

Abstract
The gain or loss of a chromosome—or aneuploidy—acts as one of the major triggers for infertility and pregnancy loss in humans. These chromosomal abnormalities affect more than 40% of eggs in women at both ends of the age spectrum, that is, young girls as well as women of advancing maternal age. Recent studies in human oocytes and embryos using genomics, cytogenetics, and in silico modeling all provide new insight into the rates and potential genetic and cellular factors associated with aneuploidy at varying stages of development. Here, we review recent studies that are shedding light on potential molecular mechanisms of chromosome missegregation in oocytes and embryos across the entire female reproductive life span.
Key Points
What is known about this topic?
Aneuploidy is a major cause of preclinical and prenatal fetal losses and congenital disorders in live born.
Aneuploidy is highly dependent upon maternal age, especially advanced maternal age.
Aneuploidy is predominantly from the mother.
What does this study add?
Aneuploidy originates from oocytes and preimplantation embryos.
Aneuploidy follows a U curve with female age.
New cellular and molecular mechanisms causing aneuploidies are discussed, including modelling approaches.
1. INTRODUCTION
We have appreciated for a long time that human conceptions are highly error‐prone in terms of whole chromosome gains and losses (aneuploidy; Table 1). Many of these originate in the germline, but there is also a substantial contribution from preimplantation embryos. Germ cells undergo a specialized cell division—meiosis—where a single round of genome duplication is followed by two consecutive chromosomal divisions resulting in haploid gametes (sperm and eggs in human). Thus, upon fertilization, the typical chromosome content is restored with one set contributed by each parent. In human sperm, meiosis lasts about 50–70 days. However, in human oocytes, meiosis is a decades‐long process involving multiple cell‐cycle starts and stops, and it is coupled to acquisition of developmental competence to support fertilization and early embryonic development. In oocyte meiosis, DNA replication and meiotic recombination are completed during fetal development and the cell arrests at the dictyate (diplotene) stage surrounded by supporting mitotic cells. At this stage, homologous chromosomes are tethered together in a bivalent configuration due to crossover recombination between homologous chromosomes and cohesion between sister chromatids. This bivalent configuration has to be maintained for decades until ovulation of the egg, when meiosis I (MI) is completed and homologous chromosomes segregate from each other reducing the genome content by one half. This extended dictyate arrest as well as vulnerable recombination configurations are two major reasons why aneuploidy in human eggs is at least an order of magnitude higher than sperm. The mature ovulated egg arrests at metaphase II and only completes the second meiotic division, where sister chromatids segregate, upon fertilization by sperm (Figure 1A). The embryo continues to develop until the blastocyst stage, when it reaches the uterus, hatches and implants.
TABLE 1.
Incidence of aneuploidy in the human germline and early development
| Genomic alteration | Eggs | Sperm | Preimplantation embryos—cleavage | Preimplantaion embryos—blastocysts | Pregnancy loss | Stillbirths | Live births |
|---|---|---|---|---|---|---|---|
| Whole chromosome aneuploidy | 30% (20%–85% pending age) | 2.5% (2.5%–7%) | Up to 70% | 56% | 50%–60% | 6.9% | 1:1000 |
| Most common aneuploidies | Young: +1–5; AMA: +13–15; +16; +21; +22 | +13; +15; +21; +22; sex chr. | +15; +16; +21; +22 | +15; +16; +21; +22 | +13; +15; +16; +18; +21; +22 | +13; +18; +21; sex chr. | +13; +18; +21; sex chr. |
| References | 1, 2, 3 | 4, 5, 6 | 7 | 8 | 9 | 10 | 11 |
FIGURE 1.
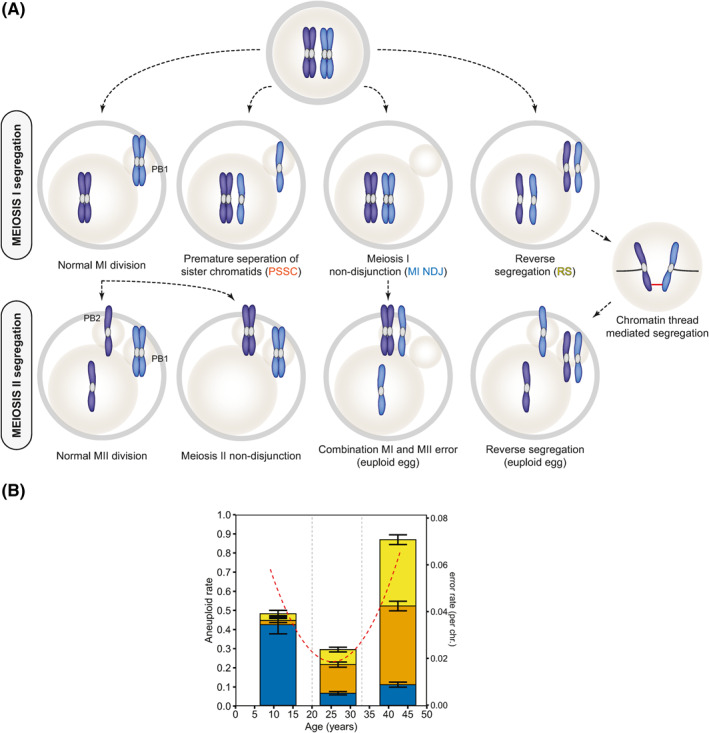
Meiosis I and meiosis II segregation errors and their age dependency in human oocytes. (A) Chromosome segregation patterns in meiosis I and meiosis II. (B) The U‐curve of aneuploidy in human oocytes (dotted red line) is a compilation of all three chromosome missegregation events—MI NDJ (blue), PSSC (orange), and RS (yellow)—and acts in an age dependent manner. MI NDJ, MI non‐disjunction; PSSC, premature separation of sister chromatids; RS, reverse segregation
Human fetal and adult oocytes have been studied for more than 60 years. Fetal oocytes are obtained from fetal ovaries (Weeks 12–24), whereas adult oocytes were originally obtained from hysterectomies (e.g., Jagiello et al. 12 ). The past decades, the success and widespread use of in vitro fertilization (IVF) has made fertility clinics the major source of adult oocytes. More recently, oocytes obtained during ovarian tissue cryopreservation have also been developed as an ex vivo source.1, 13 The large number of oocytes obtained from different sources have enabled broad conclusions to be reached, including that errors in chromosome segregation resulting in aneuploidy is a general feature of human oocytes and preimplantation embryos. Whereas aneuploidies are highly affected by maternal age, segregation errors during embryonic divisions are independent of maternal age factors. Furthermore, large screening studies of human preimplantation embryos (preimplantation genetic testing for aneuploidies [PGT‐A]) are enabling studies of aneuploidies in thousands of embryos (e.g., Franasiak et al. 14 and Girardi et al. 8 ). Such studies have revealed that meiotic errors are propagated to the blastocyst stage, where they result in vast preclinical as well as clinical losses. In contrast, mitotic errors occurring during the mitotic divisions in preimplantation development appear to be correlated with embryonic or cellular arrest and also give rise to genomically mosaic embryos.7, 15, 16 Below, we review the advances in our understanding of the origins of aneuploidies and the mechanisms that give rise to them.
1.1. The U‐curve of aneuploidy and types of meiotic segregation errors in human oocytes
In human oocytes, chromosome segregation errors during the first meiotic division are more prevalent compared to meiosis II (MII) (Table 2 and reviewed by Herbert et al. 17 ). Three different abnormal segregation patterns have been found to contribute to the high rate of human aneuploidy: premature separation of sister chromatids (PSSC), 18 reverse segregation (RS), 2 and meiosis I non‐disjunction (MI NDJ). 19 In a recent study by Gruhn et al., 1 missegregation events were identified in human oocytes spanning the entire reproductive life span (9–43 years) and were found to follow a U‐curve (Figure 1A, dotted red line) formed by a compilation of all three error patterns (Figure 1A, bar graph). More importantly, these error patterns influence aneuploidy levels in not only an age‐dependent, but also a chromosome‐dependent manner (Table 2).
TABLE 2.
Incidence of MI versus MII errors in human oocytes by chromosome (data from Gruhn et al. 1 and Zielinska et al. 23 )
| Chr. | N | MI error (%) | MII error (%) |
|---|---|---|---|
| 1 | 9 | 77.8 | 22.2 |
| 2 | 3 | 100 | 0 |
| 3 | 3 | 100 | 0 |
| 4 | 7 | 71.4 | 28.6 |
| 5 | 5 | 60.0 | 40.0 |
| 6 | 4 | 100 | 0.0 |
| 7 | 2 | 50.0 | 50.0 |
| 8 | 6 | 100 | 0 |
| 9 | 0 | 0 | 0 |
| 10 | 6 | 100 | 0 |
| 11 | 2 | 100 | 0 |
| 12 | 1 | 100 | 0 |
| 13 | 7 | 100 | 0 |
| 14 | 3 | 100 | 0 |
| 15 | 3 | 100 | 0 |
| 16 | 12 | 83.3 | 16.7 |
| 17 | 2 | 100 | 0 |
| 18 | 4 | 75.0 | 25.0 |
| 19 | 5 | 100 | 0 |
| 20 | 6 | 66.7 | 33.3 |
| 21 | 6 | 83.3 | 16.7 |
| 22 | 9 | 77.8 | 22.2 |
| X | 4 | 100 | 0 |
| Acrocentrics | 28 | 89.3 | 10.7 |
| Non‐acrocentrics | 81 | 85.2 | 14.8 |
Abbreviation: MI, meiosis I; MII, meiosis II.
PSSC was originally identified as the most common segregation error type in oocytes, primarily due to its strong positive correlation with maternal age.20, 21 PSSC, or the separation of one set of sister chromatids at MI instead of MII, leads to a 3:1 division of chromatids and the formation of two aneuploid daughter cells (Figure 1B). This segregation pattern increases linearly with age and has led to the hypothesis that premature loss of centromeric cohesion during the prolonged dictyate arrest and abnormal kinetochore attachments may directly lead to PSSC events in oocytes as maternal age increases.22, 23, 24, 25 In cases where more extreme cohesion loss occurs (i.e., weakening of both centromeric and arm cohesion in MI) or where homologous chromosomes failed to crossover, an RS event can arise where both sets of sister chromatids separate prematurely in MI.1, 2 RS was discovered to occur at an incidence nearly 100× higher than expected from two individual PSSC events,1, 2 suggesting a distinct origin from PSSC. Indeed, RS shows a dramatic increase between the mid‐ and advance maternal age groups consistent with more extensive cohesion loss along entire chromosome arms or on both sets of centromeres. Both PSSC and RS were found to primarily affect the acrocentric chromosomes (chr. 13‐15 21 & 22), thus suggesting a potential correlation between cohesion loss and aneuploidy incidence (Figure 2, discussed below). 1
FIGURE 2.
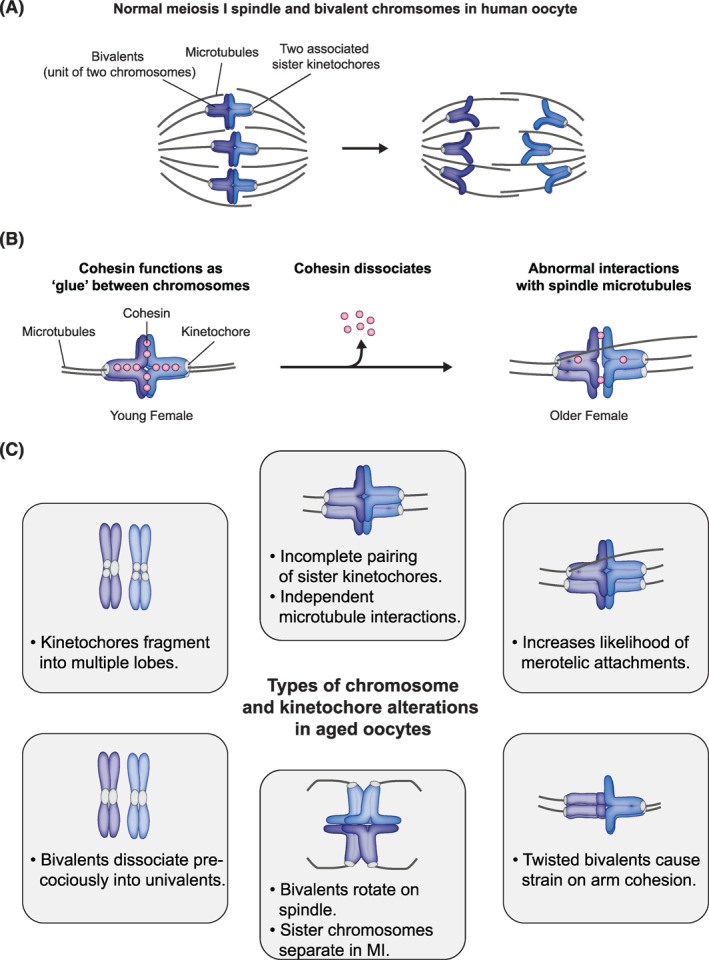
Structural changes and alternative alignments of chromosomes during meiosis I. (A) During MI, bivalents align at the metaphase plate of the meiotic spindle before chromosomes are segregated. (B) Cohesin is lost from chromosomes with advancing female age. (C) Schematic illustrating alterations in the architecture of chromosome bivalents with advancing maternal age. MI, meiosis I
Recently, however, a third pattern has re‐emerged as major error type through the inclusion of a wider age range in oocyte aneuploidy analysis. 1 MI NDJ, where the homologous chromosomes fail to separate and all four sister chromatids enter into one daughter cell (4:0), was found to be extremely prevalent at younger maternal ages (<20). Further analysis showed that not only does MI NDJ occur primarily in oocytes from young women, but the chromosomes impacted by this error type are predominantly the largest chromosomes (chr. 1–5). These chromosomes show a lower likelihood of being aneuploid in embryos28, 29 and clinically recognized pregnancies26, 27; therefore, suggesting that many of the rates seen at later developmental stages are greatly underestimating the prevalence of aneuploidy along the entire age spectrum.
Segregation errors can also occur in MII after fertilization (Table 2); however, these errors can have either a detrimental or a “beneficial” effect.30, 31 In cases where MI segregation occurs normally, the sister chromatids may undergo meiosis II non‐disjunction (MII NDJ) and both chromatids are pulled to a single daughter cell resulting in an aneuploid conception. However, there are cases where a combination of MI and MII errors result in a normal euploid oocyte 30 (Figure 1A). In addition, there is evidence that in 75% of RS events in MI—where the chromatids have no physical connection and should segregate independently—they are able to segregate correctly.1, 2 This “correct” segregation of two non‐sister chromatids in MII may be mediated by chromatin threads that connect chromosomes and were discovered recently 1 (Figure 2). Thus the impact on aneuploidy in conceptuses depends on the type of MI error and the incidence of MII error—for RS aneuploidy risk in the embryo is lower compared to PSSC (50% cause aneuploidy in embryos) and MI NDJ (all cause aneuploidy in embryos, except if a second error occurs in MII, see below).
2. UNDERSTANDING SEGREGATION ERRORS LEADING TO ANEUPLOIDY WITH MATHEMATICAL MODELLING
One limitation with direct studies of human eggs is obtaining sufficient numbers. With the improved understanding of chromosome segregation errors in human oocytes, however, it has been possible to apply mathematical modeling to expand our knowledge by analyzing large datasets generated from PGT‐A of human embryos. One limitation with direct observations in human preimplantation embryos as well as conceptuses is that only the final meiosis product (the egg) is available. This makes trisomic conceptions due to MI NDJ, PSSC, and RS indistinguishable, since any of these errors would result in a conception with two non‐homologous chromosomes originating from the egg.
In a recent study, 31 the authors constructed a mathematical model for different types of meiotic errors. Using this model, the authors formulated the proportion of resulting zygotes of chromosome segregation errors (e.g., normal, single trisomy, double trisomy, etc.) depending on the rates of MI NDJ, PSSC, and MII NDJ. By fitting the model to observed aneuploidy data from 11,157 embryos (based on PGT‐A), the best‐fitting models revealed several biological insights of female aneuploidy. For example, eggs with MI NDJ errors are more likely to have errors in MII, suggesting an association of the error mechanisms in the two meiotic stages. As observed in the several previous studies from the IVF field (e.g., Magli et al. 30 ), the model suggests that oocytes with certain MI and/or MII errors can result in a euploid embryo. For example, a combination of MI NDJ and MII NDJ can result in a euploid embryo (Figure 1A), although these account for fewer than 1% of embryos. As the meiotic error rates increase with maternal age, the proportion of euploid embryos resulting from meiotic errors increase with maternal age as well. The most common type is PSSC followed by a normal MII segregation, which has a 50% chance of resulting in a euploid embryo. In addition, the authors demonstrated the potential clinical utility of the modeling approach. By simulations based on the best‐fitting model, they determined the possibility of having at least one euploid embryo given the patient's age and the number of embryos tested. This information can potentially help clinicians and patients to estimate the expected number of IVF cycles needed to obtain a euploid conception without relying on having a diagnosis, such as specific genetic mutations that predispose to aneuploidy.
2.1. Mechanisms of aneuploidy formation: establishment and maintenance of bivalents
The segregation of entire chromosomes during MI relies on the formation of bivalents, which consist of two joined homologous chromosomes (Figure 2A). Both their establishment and their maintenance until the first meiotic division contribute towards the high incidence of aneuploidy in human eggs. Bivalents are formed during fetal development in meiotic prophase I via meiotic crossover recombination and then have to be maintained for decades, until ovulation of the egg. The sites of genetic exchange bring homologous chromosomes together and their cytological manifestation (“chiasma”) are visible due to the cohesion between the distal arm regions of the sister chromatids. Cohesion in centromeres and pericentromeres between sister chromatids is important for their co‐orientation such that sister chromatids segregate to one pole at MI.
Aberrant crossover recombination patterns—such as poor placement of exchanges or exchange‐less chromosomes—have been identified as important factors in the formation of aneuploidy in human eggs and sperm.2, 3, 4, 5, 6, 32, 33 Up to 7% of human fetal oocytes contain at least one exchange‐less chromosome, which predominantly affect the smallest chromosomes 21 and 22 33 and “programmed” inefficiency in human eggs, but not sperm, has been proposed to contribute to this high incidence. 34 Exchange‐less chromosomes may predominantly undergo RS, bi‐orienting their kinetochores to opposite spindle poles during MI.1, 23, 35
The maintenance and integrity of the bivalent during the decades‐long dictyate arrest relies on a specialized meiotic REC8‐cohesin complex. Cohesin in the chromosome arm region distal to the recombination site holds the bivalents together until ovulation and the onset of anaphase I, when cleavage of REC8‐cohesin in the arm region allows the chromosomes to segregate.36, 37 Decreased or diminishing cohesin protein levels is hypothesized as a key “hit” leading to the age dependent increase in aneuploidy. Work in mice suggests that the REC8‐cohesin complex is loaded prior to meiotic recombination, with no substantial turnover or replenishment throughout the prolonged arrest period.38, 39 As mice age, the cohesin complex is gradually lost from chromosomes and consequently, the bivalent architecture changes (Figure 2B,C).39, 40, 41, 42 The homologous chromosomes within a bivalent become separated by prominent gaps, and sometimes dissociate into univalents.43, 44 Consistent with defects in cohesin being sufficient to cause bivalent erosion with age, a meiosis‐specific cohesin component, SMC1β, is haploinsufficient in mouse. Adult oocytes from SMC1β heterozygous females show age‐dependent loss of bivalents and elevated aneuploidy rates. 45 Whether cohesin is lost in human oocytes is still unclear as studies investigating levels of individual cohesin subunits in oocytes of different ages come to different conclusions.46, 47 However, it is clear that the architecture of bivalents in human oocytes changes dramatically as women get older.23, 24, 35, 43 Bivalents become separated by gaps and dissociate into univalents, similar to what happens in mouse oocytes. This is particularly relevant for the smaller chromosomes 21 and 22 that have less arm cohesion than larger chromosomes and show elevated frequency of univalents in women of advanced maternal age. 48
2.1.1. Lack of sister kinetochore co‐orientation in meiosis I is a common source of missegregation
A dogma in the meiosis field is that sister kinetochores are fused during MI to permit their co‐segregation at anaphase I, resulting in both sister chromatids of the homolog moving towards the same spindle pole thereby completing the reductional division of meiosis.29, 49 The fusion of the sister kinetochores relies on REC8‐cohesin complexes in the centromeric and pericentromeric regions, similar to arm regions (Figure 2B).50, 51 REC8‐cohesin in peri‐ and centromeric regions is protected from cleavage during anaphase I by shugoshin proteins. 52 This protection is essential to maintain the association of sister chromatids within a chromosome at the MI to MII transition.
Interestingly, human oocytes challenge the dogma of fused sister kinetochores. Although aging also affects the fusion of sister kinetochores, young women already show a substantial fraction of kinetochores separated by large gaps.23, 24 These split sister kinetochores can interact with spindle microtubules independently and hence do not function as a single unit during the first meiotic division. Notably, the separation of sister kinetochores increases with female age. This separation of sister kinetochores favors several incorrect types of chromosome attachments to the spindle in MI. Most prominently, it favors the formation of merotelic attachments, where a kinetochore is linked to both spindle poles, instead of being linked to a single spindle pole only. 23 Moreover, it allows bivalents to rotate on the spindle, resulting in a configuration where sister kinetochores are facing opposite spindle poles like in mitosis, instead of facing to the same spindle pole. These inverted bivalents may lead to improper separation of sister chromatids during anaphase I, which may result in PSSC or RS.2, 29
The separation of sister kinetochores can also explain why prematurely dissociated univalents often align on the MI spindle like mitotic chromosomes, with sister kinetochores orienting towards opposite spindle poles.23, 43, 53, 54 Segregation of these univalents into sister chromatids during MI could be another mechanism that contributes to the RS pattern. Moreover, having four instead of two microtubule attachment sites can further cause twisting of bivalents along their axis. 23 Such twisting is likely to put additional force on chromosome arm cohesion. 23 The separation of premature separation of bivalents into univalents, as well as splitting of sister kinetochores might be further exacerbated by multidirectional pulling of spindle microtubules during the prolonged spindle assembly process in human oocytes. 55
Recent studies further revealed that centromeres and kinetochores themselves change in aged oocytes of different mammalian species, including humans. The core centromere protein Cenp‐A becomes depleted from centromeres.35, 56 Moreover, centromeric chromatin decompacts. Kinetochores built on loosened centromeric chromatin get destabilized and fragment into multiple lobes. 35 Such fragmented kinetochores are often merotelically attached on the metaphase II spindle, and may thereby contribute to aneuploidy. Fragmented kinetochores are further characterized by reduced levels of key components of the inner and outer kinetochore regions, which may further compromise kinetochore function. 35
2.1.2. The spindle assembly checkpoint, perturbed protein homeostasis, and differential mRNA expression are potential mechanisms resulting in human aneuploidy
Model organisms, such as mice, have provided much of our knowledge on genes and pathways that are important for protecting mammalian gamete euploidy. 57 Upon resumption of meiosis in the adult ovary the chromosomes are condensed, the spindles are made, and kinetochore attachments are formed. Importantly, at this stage the spindle assembly checkpoint (SAC), a complex mechanism that integrates the attachment status of the kinetochore with spindle microtubules, must be satisfied. If anaphase onset occurs when a kinetochore is unattached, an MI bivalent will fail to disjoin. Unattached kinetochores arise either from faulty spindle building, which is common in human oocytes, 55 or from Aurora kinase activity sensing an improper attachment and triggering microtubule depolymerization. 58 When kinetochores are unoccupied, MPS1 kinase initiates the SAC response and triggers recruitment of scaffold proteins that assemble the mitotic checkpoint complex (MCC). The MCC diffuses and sequesters CDC20, thereby preventing anaphase promoting complex/cyclosome (APC/C) activation and arrests the cell cycle.59, 60 In somatic cells, one unoccupied kinetochore will trigger the SAC, 61 but in mammalian oocytes, the SAC is more permissive, and can fail to prevent anaphase even in the presence of several misaligned chromosomes.62, 63, 64, 65, 66 A weaker SAC has been proposed to predispose oocytes to MI chromosome segregation errors.62, 67
Another pathway that appears to sensitize oocytes to chromosome missegregation is gene expression and control through regulated translation. 68 Oocytes complete MI and MII in the absence of transcription and instead mount a highly regulated burst of translation during pro‐metaphase I. This strategy requires proper storage of repressed maternal transcripts during oocyte growth and the prophase I arrest. 69 Upon meiotic resumption, oocytes must switch off the repression and activate their translation. Repressed messages are enriched for cell‐cycle regulators and transcriptional and epigenetic machinery encoding proteins that are required later in meiosis, fertilization, and/or embryogenesis. Regulation of translation is therefore critical to producing proteins essential for accurate chromosome segregation. Because perturbations in protein homeostasis (i.e., proteostasis) are associated with the aging process,70, 71 it is tempting to speculate that oocytes from women of advanced maternal age are uniquely sensitive to abnormal protein expression levels that could make them more prone to aneuploidy. This is further supported by findings that mRNA expression of cell cycle and DNA repair genes are differentially downregulated in aged human eggs. 72
3. GENETIC CONTRIBUTIONS AND PATHWAYS ASSOCIATED WITH ANEUPLOIDY RISK
The parental genetic contribution to aneuploid conception risk is a topic of long‐standing interest in human reproductive biology. Although increased risk of aneuploidy is strongly correlated with increasing maternal age, significant variation exists in aneuploid conception rates of IVF patients without any reproductive pathology at any given age.73, 74, 75, 76, 77 Indeed, some of the first genetic surveys of human preimplantation embryos noted that certain patients appeared to be predisposed to generating embryos with complex forms of mosaic aneuploidy (i.e., “chaotic mosaicism”), independent of maternal age. 78 Similar observations have been noted with respect to oocyte and sperm aneuploidies of meiotic origin, including in recent studies.4, 76 These observations point to the possibility that inherited genetic variation influences the fidelity of meiosis and/or early embryonic mitosis, the latter controlled by maternal gene products deposited in the oocyte.
It is worth noting that no formal estimate of the heritability of aneuploidy risk has yet been achieved, which likely owes to the challenge of defining and measuring this phenotype (generally based on rates from clinical programs of PGT‐A) in a sufficiently large cohort for quantitative genetic analysis. Such estimates generally require approximately 5000–10,000 samples to achieve substantial power for detecting modest effects, and may require even more if the true heritability is low or the phenotype is measured with error. 79 Nevertheless, the discovery of aneuploidy‐associated variation represents an outstanding goal in human reproductive genetics, with the potential to reveal mechanisms of aneuploidy formation. Even without such mechanistic knowledge, the genetic associations also hold promise for improving precision in prediction of aneuploidy risk when combined with known covariates such as maternal age. Such comprehensive risk assessment may be useful for informing reproductive decisions, in the context of IVF or natural conception.
Genome‐wide association studies (GWAS) offer a powerful approach for discovering common genetic variation influencing complex traits such as risk of aneuploid conception. This phenotype may be quantified as a proportion of aneuploid embryos produced in an IVF cycle, as measured by PGT‐A. Using parental genotype data (N maternal = ∼5000) from single‐nucleotide polymorphism (SNP) based PGT‐A, McCoy et al. 80 discovered a maternal haplotype spanning the centrosomal regulator polo‐like kinase 4 (PLK4) that is associated with complex aneuploidy of mitotic origin in day‐3 embryo biopsies. Through follow‐up analysis and intersection with time‐lapse data, they later proposed that this association is driven by a mechanism of tripolar mitosis, whereby diploid cells segregate their chromosomes on a tripolar spindle, leading to massive chromosome loss.81, 82 Interestingly, this signature is rare in data from the blastocyst stage of development at day 5, and patients with the high‐risk genotype have fewer embryos that achieve blastocyst formation—together suggesting that complex mosaic aneuploidy compromises development to the blastocyst stage.7, 82, 83 Yet depending on their timing of occurrence, even these complex forms of mosaicism may not preclude development, and indeed may be preferentially excluded from the embryo during the process of blastocyst formation.84, 85 While explaining a small fraction (∼1%) of the total variance in aneuploidy risk, these examples demonstrate how GWAS can be valuable for generating hypotheses about mechanisms of aneuploidy formation.
Only a small proportion of aneuploidies are compatible with live birth, and those that result in genetic disorders such as Down Syndrome, merit additional focus. Seeking to understand the potential maternal genetic contributions to the origins of trisomy 21, Chernus et al. 86 conducted a candidate gene association study for variants influencing trisomy 21 of MI and MII origin (N maternal = ∼700). Using SNP array genotyping data from parents and children with trisomy 21, they classified meiotic errors based on heterozygosity of pericentromeric markers, then contrasted maternal genotypes with those of the fathers, which served as a natural internal control. Genome‐wide analyses revealed no variants reaching genome‐wide significance (typically defined as p < 5 × 10−8), but several of the top suggestive associations occurred within or near genes with known meiotic functions, including AURKC, an Aurora kinase whose dysregulation is known to induce aneuploidy in mouse models.87, 88 Further focus on candidate meiosis‐related genes revealed additional potential associations for validation and follow‐up study.
In contrast to association studies, which are generally based on SNP array genotyping at sites of known variation in the human population, the discovery of rare variants that influence aneuploidy risk requires alternative approaches, such as whole genome or targeted sequencing, as well as functional validation in model organisms or human cell lines. For example, targeted sequencing of candidate genes in patients experiencing recurrent IVF failure identified loss‐of‐function variants in a primate‐specific tubulin b class VIII (TUBB8),89, 90, 91 PAT1 homolog 2 (PATL2),92, 93, 94, 95, 96, 97 and WEE2 oocyte meiosis inhibiting kinase (WEE2)98, 99, 100, 101, 102 that may predispose women to a higher incidence of oocyte and embryonic aneuploidy at younger‐than‐average ages. The products of these genes are required for essential steps in oocyte development and meiosis. On the other hand, gene variants may exist that protect euploidy and extend a woman's reproductive life span. One study identified a variant in Aurora kinase B, a protein involved in the SAC, which conferred a protective advantage in an older (39 years) IVF patient. 77
As the sequencing cost decreases, whole exome and whole genome sequencing are increasingly becoming more feasible. Comparing to the candidate gene approach, whole exome/genome studies is not limited to predetermined candidate genes and have the potential of discovering novel genetic mechanisms for aneuploidy. For example, a recent study applied exome sequencing to compare patients with high and low frequencies of aneuploid blastocysts to identify genetic factors responsible for chromosome segregation errors. 76 The power of this approach is the specific aneuploid embryo phenotype and the ability to group patients into extreme phenotype categories. The analysis of nearly 100 exomes of women who produced greater than 50% aneuploid blastocysts during IVF‐revealed variant enrichment in genes that function in biological processes such as “centriole,” “DNA repair,” and “damaged DNA binding.” In vitro assessment of one of the high‐ranking “centriole” variants (CEP120 p.Arg947His) using mouse oocytes revealed that women who are heterozygous for this allele may produce aneuploid eggs because of inefficient microtubule nucleation. 76 Despite studies like these, a comprehensive understanding of all genes contributing to embryonic aneuploidy and the relative contributions of common and rare variation to aneuploidy phenotypes is still lacking.
4. CONCLUSIONS
Our current understanding of human aneuploidies has increased dramatically the past decade, facilitated by large studies of human eggs, sperm, and embryos. Research in the areas of meiotic recombination, chromosome cohesion weakening, abnormal kinetochore structures, extended effects of maternal age, and genetic contributions to aneuploidy risk, are all moving us closer to understanding the origins of aneuploidy and therefore the potential for future clinical interventions. Once further molecular mechanisms associated with individual targets are identified, the field may improve diagnoses and genetic screening programs, as well as increase the efficacy of conceptions by developing interventions to reduce aneuploidy rates. Improved understanding of genomic mosaicism in preimplantation embryos, especially in placental lineages, may also contribute to advances in prenatal diagnostics.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENTS
The authors are part of the Origins of Aneuploidy Consortium and thank all of the other members for discussion and funding. J. Xing and K. Schindler were funded by a grant from the NIH/NICHD (R01‐HD091331). E. R. Hoffmann and J. Gruhn were funded by NNF Young Investigator Award (NNF15OC0016662), ERC Consolidator Grant (724718‐ReCAP), and support from the DNRF Center Grant (DNRF115). RCM was funded by the NIH/NIGMS (R35GM133747). L. Wartosch and M. Schuh were funded by the Max Planck Society and Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2067/1‐ 390729940 and Leibniz Prize (SCHU 3047/1‐1) to M. Schuh. The author list was generated at random and all authors contributed equally.
REFERENCES
- 1. Gruhn JR, Zielinska AP, Shukla V, et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science. 2019;365(6460):1466‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ottolini CS, Newnham LJ, Capalbo A, et al. Genome‐wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat Genet. 2015;47(7):727‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hou Y, Fan W, Yan L, et al. Genome analyses of single human oocytes. Cell. 2013;155(7):1492‐1506. [DOI] [PubMed] [Google Scholar]
- 4. Bell AD, Mello CJ, Nemesh J, Brumbaugh SA, Wysoker A, McCarroll SA. Insights into variation in meiosis from 31,228 human sperm genomes. Nature. 2020;583(7815):259‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu S, Zong C, Fan W, et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole‐genome sequencing. Science. 2012;338(6114):1627‐1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J, Fan HC, Behr B, Quake SR. Genome‐wide single‐cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150(2):402‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCoy RC, Demko ZP, Ryan A, et al. Evidence of selection against complex mitotic‐origin aneuploidy during preimplantation development. PLOS Genet. 2015;11(10):e1005601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Girardi L, Serdarogullari M, Patassini C, et al. Incidence, origin, and predictive model for the detection and clinical management of segmental aneuploidies in human embryos. Am J Hum Genet. 2020;106(4):525‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soler A, Morales C, Mademont‐Soler I, et al. Overview of chromosome abnormalities in first trimester miscarriages: a series of 1,011 consecutive chorionic villi sample karyotypes. Cytogenet Genome Res. 2017;152(2):81‐89. [DOI] [PubMed] [Google Scholar]
- 10. Reddy UM, Page GP, Saade GR, et al. Karyotype versus microarray testing for genetic abnormalities after stillbirth. N Engl J Med. 2012;367(23):2185‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kimelman D, Pavone ME. Non‐invasive prenatal testing in the context of IVF and PGT‐A. Best Pract Res Clin Obstet Gynaecol. 2020;70:51‐62. [DOI] [PubMed] [Google Scholar]
- 12. Jagiello G, Ducayen M, Fang J‐S, Graffeo J. Cytogenetic observations in mammalian oocytes. Chromosom Today. 1976;5:43‐63. [Google Scholar]
- 13. Nikiforov D, Junping C, Cadenas J, et al. Improving the maturation rate of human oocytes collected ex vivo during the cryopreservation of ovarian tissue. J Assist Reprod Genet. 2020;37(4):891‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franasiak JM, Forman EJ, Hong KH, et al. The nature of aneuploidy with increasing age of the female partner: a review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil Steril. 2014;101(3):656‐663.e1. [DOI] [PubMed] [Google Scholar]
- 15. Vanneste E, Voet T, Le Caignec C, et al. Chromosome instability is common in human cleavage‐stage embryos. Nat Med. 2009;15(5):577‐583. [DOI] [PubMed] [Google Scholar]
- 16. Levy B, Hoffmann ER, McCoy RC, Grati FR. Chromosomal mosaicism: origins and clinical implications in preimplantation and prenatal diagnosis. Prenat Diagn. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herbert M, Kalleas D, Cooney D, Lamb M, Lister L. Meiosis and maternal aging: insights from aneuploid oocytes and trisomy births. Cold Spring Harb Perspect Biol. 2015;7(4):a017970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Angell RR, Ledger W, Yong EL, Harkness L, Baird DT. Cytogenetic analysis of unfertilized human oocytes. Hum Reprod Oxf Engl. 1991;6(4):568‐573. [DOI] [PubMed] [Google Scholar]
- 19. Martin RH, Mahadevan MM, Taylor PJ, et al. Chromosomal analysis of unfertilized human oocytes. J Reprod Fertil. 1986;78(2):673‐678. [DOI] [PubMed] [Google Scholar]
- 20. Vialard F, Petit C, Bergere M, et al. Evidence of a high proportion of premature unbalanced separation of sister chromatids in the first polar bodies of women of advanced age. Hum Reprod. 2006;21(5):1172‐1178. [DOI] [PubMed] [Google Scholar]
- 21. Pellestor F, Andréo B, Arnal F, Humeau C, Demaille J. Mechanisms of non‐disjunction in human female meiosis: the co‐existence of two modes of malsegregation evidenced by the karyotyping of 1397 in‐vitro unfertilized oocytes. Hum Reprod. 2002;17(8):2134‐2145. [DOI] [PubMed] [Google Scholar]
- 22. Duncan FE, Hornick JE, Lampson MA, Schultz RM, Shea LD, Woodruff TK. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell. 2012;11(6):1121‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zielinska AP, Holubcova Z, Blayney M, Elder K, Schuh M. Sister kinetochore splitting and precocious disintegration of bivalents could explain the maternal age effect. eLife. 2015;4:e11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel J, Tan SL, Hartshorne GM, McAinsh AD. Unique geometry of sister kinetochores in human oocytes during meiosis I may explain maternal age‐associated increases in chromosomal abnormalities. Biol Open. 2015;5(2):178‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lagirand‐Cantaloube J, Ciabrini C, Charrasse S, et al. Loss of centromere cohesion in aneuploid human oocytes correlates with decreased kinetochore localization of the sac proteins Bub1 and Bubr1. Sci Rep. 2017;7(1):44001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hassold T, Jacobs P, Kline J, Stein Z, Warburton D. Effect of maternal age on autosomal trisomies. Ann Hum Genet. 1980;44(Pt 1):29‐36. [DOI] [PubMed] [Google Scholar]
- 27. Hassold T, Chiu D. Maternal age‐specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum Genet. 1985;70(1):11‐17. [DOI] [PubMed] [Google Scholar]
- 28. Konstantinidis M, Ravichandran K, Gunes Z, et al. Aneuploidy and recombination in the human preimplantation embryo. Copy number variation analysis and genome‐wide polymorphism genotyping. Reprod Biomed Online. 2020;40(4):479‐493. [DOI] [PubMed] [Google Scholar]
- 29. Capalbo A, Hoffmann ER, Cimadomo D, Maria Ubaldi F, Rienzi L. Human female meiosis revised: new insights into the mechanisms of chromosome segregation and aneuploidies from advanced genomics and time‐lapse imaging. Hum Reprod Update. 2017;23(6):706‐722. [DOI] [PubMed] [Google Scholar]
- 30. Magli MC, Grugnetti C, Castelletti E, et al. Five chromosome segregation in polar bodies and the corresponding oocyte. Reprod Biomed Online. 2012;24(3):331‐338. [DOI] [PubMed] [Google Scholar]
- 31. Tyc KM, McCoy RC, Schindler K, Xing J. Mathematical modeling of human oocyte aneuploidy. Proc Natl Acad Sci U S A. 2020;117(19):10455‐10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age‐old problem. Nat Rev Genet. 2012;13(7):493‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hassold T, Maylor‐Hagen H, Wood A, et al. Failure to recombine is a common feature of human oogenesis. Am J Hum Genet. 2021;108(1):16‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang S, Hassold T, Hunt P, et al. Inefficient crossover maturation underlies elevated aneuploidy in human female meiosis. Cell. 2017;168(6):977‐989.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zielinska AP, Bellou E, Sharma N, et al. Meiotic kinetochores fragment into multiple lobes upon cohesin loss in aging eggs. Curr Biol. 2019;29(22):3749‐3765.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kudo NR, Anger M, Peters AHFM, et al. Role of cleavage by separase of the Rec8 kleisin subunit of cohesin during mammalian meiosis I. J Cell Sci. 2009;122(15):2686‐2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kudo NR, Wassmann K, Anger M, et al. Resolution of chiasmata in oocytes requires separase‐mediated proteolysis. Cell. 2006;126(1):135‐146. [DOI] [PubMed] [Google Scholar]
- 38. Burkhardt S, Borsos M, Szydlowska A, et al. Chromosome cohesion established by Rec8‐cohesin in fetal oocytes is maintained without detectable turnover in oocytes arrested for months in mice. Curr Biol. 2016;26(5):678‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tachibana‐Konwalski K, Godwin J, van der Weyden L, et al. Rec8‐containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev. 2010;24(22):2505‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lister LM, Kouznetsova A, Hyslop LA, et al. Age‐related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol. 2010;20(17):1511‐1521. [DOI] [PubMed] [Google Scholar]
- 41. Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age‐related aneuploidy in oocytes. Curr Biol. 2010;20(17):1522‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu L, Keefe DL. Defective cohesin is associated with age‐dependent misaligned chromosomes in oocytes. Reprod Biomed Online. 2008;16(1):103‐112. [DOI] [PubMed] [Google Scholar]
- 43. Sakakibara Y, Hashimoto S, Nakaoka Y, Kouznetsova A, Höög C, Kitajima TS. Bivalent separation into univalents precedes age‐related meiosis I errors in oocytes. Nat Commun. 2015;6(1):7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yun Y, Lane SIR, Jones KT. Premature dyad separation in meiosis II is the major segregation error with maternal age in mouse oocytes. Development. 2014;141(1):199‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hodges CA, Revenkova E, Jessberger R, Hassold TJ, Hunt PA. SMC1β‐deficient female mice provide evidence that cohesins are a missing link in age‐related nondisjunction. Nat Genet. 2005;37(12):1351‐1355. [DOI] [PubMed] [Google Scholar]
- 46. Garcia‐Cruz R, Brieño MA, Roig I, et al. Dynamics of cohesin proteins REC8, STAG3, SMC1β and SMC3 are consistent with a role in sister chromatid cohesion during meiosis in human oocytes. Hum Reprod. 2010;25(9):2316‐2327. [DOI] [PubMed] [Google Scholar]
- 47. Tsutsumi M, Fujiwara R, Nishizawa H, et al. Age‐related decrease of meiotic cohesins in human oocytes. PLoS One. 2014;9(5):e96710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Angell RR. Meiosis I in human oocytes. Cytogenet Genome Res. 1995;69(3‐4):266‐272. [DOI] [PubMed] [Google Scholar]
- 49. Petronczki M, Siomos MF, Nasmyth K. Un ménage à quatre: the molecular biology of chromosome segregation in meiosis. Cell. 2003;112(4):423‐440. [DOI] [PubMed] [Google Scholar]
- 50. Watanabe Y. Geometry and force behind kinetochore orientation: lessons from meiosis. Nat Rev Mol Cell Biol. 2012;13(6):370‐382. [DOI] [PubMed] [Google Scholar]
- 51. Webster A, Schuh M. Mechanisms of aneuploidy in human eggs. Trends Cell Biol. 2017;27(1):55‐68. [DOI] [PubMed] [Google Scholar]
- 52. Marston AL. Shugoshins: tension‐sensitive pericentromeric adaptors safeguarding chromosome segregation. Mol Cell Biol. 2015;35(4):634‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hunt P, LeMaire R, Embury P, Sheean L, Mroz K. Analysis of chromosome behavior in intact mammalian oocytes: monitoring the segregation of a univalent chromosome during female meiosis. Hum Mol Genet. 1995;4(11):2007‐2012. [DOI] [PubMed] [Google Scholar]
- 54. Kouznetsova A, Lister L, Nordenskjöld M, Herbert M, Höög C. Bi‐orientation of achiasmatic chromosomes in meiosis I oocytes contributes to aneuploidy in mice. Nat Genet. 2007;39(8):966‐968. [DOI] [PubMed] [Google Scholar]
- 55. Holubcová Z, Blayney M, Elder K, Schuh M. Error‐prone chromosome‐mediated spindle assembly favors chromosome segregation defects in human oocytes. Science. 2015;348(6239):1143‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smoak EM, Stein P, Schultz RM, Lampson MA, Black BE. Long‐term retention of CENP‐A nucleosomes in mammalian oocytes underpins transgenerational inheritance of centromere identity. Curr Biol. 2016;26(8):1110‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marston AL, Wassmann K. Multiple duties for spindle assembly checkpoint kinases in meiosis. Front Cell Dev Biol. 2017;5:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vallardi G, Cordeiro MH, Saurin AT. A kinase‐phosphatase network that regulates kinetochore‐microtubule attachments and the SAC. In: Black BE, ed. Centromeres and Kinetochores: Discovering the Molecular Mechanisms Underlying Chromosome Inheritance. Cham: Springer International Publishing; 2017. p. 457‐484. [DOI] [PubMed] [Google Scholar]
- 59. Musacchio A, Salmon ED. The spindle‐assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5):379‐393. [DOI] [PubMed] [Google Scholar]
- 60. Nezi L, Musacchio A. Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol. 2009;21(6):785‐795. [DOI] [PubMed] [Google Scholar]
- 61. Kuhn J, Dumont S. Mammalian kinetochores count attached microtubules in a sensitive and switch‐like manner. J Cell Biol. 2019;218(11):3583‐3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagaoka SI, Hodges CA, Albertini DF, Hunt PA. Oocyte‐specific differences in cell‐cycle control create an innate susceptibility to meiotic errors. Curr Biol. 2011;21(8):651‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gui L, Homer H. Spindle assembly checkpoint signalling is uncoupled from chromosomal position in mouse oocytes. Development. 2012;139(11):1941‐1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kolano A, Brunet S, Silk AD, Cleveland DW, Verlhac M‐H. Error‐prone mammalian female meiosis from silencing the spindle assembly checkpoint without normal interkinetochore tension. Proc Natl Acad Sci U S A. 2012;109(27):E1858‐E1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lane SIR, Jones KT. Non‐canonical function of spindle assembly checkpoint proteins after APC activation reduces aneuploidy in mouse oocytes. Nat Commun. 2014;5(1):3444. [DOI] [PubMed] [Google Scholar]
- 66. Sebestova J, Danylevska A, Dobrucka L, Kubelka M, Anger M. Lack of response to unaligned chromosomes in mammalian female gametes. Cell Cycle. 2012;11(16):3011‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. LeMaire‐Adkins R, Radke K, Hunt PA. Lack of checkpoint control at the metaphase/anaphase transition: a mechanism of meiotic nondisjunction in mammalian females. J Cell Biol. 1997;139(7):1611‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Conti M, Franciosi F. Acquisition of oocyte competence to develop as an embryo: integrated nuclear and cytoplasmic events. Hum Reprod Update. 2018;24(3):245‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luong XG, Daldello EM, Rajkovic G, Yang C‐R, Conti M. Genome‐wide analysis reveals a switch in the translational program upon oocyte meiotic resumption. Nucleic Acids Res. 2020;48(6):3257‐3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol. 2018;217(1):51‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421‐435. [DOI] [PubMed] [Google Scholar]
- 72. Grøndahl ML, Yding Andersen C, Bogstad J, Nielsen FC, Meinertz H, Borup R. Gene expression profiles of single human mature oocytes in relation to age. Hum Reprod. 2010;25(4):957‐968. [DOI] [PubMed] [Google Scholar]
- 73. Franasiak JM, Olcha M, Shastri S, et al. Embryonic aneuploidy does not differ among genetic ancestry according to continental origin as determined by ancestry informative markers. Hum Reprod. 2016;31(10):2391‐2395. [DOI] [PubMed] [Google Scholar]
- 74. Franasiak JM, Forman EJ, Hong KH, et al. Aneuploidy across individual chromosomes at the embryonic level in trophectoderm biopsies: changes with patient age and chromosome structure. J Assist Reprod Genet. 2014;31(11):1501‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Demko ZP, Simon AL, McCoy RC, Petrov DA, Rabinowitz M. Effects of maternal age on euploidy rates in a large cohort of embryos analyzed with 24‐chromosome single‐nucleotide polymorphism–based preimplantation genetic screening. Fertil Steril. 2016;105(5):1307‐1313. [DOI] [PubMed] [Google Scholar]
- 76. Tyc KM, El Yakoubi W, Bag A, et al. Exome sequencing links CEP120 mutation to maternally derived aneuploid conception risk. Hum Reprod. 2020;35(9):2134‐2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nguyen AL, Marin D, Zhou A, et al. Identification and characterization of aurora kinase B and C variants associated with maternal aneuploidy. Mol Hum Reprod. 2017;23(6):406‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Delhanty JDA, Harper JC, Ao A, Handyside AH, Winston RML. Multicolour FISH detects frequent chromosomal mosaicism and chaotic division in normal preimplantation embryos from fertile patients. Hum Genet. 1997;99(6):755‐760. [DOI] [PubMed] [Google Scholar]
- 79. Neale BM. Relationship of LDSR Results with Sample Size. 2019. https://nealelab.github.io/UKBB_ldsc/viz_sampsize.html. Accessed September 16, 2020. [Google Scholar]
- 80. McCoy RC, Demko Z, Ryan A, et al. Common variants spanning PLK4 are associated with mitotic‐origin aneuploidy in human embryos. Science. 2015;348(6231):235‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhan Q, Ye Z, Clarke R, Rosenwaks Z, Zaninovic N. Direct unequal cleavages: embryo developmental competence, genetic constitution and clinical outcome. PLoS One. 2016;11(12).e0166398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. McCoy RC, Newnham LJ, Ottolini CS, et al. Tripolar chromosome segregation drives the association between maternal genotype at variants spanning PLK4 and aneuploidy in human preimplantation embryos. Hum Mol Genet. 2018;27(14):2573‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang Q, Li G, Zhang L, et al. Maternal common variant rs2305957 spanning PLK4 is associated with blastocyst formation and early recurrent miscarriage. Fertil Steril. 2017;107(4):1034‐1040.e5. [DOI] [PubMed] [Google Scholar]
- 84. Ottolini CS, Kitchen J, Xanthopoulou L, Gordon T, Summers MC, Handyside AH. Tripolar mitosis and partitioning of the genome arrests human preimplantation development in vitro. Sci Rep. 2017;7(1):9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McCollin A, Swann RL, Summers MC, Handyside AH, Ottolini CS. Abnormal cleavage and developmental arrest of human preimplantation embryos in vitro. Eur J Med Genet. 2020;63(2):103651. [DOI] [PubMed] [Google Scholar]
- 86. Chernus JM, Allen EG, Zeng Z, et al. A candidate gene analysis and GWAS for genes associated with maternal nondisjunction of chromosome 21. PLoS Genet. 2019;15(12):e1008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yang K‐T, Li S‐K, Chang C‐C, et al. Aurora‐C kinase deficiency causes cytokinesis failure in meiosis I and production of large polyploid oocytes in mice. Mol Biol Cell. 2010;21(14):2371‐2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Balboula AZ, Schindler K. Selective disruption of aurora C kinase reveals distinct functions from aurora B kinase during meiosis in mouse oocytes. PLoS Genet. 2014;10(2):e1004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feng R, Yan Z, Li B, et al. Mutations in TUBB8 cause a multiplicity of phenotypes in human oocytes and early embryos. J Med Genet. 2016;53(10):662‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhao L, Guan Y, Wang W, et al. Identification novel mutations in TUBB8 in female infertility and a novel phenotype of large polar body in oocytes with TUBB8 mutations. J Assist Reprod Genet. 2020;37(8):1837‐1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lanuza‐López MC, Martínez‐Garza SG, Solórzano‐Vázquez JF, et al. Oocyte maturation arrest produced by TUBB8 mutations: impact of genetic disorders in infertility treatment. Gynecol Endocrinol. 2020;36(9):829‐834. [DOI] [PubMed] [Google Scholar]
- 92. Maddirevula S, Coskun S, Alhassan S, et al. Female infertility caused by mutations in the oocyte‐specific translational repressor PATL2. Am J Hum Genet. 2017;101(4):603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chen B, Zhang Z, Sun X, et al. Biallelic mutations in PATL2 cause female infertility characterized by oocyte maturation arrest. Am J Hum Genet. 2017;101(4):609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Christou‐Kent M, Kherraf Z‐E, Amiri‐Yekta A, et al. PATL2 is a key actor of oocyte maturation whose invalidation causes infertility in women and mice. EMBO Mol Med. 2018;10(5):e8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Huang L, Tong X, Wang F, et al. Novel mutations in PATL2 cause female infertility with oocyte germinal vesicle arrest. Hum Reprod. 2018;33(6):1183‐1190. [DOI] [PubMed] [Google Scholar]
- 96. Wu L, Chen H, Li D, et al. Novel mutations in PATL2 : expanding the mutational spectrum and corresponding phenotypic variability associated with female infertility. J Hum Genet. 2019;64(5):379‐385. [DOI] [PubMed] [Google Scholar]
- 97. Liu Z, Zhu L, Wang J, et al. Novel homozygous mutations in PATL2 lead to female infertility with oocyte maturation arrest. J Assist Reprod Genet. 2020;37(4):841‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao S, Chen T, Yu M, et al. Novel WEE2 gene variants identified in patients with fertilization failure and female infertility. Fertil Steril. 2019;111(3):519‐526. [DOI] [PubMed] [Google Scholar]
- 99. Dai J, Zheng W, Dai C, et al. New biallelic mutations in WEE2: expanding the spectrum of mutations that cause fertilization failure or poor fertilization. Fertil Steril. 2019;111(3):510‐518. [DOI] [PubMed] [Google Scholar]
- 100. Sang Q, Li B, Kuang Y, et al. Homozygous mutations in WEE2 cause fertilization failure and female infertility. Am J Hum Genet. 2018;102(4):649‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang Z, Mu J, Zhao J, et al. Novel mutations in WEE2: expanding the spectrum of mutations responsible for human fertilization failure. Clin Genet. 2019;95(4):520‐524. [DOI] [PubMed] [Google Scholar]
- 102. Yang X, Shu L, Cai L, Sun X, Cui Y, Liu J. Homozygous missense mutation Arg207Cys in the WEE2 gene causes female infertility and fertilization failure. J Assist Reprod Genet. 2019;36(5):965‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]