

Abstract

Protein–protein modulation has emerged as a proven approach to drug discovery. While significant progress has been gained in developing protein–protein interaction (PPI) inhibitors, the orthogonal approach of PPI stabilization lacks established methodologies for drug design. Here, we report the systematic ″bottom-up″ development of a reversible covalent PPI stabilizer. An imine bond was employed to anchor the stabilizer at the interface of the 14-3-3/p65 complex, leading to a molecular glue that elicited an 81-fold increase in complex stabilization. Utilizing protein crystallography and biophysical assays, we deconvoluted how chemical properties of a stabilizer translate to structural changes in the ternary 14-3-3/p65/molecular glue complex. Furthermore, we explore how this leads to high cooperativity and increased stability of the complex.
1. Introduction
Protein–protein interaction (PPI) modulation has fundamentally changed drug discovery, expanding the druggable proteome.1−4 This shift in perspective has rapidly expanded the number of entry points for novel therapeutic intervention for many diseases.2,5 The development of PPI inhibitors has progressed steadily, leading to the emergence of clinically approved PPI inhibitors such as Venetoclax for chronic lymphocytic leukemia or small lymphocytic lymphoma.5 In contrast, PPI stabilization has emerged slowly, with the retrospective elucidation of the mode of action of clinically approved immunosuppressants, rapamycin,6,7 and the cytostatic paclitaxel.8,9 Successes of immunomodulators, lenalidomide, and cooperative PROTACs like AT1 provide valuable ″proofs-of-concepts″ for modulating cell homeostasis using synthetically derived PPI stabilizers.10−12 However, a systematic understanding of how to design small molecules that stabilize PPIs is urgently needed. The disconnection between these orthogonal PPI modulation approaches is primarily driven by a poor understanding of drug design rules for PPI stabilizers due to the complicated kinetics of dynamically interacting components that form the complex,13,14 shallow surface pockets, and limited or no structural information about the essential interactions that drive complex formation. A major obstacle for the development of PPI stabilizers is identifying chemical starting points, as the transient nature of complexes makes detecting ligand binding challenging. Further, affinity of a ligand to a complex is not necessarily correlated to the ligand’s stabilizing activity.
There is a significant need to develop novel drug discovery methodologies and alternative types of chemical matter to identify hit compounds for the development of PPI stabilizers. Fragment-based drug discovery has emerged as a promising approach leading to several clinically approved drugs.15,16 Since the clinical approval of ibrutinib, the stigma relating to covalent drugs and fragments has dissipated,17 resulting in a rise in the application of covalent tethering, particularly for challenging drug targets.18,19 The emergence of fragment tethering approaches using covalent chemistry has simplified the identification of hit compounds as binding fragments are easily detectable, and instrumentation is not reliant on detecting transient fragments/complex interactions to identify hits.20,21 Covalent fragment screening uses a covalent bond that is formed between a natural or engineered amino acid inside the targeted binding pocket and a reactive handle installed on the fragments.22 Recent fragment screening approaches utilizing disulfide tethering have proven successful in identifying hit fragments for PPI stabilization. Such site-directed fragment identification has recently been reported by us for stabilization of the protein 14-3-3 in complex with estrogen receptor α (ERα)23 and estrogen related receptor gamma (ERRγ)24 utilizing a disulfide tethering approach. We have also reported a new aldimine-based covalent tethering approach for the 14-3-3/p65 subunit of NF-κB PPI.25 This approach employs a dynamic imine-based tether to anchor the fragment at the interface of the composite pocket. Lysine tethering is a particularly attractive approach as the large percentage of the proteinogenic amino acids makes it amendable to a wide array of chemical probe development projects.26−29
Targeting the p65 subunit of NF-κB is of specific interest since NF-κB is a homo- and/or heterodimeric transcription factor involved in the regulation of immune responses, cell proliferation, and inflammation and therefore is connected to cancer and autoimmune diseases, among others.30−32 Attempts to directly inhibit the transcriptional activity of NF-κB have typically failed due to the inability to identify NF-κB-targeting matter.33,34 Interestingly, increased transcriptional activity of p65 has been correlated with downregulation of 14-3-3 in studies on ischemia–reperfusion and breast cancer.35,36 Also, upregulation of 14-3-3 has been shown to favor cytosolic localization of p65,37 subsequently preventing transcriptional activity. Stabilization of the 14-3-3/p65 complex could therefore furnish a novel entry point for targeting NF-κB and enabling a controlled therapeutic intervention.
Point mutational studies on p65 revealed three potential 14-3-3 binding sites surrounding the phosphorylation sites S45, S281, and S340.37 Binding affinities and structural information for two of these sites, pS45 and pS281, were reported, showing the direct physical interaction between both proteins.38 Benzaldehyde-based molecular fragments were shown to bind specifically to Lys122 of 14-3-3 via imine bond formation, thereby stabilizing the interaction with the p65 motif around phosphorylation site pS45 via hydrophobic contacts with p65.25
Here, we extend upon our initial communication reporting on an imine-based site-directed fragment approach to develop a 14-3-3/p65 molecular glue.25 Critical to the development of molecular glues is a robust understanding of molecular interactions and structural changes in the protein–protein–ligand complex that result in cooperative behavior. To understand the chemical properties that produce cooperative ligands, we employed a fragment extension design process using structural information gathered from X-ray crystallography soaking experiments and fluorescence anisotropy (FA) measurements. This enabled us to design initial fragments into molecular glues that showed stabilizing activity for the 14-3-3/p65 complex, culminating with the discovery of compound 24j that elicits an 81-fold stabilizing effect on the 14-3-3/p65 complex.
2. Results and Discussion
We have previously shown that aldimine bond formation is highly selective for Lys122 of 14-3-3 that lies at the interface between 14-3-3 and p65 in the composite binding pocket (Figure 1A). The enhanced selectivity for Lys122 is the result of a combination of the local hydrophobic character of the composite pocket, a lowered pKa of the lysine side chain, and the templating effects of p65 binding.25 Given the intrinsically disordered nature of large parts of the p65 subunit of NF-κB (Figure 1B), we utilized a 13-mer phosphopeptide representing (EGRSAG pSer45 IPGRRS) the recognition sequence of 14-3-3 to expedite chemical matter elucidation. Our initial investigation used X-ray crystal soaking experiments and a fragment library of commercially available aldehydes (34 fragments) to identify four key chemotypes that induced imine bond formation with Lys122 within the 14-3-3/p65 composite pocket (Figure 1C,D): methylsulfonyl (1 and 2), 1-nitro,3-hydroxybenzene (3), methyl acetamide (4), and five-membered N-heterocycles (5, 6, and 7). Biochemical assessment of fragments 1–7 used a fluorescence anisotropy (FA) assay. To assess the fragments’ capacity to induce a ternary complex, the fragments were titrated to a solution of 100 nM fluorescently labeled p65_45 peptide (FITC-βAla-EGRSAG pSer45 IPGRRS) and 50 μM of 14-3-3γ. The subsequent outputs are herein termed ″half maximal complex concentration (CC50)″. Results from the assay showed that these fragments did not increase complex formation. However, the lack of increase in complexation at relevant fragment concentrations was in accordance with crystal data, which indicated little to no direct contacts between the fragments and the p65 peptide. Fragments that are detectable in crystallography experiments but do not induce a detectable effect on complex formation are termed ″silent-binders″. We sought to develop a structure–activity relationship (SAR) based upon these four chemotypes with the specific goal of developing molecular glues that orthosterically engage with p65 and stabilize the 14-3-3/p65 complex.
Figure 1.

Exploring the p65/14-3-3 interface with imine forming fragments. (A) Overview of the composite binding pocket of 14-3-3 and the p65_45 peptide (PDB: 6QHL).38 (B) Crystal structure of the NF-κB subunit p65 (PDB: 1IKN,39 IκBα and p50 hidden for clarity) with known phosphorylated serine residues Ser281 and Ser45 labeled. (C) Interface of the ternary complex of 1/p65_45/14-3-3 (PDB: 6YOW).25 Indicated are three subpockets (the roof of 14-3-3, the FC-binding pocket, and the deep binding pocket), which can be exploited by fragment extension. Shown are 14-3-3 (white sticks, surface and cartoon), p65_45 peptide (red sticks), and fragment 1 (cyan sticks). (D) Fragment hits based on X-crystallography screening with benzaldehyde binding to Lys122 of 14-3-3.
To facilitate rapid optimization of the initial hit compounds into molecular glues, six focused libraries were designed based on the four chemotypes from the initial screen. Key to the development of molecular glues is understanding which specific interactions (hotspots) between the fragments and the 14-3-3/p65 complex lead to cooperative binding. Within our lab, we have developed a highly robust crystallography screening system to rapidly access X-ray crystal structures of soaked fragments. Utilizing this system, we gain unparalleled structural information that guides hit optimization.
2.1. Focused Library Development around the 3-Hydroxy,4-nitrobenzaldehyde Scaffold
Initial focused library development concentrated on extension of 3 based on crystal data, which showed complete coverage of the fragment by the electron density map, indicating a high occupancy for 3 within the composite pocket. Analysis of the X-ray crystal structure of 3 indicated that a relatively large solvent exposed pocket was present in front of the fragment formed by p65 (Figure 2A). We sought to explore this chemical space via extension of 3 from the 3-hydroxy position. To expedite exploration, focused libraries 1 and 2 were obtained from a commercial supplier and screened using X-ray crystallography (Scheme 1).
Figure 2.

Nitrobenzaldehyde structural analogues. (A) Initial fragment hit 3 covalently bound to the 14-3-3/p65_45 complex. The fragment can be extended from its meta-position (black arrow shows proposed vector for extension) (PDB: 6YOY).25 (B) Ternary structure of 9a (salmon sticks; PDB: 7NMH), p65_45 (red sticks), and 14-3-3 (white cartoon/sticks). Water molecules: red spheres; hydrogen bonds: black dashes. (C) Ternary structure of 10a/p65_45/14-3-3 (PDB: 7BJF). Details as in (B). (D) Ternary structure of 13e (left; PDB: 7BJL), 13f (middle; PDB: 7BJW), or 13l (right; PDB: 7BKH) binding to the p65_45/14-3-3 complex. Compounds are shown as yellow sticks; the rest are as described before. The 2Fo-Fc electron density map is contoured at 1σ for all.
Scheme 1. Synthesis of Focused Fragment Libraries 1 to 3.

Analysis of focused libraries 1 and 2 using X-ray crystal soaking experiments showed three sulfonate fragments (9a–c) and four ester fragments (10a, d, j, and m) bound to Lys122 in the crystal structures (Figure 2B,C; Figure S1A,B; Table S1). The various substituents elicited poor to moderate coverage by the electron density map, except for the sulfonates 9a and 9b. However, neither 9a nor 9b were active in functional FA assays (Figure S1A). For these smaller substitutes (9a and 9b), no favorable contacts with the peptide could be observed, whereas the larger substitutions resulted in a loss of electron density, indicative of a high conformational freedom of the ester side chain (9c, 10a, and 10j; Figure S1A,B).
An additional collection of fragments (focused library 3) was synthesized based on tertiary amines at the meta-position. The replacement of the phenol oxygen of 3 with a ternary amine increased the number of vectors for fragment extension, reduced conformational freedom, and enabled the exploration of a different chemical space. A one-step nucleophilic aromatic substitution using the key benzaldehyde intermediate 11 and the corresponding amine (12a–u) was employed to afford 21 analogues (13a–u) with yields ranging from 20 to 68% (Scheme 1). Focused fragment library 3 was then soaked into p65_45/14-3-3 crystals and tested in the FA assay. Of this collection, four fragments bound in crystallography experiments, with 13e, f, l, and q showing significant electron density coverage (Figure 2D, Figure S1C, Table S1). Notably, all four fragments contained six-membered saturated N-heterocyclic rings with polar functional groups that formed either direct hydrogen bonds with the backbone of p65 or water-mediated hydrogen bonds. The binding poses of these compounds were well defined, whereby the saturated N-heterocyclic ring extends toward the C-terminus of the p65 peptide. These results suggest that these fragments are shielded by the amphiphilic amino acids Pro47, Gly48, Arg49, and Arg50 of p65, which facilitate covalent tethering. Biophysical assessment of 13e, f, l, and q using FA compound titration assays showed that the fragments did not elicit significant ternary complex formation at biochemically relevant concentrations (Figure S1C). Surprisingly, 13f and 13l showed no increased complex formation, considering that both fragments engage in hydrogen bonding with p65. Given the lack of 14-3-3/p65 increased complex formation in FA compound titration assays at concentrations practical for hit optimization, we shifted focus to the five-membered N-heterocycles chemotype.
2.2. Focused Library Development around the 4-(1H-Imidazol-1-yl)benzaldehyde Scaffold
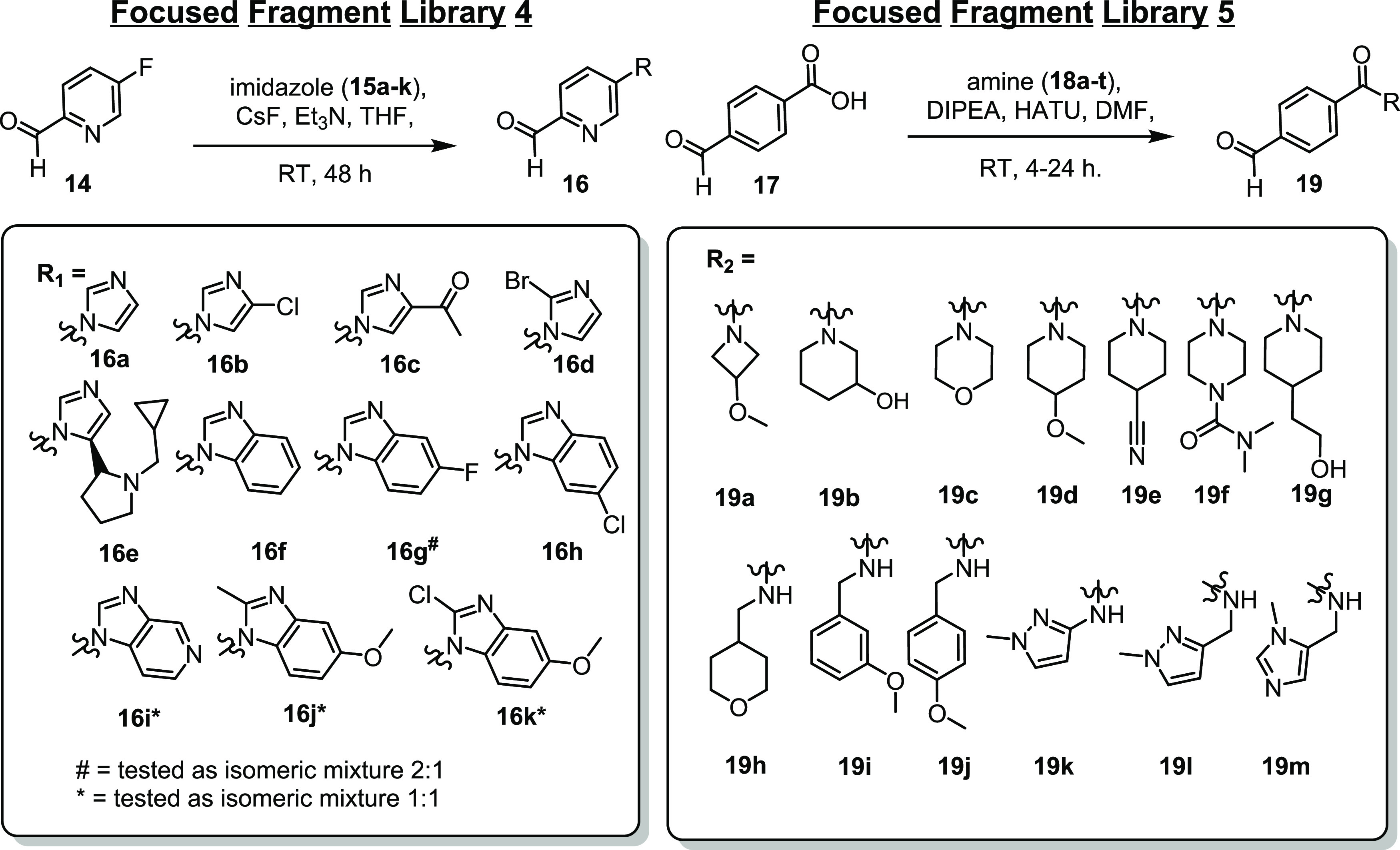
Fragment 5 was selected for fragment library development as the para-substitution proved to be more solvent exposed compared with 7 and the 1,3-substituted imidazole provided the possibility for fragment extension from three vectors of the N-heterocycle (Figure 3A). A focused library of 13 fragments (focused library 4) was synthesized using a nucleophilic aromatic substitution reaction with cesium fluoride, triethylamine, 4-fluoro-nicotinaldehyde (14), and an array of substituted imidazoles (15a–e) or benzimidazoles (15f–k) (Scheme 2). The starting reactant 4-fluoro-nicotinaldehyde (14) was used to improve the solubility of the fragments and to provide a further point for a polar interaction compared with 5–7. The resulting library was then subjected to X-ray crystal soaking experiments and FA assay. Notably, fragments 16b, c, f, and g–k were tested as mixtures of regioisomers.
Figure 3.

Imidazole-benzaldehydes are stabilizing the p65_45/14-3-3 complex. (A) The imidazole moiety of 5 (teal sticks) offers three vectors for fragment growth (arrows) (PDB: 6YP3).25 (B) Fragment induced p65_45/14-3-3γ ternary complex formation, measured with compound titrations in FA (r/mAU). The compounds were titrated to 50 μM 14-3-3γ and 100 nM p65_45. (C) Ternary structure of 16d (light blue sticks; PDB: 7NM9) binding to the p65_45/14-3-3σΔC complex. (D) Mixture of structural isomers of 16j and 16k. (E, F) Crystal structure of 16k binding to the p65_45/14-3-3σΔC complex (PDB: 7NR7). The electron density map indicates two binding poses (E). Based on the position of the methoxy substitution, the 2-chloro substitution points either toward the peptide (16k-5) or to the FC pocket (16k-6). Both binding poses engage in hydrophobic contacts with 14-3-3 and p65 (sphere representation, F). For all: 14-3-3: white cartoon, surface, sticks; p65_45: red sticks; 2Fo-Fc electron density map (contoured at 1σ): gray mesh.
Scheme 2. Synthesis of Focused Fragment Libraries 4 and 5.
Analysis of the fluorescence anisotropy assay identified that fragments 16d, e, j, and k induced an increase in anisotropy (Figure 3B); however, only 16d, j, and k were detected in the electron density map of soaking experiments. Notably, all three fragments showed a mixture of conformational poses (Figure 3C–F, Figure S2, Table S2). Both structural isomers of 16j (2-methyl-5-methoxy-benzimidazole) and 16k (2-chloro-5-methoxy-benzimidazole) were observed to bind within the composite binding pocket as result of the soaking experiment using mixtures of the structural isomers (Figure 3D–F). For instance, the chlorine atom in 16k points toward the p65 peptide, while the methoxy-substitution can be detected in the 5-position (Figure 3E). For the other binding pose, the chlorine atom in 16k points to the FC pocket, whereas the methoxy-substitution located in the 6-position is positioned above Ile46 of p65. Both structural isomers of 16k engage in hydrophobic contacts with the roof of 14-3-3 and Ill46 of p65_45 (Figure 3F). Despite their similar binding poses, 16k (CC50 = 260 μM) elicited a significantly higher increase in anisotropy compared to 16j, which induced a negligible ternary complex formation in the FA assay (Figure 3B). The replacement of a chloro-moiety for a methyl group in 16k (16j) on the imidazole ring has a significant impact on complex formation, resulting in diminished anisotropy.
2.3. Focused Library Development around the 4-Formylbenzamide Scaffold
We next turned our attention to fragment extension based upon fragment 4. Analysis of the co-crystal structure showed that the N-acetyl group of 4 probes up and toward the p65 peptide. To expediate the rapid parallel synthesis of focused library 5, the N-(4-formylphenyl)acetamide (4) was inverted to a N-substituted 4-formyl-benzamide (19a–t), improving the nucleophilicity of the amine in the corresponding amide coupling reactions as well as providing greater chemical diversity of the fragment extension (Scheme 2). Further, a previously published cluster analysis of occurring torsion (τ) angles of benzamide report a τ ≈ 30 and 150° between the benzene ring and the acetamide head group (Figure 4A).40 We hypothesized that extension of the fragment from this vector provides the opportunity to engage with the p65 peptide and enhance fragment binding (Figure 4B). Focused library 5 was synthesized using standard amide coupling conditions, 1-formylbenzoic acid (17), and amines 18a–t. In total, 13 fragments (19a–t) with amide substitution in the para-position of the aldehyde functionality were synthesized, soaked in p65_45/14-3-3σΔC crystals, and tested in FA compound titrations. All fragments from library 5 were shown to bind within the composite binding pocket using X-ray crystallography (Figure S3, Table S2). Subsequent biophysical analysis identified that all compounds were silent binders, not inducing detectable formation of a ternary complex at assay relevant concentrations. X-ray co-crystallization experiments and an assessment of the C–C–C–N torsion angles provided an explanation for the lack of activity for this series of fragments. The electron density map of the aldehydes showed highly resolved electron density for the benzaldehyde ring and the amide of the benzamide. The carbonyl of the benzamide engages in polar contacts with the water shell of 14-3-3, thereby stabilizing this orientation of the fragments (Figure 4C). The binding poses of the different R-substitutions are poorly resolved by the electron density map, suggesting a high level of conformational freedom and a high-level entropy of the substitutes, unfavorable for binding. Assessment of this library of fragments showed the R-substitution pointing toward the solution above the p65 peptide or Asp215 of 14-3-3 with limited possibilities to engage in favorable contacts. For a few fragments, the R-substitutes engage in polar contacts with the 14-3-3 water shell, exemplified by 19e (Figure 4C). However, the additional polar contacts do not translate to significant increases in ternary complex formation potentially due to entropic penalties or lack of contacts with the p65 peptide. Given the lack of cooperative ternary complex formation of this library at biochemically relevant concentration, we shifted focus to the 4-formyl benzenesulfonamide chemotype.
Figure 4.

Acetamide-benzaldehydes in complex with 14-3-3/p65_45. (A) 3D (beige sticks) and 2D structure of 4-formyl-benzamide. The C–C–C–N torsion angles (orange lines) cluster at τ ≈ 0 and 180°.40 (B) Initial fragment hit 4 can be extended from its para-position (black arrows; PDB: 6YOX).25 (C) Ternary structure of 19e (green sticks) in complex with p65_45 (red sticks) and 14-3-3σΔC (white cartoon and sticks) (PDB: 7BIY). Hydrogen bonds are indicated with green dashes; the 2Fo-Fc electron density map is shown as gray mesh, contoured at 1σ.
2.4. Focused Library Development around 4-Formyl-benzenesulfonamides
Having investigated focused libraries 1–5, we shifted our attention to development of a focused library based on fragment 1. Analysis of the crystal structure of fragment 1, specifically the torsion angle of τ = 90 ± 30° between the benzene ring and the mesyl group, proved interesting (Figure 5A).40 Extension of the fragment from the methyl group provided a potential point of reaching over the p65 peptide, trapping its binding to 14-3-3 (Figure 5B). Alternatively, we postulated that fragment extension could also result in a change in the conformation of the fragment leading to the occupation of the FC pocket and increasing 14-3-3-based affinity. To expedite fragment development, the methyl group was replaced for N-substituted amines to facilitate rapid access to a library of 25 structural analogues of 4-formyl-benzenosulfonamides. This library of sulfonamides was synthesized by conversion of sodium 2-formylbenzene-1-sulfonate (20) to 4-formyl-benzenesulfonyl chloride (21) using thionyl chloride in DMF. Subsequent coupling of 21 with N-substituted amines (22a–z) afforded fragments 23a–z.
Figure 5.

Sulfonamide-benzaldehydes are active in FA compound titrations. (A) 3D and 2D chemical structure of 4-formyl-benzenesulfonamide with a C–C–S–N torsion angle τ as indicated.40 (B) The benzenesulfonamides are extended toward the FC pocket or toward the p65 peptide (red; 14-3-3: white; both with sticks and transparent surface) (PDB: 6YOW).25 (C) FA compound titrations in the presence of 50 μM 14-3-3γ and 100 nM p65_45. The compounds are divided into weak (left), medium (middle), and high (right) activity. The legends are sorted by activity.
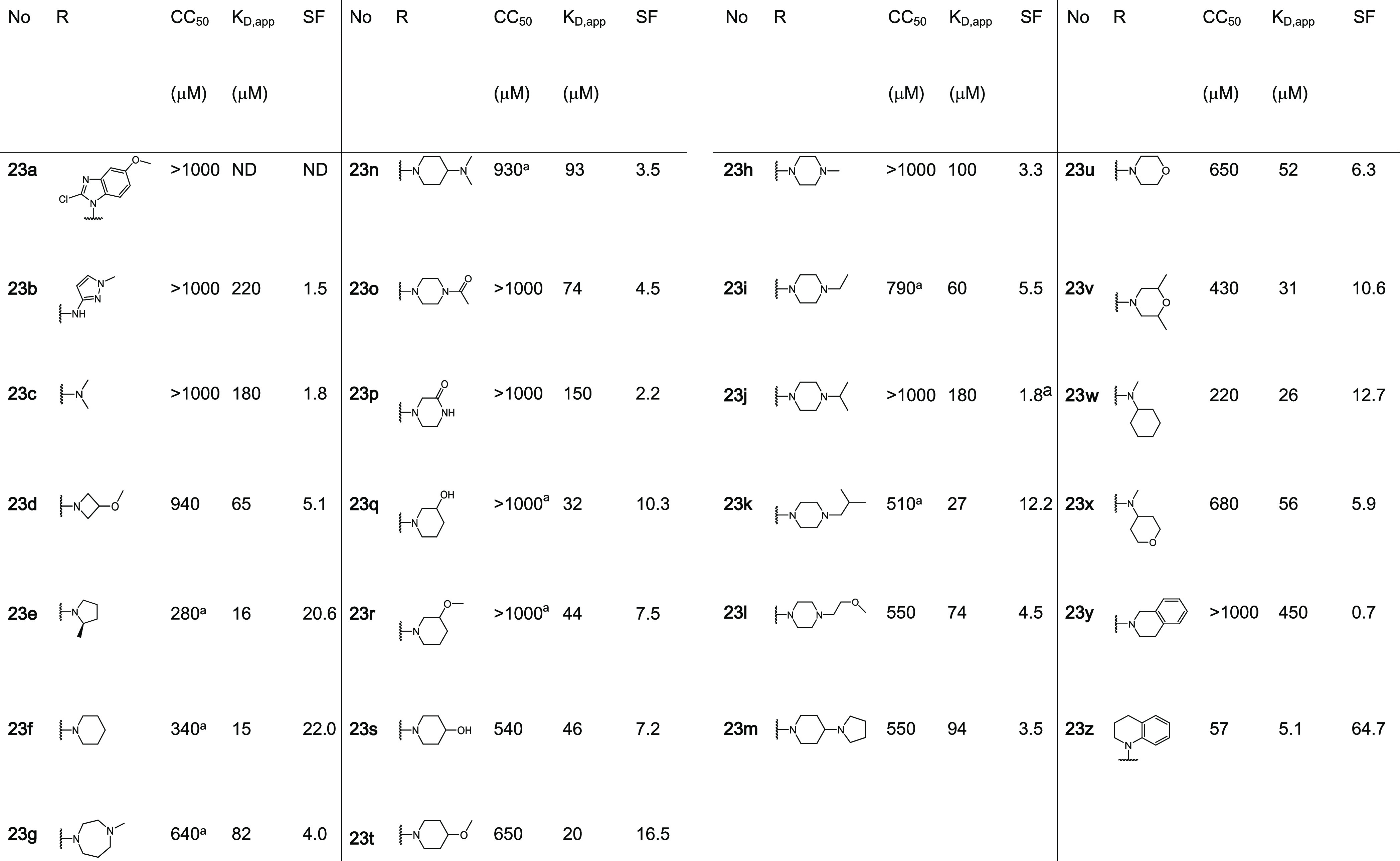
In contrast to focused libraries 1–5, most of the tested 4-formyl-benzenesulfonamide fragments showed significant stabilization in FA assays, allowing a differentiated SAR analysis (Table 1, Figure 5C). The activity ranged from a weak affinity with a small, not quantifiable increase in anisotropy to a two-digit micromolar CC50 potency in compound titrations. From the focused library 6, nine fragments elicited CC50 values ranging from 57 to 430 μM. An additional assessment of stabilization using the FA protein titration assay showed for 19 fragments a significant shift of apparent KD values in the presence of 1 mM of the fragments. Fragments 23e, f, k, t, w, and z showed KD values of <30 μM compared to the binary complex of full-length 14-3-3 and the p65_45 peptide that gave a KD = 300 ± 100 μM. Analysis of this subset of fragments showed that these shifts in KD’s (stabilization factor, SFs range from ∼12- to 65-fold). Fragment 23z, possessing a N-substituted 1,2,3,4-tetrahydroquinoline, showed the greatest SF of ∼65-fold.
Table 1. Exploration of 4-Formyl-benzenesulfonamidesb.

Fit did not converge.
CC50: values of compound titrations with 50 μM 14-3-3γ. KD,app: value of protein titrations in the presence of 1 mM of the fragment. SF: the fold-change of apparent KD in comparison to a DMSO control. ND: not determined. All fragments bound to the p65_45/14-3-3σΔC complex. For additional data, see Figure S4 and Table S3.
Comparison of X-ray crystal soaks provided an explanation for the FA assay results. Structural data showed that 2-methyl pyrrolidine (23e) and piperidine (23f) were tolerated within the composite binding pocket. Ring expansion to the N-methyl diazepane (23 g) proved to be detrimental to stabilization (CC50 = 640 μM). Notably, polar functionalities within six-membered N-heterocycles were not well tolerated; specifically, hydrogen bond accepting and donating groups (23h, n–u) elicited high CC50 values and modest SFs (6–16-fold). This can be exemplified by fragment 23f containing piperidine (app KD = 15 μM, SF =22) compared with N-methylpiperazine 23h (app KD = 100 μM, SF = 3.3) and morpholine 23u (app KD = 52 μM, SF = 6.3) (Figure 6A,B). Analysis of the X-ray structures showed that introduction of a polar functionality typically resulted in reorientation of the R-substitute of the fragment toward the FC-binding pocket. Introduction of a hydrophobic functionality such as 23k (app KD = 27 μM, SF = 12.2) and 23v (app KD = 430 μM, SF = 10.6)25 recovered stabilizing activity, with the fragments re-establishing hydrophobic contacts with the p65 peptide. Notably, large amphiphilic modifications were also not well tolerated, such as 23m (app KD = 74 μM, SF = 4.5) and 23n (app KD = 94 μM, SF = 3.5) (Figure 6C, Table 1), with both 23m and 23n occupying solvent exposed space within the composite pocket but not engaging in contacts with p65. Interestingly, fragment 23w (app KD = 26 μM, SF = 12.7) showed a significant drop in stabilization with respect to 23f. Crystal soaking experiments showed that fragment 23w engages in hydrophobic interactions with Ile46, Pro47, and Gly48 of p65 (Figure 6D). However, unlike other fragments, 23w induces a conformational change in the C-terminus of the peptide shifting toward the pS45. Analysis of soaking experiment with fragment 23y showed that the tetrahydroisoquinoline ring engages in hydrophobic contacts with the p65 peptide; however, unlike 23e and 23f, the bicyclic rings repulse Pro47 of p65 (Figure 6E). This provides an explanation for the poor stabilizing activity of 23y. In contrast to 2-tetrahydroquinoline (23y), 1-tetrahydroquinoline (23z) was highly tolerated, eliciting CC50 = 57 μM and app KD = 5.1 μM; this translated to ∼65-fold stabilization. X-ray crystallography analysis of fragment 23z provided valuable explanation for the high stabilization observed in FA assays. Specifically, the bicyclic substructure in 23z engages the p65 peptide and is positioned upwards occupying the hydrophobic roof of 14-3-3σΔC shaped by residues Ile219, Leu218, and Leu222 (Figure 6F). Further, 23z also engages in direct hydrophobic contacts with Ile46 and Pro47 of p65. Most notably, the 14-3-3/p65/23z ternary complex formation results in a reorientation of the p65 peptide leading to additional 14-3-3/p65 contacts. Fragment 23z appears to function as a template for the additional binding of p65, increasing cooperative behavior of the ternary complex. Fragment 23z facilitates additional 14-3-3/p65 contacts by enabling the C-terminus of p65 to wrap over 23z and engage in electrostatic contacts with 14-3-3. Specifically, salt bridges are observed between Arg49/Glu14 and Arg50/Asp215 of p65_45/14-3-3, respectively. These structural features translate to high activity in FA assays, with a CC50 of 57 μM and an SF of 65 (Figure 6G). The additional interactions between 14-3-3 and p65 induced by the binding of 23z lead to increased cooperativity of the ternary complex. This improved cooperative behavior directly translates into improved stabilization of the 14-3-3/p65 complex.
Figure 6.

Sulfonamide-benzaldehydes inducing complex stabilization. (A) Overlay of 23h (green sticks; PDB: 7NLE), 23k (orange sticks; PDB: 7NK5), 23f (cyan sticks; PDB: 7NJB), and 23u (yellow sticks; PDB: 7BJB) binding to p65_45 (red sticks) and 14-3-3 (white cartoon and sticks). Hydrogen bonds are shown as black dashes, and hydrophobic contacts are indicated with transparent spheres. A conserved water molecule (small sphere) is shown colored based on the related fragment color. (B) Fragments 23h, k, f, and u show complex stabilization in FA compound titrations (left; [14-3-3γ]: 50 μM, [p65_45]: 100 nM) and protein titrations (right; [fragment]: 1 mM, [p65_45]: 100 nM). (C) Overlay of 23n (brown sticks; PDB: 7NM3) and 24m (light blue sticks; PDB: 7NM1) in complex with 14-3-3σΔC/p65_45. (D) Crystal structures of 23w (blue sticks; PDB: 7NLA) binding to the composite binding pocket of p65 (red sticks) and 14-3-3 (white cartoon and sticks). The 2Fo-Fc electron density map (1σ) is displayed as gray mesh, and hydrogen bonds are shown as black dashes. (E) Overlay of 23y (green sticks; PDB: 7NK3) and 23z (yellow; PDB: 7NJ9) binding to the p65_45/14-3-3 complex. Upon binding of 23y, Pro47 changes its conformation (red sticks, as reference: Pro47 of the 23z/p65_45/14-3-3σΔC complex shown as yellow sticks). (F) Ternary structure of 23z (yellow sticks; PDB: 7NJ9) binding to the p65_45/14-3-3 complex. Details as in (D). (G) Ternary complex formation of 23x, y, and z was measured with FA compound titrations (left; [14-3-3γ]: 50 μM, [p65_45]: 100 nM) and the stabilizing activity of 23w, y, and z with protein titrations (right; [fragment]: 1 mM, [p65_45]: 100 nM).
2.5. Optimization of Fragment 23z
Encouraged by the high stabilizing effect of covalent fragment 23z, we looked to develop a library of 10 sulfonylamides based on tricyclic fragment 23z. Due to the rearrangement of the p65 peptide in the presence of 23z forming a narrow and enclosed binding pocket, this limited the sites for fragment modifications to the 5-, 6-, or 7-position of the tetrahydroquinoline ring (Figure 7A). As a result, we focused on small modifications to build additional interactions with the hydrophobic patch in the roof of 14-3-3 or extension of the fragments over Ile46 of p65. Additionally, we investigated the addition of heteroatoms in the 4-position of the tetrahydroquinoline ring to engage with the backbone carbonyl or nitrogen of Pro47 or Gly48 to establish additional polar contacts. A small library of 10 analogues was synthesized using our previously mentioned sulfonamide coupling (Table 2).
Figure 7.

Optimization of 23z. (A) Ternary structure of 23z (yellow spheres) in complex with 14-3-3σΔC (white surface) and p65_45 (red cartoon, transparent spheres) (PDB: 7NJ9). Carbons of the bicyclic head group are numbered. (B) Ternary structure of 24b (orange sticks) in complex with p65_45/14-3-3σΔC (PDB: 7BIQ). The distance between the 2-methyl and water (red spheres) is indicated with black dashes. Polar contacts are shown as black dashed lines. (C) Structure of 24e (cyan sticks) binding to the p65_45/14-3-3σΔC complex (as in A; PDB: 7BI3). Hydrophobic contacts are indicated with transparent spheres. (D) FA compound titrations with 50 μM 14-3-3γ and 100 nM p65_45. (E) FA protein titrations with 1 mM fragment and 100 nM p65_45. (F) Structure of 24j binding to the p65_45/14-3-3σΔC complex (as in A; PDB: 7BIW). A beneficial hydrogen bond is formed between the backbone carbonyl of p65s’ Pro47 and 24j (black dash).
Table 2. Exploration of structural analogs of 23zb.

Tested as an enantiomeric mixture.
CC50: values of compound titrations with 50 μM 14-3-3γ. KD,app: values of protein titrations in the presence of 1 mM of the fragment. SF: the fold-change of apparent KD in comparison to a DMSO control. ND: not determined. All fragments bound to the p65_45/14-3-3σΔC complex. For additional data, see Figure S5 and Table S4.
Structural analysis of this library showed that this series of analogues retained a similar cooperative behavior as 23z, inducing additional PPI contacts. Biophysical analysis using an FA assay provided valuable insight into the SAR around fragment 23z. Analysis of the CC50 concentration indicated that 23z showed the highest ternary complex formation, with all other analogues in this series eliciting a drop in complexation (CC50 values ranging from 110 to 310 μM). However, significant insight into complex stabilization was gained from the analysis of app KD values. Substitution of 23z for an indoline (24a) or racemic 2-methyl-tetrahydroquinoline (24b) was tolerated; however, a 2-fold reduction was observed in stabilization, with app KD values of 11 and 9.6 μM, respectively. Structural analysis of 24a showed additional space around the five-membered heterocycle potentially providing additional sites for functionalization in the future (Figure S5). Fragment 24b showed excellent electron density across the entire fragment within the X-ray crystal structure, enabling unambiguous assignment of stereochemistry. From the structure, it was identified that the R-enantiomer exclusively bound in the crystal, suggesting that the pure R-enantiomer may have significantly underestimated CC50 and app KD (Figure 7B). This site may provide an excellent point for fragment extension toward the p65 peptide. Addition of a halogen or methoxy in the 6-position resulted in a reduction in stabilization, with 6-fluoro (24c), 6-chloro (24d), or 6-methoxy (24e) substituted tetrahydroquinoline fragments affording app KD values of 6.2, 9.9, and 10 μM, respectively. Analysis of soaking experiments shows that modifications in this position resulted in unfavorable contacts with the roof of 14-3-3 (Figure 7C). Addition of an oxygen to the saturated six-membered ring (24f) proved detrimental to stabilization (app KD = 15 μM). Further addition of halogens or methoxy groups in the 6 or 7 positions did not re-establish lost stabilization, such as 6-chloro (24h), 6-methoxy (24i), or 7-fluoro (24g). Substitution of the benzomorpholine (24f) for tetrahydroquinoxaline (24j) resulted in an improvement in stabilization with 24j eliciting an app KD of 4.3 μM, translating into the highest stabilizing effect with an 81-fold stabilization (Figure 7D,E). Structural analysis showed that the introduction of a nitrogen atom in the saturated ring installs an additional hydrogen bond with the backbone carbonyl of Pro47 of p65 (Figure 7F).
To assess the selective ternary complex formation of 24j with 14-3-3/p65, the fragment was further screened in a compound titration FA assay against three well-established 14-3-3 interactions (p53, ERα, and TAZ; Figure S6). Gratifyingly, no ternary complex formation was detected for p53, ERα, or TAZ complexes, proving that 24j is not a pan-stabilizer of 14-3-3 interactions and indicating an initial selectivity of 24j toward the p65/14-3-3 interaction.
3. Conclusions
Summarizing, here we build upon our initial report of a novel covalent fragment screening approach using an aldehyde fragment library. These covalent fragments form a covalent tether with Lys122 within the 14-3-3/p65 composite binding pocket. Specifically, we describe the optimization of initial hit covalent fragments into a p65/14-3-3 molecular glue that elicited an 81-fold stabilization of the 14-3-3/p65 complex. Critical to this success was the use of X-ray crystallography and FA measurements to develop a robust understanding of the structural activity relationship. Direct hydrophobic engagement with p65 was the driving interaction that established complex stabilization. Hydrophobic contacts between 23z or 24j and p65 resulted in a conformational change in the 14-3-3/p65 interface with extended interactions, in turn increasing the cooperativity of the ternary complex. Fragment 23z or 24j might serve as a valuable starting point for optimization for the development of a chemical probe to study the effects of 14-3-3-mediated regulation of p65 in a cellular context. Particularly, the aldehyde functionality requires additional investigations to validate its activity and oxidative stability in a cellular context such as with the aldehyde-containing drug voxelotor.41 Alternatively, the aldehyde could be replaced with a more benign chemical handle that is less susceptible to oxidation, or the application of a prodrug approach to protect the aldehyde may need to be considered when moving toward cellular studies.
Considering the lack of rational methodologies to develop PPI stabilizers, here we describe a systematic approach to developing initial hit fragments into molecular glues. Observations from this research indicate that the direct engagement of molecular glues with the partner peptide is significant for stabilization. Specifically, hydrophobic contacts are effective to enhance ternary complex formation and support a cooperative binding mode. Analysis of SAR results indicates that fragments that facilitate templating of the p65 peptide and promotion of additional contacts between 14-3-3 and p65 resulted in increased complex stabilization. This research has direct applications to further development of PPI stabilizers as well as the field of PROTACs. There is a growing body of evidence showing that cooperative PROTACs, which stabilize direct PPIs, lead to improved efficacy, reducing problems associated with the hook-effect and the necessity for highly potent molecules.42−45 Additionally, research from the field of PROTACs and our findings appear to be in agreement that the molecular glue/cooperative PROTAC potency of a ligand for a complex is not the driving force for increased complex stabilization; rather, the cooperativity behavior of the ternary complex appears to be the driving force for PPI stabilization.11,42,45,46
4. Experimental Section
4.1. Protein Expression and Purification
The 14-3-3 proteins were expressed and purified using standard protocols. In short, pPROEX HTb vectors encoding the 14-3-3σΔC (truncated C-terminus ΔC17) and 14-3-3γ isoform were transformed into BL21(DE3) cells. Protein expression was initiated with 0.4 mM IPTG at a cell density OD600 = 0.8–1. The expression took place overnight at 18 °C. The cells were harvested by centrifugation (10,000g, 15 min) and resuspended in a lysis buffer (50 mM Tris/HCl pH 8, 300 mM NaCl, 12.5 mM imidazole, and 2 mM β-mercaptoethanol). The cells were lysed with a homogenizer, and the lysate was cleared via centrifugation (40,000g, 30 min). Ni-NTA columns were used to isolate the protein, which was washed with 10 CV lysis buffer and eluted with 250 mM imidazole (50 mM Tris/HCl pH 8, 300 mM NaCl, 250 mM imidazole, and 2 mM β-mercaptoethanol). The full-length 14-3-3γ was dialyzed against 25 mM HEPES pH 7.5, 100 mM NaCl, 10 mM MgCl2, and 0.5 mM Tris(2-carboxyethyl)phosphine) and stored at −80 °C. For the 14-3-3σΔC, the His6-tag was removed by the TEV protease; the TEV was removed with Ni-NTA columns. The rest of the imidazole of the 14-3-3σΔC solution was removed by size exclusion chromatography (20 mM HEPES pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol) and stored at −80 °C.
4.2. X-ray Crystallography
Binary crystals with p65_45 peptide (sequence: EGRSAG pS45 IPGRRS, C-terminus: amidation; N-terminus: acetylation)47 and 14-3-3σΔC were grown at a 14-3-3σΔC concentration of 12 mg/mL in a 1:2 ratio with the acetylated peptide in 20 mM HEPES pH 7.5, 2 mM MgCl2, and 2 mM β-mercaptoethanol. This complexation mixture was incubated overnight. In a hanging drop setup, the complexation mixture was mixed in 1:2 ratio with the precipitation buffer (95 mM HEPES pH 7.5, 27–28% PEG400, 190 mM CaCl2, and 5% glycerol). For data acquisition, crystals were directly flash-frozen in liquid nitrogen.
Fragment soaks were performed by adding compounds in DMSO stock solutions direct to fully grown crystals with a final compound concentration of 10 mM (≤1% DMSO). The soaks were incubated for 7 days prior to data acquisition. Diffraction data were measured either at the P11 beamline of PetraIII (DESY campus, Hamburg, Germany) or the i-03/i-24 beamline of the diamond light source (Oxford, UK) or in-house. The diffraction data were integrated with the xia2/DIALS pipeline48 followed by molecular replacement with MolRep.49,50 The binary p65_45/14-3-3σΔC structure was used as a search model (PDB ID: 6QHL). Model refinement took place in iterative cycles with Coot,51 Refmac5,52 and phenix.refine.53 3D structures of ligands were prepared using the fragment SMILES and elbow of the phenix suite.53 Figures were generated with PyMOL (V2.0.6, Schrodinger LLC).
4.3. Fluorescence Anisotropy Assays
Complex stabilization was measured using a fluorescently labeled p65_45 peptide (FITC-βAla-EGRSAG pS45 IPGRRS) at a concentration of 100 nM throughout all assays. During compound titrations, the 14-3-3γ concentration was constant at 50 μM and the compound was titrated in a 1:1 dilution series. In protein titrations, 14-3-3γ was titrated in a 1:1 dilution series in the presence of 1 mM compound. The plates (Corning 384 well plates, black, round bottom, low binding) were incubated for 3 h at RT prior to fluorescence anisotropy (FA) measurements with the Tecan Infinite 500 plate reader (FITC dye: excitation 485 nm and emission 535 nm). Dilution series were prepared in an FA buffer (10 mM HEPES pH 7.4, 150 mM NaCl, and 0.1% Tween20).
The activity of the hit compound 24j was measured for the TAZ-peptide23 (FITC-βAla-RSH pS89 SPASLQ), p53-peptide54 (TAMRA-Ahx-SRAHSSHLKSKKGQSTSRHKKLMFK pT387 EGPDSD-COOH), or ERα-peptide13 (FITC-O1Pen-AEGFPA pT594 V-COOH) and 14-3-3γ. Therefore, the compound was titrated in a 1:1 dilution series to a constant concentration of 14-3-3γ and 10 nM peptide. The protein concentration was adjusted to one-third of the KD of the binary protein/peptide complex (final 14-3-3γ concentration for the measurement with TAZ: 0.1 μM; with p53: 0.3 μM and with ERα: 0.1 μM). For a better comparison, the baseline of each measurement was subtracted (Δr/mAU).
All measurements were performed as single measurements. Data analysis was performed with Origin 2019 (V9.6.0.172, OriginLab Corporation) with the inbuilt ″Hill1″ function for data fitting. For data sets without an upper plateau, the Hill coefficient n was set to n = 1.
4.4. Chemistry: General Information
All commercial chemicals were used as received. Reagents were used without further purification unless otherwise noted. TLC analysis was performed on TLC aluminum sheets, silica gel layer, ALUGRAM SIL G UV254, 20 × 20 cm by MACHEREY-NAGEL. TLC plates were analyzed by UV fluorescence (254 nm). UHPLC–MS analysis was performed using the UPLC Agilent Technologies 1290 Infinity coupled with the Agilent Technologies 6120 Quadrupole LC/MS DAD detector. Column: ACQUITY UHPLC BEH C18 (1.7 μm) 2.1 mm × 50 mm. Temperature: 40 °C. Detection: DAD + MS/6120 Quadrupole. Injected volume: 1 μL. Flow: 1.2 mL/min. Solvent A: water + 0.1% formic acid. Solvent B: acetonitrile + 0.1% formic acid. Gradient: 0 min 2% B, 0.2 min 2% B, 2.0 min 98% B, 2.2 min 98% B, 2.21 min 2% B, and 2.5 min 2% B. Preparative HPLC was performed using the UPLC Agilent Technologies 1260 Infinity coupled with the Agilent Technologies 6120 Quadrupole LC/MS. Column: Waters XBridge Prep C18 5 μm OBD 19 × 150 mm. Detection: DAD + MS/6120 Quadrupole. Flow: 32 mL/min. Solvent A: water + 0.1% formic acid. Solvent B: acetonitrile + 0.1% formic acid. Gradient: 0 min 77% A/23% B, 1 min 77% A/23% B, 9 min 16% A/84% B, 9.01 min 2% A/98% B, and 11 min 2% A/98% B. The purity of the synthesized compounds is ≥95%. 1H NMR and 13C NMR spectra were recorded on a Bruker 300 MHz spectrometer at ambient temperature. The chemical shifts are listed in ppm on the δ = scale, and coupling constants were recorded in hertz (Hz). Chemical shifts are calibrated relative to the signals corresponding of the nondeuterated solvent (CHCl3: δ = 7.26 ppm for 1H and 77.16 ppm for 13C; DMSO: δ = 2.50 ppm for 1H and 39.52 ppm for 13C). Abbreviations are used in the description of NMR data as follows: chemical shift (δ = ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, bs = broad singlet, dd = doublet of doublets, td = triplet of doublets), and coupling constant (J = Hz).
For general procedures, chemical characterization, NMR spectra, and LC–MS traces, see the Supporting Information.
Glossary
Abbreviations
- app. KD
apparent KD
- CC50
half-maximal complex concentration
- FA
fluorescence anisotropy
- PPI
protein–protein interaction
- PROTAC
proteolysis targeting chimera
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00401.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under the following accession codes: 7NMH, 7BI3, 7BIQ, 7BIW, 7BIY, 7BJB, 7BJF, 7BJL, 7BJW, 7BKH, 7NJ9, 7NJB, 7NK3, 7NK5, 7NLA, 7NLE, 7NM1, 7NM3, 7NM9, 7NR7, 7NV4, 7NVI, 7NWS, 7NXS, 7NXT, 7NXW, 7NXY, 7NY4, 7NYE, 7NYF, 7NYG, 7NZ6, 7NZG, 7NZK, 7NZV, 7O34, 7O3A, 7O3F, 7O3P, 7O3Q, 7O3R, 7O3S, 7O57, 7O59, 7O5A, 7O5C, 7O5D, 7O5F, 7O5G, 7O5O, 7O5P, 7O5S, 7O5U, 7O5X, 7O6F, 7O6G, 7O6I, 7O6J, 7O6K, 7O6M, and 7O6O. Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
§ M.W., D.V., and P.J.C. contributed equally. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The research was supported by funding from the European Union through the TASPPI project (H2020-MSCA-ITN- 2015, grant number 675179), through the Eurotech Postdoctoral Fellow program (Marie Skłodowska-Curie Co-funded, grant number 754462), and through The Netherlands Organization for Scientific Research (NWO) via VICI grant 016.150.366 and via Gravity Program 024.001.035.
L.B. and C.O. are scientific co-founders of Ambagon Therapeutics.
The authors declare the following competing financial interest(s): LB and CO are co-founders of Ambagon Therapeutics.
Supplementary Material
References
- Arkin M. M. R.; Wells J. A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing towards the Dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Lu H.; Zhou Q.; He J.; Jiang Z.; Peng C.; Tong R.; Shi J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduction Targeted Ther. 2020, 5, 213. 10.1038/s41392-020-00315-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy L. G.; Grossmann T. N.; Hennig S.; Brunsveld L.; Ottmann C. Modulators of Protein-Protein Interactions. Chem. Rev. 2014, 114, 4695–4748. 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Stevers L. M.; Sijbesma E.; Botta M.; MacKintosh C.; Obsil T.; Landrieu I.; Cau Y.; Wilson A. J.; Karawajczyk A.; Eickhoff J.; Davis J.; Hann M.; O’Mahony G.; Doveston R. G.; Brunsveld L.; Ottmann C. Modulators of 14-3-3 Protein–Protein Interactions. J. Med. Chem. 2017, 3755–3778. 10.1021/acs.jmedchem.7b00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. Small Molecules, Big Targets: Drug Discovery Faces the Protein–Protein Interaction Challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Li J.; Kim S. G.; Blenis J. Rapamycin: One Drug. Many Effects. Cell Metab. 2014, 19, 373–379. 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz J.; Hall M. N. Cyclosporin A, FK506 and Rapamycin: More than Just Immunosuppression. Trends Biochem. Sci. 1993, 18, 334–338. 10.1016/0968-0004(93)90069-Y. [DOI] [PubMed] [Google Scholar]
- Wani M. C.; Taylor H. L.; Wall M. E.; Coggon P.; McPhail A. T. Plant Antitumor Agents. VI. Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus Brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- Joerger M. Metabolism of the Taxanes Including Nab-Paclitaxel. Expert Opin. Drug Metab. Toxicol. 2015, 11, 691–702. 10.1517/17425255.2015.983074. [DOI] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thomä N. H. Structural Basis of Lenalidomide-Induced CK1α Degradation by the CRL4CRBN Ubiquitin Ligase. Nature 2016, 532, 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K. H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Ciulli A. Bifunctional Chemical Probes Inducing Protein–Protein Interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. 10.1016/j.cbpa.2019.07.003. [DOI] [PubMed] [Google Scholar]
- de Vink P. J.; Andrei S. A.; Higuchi Y.; Ottmann C.; Milroy L.-G.; Brunsveld L. Cooperativity Basis for Small-Molecule Stabilization of Protein–Protein Interactions. Chem. Sci. 2019, 10, 2869–2874. 10.1039/C8SC05242E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R. Cooperativity in Macromolecular Assembly. Nat. Chem. Biol. 2008, 4, 458–465. 10.1038/nchembio.102. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Fesik S. W.; Hubbard R. E.; Jahnke W.; Jhoti H. Twenty Years on: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Davis B. J.; Jahnke W.; Box G. Perspective Fragment-Based Drug Discovery : Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 2019, 26, 9–15. 10.1016/j.chembiol.2018.10.001. [DOI] [PubMed] [Google Scholar]
- Dalton S. E.; Campos S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. ChemBioChem 2019, 1–1100. 10.1002/cbic.201900674. [DOI] [PubMed] [Google Scholar]
- Gabizon R.; Shraga A.; Gehrtz P.; Livnah E.; Shorer Y.; Gurwicz N.; Avram L.; Unger T.; Aharoni H.; Albeck S.; Brandis A.; Shulman Z.; Katz B.; Herishanu Y.; London N. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick E.; Bradley A.; Gan J.; Douangamath A.; Krojer T.; Sethi R.; Geurink P. P.; Aimon A.; Amitai G.; Bellini D.; Bennett J.; Fairhead M.; Fedorov O.; Gabizon R.; Gan J.; Guo J.; Plotnikov A.; Straub V. M.; Szommer T.; Velupillai S.; Zaidman D.; Zhang Y.; Coker A. R.; Brennan P. E.; Ovaa H.; Von Delft F.; London N. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 8951–8968. 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick E.; Bradley A.; Gan J.; Douangamath A.; Krojer T.; Sethi R.; Geurink P. P.; Aimon A.; Amitai G.; Bellini D.; Bennett J.; Fairhead M.; Fedorov O.; Gabizon R.; Gan J.; Guo J.; Plotnikov A.; Reznik N.; Ruda G. F.; Díaz-Sáez L.; Straub V. M.; Szommer T.; Velupillai S.; Zaidman D.; Zhang Y.; Coker A. R.; Dowson C. G.; Barr H. M.; Wang C.; Huber K. V. M.; Brennan P. E.; Ovaa H.; Von Delft F.; London N. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallenbeck K. K.; Davies J. L.; Merron C.; Ogden P.; Sijbesma E.; Ottmann C.; Renslo A. R.; Wilson C.; Arkin M. R. A Liquid Chromatography/Mass Spectrometry Method for Screening Disulfide Tethering Fragments. SLAS Discov. 2018, 23, 183–192. 10.1177/2472555217732072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N.; Miller R. M.; Krishnan S.; Uchida K.; Irwin J. J.; Eidam O.; Gibold L.; Cimermančič P.; Bonnet R.; Shoichet B. K.; Taunton J. Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nat. Chem. Biol. 2014, 10, 1066–1072. 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijbesma E.; Hallenbeck K. K.; Leysen S.; De Vink P. J.; Skóra L.; Jahnke W.; Brunsveld L.; Arkin M. R.; Ottmann C. Site-Directed Fragment-Based Screening for the Discovery of Protein-Protein Interaction Stabilizers. J. Am. Chem. Soc. 2019, 141, 3524–3531. 10.1021/jacs.8b11658. [DOI] [PubMed] [Google Scholar]
- Sijbesma E.; Somsen B. A.; Miley G. P.; Leijten-van de Gevel I. A.; Brunsveld L.; Arkin M. R.; Ottmann C. Fluorescence Anisotropy-Based Tethering for Discovery of Protein–Protein Interaction Stabilizers. ACS Chem. Biol. 2020, 3143–3148. 10.1021/acschembio.0c00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter M.; Valenti D.; Cossar P. J.; Levy L. M.; Hristeva S.; Genski T.; Hoffmann T.; Brunsveld L.; Tzalis D.; Ottmann C. Fragment-Based Stabilizers of Protein–Protein Interactions through Imine-Based Tethering. Angew. Chem., Int. Ed. 2020, 59, 21520–21524. 10.1002/anie.202008585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J.; Jones K.; Cheeseman M. D. Lysine-Targeting Covalent Inhibitors. Angew. Chem., Int. Ed. 2017, 56, 15200–15209. 10.1002/anie.201707630. [DOI] [PubMed] [Google Scholar]
- Pettinger J.; Carter M.; Jones K.; Cheeseman M. D. Kinetic Optimization of Lysine-Targeting Covalent Inhibitors of HSP72. J. Med. Chem. 2019, 62, 11383–11398. 10.1021/acs.jmedchem.9b01709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J.; Le Bihan Y.-V.; Widya M.; van Montfort R. L. M.; Jones K.; Cheeseman M. D. An Irreversible Inhibitor of HSP72 That Unexpectedly Targets Lysine-56. Angew. Chem., Int. Ed. 2017, 56, 3536–3540. 10.1002/anie.201611907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S. E.; Dittus L.; Thomas D. A.; Convery M. A.; Nunes J.; Bush J. T.; Evans J. P.; Werner T.; Bantscheff M.; Murphy J. A.; Campos S. Selectively Targeting the Kinome-Conserved Lysine of PI3Kδ as a General Approach to Covalent Kinase Inhibition. J. Am. Chem. Soc. 2018, 140, 932–939. 10.1021/jacs.7b08979. [DOI] [PubMed] [Google Scholar]
- Li Q.; Verma I. M. NF-ΚB Regulation in the Immune System. Nat. Rev. Immunol. 2002, 2, 725–734. 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Nakanishi C.; Toi M. Nuclear Factor-ΚB Inhibitors as Sensitizers to Anticancer Drugs. Nat. Rev. Cancer 2005, 5, 297–309. 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- Taniguchi K.; Karin M. NF-κB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- Arepalli S. K.; Choi M.; Jung J.-K.; Lee H. Novel NF-κB Inhibitors: A Patent Review (2011 – 2014). Expert Opin. Ther. Pat. 2015, 25, 319–334. 10.1517/13543776.2014.998199. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B.; Greiner J.; Kadhim H.; Kaltschmidt C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. 10.3390/biomedicines6020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Hu D. X.; Chen R. Q.; Chen X. Q.; Dong W.; Yi C. 14-3-3 Isoforms Differentially Regulate NFκB Signaling in the Brain After Ischemia-Reperfusion. Neurochem. Res. 2017, 42, 2354–2362. 10.1007/s11064-017-2255-3. [DOI] [PubMed] [Google Scholar]
- Inglés-Esteve J.; Morales M.; Dalmases A.; Garcia-Carbonell R.; Jené-Sanz A.; López-Bigas N.; Iglesias M.; Ruiz-Herguido C.; Rovira A.; Rojo F.; Albanell J.; Gomis R. R.; Bigas A.; Espinosa L. Inhibition of Specific NF-κB Activity Contributes to the Tumor Suppressor Function of 14-3-3σ in Breast Cancer. PLoS One 2012, 7, e38347 10.1371/journal.pone.0038347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera C.; Fernández-Majada V.; Inglés-Esteve J.; Rodilla V.; Bigas A.; Espinosa L. Efficient Nuclear Export of P65-IκBα Complexes Requires 14-3-3 Proteins. J. Cell Sci. 2016, 129, 2472–2472. 10.1242/jcs.192641. [DOI] [PubMed] [Google Scholar]
- Wolter M.; de Vink P.; Neves J. F.; Srdanović S.; Higuchi Y.; Kato N.; Wilson A.; Landrieu I.; Brunsveld L.; Ottmann C. Selectivity via Cooperativity: Preferential Stabilization of the P65/14-3-3 Interaction with Semisynthetic Natural Products. J. Am. Chem. Soc. 2020, 142, 11772–11783. 10.1021/jacs.0c02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxford T.; Huang D.-B.; Malek S.; Ghosh G. The Crystal Structure of the IκBα/NF-κB Complex Reveals Mechanisms of NF-κB Inactivation. Cell 1998, 95, 759–770. 10.1016/S0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- Brameld K. A.; Kuhn B.; Reuter D. C.; Stahl M. Small Molecule Conformational Preferences Derived from Crystal Structure Data. A Medicinal Chemistry Focused Analysis. J. Chem. Inf. Model. 2008, 48, 1–24. 10.1021/ci7002494. [DOI] [PubMed] [Google Scholar]
- Oksenberg D.; Dufu K.; Patel M. P.; Chuang C.; Li Z.; Xu Q.; Silva-Garcia A.; Zhou C.; Hutchaleelaha A.; Patskovska L.; Patskovsky Y.; Almo S. C.; Sinha U.; Metcalf B. W.; Archer D. R. GBT440 Increases Haemoglobin Oxygen Affinity, Reduces Sickling and Prolongs RBC Half-Life in a Murine Model of Sickle Cell Disease. Br. J. Haematol. 2016, 175, 141–153. 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 1–13. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppi V.; Hughes S. J.; Maniaci C.; Testa A.; Gmaschitz T.; Wieshofer C.; Koegl M.; Riching K. M.; Daniels D. L.; Spallarossa A.; Ciulli A. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W. D.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter M.; de Vink P.; Neves J. F.; Srdanovic S.; Higuchi Y.; Kato N.; Wilson A. J.; Landrieu I.; Brunsveld L.; Ottmann C. Selectivity via Cooperativity: Preferential Stabilization of the P65/14-3-3 Interaction with Semi-Synthetic Natural Products. J. Am. Chem. Soc. 2020, 142, 11772–11783. 10.1021/jacs.0c02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clabbers M. T. B.; Gruene T.; Parkhurst J. M.; Abrahams J. P.; Waterman D. G. Electron Diffraction Data Processing with DIALS. Acta Crystallogr. Sect. Struct. Biol. 2018, 74, 506–518. 10.1107/S2059798318007726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. A.; Vagin A. A.; Murshudov G. N. Model Preparation in MOLREP and Examples of Model Improvement Using X-Ray Data. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 33–39. 10.1107/S0907444907049839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular Replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX : A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doveston R. G.; Kuusk A.; Andrei S.; Leysen S.; Cao Q.; Castaldi P.; Hendricks A.; Chen H.; Boyd H.; Ottmann C. Small-Molecule Stabilization of the P53 – 14-3-3 Protein-Protein Interaction. FEBS Lett. 2017, 591, 2449–2457. 10.1002/1873-3468.12723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.