

Abstract
The study consists of a detailed investigation of the degradability of the emerging water contaminant-caffeine by homogeneous and heterogeneous Advanced Oxidation Processes (AOP’s), estimation of a synergy index for each hybrid operation thereof, and proposing the most plausible reaction mechanisms that are consistent with the experimental data. It also encompasses evaluation of the effect of the water matrix represented by carbonate species and humic acids, as strong scavengers of hydroxyl radicals. The results showed that single AOP’s such as sonolysis (577 kHz) and photolysis with H2O2 provided complete caffeine elimination, but they were insufficient for the mineralization of the compound. Hybrid AOP’s were considerably more effective, particularly when operated at a heterogeneous mode using commercial TiO2. The most effective hybrid process was UV-H2O2/TiO2, which provided more than 75% TOC decay at the minimum test doses of the reagent and catalyst. While the addition of ultrasound to the process significantly increased the rate of caffeine decomposition, it reduced the overall degradation of the compound to 64% in terms of TOC decay. The antagonistic effect was attributed to the formation of excess H2O2, and the presence of cavity clouds and/or high density layers that inhibited the transmission of UV light. The effect of natural water ingredients was found to reduce the reaction rates, signifying the major contribution of hydroxyl radicals to the destruction of caffeine. The proposed reaction mechanisms based on OH radical attack and the calculated energy barriers were in good agreement with the experimentally detected reaction byproducts.
Keywords: AOP’s, Cavitation, •OH, Hybrid, Synergy index, Energy barrier
1. Introduction
Caffeine is a common compound of PPCP’s and an ingredient of coffee, tea, chocolate, cocoa and beverages; while it is widely used in the production of prescription medicines and personal care products [1]. Upon consumption, the compound and/or its metabolic byproducts are readily discharged (via wash water and excretion) into sewage treatment facilities with low biodegradability. Hence, caffeine is not only detected frequently in effluents of wastewater treatment operations, but also in soil, groundwater and fresh water systems [2]. Lately, the presence of caffeine in the water environment has been a serious environmental and public concern, due to its toxic effects to aquatic organisms, and potential health threat to humans as hyperactivity and cardiovascular diseases [3]. Consequently, a lot of research has been lately focused on finding effective processes that control the discharge and/or the presence of caffeine in water supply systems.
Studies on the elimination of caffeine from water have shown that the most effective prorcesses are adsorption [4], [5], advanced biological treatment [6] and homogeneous or heterogeneous advanced oxidation processes (AOP's) using ozone, UV, Fenton’s reagent, and semiconductors such as TiO2 [7], [8], [9]. AOPs are based on the in-situ production of hydroxyl radicals (•OH) using UV irradiation and/or chemical reagents such as ozone ozone [10], [11], H2O2 [12], [13], Fenton reagents [14], [15] and TiO2 [12], [16]. A less common method of •OH generation in water is ultrasonication or sonolysis, which has been recognized as a green method due to the unique properties of ultrasound for producing radical species without the use of chemical reagents [17]. The phenomenon is based on nucleation, growth and the violent collapse of acoustic cavity bubbles that result in thermal decomposition of water and organic molecules entrapped in the bubbles [18]. Some of the radicals produced during the collapse phase recombine at the cooler bubble interface to yield water and hydrogen peroxide, which may also contribute to the oxidation of some organic chemicals [18], [19]:
| H2O + ))) → •OH + •H | (1) |
| •OH + •OH → H2O2 | (2) |
| H• + HO• → H2O | (3) |
Combined or hybrid applications of AOP’s have been lately found promising for the degradation of recalcitrant organic compounds, owing to the production of excess •OH and additional oxygenated species such as peroxyl and superoxide radicals (•–O2H, •O2–), which although more selective than •OH are also very powerful oxidizing agents [20]. Most commonly used lab-scale hybrid AOP’s for the degradation of caffeine in water are ozonation with UV and H2O2 [21], electrochemical oxidation with UV and/or ultrasound [22], and US-assisted catalytic oxidation using composites of metallic particles as TiO2, CdS and CeO2 [23], [24], [25].
The objective of this research was to assess the effect of hybrid AOP’s on the oxidation and mineralization of caffeine, which has lately been recognized as an emerging water pollutant. The test processes comprised of sonolysis at 577 kHz, UV-photolysis at 254 nm (with or without H2O2), photocatalysis with TiO2, and combinations thereof. The assessment was based on the rate of caffeine degradation and the degree of carbon mineralization, as determined by the analysis of total dissolved organic carbon (TOC). The study also covers estimation of a synergy index for each hybrid process, and modeling of •OH and H2O-mediated reactions to propose the most probable reaction mechanisms for the formation of intermediate products that are consistent with the experimentally detected ones. The work is novel, for there are no similar studies in the literature reporting the degradation of caffeine by hybrid AOPs assisted by high-frequency ultrasound, nor any others on computational modeling of the reaction mechanisms and comparison of the predicted oxidation byproducts with the experimentally detected ones.
2. Materials and methods
Caffeine was obtained from Doğa İlaç, Turkey with 99% purity and used as received. The structure and properties of the compound are as given [26], [27]:
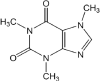 MW = 194.2 g mol−1
MW = 194.2 g mol−1
pKa = 14
Kow = 0.01
Solubility = 21.6 g L−1
EC50 (D. Magna) = 182 mg L−1
HPLC grade methanol, reagent grade HCl, NaOH, H2SO4, H2O2, NaCO3, KI, ammonium molybdate tetrahydrate and KHP were all purchased from Merck, Istanbul and used in the analysis of caffeine, pH adjustment, H2O2 analysis and TOC calibration, respectively. Aeroxide TiO2 (P25) was obtained from Evonik Industries, Germany. Technical grade humic acid sodium carbonate were obtained from Aldrich, Istanbul. Both chemicals were dissolved in ultrapure water to prepare stock solutions of 1000 mg L−1. Samples were prepared from the stocks by filtation through 0.45 µm followed by dilution.
2.1. Experimental
A stock solution of caffeine was prepared by dissolving 1.0 g of the compound in 1 L of ultrapure water (Milli-Q) via magnetic stirring for 2-h. The solution was kept at 4˚C in the dark for use in making the samples. The adsorption of caffeine on particles of TiO2 was investigated in a shaker system spiked with 1 g L−1 TiO2 and varying concentrations of caffeine cduring 1-h mixing at 250 rpm at room temperature. All single and hybrid AOP’s were carried out in a high-frequency plate-type ultrasonic reactor with a 120 W generator (operated at 90% capacity) connected to three piezoelectric transducers (22 cm2) that emitting at 577 kHz (Ultraschall/ Meinhardt, Germany). The solution temperature in the reactor was maintained at 25 ± 0.5 °C by water circulation, and the power density was kept constant at 0.23 W mL−1 (determined by calorimetry) as reported in a previous work [28]. The light source in UV-supported AOP’s was a low-pressure mercury UV-lamp emitting monochromatically at 253.7 nm and operated at an intensity of 4.7 W m−2. The lamp was placed in a quartz jacket and immersed vertically along the center of the reactor. The optimum pH in all experiments was found using three test levels as 4.0, 7.0 and 9.0, at which the degree of caffeine decay was continuously monitored throughout the reaction time. Selection of the optimum H2O2 dosage was based on monitoring the concentration of caffeine during sonolysis and photolysis in the presence of 5, 10 and 25 mg L−1 of the reagent. Catalytic AOP’s were run by adding 0.1, 0.25 and 0.5 g L−1 of a TiO2 suspension prepared by sonication of P25 particles in 20 mL of ultrapure water for 1-min using an ultrasonic horn (20 kHz). All samples were aerated for 15-min unless otherwise stated, and those exposed to ultrasound were injected with air and/or argon during reaction to enhance the formation of cavitation nuclei. The ultrasonic generator was shut down during US-free processes.
Humic acid (HA) and NaCO3 were added (at 5 mg L−1 and 5 µM, respectively) to the sample solutions containing 5 mg L−1 caffeine to assess the impact of water matrix on the efficiency of the applied processes. The effect of mixtures of ingredients was also tested by spiking a sample solution with a mixture of HA and NaCO3 followed by processing it again. Additionally, the effect of mixing vs sonication was tested by using a mechanical agitator (operated consecutively at 100 and 1000 rpm) to stir the sample solutions containing 5 mg L−1 caffeine.
The list of single and hybrid AOP’s tested with caffeine at various conditions is given in the following (the symbols “US” and “UV” stand for 577 kHz ultrasound and 254 nm UV irradiation, respectively):
Single-Homogeneous: US (P1); UV/H2O2 (P2)
Hybrid-Homogeneous: US/UV (P3); US/UV-H2O2 (P4)
Hybrid-Heterogeneous: US/TiO2 (P5); UV/TiO2 (P6); US/UV/TiO2 (P7); UV-H2O2/TiO2 (P8); US/UV-H2O2/TiO2 (P9).
2.2. Analytical
Caffeine was analyzed by HPLC using a Shimadzu LC-20AT HPLC with a 20A UV–vis photo diode array detector set at 272 nm, and equipped with a Hyperpack Basic ODS (5 µm, 15x0.46). The mobile phase consisted of 40:60 methanol flowing at 1 mL min−1. Sample injection volume and retention times were set at 20 µL and 3.207 min, respectively. The compound was also analyzed spectrophotometrically at 273 nm, at which it had a maximum absorption, as also reported in the literature [29], [30]. The analysis of soluble TOC was carried out by a Shimadzu TOC-V CSH analyzer, following filtration of the samples through a 0.45 µm PDFE filter.
The oxidation byproducts were identified using a LC-MS system (LCMS 2020, Shimadzu, Japan) with a C-18 (150 × 4.6 mm) column. The method was gradient using acetonitrile and deionized water mixed with 0.05% trifluoro-acetic acid. The protocol was as the following: LC: 10%, 10%, 95%, 95% and 100% acetonitrile at t = 0, 2, 10, 12 and 14 min, respectively; MS: positive scan at 100–800 m/z. The effluent flow rate in both analyses was 0.5 mL min−1.
2.3. Computational
The reactions were modeled using a quantum chemical computational method (Density Functional Theory) incorporated in the Gaussian 16 software [31]. The functional, B3LYP, and the 6-311G (d, p) basis sets were used for the conformational search of each stationary point [32], [33], [34]. Transition state structures were characterized by a single imaginary frequency, and reactions were modeled in water (ε = 78.36) at 298 K using the Integral Equation Formalism Polarizable Continuum Model [35]. The intrinsic reaction coordinates (IRC) were calculated to verify the relation of each transition state with the corresponding reactants and products. The oxidation reactions were modeled based on the attack of the OH radical to the reactant, while those occurring in water without AOP’s (as verified by the peaks depicted in LCMS chromatograms at t = 0) were modeled assuming water as the attacking reactant. The energy barrier for the formation of each byproduct was referred to as activation Gibbs free energy (ΔG‡), and found by subtracting the Gibbs free energy of the transition state from that of the corresponding reactants.
3. Results and discussion
3.1. Single processes: effects of pH, bubbling gas and H2O2
It was found that caffeine was easily decomposed by high-frequency ultrasound, and no decomposition was observed by magnetic or mechanical agitation of the sample (data not presented). The rate of caffeine decay by sonolysis alone was pseudo-first order, but the rate constant with decreases in pH is most likely due to hydrophobic enrichment of the compound that facilitates its migration towards the bubble–liquid interface, where the concentration of OH radicals is much higher than that in the bulk solution [36]. Fig. 1 also shows that acidic pH was more favorable than neutral or alkaline pH, as it provided a higher degree of concentration decay and a faster reaction. Note however that at neutral pH, the reaction was slow during the first 10-min, but faster after 15-min, by which the pH of the solution was dramatically reduced to 2.55.
Fig. 1.

The drop in pH during sonolysis of caffeine (a), and the variations in the the apparent reaction rate constant within 15 and 30-min intervals by the initial solution pH (b). The buffering agent was sodium phosphate (0.1 M; pH 7.0), pH adjustment was made with NaOH (0.1 M), the initial values of pH and concentration were 4.0 and 5 mg L−1, respectively.
The decrease in pH during sonolysis has also been reported in the literature for air-saturated and/or air-injected solutions, and explained by the reactions of nitrogen gas to form nitrous oxide and nitric acid [37], [38]:
| N2 + O → NO + N | (4) |
| N + O2 → NO + O | (5) |
| NO + •OH → HNO2 | (6) |
| HNO2 → H+ + NO2– | (7) |
| HNO2 + H2O2 → HNO3 + H2O | (8) |
However, the dramatic acidification observed in this study is not solely explainable by the applied aeration and the above reactions, but more likely by the formation of highly acidic and hydrophilic intermediates via OH radical-mediated oxidation of caffeine. This argument was justified by the insignificant degree of TOC decay after 30 and 60-min sonication of the sample (0.91% and 1.67%, respectively).
It was also found that at t = 0, the observed value of C0 was considerably lower than the input quantity (5.0 mg L−1), except when no pH adjustment was made (pH 4.0). The data are presented in Table 1 for each sample, as well as that with a phosphate buffer. The instant loss of mass upon pH adjustment and/or buffer addition can be attributed to alkalinization of the compound, leading to hydrolysis and/or fragmentation of the molecule. The argument was justified by the LCMS analysis of the compound at time zero, displaying a series of major fragment ions (to be discussed in the next section). Note that a similar finding has been reported in the literature using the ESI analysis of caffeine, which revealed fragment ions at time zero upon protonation of the compound, followed by the loss of a methyl group [39].
Table 1.
The influence of pH adjustment and buffering on the observed concentration of caffeine at t = 0. The actual quantity was 5.0 mg L−1.
| C0 (mg L−1) | |||
|---|---|---|---|
| pH=4.0 | pH=7.0 | pH=9.0 | |
| no buffer | with buffer | ||
| 4.85±0.15 | 4.30±0.29 | 4.49±0.59 | 3.98±0.50 |
The second operation parameter of interest was the bubbling gas. It has been shown that injection of a gas during sonolysis of aqueous solutions enhances the number of cavity nuclei, leading to increased incidence of bubble collapse, and thus enhanced generation of OH radicals [40], [41]. Comparing the effects of air and argon on the rate of caffeine degradation showed that the reaction kinetics followed pseudo-first order rate law, and the reaction rate was higher in the presence of Ar than air, as demonstrated in Fig. 2. It was also found that the reaction proceeded practically at the same rate in air-injected and the control sample (sample with no gas injection). A faster reaction in the presence of argon bubbles is due to a higher polytrophic gas ratio of the gas (than that of air), which is responsible for a higher collapse temperature and thus more violent and intense reactions [42]. This is significant, because as the hydrophobicity of caffeine is increased with decreasing pH, it moves away from the bulk solution towards the bubble–liquid interface, where it reacts effectively with the excess •OH. On the other hand, some researchers have reported higher rates of decomposition of organic compounds by sonolysis in the presence of air bubbles than argon, and explained their finding by the production of additional radical species (e.g. NO2• and NO3•) that may react with the target compound [43]. The opposite trend in our case implies the presence of too many air bubbles, leading to a high rate of •OH consumption by the excess of nitrogen species, as represented by Eq. (5) and Eq. (8) [38], [43], describing respectively the oxidation of NO to yield HNO2, and that of HNO2 to yield nitric acid:
| HNO2 + •OH → HNO3 + •H+ | (9) |
Fig. 2.

The pseudo-1st order rate of caffeine decay and the corresponding rate constants by 60-min sonolysis of the compound under air and argon atmospheres (“control” refers to sonolysis without gas injection). Initial conditions were C0 = 5 mg L−1, pH0 = 4.0, gas flow rate = 1.5 L min−1.
The second single process was photolysis at 254 nm, which was found totally ineffective (regardless of the initial pH), signifying the disability of caffeine to undergo photolytic decay at the applied UV conditions. The performance of the process was considerably improved by the addition of H2O2, as depicted in Fig. 3. Unlike sonolysis, the reaction rate during UV/H2O2 process was not affected by the initial pH of the solution. It was found that caffeine was completely eliminated within 20-min exposure to the process, owing to the photolysis of H2O2 to produce two OH radicals per mole. As such, the rate of reaction was found to be directly proportional to the amount of H2O2 added (data not given). On the other hand, the process was ineffective for C-mineralization, as the percentage of TOC decay after 1-h was only 12%. Such a low efficiency can be explained by the depletion of H2O2, i.e. termination of the •OH production route after longer reaction time.
Fig. 3.

Comparison of single processes for the rate of caffeine decay during 1-h reaction at pH 4, H2O2 = 10 mg L−1 and C0 = 5 mg L−1. No gas injection was applied during any of the processes, but all samples were air-saturated prior to the experiments.
To assess the role of molecular hydrogen peroxide on the overall degradation of caffeine, we added the same quantity of H2O2 to the samples before sonolysis, and found that neither the rate of reaction, nor the degree of TOC decay were increased. The result is a clear indication of the oxidation path, i.e. the reactions were governed mainly by the attack of hydroxyl radicals to the parent compound and the oxidation byproducts. The data are added to Fig. 3 to allow comparison of the all single AOP’s for the rate of caffeine decay.
3.2. Hybrid processes
3.2.1. Homogeneous
3.2.1.1. US/UV and US/UV-H2O2
Hybrid operation of UV with ultrasound did not improve the rate of caffeine decay as expected, but enhanced the mineralization of the compound from 1.67% by US alone to 11.8% at pH 4. The explanation for the lack of synergy in caffeine degradation is that at high-frequency irradiation, too many bubbles are formed and the coalescence of these bubbles inhibit the transmission of UV light and attenuation of it [44]. On the other hand, the improvement in TOC mineralization after 60-min reaction is most likely due to the accumulation and partial photolysis of US-generated H2O2 (despite the inhibition of light transmission by bubble coalescence), leading to the production of excess •OH. Normalized plots of concentration vs. time for two pH test levels are presented in Fig 4. with an inset showing fractions of TOC decay after 30 and 60-min reaction. Moreover, some of the oxidation byproduct and the supercritical water formed by ultrasound may undergo direct photolysis, the latter leading to the generation of excess radical species [45], [46].
Fig. 4.

The effect of pH on the rate of caffeine oxidation (C0 = 5 mg L−1) by 30-min sonolysis in the presence of UV-irradiation. The data referred to as “Control” belong to singly applied US at pH 4. The rate constants of the hybrid process were 0.065 min−1 and 0.051 min−1, at pH 4 and 7, respectively.
A further improvement in the efficiency of US/UV hybrid system was obtained by adding consecutively three different doses of H2O2 to three fresh caffeine samples and monitoring the concentration of caffeine with time. Relative impacts of 5, 10 and 25 mg L−1 H2O2 (corresponding respectively to caffeine/peroxide mole ratios of 0.043, 0.087 and 0.217) on the rate of caffeine decay are presented in Fig 5. The data show that mineralization of the compound was directly related to the dose of H2O2, reaching a maximum at the highest dose of the reagent. We selected the optimal dose as 10 mg L−1, as it provided 28-fold enhancement in the rate of reaction, and 2-fold enhancement in the mineralization of the compound The synergy obtained is the result of excess •OH generation upon photodecomposition of H2O2.
Fig. 5.

The effect of H2O2 dose on the rate of caffeine (C0 = 5 mg L−1) oxidation by 30-min operation of US/UV. Numbers in parenthesis represent the estimated apparent reaction rate constants at the applied H2O2 dose.
3.2.2. Heterogeneous
3.2.2.1. UV/TiO2 and US/TiO2
Heterogeneous processes with TiO2 were in general more effective than homogeneus ones operated singly or in hybrid mode. UV/TiO2 operation, for example, provided the same rate of caffeine decay as that obtained by UV-H2O2 (k = 0.025 ± 0.02 min−1), but nearly a 2-fold higher degree of C-mineralization (61% vs 35%). Improved performance for TOC decay must be due to the oxidation of the reaction byproducts on the heterogeneous surface, which was enriched with reactive oxygen species upon excitation. On the other hand, the presence of TiO2 under sonolysis (without UV) was not as effective, but the operation provided a larger fraction of TOC decay than that obtained by the sonolysis with UV (20% vs 11.8%). The result can be attributed to the presence of heterogeneous surfaces, which led consecutively to the formation of additional cavity nuclei, additional bubble collapse, and additional OH radicals. Moreover, the sonoluminescence created by cavitational collapse on the surface of TiO2 is made of a wide wavelength of UV range, and those wavelengths shorter than 375 nm may excite the surface of the semiconductor, just like in photocatalytic processes [47].
Relative performances of TiO2-catalyzed photolysis and sonolysis are presented in Fig. 6. It was found that the decline in caffeine concentration with time followed a rate expression that slightly deviated from the pseudo-1st order rate law as:
| (10) |
where: C/C0 is the fraction of caffeine removed at time t, k is the reaction rate constant (min−1) and P is a non-zero plateau (mg L−1). Note that the expression is typical of adsorption-mediated reaction rates, which are limited by the degree of adsorption on a reactive or non-reactive heterogeneous surface, and the quantity of reactive radical species it holds.
Fig. 6.

Photo- and sonocatalytic decay of caffeine (C0 = 5 mg L−1) and its oxidation byproducts by 30-min sonolysis and photolysis of the compound in the presence of 0.1 g L−1 TiO2 at pH 4. The solid lines are the fit of Eq. (9) to the data, with estimated reaction rate constants of 0.19 and 0. 0.05 min−1 for UV/TiO2 and US/TiO2, respectively.
3.2.2.2. US/UV/TiO2 and US/UV-H2O2/TiO2
The addition of TiO2 to US/UV homogeneous system provided a significant enhancement on both the rate of caffeine decomposition and the degree of TOC decay, as depicted in Fig. 7. The data also reveal that mineralization was directly related to the quantity of TiO2 added, while the rate of caffeine decay remained nearly constant at all test concentrations of the catalyst (k = 0.26 ± 0.05 min−1, 0.27 ± 0.02 min−1, 0.25 ± 0.01 min−1). Kinetic analysis of the data showed that the decomposition of caffeine followed a two-phase decay pattern, in which the rate was very fast during the first five min, and slowed down thereafter with apparent rate constants of similar magnitudes at each test concentration of TiO2. A constant rate of caffeine decay regardless of the increases in the concentration of TiO2 is related to its poor adsorption capacity on the surface of the catalyst. Batch experiments testing the adsorption of caffeine on P25 showed that it adsorbed weakly on the surface of TiO2, as depicted in Fig. 8. Hence, the compound remains in the bulk solution during the operation of US/UV-TiO2, reacting only with the OH radicals that are ejected to the solution from collapsing bubbles and generated by the photolysis H2O2. On the other hand, the enhancement in carbon mineralization by increasing doses of the catalyst is an indication of a higher adsorption capacity of the oxidation byproducts on the surface of TiO2, which was enriched with reactive oxygen species upon excitation. Consequently, mineralization reactions are facilitated as the number of particles increase with increasing doses of TiO2, at which the incidence of bubble collapse and •OH production also increase. Note also that the “hot spots” and sonoluminescene generated on the surface of the catalyst during bubble collapse may excite the catalyst, leading to the production of excess reactive species on its surface. Finally, ultrasound overcomes the typical disadvantages of TiO2-photocatalysis (surface corrosion and fouling) via cleaning and disintegrating of the surface, thus producing fresh adsorption sites with smaller surface areas.
Fig. 7.

The impact of TiO2 concentration on the rate of caffeine decay and the degree of C-mineralization by 15 and 60-min photocatalysis, respectively in the presence of ultrasound. Initial conditions were pH = 4, C0 = 5 mg L−1.
Fig. 8.

The adsorption equilibrium of caffeine (5 mg L−1) during 60-min shaking in the presence of four different concentrations of TiO2.
In the second heterogeneous hybrid process, we ran sono-photocatalysis (US/UVTiO2) in the presence of 10 mg L−1 H2O2 and the lowest test dose of the catalyst, (0.1 g L−1) and found that while the rate of reaction increased significantly (k = 0.39 min−1), the degree of mineralization was reduced from 75.4% without H2O2 to 63.6% with it. The explanation for the rate enhancement is the strong adsorption of H2O2 on the surface of the catalyst [48]. When such a surface is irradiated by UV, the photo-generated electrons react with H2O2 leading to the formation of excess •OH via reductive dissociation [49]. It was also found that molecular H2O2 is attached only on the oxygen vacancies at the surface of TiO2 forming Ti–O–O–Ti peroxides [48]. Once all peroxides disappear from the surface by reductive dissociation, water molecules adsorb on Ti atoms and remain intact there [49]. Hence, the antagonistic effect of H2O2 to the mineralization of caffeine is due to the occupancy of all oxygen vacancies, which eventually deplete together with the attached H2O2, leading to termination of radical generation reactions.
3.3. Comparative evaluation of all processes
Application of AOPs for the destruction of recalcitrant organics is based on their potential for the in-situ production of hydroxyl radicals [50]. Hybridization of these processes with ultrasound has lately received much attention, owing to the excess •OH produced by the pyrolytic cleavage of water molecules entrapped in the collapsing cavity bubbles, as depicted by the chemical reaction given in Eq. (1). The hydroxyl radicals ejected from the bubbles combine at the outer surface or the bulk liquid to form hydrogen peroxide as shown in Eq. (2). Hence, it has been a common practice in sonochemistry to monitor the concentration of H2O2 with time to get a fairly accurate idea about the rate of •OH production by ultrasound [51]. Changes in the concentration of H2O2 with time during sonolysis of ultrapure water at 577 kHz is presented in Fig. 9. We also plotted in the same figure the concentration of the reagent during US/UV, US/TiO2 and US/UV/TiO2 processes applied to ultrapure water to elucidate the changes in the rate of production and depletion of the compound.
Fig. 9.

The rate of H2O2 production during 30-min sonolysis, sono-photolysis, sono-catalysis and sonophotocatalysis of ultrapure water.
The data show that H2O2 increases linearly during single sonolysis; fluctuates slightly in the first 15-min and increases thereafter during sonophotolysis; and fluctuates dramatically in the heterogeneous processes, particularly in US/UV/TiO2. It is also interesting that after 20-min sonolysis in the presence of UV and TiO2, the concentration of H2O2 reaches a value close to zero, and does not increase anymore during further reaction. The explanation for the depletion of the reagent after 15-min is based on the strong adsorption tendency of H2O2 on the surface of TiO2 particles and its reductive dissociation by photo-generated electrons, as explained previously. It is also possible that photo-generated holes react with H2O2 − H2O molecules on the surface of the catalyst, causing dissociation of water into H+ and •OH radicals, leaving the peroxide intact [49].
Relative performance of each test process for the rate of caffeine oxidation and the degree of TOC decay is summarized in Table 2. Note that although P9 was found to provide the highest rate of reaction as indicated above, P8 was the most effective combination, regarding TOC decay.
Table 2.
Comparison of all processes for their performance in the oxidation and mineralization of caffeine (C0 = 5 mg L−1) after 20 and 60-min reaction, at pH 4, respectively.
| Process (Symbol) | k′ (min−1) | %TOC decay |
|---|---|---|
| Homogeneous | ||
| US (P1) | 0.05 | 1.67 |
| UV-H2O2 (P2) | 0.26 | 21.66 |
| US/UV (P3) | 0.08 | 11.83 |
| US/UV-H2O2 (P4) | 0.25 | 35.36 |
| Heterogeneous | ||
| US/TiO2 (P5) | 0.05 | 20.43 |
| UV/TiO2 (P6) | 0.19 | 60.94 |
| US/UV/TiO2 (P7) | 0.25 | 66.11 |
| UV-H2O2/TiO2 (P8) | 0.26 | 75.49 |
| US/UV-H2O2/TiO2 (P9) | 0.39 | 63.58 |
The data and the discussion so far have shown that combined operations, particularly those in the heterogeneous mode are significantly effective in improving the overall degradation of caffeine, expressed by the percentage of TOC decay. To quantify and compare the synergy of hybrid operations, we used the following equation adopted from the work of Choi et al., 2020 [52]:
| (11) |
where: S is the synergy index, TOCHP is the fraction of TOC decay obtained by the hybrid process of concern, and TOCSP1,SP2,SPn are fractions of TOC decay obtained by single processes that comprise the hybrid. Calculated values of S using fractions of TOC decay observed after 60-min reaction are presented in Table 3. As noted in the last paragraph of the previous section, the hybrid process-P9 provided the largest reaction rate constant, but the synergy index was very low. The most rational explanation for this is the formation of cavitation clouds via the phenomenon called “bubble coalescence”, which blocks the transmittance of UV light into solution [53]. As such, not only less •OH were produced due to inhibition of H2O2 photolysis, but excess of them were consumed via the well-known reaction of molecular H2O2 with •OH. Moreover, it has been reported that during sonication of TiO2 slurries by high frequency ultrasound, the transmittance of UV light is also reduced via the formation of high-density particle layers [54]. Hence, the insignificant synergy obtained by adding ultrasound to UV/TiO2 photocatalysis is due to the presence of these high-density particle layers, that inhibited the transmission of UV-light and thus the photolysis of US-generated H2O2. On the other hand, the antagonism observed in US/UV-H2O2/TiO2 is due solely to the excess H2O2 that accumulated and reacted readily with •OH, the production of which was inhibited or even terminated via the presence of cavity clouds and high-density particle layers. Moreover, the competition of the oxidation byproducts for •OH may further increase the antagonism, as also reported in the literature for the sono-photocatalytic degradation of nonylphenol in the presence of immobilized TiO2 [55]. Last but not least, we have reported in an earlier study that some of the TiO2 particles accumulate on the surface of the transducer, thus reducing the transmission of ultrasonic energy to the liquid [56].
Table 3.
Comparative synergy indices of the hybrid processes for the overall degradation of caffeine (C0 = 5 mg L−1) after 60-min reaction at pH 4.
| HHybrid Process (Symbol) | S |
|---|---|
| UUS/UV (P2) | 0.21 |
| UUS/UV-H2O2 (P4) | 0.25 |
| UUV/TiO2 (P6) | 3.72 |
| UUS/UV/TiO2 (P7) | 4.47 |
| UUV-H2O2/TiO2 (P8) | 65.11 |
| UUS/UV-H2O2/TiO2 (P9) | 2.82 |
Finally, Table 4 presents a comparative evaluation of our findings for caffeine degradation with those reported in a limited number of publications of or lab-scale application of similar AOPS at varying experimental conditions. The table reveal that the processes and the selected experimental conditions described in this work are considerably more effective than those in the literature, regarding the short reaction times and low reagent doses a used, as well as the TOC decay fractions observed.
Table 4.
Comparison of this work at the applied test conditions with those reported in literature for the efficiency of AOPs in eliminating caffeine and TOC. The UV source in this work and that of Afonso-Olivares et al., [13] was a low-pressure Hg lamp at 254 nm, and the one used in the work of Carotenuto et al., [16] was a solar lamp. Note that no data are available in the cited literature for the extent of carbon mineralization.
| Process | Experimental conditions |
Removal (%) |
Source | |||||
|---|---|---|---|---|---|---|---|---|
| C0 (mg L−1) | H2O2 (mg L−1) | TiO2 (mg L−1) | pH | Freq (kHz) | Caffeine | TOC | ||
| US/H2O2 | 5.0 | 10 | 4.07.0 | 577 | 8496 | 9624.3 | This work | |
| UV-H2O2 | 5.01.4 | 10 15 25 | 100 88a 87a | 47.1 - | This work [13], [13] | |||
| UV/TiO2 | 5.0 | 0.10 | 4.0 | 577 | 100 | 60.9 | This work | |
| US/UV/TiO2 | 5.0 | 0.10 0.25 0.50 | 4.0 | 577 | 100 66 100 | 100 82.2 78.6 | This work This work This work | |
| UV-H2O2/ TiO2 | 5.0 | 10 | 0.10 | 4.0 | 100 | 75.5 | This work | |
| US/UV-H2O2 | 5.0 | 10 25 | 4.0 | 577 | 100 100 | 21.7 38.7 | This work This work | |
| US/UV-H2O2/TiO2 | 5.0 | 10 | 0.10 0.25 | 4.0 | 577 | 100 100 | 63.6 70.5 | This work This work |
| UV/TiO2 | 5.0 | 0.10 | 70b | – | [16] | |||
| 8.0 | 0.15 | 80c | – | [16] | ||||
t = 75 min
t = 15 min
t = 5 min
3.4. The effect of water matrix or the presence of OH radical scavengers
It is of common knowledge that the results obtained in the laboratory with pure water spiked with a single contaminant are not replicable in real water containing mixtures of contaminants and natural ingedients, as carbonate species and humic substances. Basically in such complex mixtures, compounds may interfere, react or compete for the radical species and/or the adsorption sites added to the contaminated water, thus leading to reduced reaction rates. To check the replicability of our data in real water, we made some additional experiments by adding CO32– and humic acid (HA) to the sample solutions and running sonolysis and sonocatalysis. The selction of CO32– and HA as natural water ingredients was based on their high reactivity with •OH as depicted by the rate constants of the following reactions [57], [58]:
| •OH + CO32– → •CO3– k = 3.8 × 108 M s−1 | (12) |
| •OH + RH2 → •RH + H2O k = 2.8 × 107 M s−1 | (13) |
Note that due to the complicated structure of humic acid, the formula is represented by RH2 and the rate constant is given in units of the molar concentration of carbon per second.
The bimolecular rate constants given in Eqs. (12), (13) show that carbonates sand humic acids are both strong scavengers of OH radicals in water. As such, we found that the presence of these substances reduced the rate of caffeine decay to almost half its value obtained by sonolysis alone, while they were considerably more competitive during sonophotocatalysis with TiO2. The apparent reaction rate constants observed in the presence of CO3–, HA and both are listed in Table 5. Note that a more severe rate reduction by HA during sono-photocatalysis must be due to the reactivity of humic substances with oxygen species, which are generated on the surface of TiO2 by UV irradiation [59]. Most importantly, the data reveal that the degradation of caffeine was directly related to the quantity of OH radicals in the reactor.
Table 5.
The effect of water matrix (CO32– and humic acid as •OH scavengers) on the rate of caffeine decay (C0 = 5.0 mg L−1) by US and US-assisted photocatalysis. Control1 and Control2 refer respectively to single ultrasound and ultrasound-assisted photocatalysis with 0.1 g L−1 TiO2.
| Process | k′ × 10−3 s−1 |
|---|---|
| Control1 | 1.05 ± 0.09 |
| Control1 + CO3 | 0.63 ± 0.03 |
| Control1 + HA | 0.62 ± 0.01 |
| Control1 + CO3 + HA | 0.55 ± 0.01 |
| Control2 | 6 0.11 ± 0.20 |
| Control2 + CO3 | 2.33 ± 0.11 |
| Control2 + HA | 1.42 ± 0.13 |
| Control2 + CO3 + HA | 1.07 ± 0.09 |
3.5. An overview of the oxidation byproducts and reaction mechanisms
LCMS analyses of the reactor samples collected at t = 0, 30 and 60-min during single and hybrid processes revealed numerous fragments and byproducts, most of which were detected at retention times of 7.80 and 8.33 min. Moreover, all samples of time zero showed not only a major peak (m/z 194) corresponding to the mother compound (caffeine), but also a variety of others (e.g. m/z 60, 83, 114, 123, 165, 187, 195, 210, 236), indicating fragmentation of the molecule at acidic pH, and/or reaction with water molecules. A summary of the most common fragments/molecules and their m/z ratios, detected wıth high-to-low intensities is presented in Table 6.
Table 6.
The most commonly detected m/z ratios (at r = 7.80 and 8.33 min) showing the likely oxidation byproducts of caffeine. The operational values of H2O2 and TiO2 were 0.5 mg L−1 and 0.1 g L−1, respectively, unless marked with one or two symbols (*). P1-P9 refer to the test processes as defined in Table 2. All samples were initially at pH 4.0.
| m/z | Process |
|---|---|
| 77, 86 | P8 (r = 7.8) |
| 105 | P1, P2, P3, P4, P5, P6, P7, P8, P9 |
| 125 | P0 |
| 130 | P1, P4 (r = 12.8), P7 (r = 10.7), P8 (r = 12.9), P9 (r = 10.7) |
| 142–3 | P3, P7*(r = 12.9) |
| 155 | P8 (r = 12.9) |
| 210–213 | P0, P1, P2, P3, P4, P6, P7, P8 (r = 12.9), P9 |
| 228–229-231 | P1, P2, P3, P4, P5, P6, P7, P8, P9 |
| 252–254 | P4, P6**, P7 |
| 273–275 | P6-P9, (P7**) |
P0 refers to t = 0, P6** and P7*refer to TiO2 concentration of 0.25 g L−1, P7* refers to TiO2 concentrations 0.50 g L−1.
The most probable reaction mechanisms and the corresponding free energies predicted for the samples at t = 0 (no processing) and pH 4 are presented in Fig. 10 (a, b). The reaction products P1 and P2 in the proposed pathways are consistent with the experimentally detected fragments (in samples of t = 0) with m/z ratios of 210 and 84.
Fig. 10.

The reaction pathways and Gibbs free energies (kcal mol−1) of caffeine decomposition in acidic water (pH 4), leading to the formation of P1 (a) and P2 (b).
The first mechanism (a) shows the reaction of protonated caffeine with water leading to the rupture of C1-C14 bond to yield an intermediate P1 (MW = 213) with a high energy barrier (ΔG‡=42.4 kcal mol−1, ΔGrxn = 16.6 kcal mol−1). The reaction of P1 with another molecule of water yields two intermediates with MWs of 98 and 117 (ΔGrxn = 1.5 kcal mol−1). The former is in protonated form (MW = 99) and undergoes demethylation to produce another P1 (ΔG‡=38.2 kcal mol−1, ΔGrxn = 6.5 kcal mol−1) with a high energy barrier, as also reported by Dalmazio et al, for demethylation of caffeine in protonated water [39]. The second mechanism (b) shows the formation of P2 (MW = 125) via the reaction of caffeine with water to yield an intermediate (MW = 140) through bond rupture, followed by demethylation (ΔG‡ =54.5 kcal mol−1, ΔGrxn = 32.9 kcal mol−1). In general, high energy barriers of bond rupture and demethylation reactions are well known and reported in the literature [60], [61].
The most probable reaction mechanisms for the degradation of caffeine by •OH-mediated oxidation are presented in Fig. 11. The first one (a) shows the formation of P3 (MW = 221) and P4 (MW = 228), which were both the experimentally byproducts. Accordingly, caffeine is first attacked by an OH radical at the bridge-side, leading to the formation of P3 (ΔG‡=3.1 kcal/mol, ΔGrxn = −3.3 kcal mol−1), which most likely reacts spontaneously with another •OH via electron-coupling to yield exothermically the product P4 (ΔGrxn = −51.7 kcal mol−1). On the other hand, •OH may also attack the relatively positive carbonyl group of caffeine at C1 to produce P32 with a relatively low barrier (ΔG‡=23.7 kcal mol−1, ΔGrxn = 7.5 kcal mol−1), which reacts readily with a very low barrier (2.1 kcal mol−1) to produce an intermediate with the same MW as P3, referred in the scheme as P33. The compound is also very reactive and is attacked spontaneously by an •OH via electron coupling (ΔGrxn = −95.7 kcal mol−1) to yield the enolic structure P42, which is readily converted to P43 via keto-enol tautomerism (ΔG‡=23.4 kcal mol−1, ΔGrxn = −7.3 kcal mol−1). The reaction of P43 with •OH gives an intermediate with MW 245, which undergoes bond rupture through a highly exothermic reaction (ΔGrxn = −36.4 kcal mol−1) to produce two intermediates with MWs of 120 and 126. The latter is a radical, and reacts barrierlessly with •OH to produce exothermically two intermediates with MWs of 119 and 143 (ΔGrxn = −41.7 kcal mol−1). The former (MW 119) is demethylated via reaction with a water molecule, leading to the formation of P5 (MW = 105), which was one of the most commonly detected byproducts, as it was found in all process effluents. Finally, P5 undergoes reductive decomposition and is converted to P6 (MW = 86), which was also detected experimentally. Reductive cleavage is a likely reaction in all sample solutions, due to the presence of excess air bubbles and nitrogen species, as NOx. Moreover, thermolysis of water results in the formation of •OH as well as •H, which may also promote redox reactions.
Fig. 11.

•OH-mediated reaction pathways and Gibbs free energies (kcal mol−1) for the oxidative degradation of caffeine at pH 4.0 to produce P3, P4 (a), P32, P33, P42, P5 and P6 (b).
The reaction mechanisms proposed in Fig. 11 are in good agreement with the corresponding LCMS analyses of the samples. The intermediate product P1 forms with a reasonable barrier by the reaction of caffeine with water. The product was also detected in all process samples at high intensity, indicating the resistance of the compound to oxidation, most likely due to its unique cyclic structure. The formation of P3 and P4 via reaction with •OH was much easier, as they proceeded with lower energy barriers. Moreover, these products were detected in all samples except those at t = 0 at lower intensities than that of P1, signifying their ease of oxidation by reaction with •OH. More significantly, the intensity of both products was reduced in hybrid operations of AOPs, by which the abundance of OH radicals was increased via additional routes and/or sources of generation.
4. Conclusions
The study described herein has shown that the emerging water contaminant caffeine is easily decomposed by singly applied ultrasonic irradiation (577 kHz) and photolysis with H2O2 at optimized conditions, but with insufficient mineralization. The efficiency of the degradation reactions was significantly improved via hybrid applications, particularly when operated at a heterogeneous mode using commercial TiO2 (P25) as the catalyst. It was found that under equivalent conditions, the percentage of TOC decay increased from 21.7 by single photolysis (UV-H2O2) to 35.4 by US-assisted photolysis. The result was attributed to the formation of excess •OH and H2O2 (via cavitation collapse), and enhanced mixing of the solution, that facilitated the transmission of UV-light. Similarly, mineralization increased from 60% by TiO2-photocatalysis to 66% by US-assisted photocatalysis, owing mostly to the mechanical actions of ultrasound for cleaning and disintegrating of solid particles, thus increasing the number of fresh adsorption sites. Assessment of all hybrid processes for their synergy in TOC decay showed that the best was UV-H2O2/TiO2 with a synergy index of 65 (as opposed to an index of 0.25 in US/UV hybrid). It was also found that further hybridization by adding ultrasound to the above process drastically reduced the synergy to 2.8, mostly due to the presence of excess H2O2 that scavenged some of the OH radicals, and the formation of cavity clouds and high density particle layers that inhibited the transmission of UV light. Finally, the addition of strong OH radical scavengers (as carbonates and humic acid) sto the sample solutions considerably reduced the rate of caffeine degradation, signifying the role of OH radicals in the elimination of the compound. The reaction mechanisms proposed using the computational technique described in the text were consistent with the experimental data, as many of the byproducts predicted were detected by LCMS analysis of the samples.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The research was funded by Boğazici University Research Fund through Project 14862. The authors are grateful to Sedef Kutlu Özkaya at Life Sciences and Technologies Center (Boğazici University) for the LCMS analysis of the samples and for her kind assistance in interpretation of the chromatograms. Computiational resources were partially funded by the National Center for High Performance Computing of Turkey (UheM) with grant number 5005832018.
References
- 1.Załęska-Radziwiłł M., Affek K., Rybak J. Ecotoxicity of chosen pharmaceuticals in relation to micro-organisms-risk assessment. Desalin. Water Treat. 2014;52(19-21):3908–3917. doi: 10.1080/19443994.2014.887503. [DOI] [Google Scholar]
- 2.Bruton T., Alboloushi A., De La Garza B., Kim B.O., Halden R.U. Fate of caffeine in the environment and ecotoxicological considerations. ACS Symp. Ser. 2010 [Google Scholar]
- 3.Gardinali P.R., Zhao X.u. Trace determination of caffeine in surface water samples by liquid chromatography–atmospheric pressure chemical ionization–mass spectrometry (LC-APCI-MS) Environ. Int. 2002;28(6):521–528. doi: 10.1016/s0160-4120(02)00080-6. [DOI] [PubMed] [Google Scholar]
- 4.Sotelo J.L., Ovejero G., Rodríguez A., Álvarez S., Galán J., García J. Competitive adsorption studies of caffeine and diclofenac aqueous solutions by activated carbon. Chem. Eng. J. 2014;240:443–453. [Google Scholar]
- 5.J.L. Sotelo, G. Ovejero, A. Rodríguez, S. Álvarez, J. García, Study of Natural Clay Adsorbent Sepiolite for the Removal of Caffeine from Aqueous Solutions : Batch and Fixed-Bed Column Operation, (2013). DOI:10.1007/s11270-013-1466-8.
- 6.M. Kim, P. Guerra, A. Shah, M. Parsa, M. Alaee, S.A. Smyth, Removal of pharmaceuticals and personal care products in a membrane bioreactor wastewater treatment plant, Water Sci. Technol. 69 (2014). [DOI] [PubMed]
- 7.O. Ganzenko, C. Trellu, S. Papirio, N. Oturan, D. Huguenot, E.D. van Hullebusch, G. Esposito, M.A. Oturan, Bioelectro-Fenton: evaluation of a combined biological—advanced oxidation treatment for pharmaceutical wastewater, Environ. Sci. Pollut. Res. 25 (2018) 20283–20292. DOI:10.1007/s11356-017-8450-6. [DOI] [PubMed]
- 8.S. Miralles-Cuevas, I. Oller, A. Ruíz-Delgado, A. Cabrera-Reina, L. Cornejo-Ponce, S. Malato, EDDS as complexing agent for enhancing solar advanced oxidation processes in natural water: Effect of iron species and different oxidants, J. Hazard. Mater. 372 (2019) 129–136. DOI:10.1016/j.jhazmat.2018.03.018. [DOI] [PubMed]
- 9.Shu Z., Singh A., Klamerth N., McPhedran K., Chelme-Ayala P., Bolton J.R., Belosevic M., El Din M.G. Application of the UV/H 2 O 2 advanced oxidation process for municipal reuse water: bench- and pilot-scale studies. Water Pollut. XIII. 2016;1:233–244. doi: 10.2495/wp160211. [DOI] [Google Scholar]
- 10.Rosal R., Rodríguez A., Perdigón-Melón J.A., Petre A., García-Calvo E., Gómez M.J., Agüera A., Fernández-Alba A.R. Degradation of caffeine and identification of the transformation products generated by ozonation. Chemosphere. 2009;74(6):825–831. doi: 10.1016/j.chemosphere.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Tezcanli-Güyer G., Ince N.H. Individual and combined effects of ultrasound, ozone and UV irradiation: a case study with textile dyes. Ultrasonics. 2004;42:603–609. doi: 10.1016/j.ultras.2004.01.096. [DOI] [PubMed] [Google Scholar]
- 12.Rendel P.M., Rytwo G. Degradation kinetics of caffeine in water by UV/H2O2 and UV/TiO2. Desalin. Water Treat. 2020;173:231–242. doi: 10.5004/dwt.2020.24693. [DOI] [Google Scholar]
- 13.Afonso-Olivares C., Fernández-Rodríguez C., Ojeda-González R.J., Sosa-Ferrera Z., Santana-Rodríguez J.J., Rodríguez J.M.D. Estimation of kinetic parameters and UV doses necessary to remove twenty-three pharmaceuticals from pre-treated urban wastewater by UV/H2O2. J. Photochem. Photobiol. A Chem. 2016;329:130–138. doi: 10.1016/j.jphotochem.2016.06.018. [DOI] [Google Scholar]
- 14.Trovó A.G., Silva T.F.S., Gomes O., Machado A.E.H., Neto W.B., Muller P.S., Daniel D. Degradation of caffeine by photo-Fenton process: optimization of treatment conditions using experimental design. Chemosphere. 2013;90:170–175. doi: 10.1016/j.chemosphere.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 15.Klamerth N., Miranda N., Malato S., Agüera A., Fernández-Alba A.R., Maldonado M.I., Coronado J.M. Degradation of emerging contaminants at low concentrations in MWTPs effluents with mild solar photo-Fenton and TiO2. Catal. Today. 2009;144(1-2):124–130. doi: 10.1016/j.cattod.2009.01.024. [DOI] [Google Scholar]
- 16.M. Carotenuto, G. Lofrano, A. Siciliano, F. Aliberti, M. Guida, TiO2 photocatalytic degradation of caffeine and ecotoxicological assessment of oxidation by-products, Glob. Nest J. 16 (2014) 463–473. DOI:10.30955/gnj.001346.
- 17.N.H. Ince, A. Ziylan, Single and Hybrid Applications of Ultrasound for Decolorization and Degradation of Textile Dye Residuals in Water, in: Sanjay K. Sharma (Ed.), Green Chem. Dye. Remov. from Wastewater, 1st ed., Scrivener Publishing LLC, 2015: pp. 261–293.
- 18.Ince N.H., Tezcanli G., Belen R., Apikyan I. Ultrasound as a catalyzer of aqueous reaction systems: the state of the art and environmental applications. Appl. Catal. B Environ. 2001;29:167–176. [Google Scholar]
- 19.Goncharuk V.V., Malyarenko V.V., Yaremenko V.A. Use of ultrasound in water treatment. J. Water Chem. Technol. 2008;30(3):137–150. doi: 10.3103/S1063455X08030028. [DOI] [Google Scholar]
- 20.Wang J., Wang S. Reactive species in advanced oxidation processes: formation, identification and reaction mechanism. Chem. Eng. J. 2020;401 doi: 10.1016/j.cej.2020.126158. [DOI] [Google Scholar]
- 21.Souza F.S., Féris L.A. Degradation of caffeine by advanced oxidative processes: O3 and o3/UV. Ozone Sci. Eng. 2015;37(4):379–384. doi: 10.1080/01919512.2015.1016572. [DOI] [Google Scholar]
- 22.de Vidales M.J.M., Sáez C., Pérez J.F., Cotillas S., Llanos J., Cañizares P., Rodrigo M.A. Irradiation-assisted electrochemical processes for the removal of persistent organic pollutants from wastewater. J. Appl. Electrochem. 2015;45(7):799–808. doi: 10.1007/s10800-015-0825-0. [DOI] [Google Scholar]
- 23.Ziylan-Yavas A., Mizukoshi Y., Maeda Y., Ince N.H. Supporting of pristine TiO2 with noble metals to enhance the oxidation and mineralization of paracetamol by sonolysis and sonophotolysis. Appl. Catal. B Environ. 2015;172–173:7–17. doi: 10.1016/j.apcatb.2015.02.012. [DOI] [Google Scholar]
- 24.A.K. Nur Fadzeelah, A.Z. Abdullah, N.A. Zubir, A.H. Abd Razak, N.A. Azha, Sonocatalytic degradation of caffeine using CeO2 catalyst: Parametric and reusability studies, J. Phys. Conf. Ser. 1349 (2019). DOI:10.1088/1742-6596/1349/1/012147.
- 25.Huang Y., Wang G., Zhang H., Li G., Fang D., Wang J., Song Y. Hydrothermal-precipitation preparation of CdS@(Er3+:Y3Al5O12/ZrO2) coated composite and sonocatalytic degradation of caffeine. Ultrason. Sonochem. 2017;37:222–234. doi: 10.1016/j.ultsonch.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Kamaya Y., Fukaya Y., Suzuki K. Acute toxicity of benzoic acids to the crustacean Daphnia magna. Chemosphere. 2005;59(2):255–261. doi: 10.1016/j.chemosphere.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Hansch C., Leo A. ACS Professional Reference Book; 1995. Fundamentals and Applications in Chemistry and Biology. [Google Scholar]
- 28.Ziylan-Yavas A., Ince N.H. Single, simultaneous and sequential applications of ultrasonic frequencies for the elimination of ibuprofen in water. Ultrason. Sonochem. 2018;40:17–23. doi: 10.1016/j.ultsonch.2017.01.032. [DOI] [PubMed] [Google Scholar]
- 29.A. Belay, K. Ture, M. Redi, A. Asfaw, Food Chemistry Measurement of caffeine in coffee beans with UV / vis spectrometer, 108 (2008) 310–315. DOI:10.1016/j.foodchem.2007.10.024.
- 30.S.A. Bhawani, S.S. Fong, M. Nasir, M. Ibrahim, Spectrophotometric Analysis of Caffeine, 2015 (2015). [DOI] [PMC free article] [PubMed]
- 31.Frisch M.J., Trucks G.W., Schlegel H.B. Gaussian Inc.; Wallingford CT: 2004. Gaussian 03, Revision C. [Google Scholar]
- 32.Lee C., Yang W., Parr R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 33.Becke A. Density-functional thermochemistry. J. Chem. Phys. 1993;5648–5652 [Google Scholar]
- 34.Becke A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993;98(2):1372–1377. [Google Scholar]
- 35.Onsager L. Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936;58(8):1486–1493. [Google Scholar]
- 36.Ince N.H., Gültekin I., Tezcanli-Güyer G. Sonochemical destruction of nonylphenol: effects of pH and hydroxyl radical scavengers. J. Hazard. Mater. 2009;172(2-3):739–743. doi: 10.1016/j.jhazmat.2009.07.058. [DOI] [PubMed] [Google Scholar]
- 37.R.A. Torres-Palma, E.A. Serna-Galvis, Sonolysis, Elsevier Inc., 2018. 10.1016/B978-0-12-810499-6.00007-3.
- 38.Wakeford C.A., Blackburn R., Lickiss P.D. Effect of ionic strength on the acoustic generation of nitrite, nitrate and hydrogen peroxide. Ultrason. Sonochem. 1999;6(3):141–148. doi: 10.1016/S1350-4177(98)00039-X. [DOI] [Google Scholar]
- 39.Dalmázio I., Santos L.S., Lopes R.P., Eberlin M.N., Augusti R. Advanced oxidation of caffeine in water: on-line and real-time monitoring by electrospray ionization mass spectrometry. Environ. Sci. Technol. 2005;39:5982–5988. doi: 10.1021/es047985v. [DOI] [PubMed] [Google Scholar]
- 40.Gültekin I., Ince N.H. Ultrasonic destruction of bisphenol-A: the operating parameters. Ultrason. Sonochem. 2008;15(4):524–529. doi: 10.1016/j.ultsonch.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Sivasankar T., Moholkar V.S. Physical insight into the sonochemical degradation of 2,4-dichlorophenol. Environ. Technol. 2010;31(14):1483–1494. doi: 10.1080/09593331003777136. [DOI] [PubMed] [Google Scholar]
- 42.Hua I., Hoffmann M.R. Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol. 1997;31(8):2237–2243. doi: 10.1021/es960717f. [DOI] [Google Scholar]
- 43.Méndez-Arriaga F., Torres-Palma R.A., Pétrier C., Esplugas S., Gimenez J., Pulgarin C. Ultrasonic treatment of water contaminated with ibuprofen. Water Res. 2008;42(16):4243–4248. doi: 10.1016/j.watres.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 44.Son Y., Lim M., Ashokkumar M., Khim J. Geometric optimization of sonoreactors for the enhancement of sonochemical activity. J. Phys. Chem. C. 2011;115(10):4096–4103. doi: 10.1021/jp110319y. [DOI] [Google Scholar]
- 45.Muruganandham M. Photochemical oxidation of reactive azo dye with UV–H2O2 process. Dye. Pigment. 2004;62(3):269–275. doi: 10.1016/j.dyepig.2003.12.006. [DOI] [Google Scholar]
- 46.Patidar R., Srivastava V.C. Evaluation of the sono-assisted photolysis method for the mineralization of toxic pollutants. Sep. Purif. Technol. 2021;258 doi: 10.1016/j.seppur.2020.117903. [DOI] [Google Scholar]
- 47.Shimizu N., Ogino C., Dadjour M.F., Murata T. Sonocatalytic degradation of methylene blue with TiO2 pellets in water. Ultrason. Sonochem. 2007;14(2):184–190. doi: 10.1016/j.ultsonch.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Kang J.S., Sohn S.D., Shin H.J., Shin H.J. Dissociative adsorption of H2O2on the TiO2(110) surface for advanced oxidation process. J. Phys. Chem. C. 2020;124:11930–11934. doi: 10.1021/acs.jpcc.0c02143. [DOI] [Google Scholar]
- 49.Harir M., Gaspar A., Kanawati B., Fekete A., Frommberger M., Martens D., Kettrup A., El Azzouzi M., Schmitt-Kopplin P.h. Photocatalytic reactions of imazamox at TiO2, H2O2 and TiO2/H2O2 in water interfaces: Kinetic and photoproducts study. Appl. Catal. B Environ. 2008;84(3-4):524–532. doi: 10.1016/j.apcatb.2008.05.010. [DOI] [Google Scholar]
- 50.Ince N.H. Ultrasound-assisted advanced oxidation processes for water decontamination. Ultrason. Sonochem. 2018;40:97–103. doi: 10.1016/j.ultsonch.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 51.Ziylan-Yavas A., Ince N.H. Enhanced photo-degradation of paracetamol on n-platinum-loaded TiO 2: The effect of ultrasound and OH / hole scavengers. Chemosphere. 2016;162:324–332. doi: 10.1016/j.chemosphere.2016.07.090. [DOI] [PubMed] [Google Scholar]
- 52.Choi Y., Lee D., Hong S., Khan S., Darya B., Lee J.-Y., Chung J., Cho S. Investigation of the synergistic effect of sonolysis and photocatalysis of titanium dioxide for organic dye degradation. Catalysts. 2020;10:500. [Google Scholar]
- 53.Son Y., Lim M., Khim J., Ashokkumar M. Attenuation of UV light in large-scale sonophotocatalytic reactors: the effects of ultrasound irradiation and TiO 2 concentration. Ind. Eng. Chem. Res. 2012;51:232–239. doi: 10.1021/ie202401z. [DOI] [Google Scholar]
- 54.Adewuyi Y. Sonochemistry: environmental science and engineering applications. Ind. Eng. Chem. Res. 2001;40:4681–4715. doi: 10.1021/ie010096l. [DOI] [Google Scholar]
- 55.Anandan S., Ashokkumar M. Sonochemical synthesis of Au-TiO2 nanoparticles for the sonophotocatalytic degradation of organic pollutants in aqueous environment. Ultrason. Sonochem. 2009;16:316–320. doi: 10.1016/j.ultsonch.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 56.Eren Z., Ince N.H. Sonolytic and sonocatalytic degradation of azo dyes by low and high frequency ultrasound. J. Hazard. Mater. 2010;177(1-3):1019–1024. doi: 10.1016/j.jhazmat.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 57.Gultekin I., Ince N.H. Degradation of reactive azo dyes by UV/H 2 O 2: impact of radical scavengers. J. Environ. Sci. Heal. Part A. 2004;39:1069–1081. doi: 10.1081/ESE-120028414. [DOI] [PubMed] [Google Scholar]
- 58.Wang G., Hsieh S., Hong C. Destruction of humic acid in water by UV lightðcatalyzed oxidation with hydrogen peroxide. Water Res. 2000;34:3882–3887. [Google Scholar]
- 59.Yang J.-K., Lee S.-M. Removal of Cr(VI) and humic acid by using TiO2 photocatalysis. Chemosphere. 2006;63(10):1677–1684. doi: 10.1016/j.chemosphere.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 60.Arslan E., Hekimoglu B.S., Cinar S.A., Ince N., Aviyente V. Hydroxyl radical-mediated degradation of salicylic acid and methyl paraben: an experimental and computational approach to assess the reaction mechanisms. Environ. Sci. Pollut. Res. 2019;26:33125–33134. doi: 10.1007/s11356-019-06048-3. [DOI] [PubMed] [Google Scholar]
- 61.Okovytyy S., Kholod Y., Qasim M., Fredrickson H., Leszczynski J. The mechanism of unimolecular decomposition of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane. A computational DFT study. J. Phys. Chem. A. 2005;109(12):2964–2970. doi: 10.1021/jp045292v. [DOI] [PubMed] [Google Scholar]