
Flow cytometry
PBMCs were isolated from whole blood of LRBA carriers and non-carrier controls by centrifugation in BD Vacutainer CPT tubes. Cells were cultured in RPMI complete (10% HI-FBS, 100 U/mL penicillin/streptomycin, 2 mM L-glutamine) and stimulated with 2.5 μg/mL PHA-L for 96 hours. On the third day of stimulation, 3 replicates from each individual were treated with 100 μM chloroquine for 18 hours.
On day 4, 500,000 cells were collected and stained for viability with Live/Dead Fixable Near-IR Dead Cell Stain for 30 min at room temperature. Extracellular markers were stained for 20 minutes at 4°C with eBioscience CD45-eFluor506 (5 μL/test), CD4-SB645 (2.5 μL/test), CD25-SB600 (5 μL/test), CD127-APC (5 μL/test), and CTLA4-FITC (5 μL/test). Following fixation and permeabilization for 30 minutes at room temperature, cells were stained for intracellular markers for 60 minutes at room temperature with FoxP3-eFluor450 (eBioscience, 5 μL/test) and LRBA (Sigma, 1:400). Subsequently, cells were incubated with BD BioSciences anti-LRBA secondary antibody for 30 minutes at 4°C (1:10).
All samples were collected with the Attune Nxt Flow Cytometer (Invitrogen) and analyzed with FCS Express 7 (De Novo Software). Mononuclear lymphocytes were gated using FSC-A vs. SSC-A. Living cells were gated based on negative staining for the viability dye and separated based on extracellular markers. Arithmetic mean fluorescence intensity of CTLA4 and LRBA was analyzed from the CD25+CD127- gate.
Droplet-based single cell RNA-sequencing
The 10x Chromium v2 gene expression kit was used with uninflamed and inflamed biopsies obtained from the colonic region during standard colonoscopies at Mount Sinai Hospital from 4 TNF-a naïve UC patients of European ancestry. Cells were treated as in Martin et al.29, except that cell removal involved a single 30 minute EDTA treatment to keep the epithelial cell fraction. Libraries were sequenced using Illumina NextSeq 500, and clustering was performed using Seurat 3.030. Data available on GEO (accession GSE150516).
Results
IBD prevalence in BioMe Biobank populations mirrors population-based prevalence estimates
The BioMe Biobank cohort used in this study included 29,358 total individuals of European, AJ, African American, or Hispanic ancestries, each of whom were recruited from primary care clinics without bias toward IBD status. Across all 29,358 individuals, 339 had IBD (1.13%), which aligns with our expectations based on 2015 census data which reported that 1.3% of US adults had been diagnosed with IBD31. European ancestry cohorts demonstrated a higher prevalence than non-Europeans, with African-American and Hispanic cohorts demonstrating 0.77% and 0.72% prevalence, respectively. Among European ancestry cohorts, AJ populations showed a higher prevalence (2.55%), compared to the non-Jewish European cohort (1.67%). A Biobank population representative of prior estimates indicates that our results should be useful in developing general models for polygenic risk and rare variant penetrance estimates.
Calculation of polygenic risk scores across populations
To maximize IBD risk prediction within each ancestry group we used a two-part analytic process. First, PRS were calculated and assessed for each racial group using association data from different case-control cohorts individually or from a single set of meta-GWAS statistics, then risk scores were combined and assessed using regression modelling. Initial risk scores were calculated using summary statistics from large European GWAS2, African American GWAS16, and AJ exome chip association15 studies (Supplementary Table S3) for 19,541 out of 29,358 total individuals in the BioMe BioBank cohort (Table 1) based on stringent case/control definitions. Scores were also calculated from a meta-analysis using the program METAL17, which combined association information from all three population-specific studies. SNPs were selected for inclusion in risk scores using pruning and thresholding approaches and by using the program PRSice232, which was shown to be as accurate as LDpred33 while having substantially improved run times. SNPs were weighted according to their IBD-association log(OR) using the 3 association datasets as well as the meta-GWAS data from METAL, then used to calculate PRS for each of the 4 primary BioMe populations. An overview of the risk score calculation and evaluation pipeline can be seen in Figure 1.
Table 1:
BioMe Biobank composition and IBD rate
| Primary Ancestry | IBD cases | CD cases | UC cases | Controls | Total individuals | Percentage with IBD |
|---|---|---|---|---|---|---|
| All populations | 339 | 311 | 65 | 19,202 | 29,358 | 1.15 |
| European – non Jewish | 121 | 113 | 24 | 5,033 | 7,254 | 1.67 |
| Ashkenazi Jewish | 76 | 71 | 7 | 2,085 | 2,982 | 2.55 |
| African American | 61 | 54 | 12 | 5,022 | 7,876 | 0.77 |
| Hispanic | 81 | 73 | 22 | 7,062 | 11,246 | 0.72 |
Figure 1: Polygenic risk score calculation.
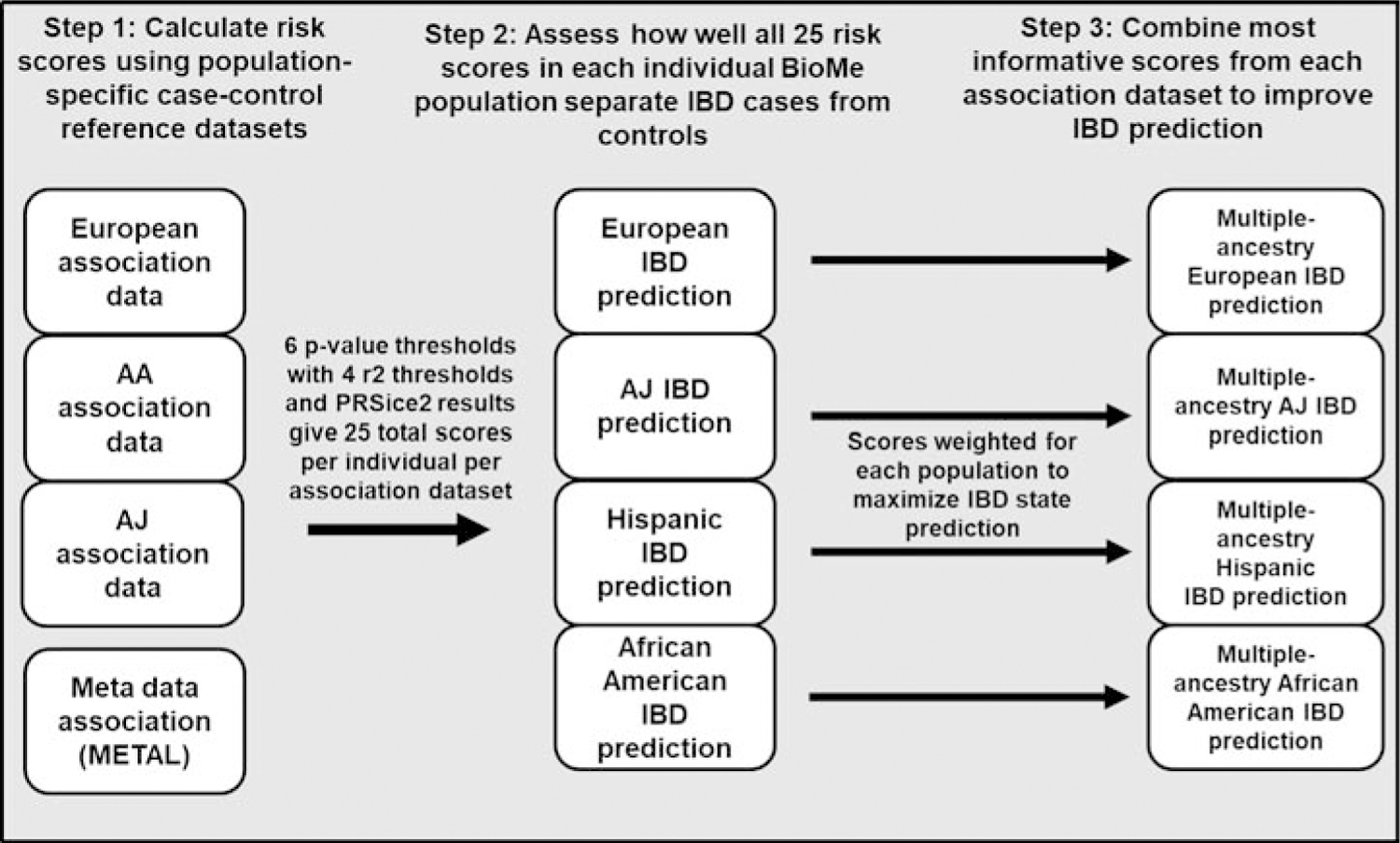
Polygenic risk scores were generated and evaluated for each individual based on their ability to predict IBD status (25 from each association dataset). Combined models using the most predictive scores from each individual-population dataset were then used to assess improvements to prediction using multi-racial scores.
Evaluation of risk scores within populations
To evaluate IBD prediction a series of logistic regression models were applied within each BioMe population to calculate the partial R2 values associated with each risk score and plotted using the R package ggplot234(p2) (Figure 2a, one risk score is included in each model). In addition, 100 iterations were run using randomly selected halves of the total available cases and of the controls from each population, and the IBD prediction p-value was calculated across all 100 runs to ensure consistency (the most significantly predictive risk scores are shown in Supplementary Figure S1 and highlighted in Supplementary Table S4). Prediction in non-Jewish European ancestry populations using association data from Liu et al.2 (primarily European, non-Jewish) to calculate SNP weights demonstrated maximal prediction capacity with p-value thresholds between 5 x 10−4 and 5 x 10−8. This underscores study design differences between the hypothesis-testing of GWAS, where genome-wide significance is defined as p < 5 x 10−8, and prediction, where inclusion of more genetic markers often improves performance35. For each predictive p-value significance threshold, the most stringent pruning threshold, namely r2 = 0.2 (where all pairs of markers with r2 > 0.2 between each other are pruned, resulting in fewer markers per locus) provided the best prediction in the non-Jewish European ancestry cohort. In contrast, while AJs demonstrated high risk prediction comparable to non-Jewish European ancestry cohorts with the Liu et al. data2 (Figure 2a), this cohort did not as consistently mirror improvement with stricter pruning thresholds, reflecting the distinct linkage disequilibrium patterns between Jewish and non-Jewish European ancestry cohorts. Finally, risk prediction in African-American cohorts was generally poor, using both the large, European ancestry cohort2 and the smaller African-American16 ancestry cohort.
Figure 2: Risk score prediction of disease is most accurate when including association data from multiple racial groups.
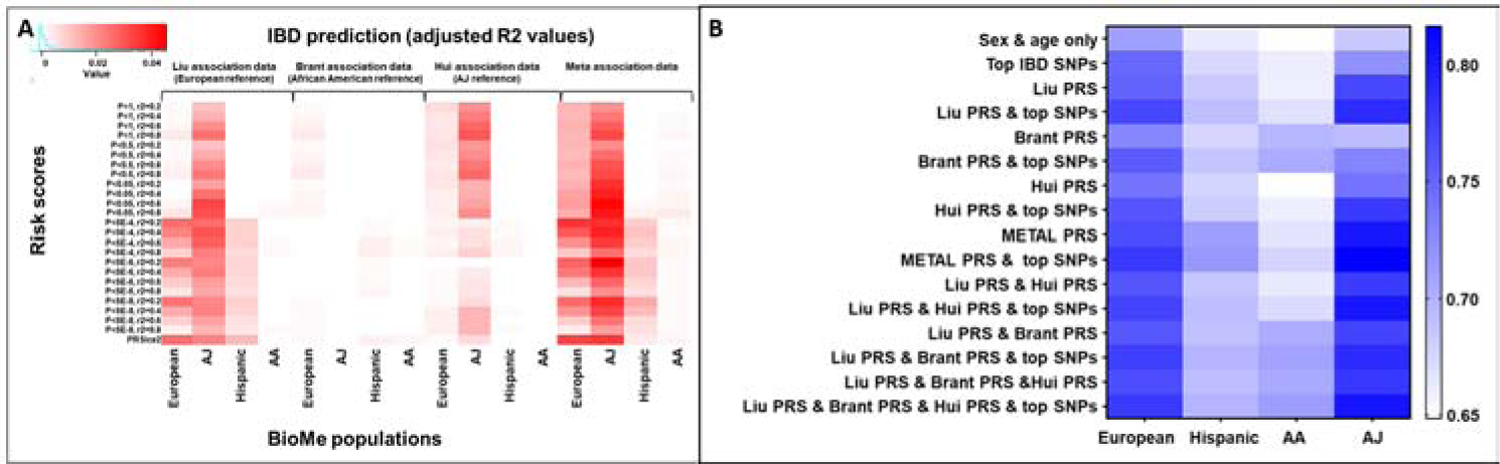
A. Single-population association risk scores were assessed using logistic regression models, and the significance of the risk score as a predictor of IBD state is shown. B. AUC values calculated using logistic regression models which predicted IBD status using age, sex, and the combination of predictors on the x-axis. Cross-validation was also performed to test for overfitting. Association data used for risk scores: Liu included European individuals, Brant included African American individuals, and Hui included AJ individuals.
Cross-population analyses improve prediction in multiple IBD population cohorts
We next sought to determine whether combining information across populations could improve prediction. First, the set of risk scores from each association dataset which were individually most predictive of IBD status in each BioMe population were combined in logistic regression models along with age, sex, and smoking status to predict IBD. Prediction using each combination of scores was evaluated using AUC analysis (Figure 2b). Another set of models were used which included minor allele counts for top IBD SNPs2,11 as additional predictors. In every BioMe population, predictive power was maximized by using scores from a combination of association datasets2,15,16 along with additional weighting for top IBD-associated SNPs (listed in Supplementary Table S5), despite differences in effect between CD and UC. The improvement was more significant in Europeans and AJs, since these were the populations in which these SNPs were identified. In every population except African Americans the predictive power of the meta-analysis using METAL was at least moderately superior, while in African Americans the combination of risk scores from each independent GWAS within the logistic regression model was slightly more predictive, though not significantly different (European Liu only AUC vs. combined model AUC p-value = 0.042, African American Brant only vs. combined model AUC p-value = 0.224, AJ Hui only vs. combined model AUC p-value = 0.008, Hispanic Liu only vs. combined model AUC p-value = 0.022). IBD in non-Jewish Europeans was approximately equally well predicted using meta-association risk scores or risk scores from all three association datasets. In AJ and Hispanic individuals, IBD was best predicted using meta-association risk scores. Conversely, IBD in AA individuals was best predicted using risk scores from all three association datasets separately (Figure 2b, Supplementary Table S6). The change in relative predictive power for African Americans is likely related to how each approach weights individual association statistics. METAL was used to weight association statistics according to sample size, effect size, and allele frequency while the regression modelling approach weights each risk score only according to predictive power, giving the association data from Brant et al. more impact despite it having less significant association values. The overall accuracy of IBD prediction was highest in AJ individuals, followed by non-Jewish Europeans and Hispanic individuals. Prediction among African American individuals was the least accurate, and not greatly improved through integration of data from other races. To ensure that overfitting was not occurring, k-fold cross validation using 3 folds was run 100 times for each model using different seed values for fold selection and average mean squared error values were found to be the lowest for combined models.
Prevalence estimates in BioMe populations based on PRS percentiles
To assess the clinical impact of IBD prediction we looked at the percentage of individuals who had IBD within the top 50%, 20%, 10% and 5% of risk scores. These were evaluated first using only the risk scores built from relevant single-population association data along with age and sex (European association data was also used for Hispanics), followed by the most predictive combination of scores for each population (Figure 3a). Such empiric estimates across populations may provide a foundation for designing early intervention and/or preventive studies in IBD. Under most conditions tested (by population and percentile), the estimates of IBD frequency or penetrance were highest for the combined model. As might be expected for the population with the highest prevalence, AJs demonstrated higher penetrance than non-Jewish European ancestry individuals, although this difference was minimal at the top 5th percentile of PRS scores, with IBD rates of 18.5% and 16.2% observed, respectively. In contrast, for Hispanic and African-American cohorts, even at the top 5th PRS percentile, only 7.2% and 3.0% of individuals had IBD (comparison between each risk score percentile and the background IBD rate in the population are shown in Supplementary Table S7).
Figure 3: Combination of risk scores improves IBD prediction across BioMe racial populations.
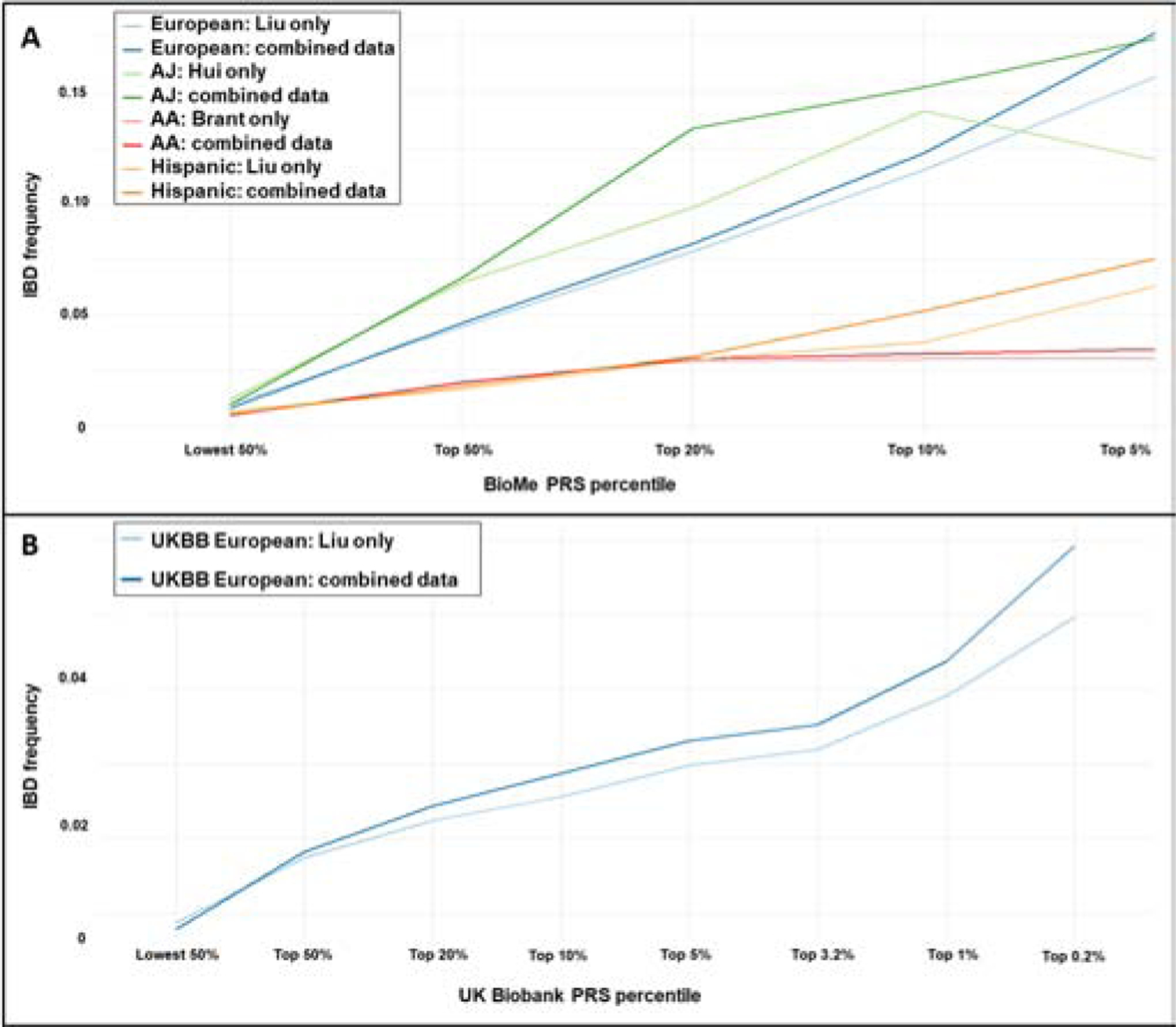
A. BioMe individuals with higher risk scores have a higher rate of IBD regardless of ancestry. Integration of association data from multiple populations also increases IBD prediction in every population, though IBD prediction is more challenging in African Americans. B. Replication in UK Biobank individuals of European ancestry shows similar improvement.
The IBD prevalence estimates for BioMe (Figure 3a) are likely over-estimated due to the modest absolute case sample size in this multi-ethnic cohort. We therefore replicated our findings among European ancestry individuals in the UK Biobank (5,635 IBD cases and 424,835 controls, Table S3) to ensure that the improvements to predictive power when using association information from multiple ancestries held true in this larger, primarily European ancestry, population-based cohort. Importantly, we found that IBD prediction was similarly improved using a combination of scores rather than scores based only on European summary statistics (Figure 3b, DeLong ROC comparison test p-value < 2.2e-16), and that the improvement was greatest at the highest risk score percentiles. We also calibrated our results by comparing the prevalence of IBD across PRS scores to the background rate of IBD within both the BioMe and UK Biobank European-ancestry cohorts (Supplementary Figure S2). After calibration both cohorts showed similar levels of relative IBD risk at each PRS percentile compared, though having a lower number of total cases in BioMe increased variance.
Penetrance of mutations in VEO-IBD-association genes
We next sought to explore the penetrance of genes generally believed to confer IBD risk, but which have been reported primarily in very early onset populations (VEO-IBD genes23–27). Many of these cases have been implicated in an autosomal recessive manner, often in consanguineous families. Genetic penetrance values, based on both recessive and dominant inheritance patterns and considering CD and UC, were calculated for each of the SNPs in 61 genes used to test for VEO-IBD risk (Supplementary Table S8)23–27 using exome sequence data from randomly ascertained European, AJ, African American, and Hispanic populations recruited to the BioMe Biobank (Table 1). Autosomal dominant penetrance values for SNPs with at least 10 total heterozygotes and more than one heterozygous case are represented in Figure 4, while the most highly penetrant SNPs in each population are shown in Supplementary Table S9 and the full set of penetrance results are located in Supplementary Table S10. The top result for Europeans using a recessive model was the well-documented NOD2 SNP rs2066844 (Supplementary Figure S3 and Supplementary Table S10).
Figure 4: VEO-IBD missense SNP penetrance.
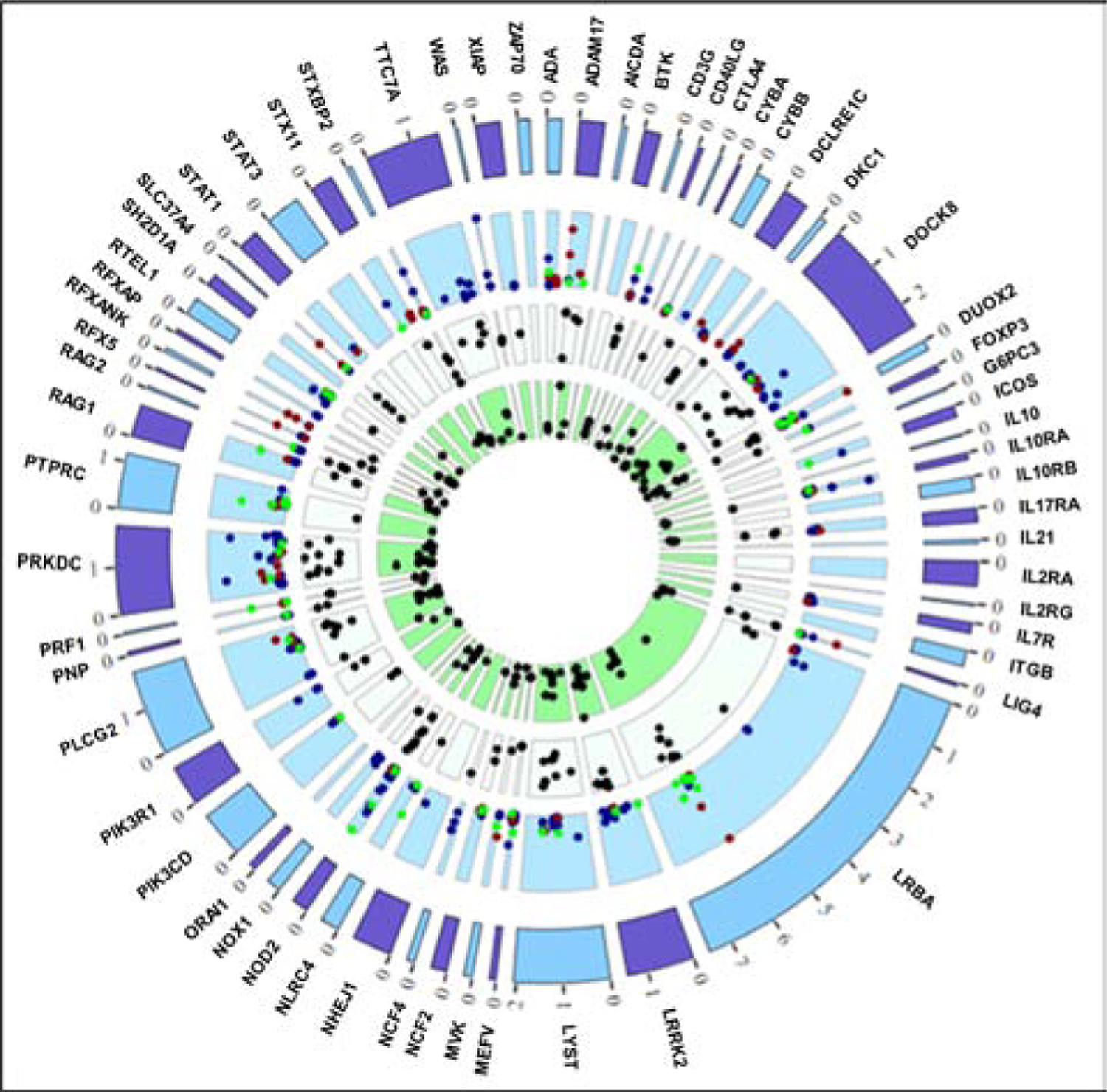
Penetrance dot colors (within light blue layer) reflect ancestry: blue = European, red = AA, green = Hispanic. AJ individuals are included as European-ancestry. Penetrance points plotted if TotalHet # > 10, HetCases > 1, and penetrance > 1. Max penetrance values for the light blue layer: Eur = 21.43, Afr = 10.53, His = 9.09. White layer = GERP scores > 3 and green layer = CADD scores > 10. Outer ticks represent 100kb in gene region.
When using a dominant SNP model, the top results for Europeans included SNPs in WAS (Wiskott-Aldrich Syndrome protein encoding), IL10RA (IL-10 receptor α), PRKDC (DNA-dependent protein kinase catalytic subunit), and NOD2 (nucleotide binding oligomerization domain containing 2). We also tested for replication in an independent European ancestry whole-genome sequence case-control cohort (6404 CD cases and 11,761 controls)3, and observed nominal evidence for replication (odds ratio 1.72) for the p.Pro295Leu variant in IL10RA.
Hispanic individuals tended to have lower SNP penetrance values compared to the European and African American individuals (Figure 4, green symbols), with the most highly penetrant SNPs being in NOD2, PTPRC (protein tyrosine phosphatase receptor type C), RAG1 (recombination activating 1), and PRF1 (perforin 1). Since these variants are so rare, we also used the European whole-genome sequence IBD case-control dataset3 to check for replication, but were only able to replicate the NOD2 alleles.
Of great interest were the rare variant penetrance analyses in African-Americans. We observed two carriers of p.Tyr185Cys in DUOX2 (dual oxidase 2), among UC cases in BioMe. This variant has an extremely high Combined Annotation Dependent Depletion (CADD) score (26: values above 20 are in the top 0.1% of predicted deleteriousness36). Even among the 3380 African-Americans in our replicative case-control cohort, no carriers were observed. In contrast, the p.Val673Ile variant in ADAM17 (ADAM metallopeptidase 17), present in three BioMe Crohn’s disease cases, demonstrated some evidence for replication in Crohn’s disease (odds ratio of 1.76). ADAM17 is a compelling candidate, given its role in proteolyzing membrane-bound TNF and in epithelial dysfunction that influences intestinal permeability when deficient37. Finally, we observed association trends for two distinct alleles, p.Thr1251Ala and p.Val737Ile in LRBA (lipopolysaccharide-responsive beige-like anchor protein) in both BioMe and our case-control cohort (odds ratio 4.1). In particular, nearly one percent of African-American IBD cases are heterozygous carriers of the p.Val737Ile variant, which has a high CADD score (23.6).
Heterozygous carriers of mutations in LRBA display functional differences in immune subsets
To determine whether heterozygous variants in LRBA are sufficient to cause functional differences, flow cytometry was performed on stimulated PBMCs from non-IBD heterozygous carriers (n = 2, both with the p.Val737Ile variant) and wild-type (WT) controls (n = 10) (Supplementary Table S11). Baseline levels of intracellular LRBA were significantly reduced in the carriers (p = 7.8x10−7), indicating that even in individuals without IBD, one copy of the p.Val737Ile mutation substantially lowers protein expression (Figure 5A). To assess the functional effects of reduced LRBA, we quantified the expression of CTLA-4 (cytotoxic T lymphocyte associated protein 4) at the cell surface. LRBA encodes a transmembrane protein found in organelles with various roles in vesicle trafficking; defects in LRBA have been shown to affect the recycling of CTLA-4 to the cell surface, contributing to autoimmune inflammation38,39. At baseline, levels of CTLA-4 at the cell surface were significantly lower in carriers compared to WT controls (p = 2.2x10−9) (Figure 5B). Treatment with chloroquine phosphate in vitro has been showed to increase CTLA-4 expression in patients with LRBA deficiency, primarily via lysosome acidification and subsequent reduction in CTLA-4 degradation39. We observed significant increases in CTLA-4 expression in both carriers and controls after 18h chloroquine treatment (Figure 5B). By inhibiting lysosomal degradation with chloroquine, CTLA-4 expression may be increased to meet a threshold required for normal immunoregulatory function in vivo.
Figure 5. Expression of LRBA and CTLA-4 in heterozygous carriers and controls.
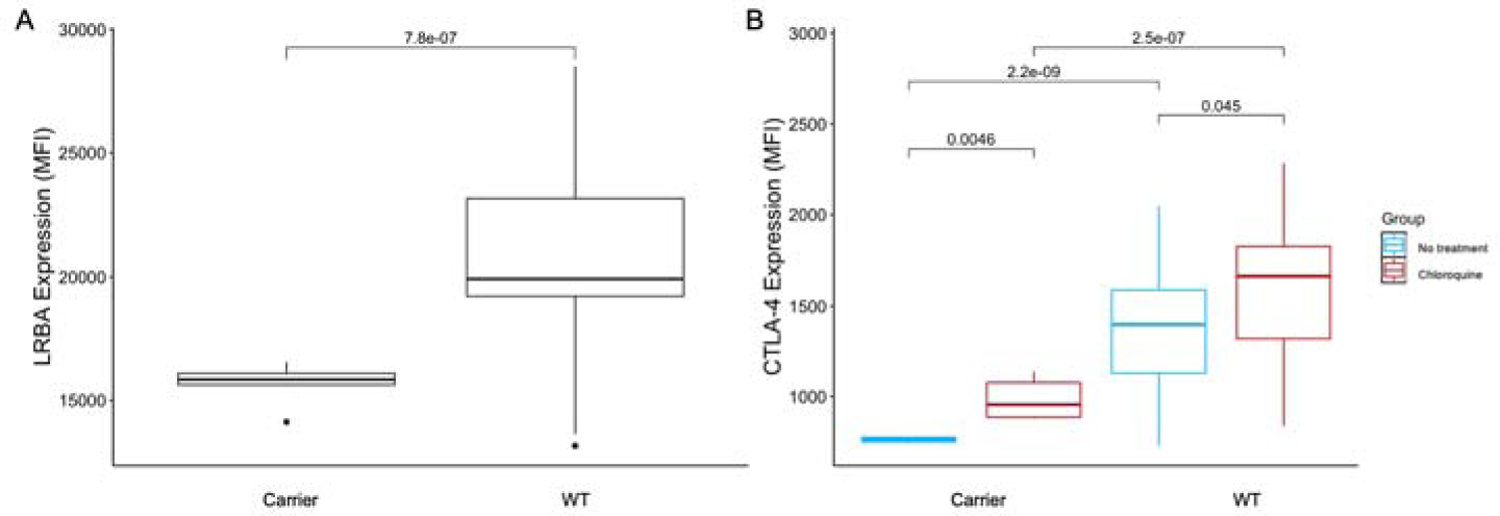
A. Mean intracellular expression of LRBA. B. Mean extracellular expression of CTLA-4, with and without 18-hour chloroquine treatment. All conditions were performed in triplicate. P-values calculated using Welch’s t-test. MFI, mean fluorescence intensity; WT, wild type.
Expression of VEO-IBD genes in single cell data
Expression of each of the VEO-IBD genes was assessed in single cell data collected from UC patient biopsies. Many of the genes had increased overall expression in T cells and inflammatory macrophages (Figure 6). Several genes that are highly expressed in the T cell subpopulations represented have immunoregulatory functions, such as IL10RA, IL10RB, and LRBA; IBD is a known clinical feature of mutations in these genes26. Expression of LRBA is highest in regulatory T cells (Tregs) and type 3 innate lymphoid cells (ILC3). In addition to LRBA IL2RA (IL-2 receptor α), CTLA4, FOXP3 (forkhead box P3 transcription factor), IL21, and IL10 (immunoregulatory cytokines) are all expressed in Tregs with very weak signals in other cell types. The expression signals of these genes are among the strongest across all of the VEO-IBD genes analyzed, defining a clear Treg signature. This may implicate that deficient regulatory or suppressive immune function underlies much of monogenic IBD in adult populations.
Figure 6: VEO-IBD gene expression in intestinal single-cell RNA-seq.
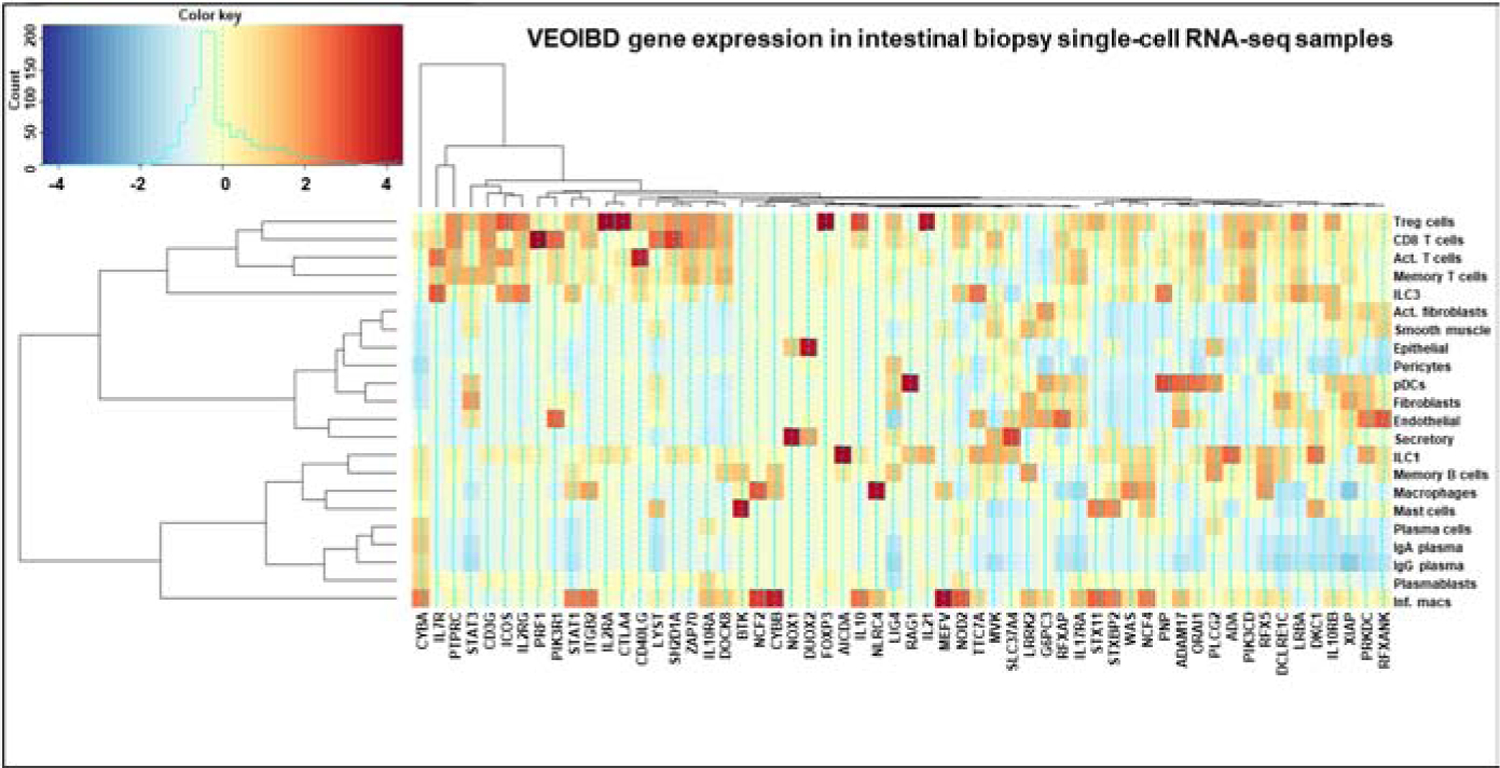
Average expression across each cell type cluster is shown, based on sorting using Seurat 3.0. Genes and cell types clustered based on similarity of expression among the VEO-IBD genes plotted.
Others genes with expression across multiple T cell subsets (Tregs, CD8+ T cells, activated T cells, memory T cells) play a role in adaptive immune cell maturation, such as ZAP70 (ζ chain of T cell receptor associated protein kinase 70), IL7R (IL-7 receptor), ADA (adenosine deaminase), and CD3G (CD3 γ chain of T cell receptor CD3 complex). Variants in these genes can have severe impacts on cell mediated immunity and T cell signaling26. Approximately 1/3 of genes were highly expressed in inflammatory macrophages, in contrast to inactivated macrophages. Epithelial cells mainly expressed DUOX2 and NOX1 (NADPH oxidase 1). Missense mutations in these genes compromise the innate immune defenses mounted by the generation of reactive oxygen species, underscoring the critical role of both genes in the maintenance of a healthy intestinal barrier40. Our data also demonstrate little expression in plasma cells and plasmablasts, indicating that they may play a lesser role in the pathology of VEO-IBD.
There may be distinct cellular drivers of IBD pathology impacted by rare variants that are not captured by common variant GWAS. The prediction of IBD status by multi-racial risk scores emphasizes the importance of diverse patient cohorts for future studies of IBD genetics, and the potential for therapeutic avenues that take advantage of the distinct pathophysiology conferred by variants that drive monogenic IBD41.
Conclusions
Our study highlights the value of integrating association data from diverse racial populations when using genetic data to predict IBD risk. Integration of both common and rare variant analyses can improve disease risk prediction and potentially assist with more targeted treatment in the future. For every BioMe population assessed (European Americans, AJ individuals, African Americans, and Hispanic Americans) predictive accuracy was maximized when association data from European, African American, and AJ-based GWAS studies were integrated, whether that integration occurred during a meta-GWAS analysis prior to PRS calculation or through integration of multiple individual PRS using regression modelling. In the past, most IBD research has focused on European cohorts, but our results highlight the need for more non-European studies – particularly in African Americans.
IBD prediction in African Americans was the least accurate among the BioMe populations assessed. This is likely due to increased overall genetic diversity in African populations, but also because the sample sizes for IBD association studies that included individuals with African ancestry are much lower than those of European ancestry. Because IBD is increasing in prevalence in non-European individuals42 it is increasingly important to understand the genetic differences that underlie disease risk which may not be measured in European ancestry association studies. African populations have shorter linkage disequilibrium blocks, favorable for fine-mapping and genome-wide prediction. In our METAL analyses (Figure 2a), we saw very modest improved prediction at the highest significance threshold (5 x 10–8) for the strictest pruning (r2 = 0.2) compared to other pruning thresholds, but very little concordance for stricter pruning thresholds at lower overall significance thresholds (Figure 2a, P-values between 5 x 10–4, 5 x 10–6). Our demonstration of improved prediction by combining European and African cohorts despite substantially asymmetric sample sizes (Figure 2b), combined with an evolving understanding that most of the genome likely contributes to complex, polygenic traits43,44, highlights the substantial, potential opportunities for a more complete dissection of IBD architecture by substantially increasing African-American IBD collections.
As a relatively early onset disease (often before reproductive age), it might be speculated that IBD risk alleles of high effect would be evolutionarily selected against (with NOD2, TNFSF15 and the MHC region reflecting exceptions to this rule). The markedly greater genetic diversity present in African populations includes rare (0.2–1%) and often deleterious variants, which may also confer risk in heterozygous form. In this study, we have identified several rare VEO-IBD variants with high genetic penetrance in a health-system-based biobank, then replicated results in large case/control African American and European datasets3. One of the variants with the highest genetic penetrance located in the gene LRBA was predicted to result in a deleterious change to the amino acid structure. Reduced expression of CTLA-4 secondary to the variants we identified in LRBA may result in auto-inflammation that contributes to IBD. It has been shown that 63% of all known cases of LRBA deficiency have enteropathy, with 27% having chronic diarrhea is the presenting symptom45. Targeting reduced CTLA-4 expression is an exciting treatment avenue, as expression of CTLA-4 has been shown to be increased by chloroquine treatment in vitro39. This finding was validated by our study, where treatment with chloroquine significantly increased extracellular CTLA-4 expression in both carriers of highly penetrant heterozygous variants in LRBA (p = 0.0046) and in WT controls (p = 0.045). Hydroxychloroquine (functionally the same as chloroquine but better tolerated by patients) is a potent disease modifying agent in rheumatologic disease46 that has previously been shown to have some effect in treating IBD47,48. Future studies of hydroxychloroquine in the treatment of IBD in carriers of LRBA mutations present a promising therapeutic opportunity for the application of personalized medicine in an ancestry-cognizant manner.
Limitations of this study include a primarily CD focus (311 of our 339 total BioMe Biobank IBD patients have CD while only 65 have UC, many of the SNPs which were given additional weight had CD-specific associations, and the AJ and African American association datasets focused on CD cases rather than UC) as well as relatively low sample size and power for the African American and Ashkenazi Jewish association datasets included. Upcoming IBD genetic studies must focus on the recruitment, enrollment, and retention of African American individuals to improve prediction and outcomes for these and all IBD patients.
Supplementary Material
“What You Need to Know”.
BACKGROUND AND CONTEXT
Polygenic risk scores (PRS) may soon be used to predict inflammatory bowel disease (IBD) risk in prevention efforts, so it is important to understand how predictive power varies within different populations with IBD.
NEW FINDINGS
We identify population-specific effects of common and rare IBD variants on disease prediction and pathophysiology and show that heterozygous carriers of a rare LRBA allele that we identified to have high genetic penetrance have lower expression of both LRBA and CTLA4.
LIMITATIONS
Currently African American and Hispanic IBD association datasets include far fewer individuals than European studies, making prediction challenging. In this study we test predictive power in a multi-ethnic Biobank population recruited from primary care clinics, which allows for inclusion of high numbers of African American and Hispanic individuals but limits the number of IBD cases.
IMPACT
This study shows how important it will be to increase the sample size and power of non-European IBD association studies in the future, both to improve prediction within each population and to gain information about variants which may be very rare
Grant support:
R01 DK123530-01, 5 U24 DK062429-18, 5 U01 DK062422-18, R01 DK106593, Helmsley Charitable Trust (VEO-IBD Consortium)
Abbreviations:
- PRS
Polygenic risk score
- IBD
inflammatory bowel disease
- AJ
Ashkenazi Jewish
- VEO-IBD
very early onset inflammatory bowel disease
- CD
Crohn’s disease
- UC
ulcerative colitis
- GWAS
genome-wide association study
- SNP
single nucleotide polymorphism
- QC
quality control
- AB
allelic balance
- ROC
receiver operating characteristic
- AUC
Area under the ROC Curve
- MFI
mean fluorescence intensity
- CADD
Combined Annotation Dependent Depletion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: CAA is a paid consultant for Genomics plc and BridgeBio, RGC completed the whole exome and chip genotyping for this project
GEO accession (UC biopsies single cell expression): GSE150516
References
- 1.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JZ, Sommeren S van, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo Y, Lange KM de, Jostins L, et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet 2017;49:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adeyanju O, Okou DT, Huang C, et al. Common NOD2 risk variants in African Americans with Crohn’s disease are due exclusively to recent Caucasian admixture. Inflamm Bowel Dis 2012;18:2357–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaahl MG, Winter T, Warnich L, et al. Analysis of the three common mutations in the CARD15 gene (R702W, G908R and 1007fs) in South African colored patients with inflammatory bowel disease. Mol Cell Probes 2005;19:278–281. [DOI] [PubMed] [Google Scholar]
- 6.Gao M, Cao Q, Luo L, et al. [NOD2/CARD15 gene polymorphisms and susceptibility to Crohn’s disease in Chinese Han population]. Zhonghua Nei Ke Za Zhi 2005;44:210–212. [PubMed] [Google Scholar]
- 7.Duncan L, Shen H, Gelaye B, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 2019;10:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tukey M, Pleskow D, Legnani P, et al. The utility of capsule endoscopy in patients with suspected Crohn’s disease. Am J Gastroenterol 2009;104:2734–2739. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber S, Reinisch W, Colombel JF, et al. Subgroup analysis of the placebo-controlled CHARM trial: increased remission rates through 3 years for adalimumab-treated patients with early Crohn’s disease. J Crohns Colitis 2013;7:213–221. [DOI] [PubMed] [Google Scholar]
- 10.Farh KK-H, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, Fang M, Jostins L, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017;547:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serra EG, Schwerd T, Moutsianas L, et al. Somatic mosaicism and common genetic variation contribute to the risk of very-early-onset inflammatory bowel disease. Nat Commun 2020;11:995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manichaikul A, Mychaleckyj JC, Rich SS, et al. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hui KY, Fernandez-Hernandez H, Hu J, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brant SR, Okou DT, Simpson CL, et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans With Inflammatory Bowel Disease. Gastroenterology 2017;152:206–217.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang D A Coefficient of Determination for Generalized Linear Models. The American Statistician 2017;71:310–316. [Google Scholar]
- 20.R Core Team (2013). R: A Language and Environment for Statistical Computing. 2013. Available at: https://www.r-project.org/ [Accessed March 12, 2020].
- 21.Márquez-Luna C, Loh P-R, South Asian Type 2 Diabetes (SAT2D) Consortium, et al. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol 2017;41:811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Candotti F Advances of gene therapy for primary immunodeficiencies. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi ZH, Wei W, Bu DF, et al. Targeted high-throughput sequencing technique for the molecular diagnosis of primary immunodeficiency disorders. Medicine (Baltimore) 2018;97:e12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi AY, Iyer VN, Hagan JB, et al. Incidence and temporal trends of primary immunodeficiency: a population-based cohort study. Mayo Clin Proc 2009;84:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picard C, Al-Herz W, Bousfiha A, et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol 2015;35:696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol 2017;139:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin JC, Chang C, Boschetti G, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019;178:1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anon. Data and Statistics. 2019. Available at: https://www.cdc.gov/ibd/data-statistics.htm [Accessed March 26, 2020].
- 32.Choi SW, O’Reilly PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019;8. [DOI] [PMC free article] [PubMed]
- 33.Vilhjálmsson BJ, Yang J, Finucane HK, et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet 2015;97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickham H ggplot2: elegant graphics for data analysis. Second edition. Cham: Springer; 2016. [Google Scholar]
- 35.The International HapMap Consortium. A haplotype map of the human genome. Nature 2005;437:1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut 2013;62:1795–1805. [DOI] [PubMed] [Google Scholar]
- 38.Martínez Jaramillo C, Trujillo-Vargas CM. LRBA in the endomembrane system. Colomb Med 2018;49:236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo B, Zhang K, Lu W, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–440. [DOI] [PubMed] [Google Scholar]
- 40.Hayes P, Dhillon S, O’Neill K, et al. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol 2015;1:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlig HH, Schwerd T, Koletzko S, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014;147:990–1007.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 2020;5:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017;169:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010;42:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habibi S, Zaki-Dizaji M, Rafiemanesh H, et al. Clinical, Immunologic, and Molecular Spectrum of Patients with LPS-Responsive Beige-Like Anchor Protein Deficiency: A Systematic Review. J Allergy Clin Immunol Pract 2019;7:2379–2386.e5. [DOI] [PubMed] [Google Scholar]
- 46.Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 2020;16:155–166. [DOI] [PubMed] [Google Scholar]
- 47.Goenka MK, Kochhar R, Tandia B, et al. Chloroquine for mild to moderately active ulcerative colitis: comparison with sulfasalazine. Am J Gastroenterol 1996;91:917–921. [PubMed] [Google Scholar]
- 48.Nagar J, Ranade S, Kamath V, et al. Therapeutic potential of chloroquine in a murine model of inflammatory bowel disease. Int Immunopharmacol 2014;21:328–335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.