

Abstract
A well-hydrated counterion can selectively and dramatically increase retention of a charged analyte in hydrophilic interaction chromatography (HILIC). The effect is enhanced if the column is charged, as in electrostatic repulsion-hydrophilic interaction chromatography (ERLIC). This combination was exploited in proteomics for the isolation of peptides with certain post-translational modifications (PTMs). The best salt additive examined was magnesium trifluoroacetate. The well-hydrated Mg+2 ion promoted retention of peptides with functional groups that retained negative charge at low pH, while the poorly-hydrated trifluoroacetate counterion tuned down the retention due to the basic residues. The result was an enhancement in selectivity ranging from 6- to 66-fold. These conditions were applied to a tryptic digest of mouse cortex. Gradient elution produced fractions enriched in peptides with phosphate, mannose-6-phosphate, N- and O-linked glycans. The numbers of such peptides identified either equaled or exceeded the numbers afforded by the best alternative methods. This method is a productive and convenient way to isolate peptides simultaneously that contain a number of different PTMs, facilitating study of proteins with “crosstalk” modifications.
The fractions from the ERLIC column were desalted prior to C-18-reversed phase (RP) LC-MS/MS analysis. Between 47-100% of the peptides with more than one phosphate or sialyl- residue or with a mannose-6 phosphate group were not retained by a C-18 cartridge but were retained by a cartridge of porous graphitic carbon. This finding implies that the abundance of such peptides may have been significantly underestimated in some past studies.
Keywords: electrostatic repulsion-hydrophilic interaction chromatography (ERLIC), post-translational modification (PTM) analysis, phosphopeptide, glycopeptide, PTM crosstalk, counterion
Graphical Abstract
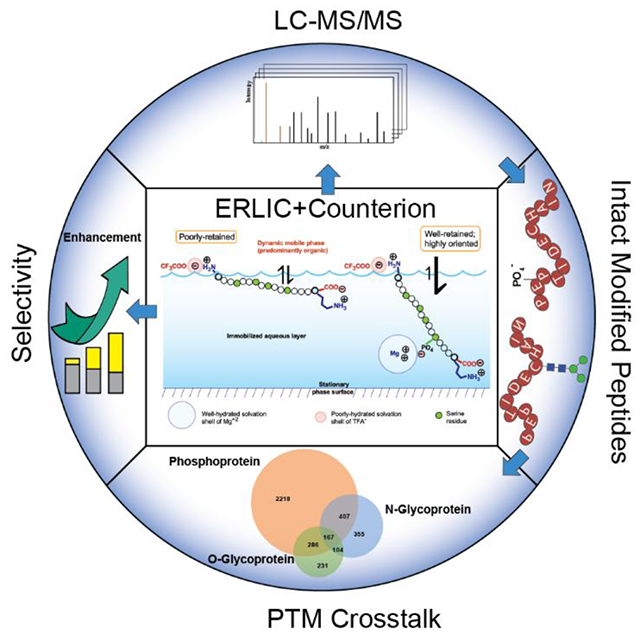
INTRODUCTION
Advances in bottom-up proteomics depend on the identification of peptides with a variety of post-translational modifications (PTMs). Usually, an affinity chromatography method is used to isolate peptides with one type of PTM. Titanium dioxide or immobilized metal affinity chromatography (IMAC) is used for phosphopeptides, sometimes sequentially,1,2 and can also be used for sialylated and phosphorylated glycopeptides.3,4 Glycopeptides can be isolated using lectins or affinity materials based on boronic acids or esters.5 These affinity methods all have shortcomings. Lectins and boronates only retain subsets of glycans. Titanium dioxide and IMAC also have difficulty either with retention or elution of all phosphopeptides and glycopeptides (especially regarding titanium dioxide and multiphosphorylated peptides). General-purpose methods tend to lack the selectivity necessary for effective identification of modified peptides of low abundance. Examples are attempts to isolate tryptic phosphopeptides using either conventional anion-exchange chromatography6 or hydrophilic interaction chromatography (HILIC).7 Neither method is selective enough to offer a satisfactory enrichment factor for phosphopeptides. An advance was introduced by Ding et al.8 by using trifluoroacetic acid (TFA) as an ion pairing agent in HILIC. Basic residues are normally the most hydrophilic residues in peptides.9 TFA forms relatively hydrophobic ion pairs tenaciously with basic residues, which tunes down their contribution to retention in HILIC. The result is to render more important the contribution of the rest of the peptide to retention. In Ding’s case this involved the glycan portion of glycopeptides. While the selectivity was appreciably lower than that associated with an affinity material, it was still good enough to produce a clean separation of glycopeptides from non-glycopeptides on a neutral HILIC column. The weakness of the affinity for the glycan portion is actually an advantage; since the affinity interaction does not dominate the chromatography, then there is a wide dynamic range in the selectivity window. A HILIC column can separate glycopeptides with high resolution.10
The electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) variation of HILIC was introduced in 2008.11 In the case of peptide separation, this usually involves an anion-exchange column being operated under HILIC conditions at a pH low enough to uncharge the carboxyl-residues of Asp- and Glu- residues. Peptides then have a net positive charge and are repelled by the stationary phase electrostatically. With sufficient organic solvent the hydrophilic interaction can balance the repulsion, causing the peptides to elute in or near the void volume. An exception occurs with peptides containing groups that retain some negative charge at the low pH. This includes phosphate groups (pKa ~ 2.1), sialyl- groups (pKa ~ 2.6) and isoaspartyl-groups (pKa ~ 3.1) resulting from deamidation of Asn-. These groups confer electrostatic attraction in addition to the hydrophilic interaction, and peptides containing these groups can be pulled away from the rest of the peptides in a complex mixture, such as a digest. ERLIC being a variant of HILIC, peptides that lack PTMs but that are particularly polar will also be well retained. This applies as well for nonsialylated glycopeptides. While ERLIC was initially used for selective isolation of phosphopeptides, the well-retained fractions were soon found to be enriched in both phosphopeptides and glycopeptides12-15. Both glycosylation and phosphorylation of proteins have been implicated in various human diseases, including diabetes, Alzheimer’s disease, autoimmunity and cancer. There is complex crosstalk between the two PTMs, including competitive or noncompetitive occupancy of the same or proximal sites, glycosylation of kinases and phosphorylation of glycosylation-related enzymes. The simultaneous identification of peptides with different PTMs provides an opportunity to identify those crosstalk interactions, in which toggling between two (or more) modifications on nearby residues controls physiological responses3,16,17. Other PTMs have been implicated in crosstalk as well, including acetylation, methylation, and SUMOylation18,19.
A recent study20 has demonstrated that the hydration of counterions plays a key role in the retention of charged analytes in HILIC. This permits manipulation of retention and selectivity by the choice of the salt used in the mobile phase. The effect is even more extreme in ERLIC. Retention and selectivity for peptides with negatively charged groups, such as phosphopeptides, increases severalfold when Na+ or NH4+ is replaced by the well-hydrated Mg+2 in ERLIC. In the present study, this increase in retention of peptides with negatively charged PTMs is combined with the use of the poorly-hydrated trifluoroacetate anion to tune down retention due to the rest of the peptide, as per Ding et al. The result is an unprecedented increase in selectivity for peptides with a number of important PTMs, to a degree that compares favorably with or exceeds the enrichment factors conferred by other methods in the literature, providing a convenient tool for PTM crosstalk analysis.
EXPERIMENTAL
Offline ERLIC fractionation:
Sample preparation followed previously described protocols with moderate adjustments14. The details of chromatography materials, mobile phase preparation, cortex tryptic digest preparation, and LC-MS/MS analysis are provided in Supporting Information.
1 mg mouse cortex tryptic digest was fractionated by either a PolyWAX LP® or PolySAX LP™ column (200 mm × 2.1 mm, 5 μm, 300 Å; PolyLC, Columbia, MD). Three sets of binary mobile phase combinations were used in a total of six fractionation experiments: (1) MP A1: 20 mM sodium methylphosphonate with 80% ACN, pH = 2.5; MP B1: 300 mM triethylammonium phosphate with 10% ACN, pH=2.5; (2) MP A2: 20 mM magnesium trifluoroacetate with 80% ACN, pH = 2.5; MP B2: 300 mM triethylammonium trifluoroacetate with 10% ACN, pH = 2.5; (3) MP A3: 20 mM ethylenediamine trifluoroacetate with 80% ACN, pH = 2.5; MP B3: 300 mM ammonium trifluoroacetate, 10% ACN, pH = 2.5. The gradient was as follows: 0-5 min, 0% B; 5-48 min, 0-100% B; 48-53 min, 100% B. The column was re-equilibrated at 100% A for 15 min between runs. Eluent was collected with a FC-4 fraction collector (Rainin Dynamax) at 1-min intervals as initial fractions. Between 6-30 min (MP1) and 7-30 min (MP2 and MP3), adjacent 2-min eluent were pooled into the first 12 fractions. Eluent between 30-42 min and 42-53 min was pooled into the last 2 fractions 13 and 14. Each fraction was dried under vacuum.
Solid phase extraction/desalting:
Every peptide fraction from the 6 experiments were desalted with OMIX C18 tips (Agilent, Santa Clara, CA). For the PTM crosstalk analysis using mobile phase combination 2, C18 SPE flow-through was collected and run through an additional HyperCarb SPE (TopTips; PolyLC) procedure per the manufacturer’s instructions. Eluates from SPE were dried and stored at −80 °C prior to mass spectrometry analysis.
Data analysis:
Raw files were processed with the Byonic search engine (Protein Metrics Inc, San Carlos, CA) embedded within Proteome Discoverer 2.1 (Thermo Fisher Scientific, San Jose, CA). Spectra were searched against the SwissProt Mus musculus proteome database (August 13, 2016; 24903 entries). Trypsin digestion missed cleavages was set at < 3. The parent mass error tolerance was 10 ppm and fragment mass tolerance was 0.01 Da. Fixed modifications were specified as carbamidomethylation (+57.02146 Da) on C residues. Dynamic modifications included oxidation of M (+15.99492 Da, rare1), deamidation (+0.984016 Da, rare1) of N or Q, N-glycosylation (common1), O-glycosylation (common1), and phosphorylation (common4). Glycan modifications were specified as Byonic embedded mammalian N-glycan database (309 entries) expanded with typical mannose-6-phosphate glycans including HexNAc (2) Hex (4-9) Phospho (1-2) modification, HexNAc (3-4) Hex (4-9) Phospho (1-2), HexNAc (2) Hex (3-4) Phospho (1) and HexNAc (3) Hex (3-4) Phospho (1)). Identifications were filtered to 1% protein false discovery rate (FDR). Typical mannose-6-phosphate glycans including HexNAc (2) Hex (4-9) Phospho (1-2) modification, HexNAc (3-4) Hex (4-9) Phospho (1-2), HexNAc (2) Hex (3-4) Phospho (1) and HexNAc (3) Hex (3-4) Phospho (1)) and O-glycosylation (Byonic embedded mammalian O-glycan database with 89 O-glycans) were set as common dynamic modifications and searched separately.
Confident crosstalk interaction analysis relies on confident identification of PTM sites. To minimize false discoveries of modified peptides, post-Byonic search filtering was applied with in-house written scripts. Briefly, identifications were filtered to 1% PSM FDR. For method comparison, a Byonic score cut-off >100 was used. More stringent filtering of N-glycopeptides and phosphopeptides used a Byonic score cut-off >300, delta Byonic score>50, ∣log10 Prob∣>4 (the absolute value of the log10 posterior error probability). For the glycan-protein network, N-glycopeptides were exclusively categorized into six glycan type categories based on glycan composition: 1) mannose-6-phosphate (containing phosphate on glycan), 2) sialic acid (containing NeuAc/NeuGc), 3) fucose (containing Fucose), 4) complex/hybrid (>2 HexNAc), 5) high-mannose (2 HexNAc and >5 Hex), 6) paucimannose (2 HexNAc and <5 Hex). Figures were created in R 3.6.0 using the ggplot2, igraph, networkD3 and ggnetwork library.
RESULTS
Method Development with Peptide Standards
Fig. S1 depicts the partitioning of a tryptic phosphopeptide and an unmodified tryptic peptide into the semi-immobilized aqueous layer in HILIC. A well-hydrated cation, such as Mg+2, promotes retention of peptides through negatively charged counterions such as phosphate. A well-hydrated anion - here, methylphosphonate – does the same through the positively charged residues. Fig. S2 depicts the situation when a poorly hydrated anion such as trifluoroacetate is used in place of methylphosphonate. While the phosphopeptide remains well-retained via the association with the Mg+2 ion, no counterion confers similar retention upon the unmodified peptide. Fig. 1A compares retention of synthetic peptides, with sequences similar to peptides resulting from digestion with trypsin, using various salt additives in the ERLIC mode. Substitution of magnesium trifluoroacetate for magnesium methylphosphonate results in a modest decrease in retention of the singly phosphorylated peptide but a much more dramatic decrease in retention of the unmodified peptide. The result is an increase in the selectivity ratio from 3.5:1 to 13:1 (the ratio of retention of a phosphopeptide:retention of the corresponding peptide with an Asp residue in place of the pSer). Ethylenediamine is perhaps the most hydrophilic volatile amine,21 and so it was substituted for Mg+2 to implement the separation with a volatile mobile phase. The result was a decrease in the selectivity ratio to 9:1. Subsequent study determined that ethylenediammonium trifluoroacetate was insufficiently volatile for use in mass spectrometry. In retrospect this is not surprising, since triethylammonium trifluoroacetate is an ionic liquid.22
Fig. 1.

(A) Effect of counterions on ERLIC of phosphopeptides. Column: PolyWAX LP, 100x4.6-mm; 3-μm, 300-Å. Mobile phase: 20 mM salt as noted, pH 2.5, with 75% ACN. Flow rate: 1 ml/min. Detection: 280 nm. The ratio refers to retention of the last phosphopeptide to retention of the Asp-peptide. (B) HILIC vs. ERLIC of phosphopeptides. HILIC column: PolyHYDROXYETHYL A, 100x4.6-mm; 3-μm, 300-Å. Salt: EDA-TFA (concentration as noted). All other variables as in (A).
In two chromatograms in Fig. 1A, positional isomers were separated that had the phosphate group on different serine residues [the order of elution was the same in both runs]. With three of the isomers, the further the phosphate group was from the C-terminus, the earlier the elution. This is consistent with an earlier study23 that showed that tryptic peptides tend to be oriented on an anion-exchange material with the C-terminus facing the stationary phase and the N-terminus facing away. Thus, the more remote the location of a phosphate group from the C-terminus, the less it can interact with the stationary phase, reducing its contribution to retention. The least well-retained positional isomer is an exception to this trend. In this case, the phosphoserine followed a proline residue in the primary sequence and was probably sequestered by it.
Fig. 1B compares the separation of these peptide standards by both ERLIC and HILIC. ERLIC clearly affords better retention and selectivity of phosphopeptides than HILIC. It is also more sensitive to positional isomers, since HILIC does not feature the high degree of orientation of peptides that can be conferred by their interaction with a charged surface.
Complex tryptic digests contain a significant number of peptides with three or more Asp- and Glu- residues. Phosphopeptides are of much lower abundance. They must be physically separated from the peptides with three or more ordinary acidic residues, which would otherwise tend to suppress their ionization in mass spectrometry.6 Fig. 2 shows that the conditions being introduced here have the necessary selectivity. Even the least well-retained singly phosphorylated peptide standard is separated from an analogous peptide in which the four serine residues have all been replaced by Asp- residues. This comfortable separation of singly phosphorylated peptides from unmodified acidic peptides helps to address potential concern about the utility of ERLIC24. A gradient to 1% unbuffered TFA and 30% ACN tunes down both the electrostatic attraction and hydrophilic interaction and permits the elution of peptides with 2, 3 or 4 phosphate residues.
Fig. 2.

ERLIC of phosphopeptides and acidic analogues. Column, flow rate and detection: As in Fig. 1A. Mobile phase A: 20 mM EDA-TFA, pH 2.1, with 77.5% ACN. Mobile phase B: 1% TFA with 30% ACN. Gradient: 12’ delay, then 0-100% B in 30’.
The excellent selectivity for phosphopeptides enabled by ERLIC motivated us to compare it with the selectivity conferred by affinity materials. Solid-phase extraction (SPE) cartridges of titanium dioxide are frequently used for isolation of phosphopeptides from complex digests. We packed an HPLC column with titanium dioxide and evaluated it with gradient elution of our peptide standards. The results are shown in Fig. S3. A peptide standard with no acidic residues was not retained. Addition of a single Asp- residue resulted in a modest degree of retention with elution in a broad peak, since carboxyl- groups are weak Lewis bases and so interact weakly with titanium dioxide. The interaction of titanium dioxide with phosphate groups is much stronger since they are strong Lewis bases. It was necessary to run a gradient to ammonium hydroxide to elute any phosphopeptides. Both singly phosphorylated isomeric peptide standards then coeluted, along with the peptide standards with two, three or four Asp- residues. With continuation of the gradient, a peptide standard with two phosphate groups was resolved from the other acidic peptides. The interaction of titanium dioxide with Lewis bases is so strong that it blinds the material to any other aspect of the peptide’s composition. These results are consistent with those from the only paper we could find in the literature where a titanium dioxide HPLC column was used with a similar gradient for peptides25. Peptides eluted in very wide peaks, and the best-retained peptides were difficult to elute at all. This accounts to some extent for the “lossy” nature of titanium dioxide regarding multiply phosphorylated peptides, cf. Fig. 2 in ref. 13. Titanium dioxide has also been shown to be inferior to HILIC for isolation and identification of N-glycopeptides26,27. Phosphopeptides eluted more cleanly from an HPLC column packed with an Fe-IMAC material, but all in a single sharp peak within the gradient25.
Application of ERLIC Fractionation to the Mouse Cortex Phospho- and Glycoproteome
Proteins were extracted from mouse cortex and digested with trypsin, per Experimental. The digest was fractionated via ERLIC using gradients with three alternative mobile phase (MP) combinations:
MP 1: Sodium methylphosphonate to triethylammonium (TEA) phosphate (TEAP).
MP 2: Magnesium trifluoroacetate (Mg-TFA) to TEA-TFA.
MP 3: Ethylenediammonium-TFA (EDA-TFA) to ammonium-TFA.
MP #1 was introduced in ref. 11 and has since been widely used for the isolation of phospho- and glycopeptides. In ref. 20 it was demonstrated that substitution of the Na+ ion by the better-hydrated Mg+2 ion as a counterion to phosphate groups resulted in much better retention of phosphopeptides in HILIC or ERLIC, while substitution with the non-hydrated tetramethylammonium ion resulted in a significant decrease in retention of phosphopeptides. This informed the composition of MP 2, where combining Mg+2 and trifluoroacetate maximizes ERLIC selectivity for phospho- and glycopeptides over nonmodified counterparts. MP 3 was an attempt to develop a totally volatile mobile phase that would permit direct ERLIC-MS analysis of peptides. In addition to the salt gradients, all three MP’s featured a decreasing gradient of acetonitrile in order to tune down hydrophilic interaction.
Fig. S4 compares the chromatograms obtained from 1 mg of mouse cortex digest with the three MP’s. Ideally, in ERLIC the unmodified peptides elute in or near the void volume while the modified peptides are well-retained and elute during the gradient. The profile associated with MP 2 appears to be the most promising in that regard. This seems to be substantiated in Fig. 3, which compares the number of unmodified peptides identified using all three MP’s with both the PolyWAX LP and PolySAX LP columns. With PolyWAX LP, the fewest identifications of unmodified tryptic peptides were obtained using MP 2. If most of the unmodified peptides do elute in the same early fractions, then they would interfere with each other’s ionization at the MS stage, reducing identifications.
Fig. 3.

Peptide identification from 1 mg of mouse cortex after ERLIC fractionation and LC-MS/MS. Number of (A) total peptides; (B) mannose-6-phosphate glycopeptides; (C) phosphopeptides; (D) N-glycopeptides; (E) O-glycopeptides. Only fractions desalted with C-18 were analyzed, and redundant identifications were eliminated (i.e., if a peptide eluted in two adjacent fractions, then it was counted only once). The Byonic cut-off filter was 100.
Fig. 3 also compares the two column materials and three mobile phase combinations with peptides with PTMs. With MP 2 or 3, 5-6x more N- and O-linked glycopeptides were identified than with the MP 1 that has routinely been used with ERLIC. With phosphorylated peptides a similar 6-fold increase in identifications was obtained when comparing MP2 to MP1 but not MP3. For peptides with mannose-6-PO4 (M-6-P) groups, the number of identifications using MP 2 was 66-fold greater than with MP 1, a remarkable increase. In most cases PolyWAX LP outperformed PolySAX LP. In view of this data, all further work was performed using a PolyWAX LP column and MP 2. Redundant identifications of the same peptide in more than one fraction were deleted from the totals unless stated otherwise.
Fig. S5B shows the distribution of modified and unmodified peptides among the 14 fractions collected during the gradient with the PolyWAX LP column. The tallies of peptides of a given type from all 14 segments are numbers larger than those in Fig. 3. This is because the numbers are the combined totals of the C-18 desalted and PGC-desalted peptide sets. Peptides identified in both sets were only counted once. The uniform distribution of peptides among the fractions in Fig. S5B is an advantage of ERLIC12. Fig. 4 focuses on the number of peptides with a given number of phosphate or sialyl- residues, or a M-6-P group, in each fraction. Peptides with more phosphate and sialyl- residues tend to elute later, as expected in HILIC in general. Also, this being a variant of HILIC, then in addition to peptides with PTMs, the later fractions also contain a number of nonmodified peptides with numerous polar residues. These include SGGNGGGGGGGGGGGGGYGGSGGGGGGAGVPSEGAAK, SAAGSEKEEEPEEEEEEEEEYDEEEEEEDDDRPPK, and other extreme sequences.
Fig. 4.

Distribution of modified peptide identifications among ERLIC fractions using MP2. Results from fractions desalted with C18 and with PGC are graphed separately except for M-6-P peptides. Redundant identifications were eliminated within each materials’ dataset but not between the C-18 and PGC datasets, since the characteristic of interest was the overall elution pattern rather than the absolute numbers. The Byonic cut-off filter was 100. The figures for each segment are listed in Tables S1-S4.
The Virtues of Porous Graphitic Carbon (PGC)
PGC retains particularly small or polar peptides that are poorly retained by C-18 material28. It has also been shown to retain some tryptic phosphopeptides that are not retained by C-18 material29. Accordingly, when the ERLIC fractions were individually desalted with C-18 cartridges, the filtrates were also collected and passed through PGC cartridges. This resulted in a significant increase in peptide identifications. Fig. 4 shows the number and distribution of peptides with various modifications among the 14 fractions, with peptides identified from the C-18-desalted fractions tallied in separate graphs from peptides identified from PGC-desalted fractions. Peptides with M-6-P groups are an exception, being compared in the same graph in stacked segments. Surprisingly, almost half of the peptides with this PTM were not retained by C-18 and so were identified from the PGC-desalted fractions. Most of these peptides eluted in late fractions in ERLIC. This suggests that they were unusually polar, which could account for the lack of retention by C-18 material. Similarly, peptides with > 1 phosphate and > 1 sialyl- residue tended to elute in later fractions, with about half retained only by PGC. The relative distribution is show in more detail in Fig. S5A. This is consistent with ref. 30, which showed separation of sialic acid isomers using PGC-LC-MS.
In a search for properties that might account for some of these trends, the average number of residues per peptide was graphed for each ERLIC fraction. An interesting trend of note (Fig. S6) was that peptides retained only by PGC tended to be 3-6 residues shorter than peptides that were retained from the same fraction by C-18.
PTM Crosstalk
Since the salt-enhanced ERLIC method described here enables simultaneous detection of peptides with four different types of PTMs with good coverage, then one can search for crosstalk between different modifications. MP2 provides the best performance in terms of PTM selectivity and coverage, and PGC enrichment complements C-18 for PTM identification. The crosstalk analysis is based on data acquired with ERLIC fractionation using MP2, followed by post-fractionation desalting and enrichment with both C-18 and PGC material. With this method and with a Byonic score cut-off of 100, we have identified 12,492 non-redundant phosphopeptides, 10,218 phosphorylation sites in 3078 proteins, 3819 N-glycopeptides, 1246 N-glycosylation sites in 788 proteins, 2730 O-glycopeptides, and 1979 O-glycosylation sites in 1033 proteins. M6P peptides PSMs were manually inspected and spectra without phospho-oxonium ions were filtered, after which 145 unique M6P peptides were identified. Fig. S11A shows the distribution of phosphor-oxonium ion containing M6P peptide PSMs. In Fig. S7, 46% of the phosphorylation sites identified have been reported in the Uniprot database while the rest are novel. 50% of the N-glycosylation sites identified here have been annotated in Uniprot. However, few of the O-glycosylation sites are listed in Uniprot; 99% of them are novel. Among the 4697 Uniprot-annotated phosphosites, most (67%) were assigned using “combinatorial evidence,” which comes from large-scale proteomics data or protein 3D structure data without manual curation; 32% was assigned using “sequence similarity,” which means the evidence is propagated from related proteins with similar sequences. For 627 annotated N-glycosites, 77% were only assigned via “sequence analysis”, which means the glycosites were predicted from the presence of the N-X-S/T sequon rather than by experimental observation. This study has provided substantiating experimental evidence for those annotated sites, as well as the novel PTM sites not annotated in Uniprot. We present one example: Neural cell adhesion molecule 1. Fig. 5A displays the complex PTM profile of this protein, depicting the PTMs this study identified in the protein as well as the ones already listed in Uniprot. Compared with the PTM sites reported in Uniprot,
Fig. 5.

(A) PTMs in Neural Cell Adhesion Molecule 1 (Uniprot identifier P13595) [Gene: Ncam1]. A Byonic filter score of 300 was used. (B) Venn diagram showing co-existence of phosphorylation, N-glycosylation and O-glycosylation at the protein level; (C) Heatmap of the correlation of number of PTM sites and number of different glycans by protein. Only co-modified proteins were included (for example, row 5, column 3 means the correlation between number of N- and O-glycosites for proteins that are both N- and O-glycosylated is 0.36). (D)(E) Scatterplots of number of modified sites within proteins that contain the two PTMs. The accessions of hypermodified proteins are labeled.
Four out of six reported N-glycosylation sites were detected here;
Seven out of eleven reported phosphorylation sites were detected here (S890, T929, T1001, and T1030 were not detected).
The following modifications were also detected that have not been reported in Uniprot:
An O-glycosylation site;
Eight additional phosphorylation sites (S788, S884, S885, S886, S950, S952, S1016, and S1113);
A novel mannose-6-phosphate site.
With such extensive PTM data available at the proteome level from the same sample, this method will be valuable in dissecting PTM characteristics and their interplay efficiently. Site-specific microheterogeneity is an important aspect of glycosylation and significantly complicates the analysis of glycoproteins.31,32 This information is lost when glycans and deglycosylated peptides are analyzed separately but is retained in the present method since the glycopeptides are analyzed intact. For example, 384 proteins identified here as O-glycosylated contained more than one O-glycosite on the protein, with 331 O-glycosites modified by two or more alternative glycans. Similarly, 233 N-glycoproteins were ascertained to have more than one N-glycosite, with 592 N-glycosites being modified with two or more alternative glycans. According to the simple linear regression model per Fig. S8A, N-glycosylation has a higher degree of microheterogeneity than O-glycosylation, with an average of three alternative N-glycans per site. Recently, Riley et al. reported a visualization method for large-scale proteomics data.24 Here, an N-glycan-protein network is plotted in Fig. S8B, which maps different types of glycans attached to identified glycoproteins. Sialylated glycans comprise the largest fraction of the N-glycans identified. The network also shows that sialylated, fucosylated, and complex/hybrid glycans occur more frequently on proteins with multiple glycosylation sites, while high-mannose, paucimannose and M-6-P glycans are more uniformly distributed. Fig. 5B plots the overlap of protein substrates of phosphorylation and glycosylation. 574 proteins contain both phosphorylation and N-glycosylation sites, 453 proteins contain phosphorylation and O-glycosylation, 271 proteins contain both N- and O-glycosylation, and 167 proteins contain all three PTMs. Among the co-modified proteins, there is positive correlation between phosphorylation and O-glycosylation sites and between N- and O-glycosylation sites per protein. However, such correlation was not observed between phosphorylation and N-glycosylation, as in the heatmap of Fig. 5C. Figs. 5D and 5E visualize the site numbers of different PTMs on the same protein directly, with some hypermodified proteins’ accessions labeled.
PTMs are known to alter protein functions. When there are multiple PTMs on the same protein, then their interplay can be quite complex. Crosstalk between O-glycosylation and phosphorylation has been reported, since they both target protein S/T residues.17,33 In general, Wang et al. described four potential categories of crosstalk between O-GlcNAcylation and phosphorylation.34 Canvassing the present data for these four categories (Fig. 6):
Fig. 6.

Sankey diagram of PTM crosstalk network at the modification site-level and protein level. Numbers in black represent node sizes, numbers in pink represent link sizes. The complete sizes (numbers) of nodes and links are summarized in Tables S5-S6. Abbreviations include PhosP (phosphoprotein), OGP (O-glycoprotein), NGP (N-glycoprotein).
Direct competition for occupancy of a single site. In the present study, 154 O-glycosylation sites were also found to be phosphorylated;
Competition via steric hindrance by reciprocal modification at proximal sites in a polypeptide. Considering sites within ± 10 amino acid residues as proximal, then in this study 4728 (46%) phosphorylation sites have proximal phosphorylation, 123 (1.2%) have proximal N-glycosylation, and 331 (3.2%) have proximal O-glycosylation. Similarly, 73 (5.9%) N-glycosylation sites have proximal phosphorylation, 55 (4.4%) have proximal N-glycosylation, and 200 (16%) have proximal O-glycosylation. For O-glycosylation sites the figures are 248 (13%), 241 (12%), and 509 (26%) for proximal phosphorylation, N-glycosylation, and O-glycosylation, respectively;
Regulation of O-glycosylation cycling enzymes by phosphorylation of their catalytic subunits or of their regulatory/targeting subunits;
Regulation of kinases or phosphatases by GlcNAcylation.
Regarding categories (3) and (4), by querying the present data for gene ontology molecular function annotation, 226 phosphoproteins, 94 O-glycosylated proteins, and 52 N-glycosylated proteins were annotated with phosphatase activity, and 223 phosphoproteins, 64 O-glycosylated proteins, and 44 N-glycosylated proteins had kinase activity. This suggests that phosphorylation cycling enzymes can be self-phosphorylated or glycosylated. Conversely, 164 phosphoproteins, 57 O-glycosylated proteins, and 35 N-glycosylated proteins were involved in protein glycosylation processes.
Aberrant protein phosphorylation and glycosylation have been associated with neurodegenerative diseases.35,36 Interestingly, 43 phosphoproteins, 29 O-glycosylated proteins, and 15 N-glycosylated proteins in the present data were annotated as related to Alzheimer’s disease (AD) by the KEGG database (Supporting Table S1). Those key modified proteins are potential targets for studying the role of PTM regulation in AD progression.
The Cutoff Score in Byonic
It has been suggested that the cutoff score for the Byonic filter be set at 300 for nonmodified peptides and in the range 50-200 for N-glycopeptides as a compromise between accuracy and coverage.37 A cutoff score of 150 was used in a recent study of the N-glycoproteome.24 In the present study, the data in Figs. 5 and S6 was obtained using the stringent cutoff score of 300, while a cutoff score of 100 was used in Figs. 3, 4, and S5. In order to assess the impact of the stringency of the cutoff filter score, Fig. S9 shows the number of peptides identified using Byonic cutoff scores of 0, 100, and 300. When a specific peptide was identified in more than one fraction (“redundancy”), then it was counted only once. A peptide was also counted only once if it was identified in oxidized or deamidated forms in addition to the original form. However, glycosylation variants of a peptide were counted individually. A change in the level of stringency tended to have the greatest effect on identification of peptides with the greatest number of modifications. Such peptides tend to be of low abundance and may also ionize less well compared to unmodified peptides. The impact was greatest on O-glycopeptides; identification of the nonsialylated peptides with Byonic scores > 300 was only 14% of the unfiltered identifications.
DISCUSSION
Attachment of phosphate and sialyl- residues increases the polarity of peptides. PGC was more effective than a conventional C-18 material at capturing peptides decorated with more than one such residue. This raises the disturbing possibility that prior studies of peptides with these PTMs may have significantly underestimated the abundance of peptides with > 1 phosphate residue, > 1 sialyl residue, or any mannose-6-phosphate residues if they involved a desalting step with C-18 material only. Supplementary Table 1 from ref. 25 presents a relevant example. A tryptic digest of human epidermoid cells was desalted on a C-18 cartridge prior to affinity isolation of phosphopeptides and subsequent fractionation. Out of 5009 phosphopeptides identified, only ten were triphosphopeptides. With our method, one might expect to identify 75-90 triphosphopeptides from brain tissue extract from a phosphopeptide set of that size.
If PGC complements the selectivity of C-18 material for binding peptides, then why not mix both materials together in a single SPE desalting step? The answer is that while PGC binds many polar peptides that do not bind well to C-18 material, it also binds many of the peptides that do bind to C-18 material, and its affinity for such peptides may be greater than for the peptides with PTMs. That would saturate the binding capacity of the PGC, leaving it unable to bind the peptides that do not bind to C-18 material. It is prudent to first deplete the sample of peptides that C-18 does bind. A similar situation was described in ref. 1. There, a single IMAC cartridge bound mostly doubly phosphorylated peptides. The filtrate was then passed through an identical cartridge, and this time the singly phosphorylated peptides were bound since there were no multiply phosphorylated peptides to displace them.
CONCLUSIONS
The number of modified peptides identified in this study compares favorably with or exceeds the number reported using other methods. No specialized materials are required. This optimized version of ERLIC should be considered as a viable alternative to affinity enrichment methods involving titanium dioxide, IMAC, lectins, or boronic acids. Since peptides with different modifications can be enriched and identified simultaneously, this approach facilitates study of PTM crosstalk and protein network interactions that are not convenient or possible to perform using the materials with higher degrees of affinity.
Supplementary Material
ACKNOWLEDGEMENTS
PolyWAX LP and PolySAX LP are trademarks of PolyLC Inc. TopTip is a trademark of Glygen Corp. (Columbia, MD, USA). HyperCarb is a trademark of ThermoFisher Scientific. Sachtopore is a trademark of Sachtleben Chemie GmbH (now part of Venator Materials PLC).
This research was supported in part by the National Institutes of Health grants U01CA231081, R01 DK071801, and RF1 AG052324 (to LL). The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531 to LL) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. LL acknowledges a Vilas Distinguished Achievement Professorship and a Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website:
Expanded experimental details of protein digestion and LC-MS/MS analysis; illustration of peptide retention with counterions added in HILIC; figures of chromatograms; figure of the distribution of modified peptides captured by C-18 material vs. PGC material; figure of residue number per modified peptide analysis; figure of comparison of modification sites identified in this study and Uniprot reported evidence; figure of glycan heterogeneity analysis; figure of Byonic cut-off score optimization analysis; chromatogram before and after column passivation; figure of M6P peptide spectra manual inspection results; figure of reproducibility analysis; figure of literate comparisons; figure of example MS spectra; figure of EThcD O-glycopeptide analysis; tables of identified modified peptide distribution in ERLIC fractions; tables of link sizes of Figure 9; table of proteins associated with Alzheimers’ Disease and their post-translational modifications identified in this study.
Data Availability:
The mass spectrometry proteomics data that support the findings of this study have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the accession code PXD022988.
Reviewer account details:
Username: reviewer_pxd022988@ebi.ac.uk
Password: uHpwzVWA
REFERENCES
- 1).Zhang X; Ye J; Jensen ON; Roepstorff P Highly efficient phosphopeptide enrichment by calcium phosphate precipitation combined with subsequent IMAC enrichment. Mol. Cell. Proteom 2007, 6, 2032–2042. [DOI] [PubMed] [Google Scholar]
- 2).Yue X; Schunter A; Hummon AB Comparing multi-step IMAC and multi-step TiO2 methods for phosphopeptide enrichment. Anal. Chem 2015, 87, 8837–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Glover MS; Yu Q; Chen Z; Shi X; Kent KC; Li L Characterization of intact sialylated glycopeptides and phosphorylated glycopeptides from IMAC enriched samples by ETHcD fragmentation: Toward combining phosphoproteomics and glycoproteomics. Int. J. Mass Spectrom 2018, 427, 35–42. [Google Scholar]
- 4).Čaval T; Zhu J; Tian W; Remmelzwaal S; Yang Z; Clausen H; Heck AJR Targeted Analysis of Lysosomal Directed Proteins and Their Sites of Mannose-6-phosphate Modification. Mol. Cell. Proteom 2019, 18, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Xiao H; Chen W; Smeekens JM; Wu R An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycopeptides. Nat. Commun 2018, 9, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Alpert AJ; Hudecz O; Mechtler K Anion-Exchange Chromatography of Phosphopeptides: Weak anion exchange versus strong anion exchange and anion-exchange chromatography versus electrostatic repulsion–hydrophilic interaction chromatography. Anal. Chem 2015, 87, 4704–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).McNulty DE; Annan RS Hydrophilic-interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteom 2008, 7, 971–980. [DOI] [PubMed] [Google Scholar]
- 8).Ding W; Nothaft H; Szymanski CM; Kelly J Identification and quantification of glycoproteins using ion-pairing normal-phase liquid chromatography and mass spectrometry. Mol. Cell. Proteom 2009, 8, 2170–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Alpert AJ Hydrophilic interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [DOI] [PubMed] [Google Scholar]
- 10).Thannhauser TW; Shen M; Sherwood R; Howe K; Fish T; Yang Y; Chen W; Zhang S A workflow for large-scale empirical identification of cell wall N-linked glycoproteins of tomato (Solanum lycopersicum) fruit by tandem mass spectrometry. Electrophoresis 2013, 34, 2417–2431 [cf. Fig. 2]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Alpert AJ Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal. Chem 2008, 80, 62–76. [DOI] [PubMed] [Google Scholar]
- 12).Zhang H; Guo T; Li X; Datta A; Park JE; Yang J; Lim SK; Tam JP; Sze SK Simultaneous characterization of glyco- and phosphoproteomes of mouse brain membrane proteome with electrostatic repulsion hydrophilic interaction chromatography. Mol. Cell. Proteom 2010, 9, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).HuyenTran T; Hwang I; Park J-M; Kim JB; Lee H An application of electrostatic repulsion hydrophilic interaction chromatography in phospho- and glycoproteome profiling of epicardial adipose tissue in obesity mouse. Mass Spectrom. Lett 2012, 3, 39–42. [Google Scholar]
- 14).Cui Y; Yang K; Tabang DN; Huang J; Tang W; Li L Finding the sweet spot in ERLIC mobile phase for simultaneous enrichment of N-glyco and phosphopeptides. J. Am. Soc. Mass Spectrom 2019, 30, 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Chen R; Williamson S; Fulton KM; Twine SM; Li J Simultaneous analysis of phosphopeptides and intact glycopeptides from secretome with mode switchable solid phase extraction. Anal. Methods 2019, 11, 5243–5249. [Google Scholar]
- 16).Zeidan Q; Hart GW The intersections between O-GlcNacylation and phosphorylation: implications for multiple signaling pathways. J. Cell Sci 2010, 123, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Leney AC; El Atmioui D; Wu W; Ovaa H; Heck AJR Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. USA 2017, 114, E7255–E7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Grimes M; Hall B; Foltz L; Levy T; Rikova K; Gaiser J; Cook W; Smirnova E; Wheeler T; Clark NR; Lachmann A; Zhang B; Hornbeck P; Ma’ayan A; Comb M Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci. Signal 2018, 11, eaaq1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Hendriks IA; Lyon D; Young C; Jensen LR; Vertegaal ACO; Nielsen ML Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol 2017, 24, 325–336. [DOI] [PubMed] [Google Scholar]
- 20).Alpert AJ Effect of salts on retention in hydrophilic interaction chromatography. J. Chromatogr. A 2018, 1538, 45–53. [DOI] [PubMed] [Google Scholar]
- 21).Cohen RD; Liu Y; Gong X Analysis of volatile bases by high performance liquid chromatography with aerosol-based detection. J. Chromatogr. A 2012, 1229, 172–179 [Fig. 5]. [DOI] [PubMed] [Google Scholar]
- 22).Shmukler LE; Gruzdev MS; Kudryakova NO; Fadeeva Yu. A.; Kolker AM; Safonova LP Thermal behavior and electrochemistry of protic ionic liquids based on triethylamine with different acids. RSC Adv. 2016. (6) 109664–109671. [Google Scholar]
- 23).Alpert AJ; Petritis K; Kangas L; Smith RD; Mechtler K; Mitulovic G; Mohammed S; Heck AJR Peptide orientation affects selectivity in ion-exchange chromatography. Anal. Chem 2010, 82, 5253–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Riley NM; Hebert AS; Westphall MS; Coon JJ Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun 2019, 10, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Ruprecht B; Kock H; Medard G; Mundt M; Kuster B; Lemeer S Comprehensive and reproducible phosphopeptide enrichment using iron immobilized metal ion affinity chromatography (Fe-IMAC) columns. Mol. Cell. Proteom 2015, 14, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Fang P; Wang X-J; Xue Y; Liu M-Q; Zeng W-F; Zhang Y; Zhang L; Gao X; Yan G-Q; Yao J; Shen H-L; Yang P-Y In-depth mapping of the mouse brain N-glycoproteome reveals widespread N-glycosylation of diverse brain proteins. Oncotarget 2016, 7, 38796–38809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Zhang C; Ye Z; Xue P; Shu Q; Zhou Y; Ji Y; Fu Y; Wang J; Yang F Evaluation of Different N-Glycopeptide Enrichment Methods for N-Glycosylation Sites Mapping in Mouse Brain. J. Proteome Res 2016, 15, 2960–2968. [DOI] [PubMed] [Google Scholar]
- 28).Piovesana S; Montone CM; Cavaliere C; Crescenzi C; La Barbera G; Laganà A; Capriotti AL Sensitive untargeted identification of short hydrophilic peptides by high performance liquid chromatography on porous graphitic carbon coupled to high resolution mass spectrometry. J. Chromatogr. A 2019, 1590, 73–79. [DOI] [PubMed] [Google Scholar]
- 29).Alpert AJ; Gygi SP; Shukla AK Desalting phosphopeptides by solid-phase extraction. ASMS; 2007, poster MP 438. [Google Scholar]
- 30).Chen R; Stupak J; Williamson S; Twine SM; Li J Online porous graphic carbon chromatography coupled with tandem mass spectrometry for post-translational modification analysis. Rapid Commun. Mass Spectrom 2019, 33, 1240–1247. [DOI] [PubMed] [Google Scholar]
- 31).An HJ; Froehlich JW; Lebrilla CB Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr. Opin. Chem. Biol 2009, 13, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Cao L; Qu Y; Zhang Z; Wang Z; Prytkova I; Wu S Intact glycopeptide characterization using mass spectrometry. Exp. Rev. Proteomics 2016, 13, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Wang Z; Gucek M; Hart GW Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 13793–13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Yao H; Li A; Wang M Systematic Analysis and Prediction of In Situ Cross Talk of O-GlcNAcylation and Phosphorylation. Biomed. Res. Int 2015, 279823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Kanninen K; Goldsteins G; Auriola S; Alafuzoff I; Koistinaho J Glycosylation changes in Alzheimer’s disease as revealed by a proteomic approach. Neurosci. Lett 2004, 367, 235–240. [DOI] [PubMed] [Google Scholar]
- 36).Barykin EP; Mitkevich VA; Kozin SA; Makarov AA Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet 2017, 8, article 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Lee LY; Moh ESX; Parker BL; Bern M; Packer NH; Thaysen-Andersen M Toward automated N-glycopeptide identification in glycoproteomics. J. Proteome Res 2016, 15, 3904–3915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data that support the findings of this study have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the accession code PXD022988.
Reviewer account details:
Username: reviewer_pxd022988@ebi.ac.uk
Password: uHpwzVWA