

SUMMARY
Beta-amyloid (Aβ) depresses excitatory synapses by a poorly understood mechanism requiring NMDA receptor (NMDAR) function. Here, we show that increased PSD-95, a major synaptic scaffolding molecule, blocks the effects of Aβ on synapses. The protective effect persists in tissue lacking the AMPA receptor subunit GluA1, which prevents the confounding synaptic potentiation by increased PSD-95. Aβ modifies the conformation of the NMDAR C-terminal domain (CTD) and its interaction with protein phosphatase 1 (PP1), producing synaptic weakening. Higher endogenous levels or overexpression of PSD-95 block Aβ-induced effects on the NMDAR CTD conformation, its interaction with PP1, and synaptic weakening. Our results indicate that increased PSD-95 protects synapses from Aβ toxicity, suggesting that low levels of synaptic PSD-95 may be a molecular sign indicating synapse vulnerability to Aβ. Importantly, pharmacological inhibition of its depalmitoylation increases PSD-95 at synapses and rescues deficits caused by Aβ, possibly opening a therapeutic avenue against Alzheimer’s disease.
Graphical Abstract
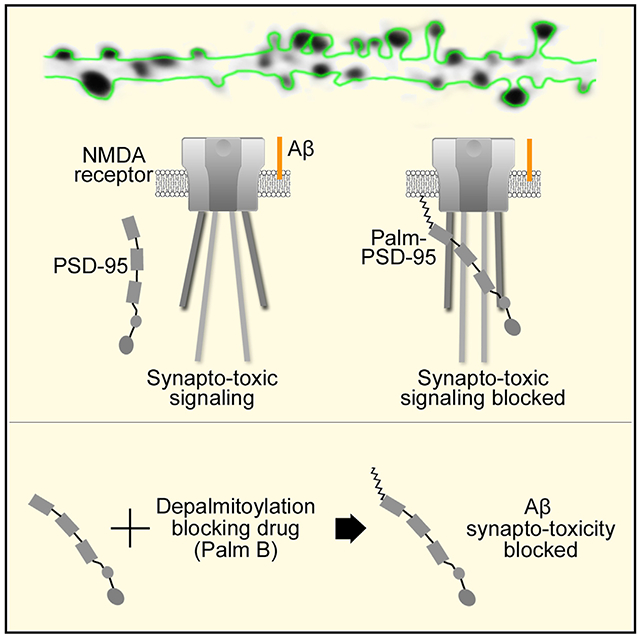
In brief
The molecular mechanisms by which beta-amyloid (Aβ) damages synapses are poorly understood. Dore et al. show that increasing PSD-95 synaptic contentprotects synapses from Ab-induced synaptic deficits by reducing NMDA receptor metabotropic function.
INTRODUCTION
Elevated levels of the beta-amyloid (Aβ) peptide have been strongly implicated in the pathophysiology of Alzheimer’s disease (AD) (Hardy and Selkoe, 2002). One of the earliest pathological changes in the brains of patients with AD is the loss of synapses (DeKosky and Scheff, 1990; Masliah et al., 2001). Elevated Aβ levels can lead to loss of synapses, requiring a number of intracellular signaling steps (Chen et al., 2013; Ehrlich et al., 2007; Olsen and Sheng, 2012; Wu et al., 2010). Notably, the effects of Aβ on synapses can be prevented by NMDA receptor (NMDAR) blockade (Hsieh et al., 2006; Kamenetz et al., 2003; Kessels et al., 2013; Shankar et al., 2007), although the mechanisms are not well understood. Elucidating how Aβ weakens synapses and preventing such weakening are important objectives in the field.
PSD-95 is a critical synaptic protein that binds to the NMDAR C-terminal domain (CTD) (Kornau et al., 1995; Niethammer et al., 1996), requires palmitoylation to remain at synapses (El-Husseini et al., 2002; Topinka and Bredt, 1998), and controls synaptic transmission and plasticity (Béïque et al., 2006; Ehrlich and Malinow, 2004; El-Husseini et al., 2000; Stein et al., 2003; Xu et al., 2008). Overexpression of PSD-95 increases synaptic transmission (Ehrlich and Malinow, 2004; Xu et al., 2008) and blocks ion-flux-independent long-term depression (LTD) (Dore and Malinow, 2021). Importantly, PSD-95 is reduced in brain tissue from AD mouse models (Shao et al., 2011), in neural tissue exposed to Aβ (Almeida et al., 2005), and in brain tissue from individuals with AD (Gylys et al., 2004; Shao et al., 2011). Given these findings regarding PSD-95 and Aβ, we reasoned that increased levels of PSD-95 might interfere with the synaptic depression produced by Aβ.
We recently studied NMDAR function and signaling during ion-flux-independent LTD by measuring fluorescence resonance energy transfer (FRET) between genetically encoded fluorescent proteins positioned at the cytoplasmic terminus of the GluN1 subunits of the NMDAR (Dore et al., 2015). We found that ligand binding to the extracellular domain of the NMDAR produced conformational movement of its CTD in the absence of ion flow through the channel. This was measured as a reduction in FRET between the GluN1 cytoplasmic tails (Dore et al., 2015). Such conformational changes triggered cytoplasmic signaling by molecules bound to the NMDAR CTD, such as protein phosphatase 1 (PP1), which produce synaptic depression (Aow et al., 2015).
Here, we find that overexpression of PSD-95 blocks an Aβ-induced NMDAR CTD conformational change and associated signaling by PP1 as well as synaptic depression. Moreover, Aβ reduced endogenous PSD-95 levels and FRET in NMDAR CTD, specifically in smaller dendritic spines. Accordingly, bigger spines, which contain synapses with more endogenous PSD-95 (Aoki et al., 2001), are protected from the effects of Aβ. Importantly, increasing synaptic PSD-95 with an exogenously applied depalmitoylation inhibitor, Palmostatin B (Palm B) rescued Aβ-induced synaptic depression in tissue from wild-type (WT) animals, but it had no effect in tissue from PSD-95 knockout (KO) mice. Aβ effects on the NMDAR conformation, as well as Aβ reduction of spine density, were also reversed by Palm B treatment. These findings suggest a novel avenue with therapeutic potential in treatment of AD.
RESULTS
PSD-95 protects synapses from Aβ independently of synaptic potentiation
Elevated Aβ reduces synaptic transmission (Kamenetz et al., 2003) and synaptic PSD-95 (Almeida et al., 2005) and destroys synapses (Hsieh et al., 2006; Shankar et al., 2007). Initially, we tested whether PSD-95 overexpression can protect synapses from Aβ. We sparsely infected organotypic hippocampal slices with a Sindbis virus driving expression of CT100 (the beta-secretase cleavage product of amyloid-precursor protein [APP] that produces Aβ [discussed later] along with GFP) and, 18–24 h later, obtained whole-cell recordings simultaneously from two neurons, one infected and one non-infected (Kamenetz et al., 2003). As expected (Kamenetz et al., 2003), neurons expressing CT100 displayed depressed synaptic transmission compared to non-infected control neurons (control: 69 ± 9 pA; CT100: 48 ± 5 pA; p < 0.05; n = 17) (Figures 1A-1C). Notably, neurons infected with a virus driving expression of both CT100 and PSD-95 displayed increased synaptic transmission compared to non-infected control (2.2-fold in neurons expressing CT100 + PSD-95), which was as great as the potentiation produced by overexpression of PSD-95 and CT84, an inactive fragment of APP (1.7-fold in neurons expressing CT84 + PSD-95; Figures 1B and 1C). This result indicates that PSD-95 overexpression can potentiate synapses (Ehrlich and Malinow, 2004; Stein et al., 2003) even in the presence of Aβ.
Figure 1. PSD-95 protects synapses from Aβ independently of synaptic potentiation.

(A) Left and middle: hippocampal organotypic slices used for dual-patch whole-cell recordings from an infected and neighboring uninfected CA1 neuron. Right: representative traces (mean of 40 consecutive trials) of evoked AMPA-receptor-mediated responses.
(B) Dot plot of excitatory post-synaptic current (EPSC) dual-patch recordings from WT tissue; infected neurons expressing indicated constructs; group average indicated by larger black outlined symbols. Error bars indicate SEM here and throughout.
(C) Bar graph of dual-patch recordings for indicated groups; responses normalized to uninfected controls (gray). *p < 0.05; **p < 0.001, paired t test; n ≥ 16 for each group.
(D) Same as in (B), but in slices made from GluA1 knockout animals.
(E) Same as in (C), but in slices made from GluA1 knockout animals; *p < 0.05, paired t test. n = 11 for each group.
We wanted to test whether PSD-95 was blocking the effects of Aβ, or whether overexpression of PSD-95 was simply potentiating transmission (Ehrlich and Malinow, 2004; Stein et al., 2003), and thereby compensating for the depressive effects of Aβ (Kamenetz et al., 2003). To distinguish between these two possibilities, we repeated the dual-patch recordings in tissue prepared from GluA1 knockout animals (Figures 1D and 1E). Interestingly, in tissue from those animals, PSD-95 overexpression produced no significant potentiation (control: 48 ± 5 pA; CT84 + PSD-95: 51 ± 5 pA; p = 0.6, n = 11) (Figure 1D), showing that PSD-95-induced synaptic potentiation requires GluA1. CT100 still produced a similar level of synaptic depression (control: 66 ± 9 pA; CT100: 40 ± 3 pA; p < 0.05; n = 11) (Figure 1D), indicating that Aβ-induced depression does not require GluA1, in accordance with a previous study (Reinders et al., 2016). Importantly, neurons expressing CT100 and PSD-95 displayed no significant depression (control: 45 ± 6 pA; CT100+PSD-95: 51 ± 5 pA; p = 0.5; n = 11) (Figure 1D), indicating that overexpression of PSD-95 blocks Aβ-induced depression. Therefore, PSD-95 overexpression prevents Aβ-induced depression independently of its ability to potentiate synaptic transmission.
Aβ drives NMDAR CTD conformational change
We next examined the impact of Aβ on NMDAR signaling. We infected dissociated cultured neurons with a Sindbis virus expressing CT100 (Kessels et al., 2013) and mCherry containing a nuclear localization sequence, nuc-mCherry, (see STAR Methods), permitting identification of infected neurons (Figure 2A). We confirmed that neurons infected with CT100 were, indeed, producing CT100 and elevated Aβ using SDS-PAGE and immunoblotting with antibody 6E10, which recognizes the N termini of Aβ and CT100 (Figure 2B). As expected, incubation of infected neurons with a gamma secretase inhibitor (GSI; L685-458; see STAR Methods) blocked Aβ production and increased the amount of CT100 fragment detected. Neurons infected with a control Sindbis virus, expressing CT84, which lacks the N terminus of Aβ, did not show any signal with antibody 6E10 (Figure 2B). Importantly, as in brain slices (Kamenetz et al., 2003), elevated Aβ (produced by CT100 expression) in dissociated cultured neurons reduced synaptic transmission that was blocked by D-2-amino-5-phosphonopentanoic acid (APV) (Figure S1).
Figure 2. PSD-95 blocks Aβ-driven conformational movement of the NMDA C-terminal domain.

(A) Diagram of neurons transfected with NMDAR FRET probes and infected with a virus expressing APP fragments along with nuclear-targeted mCherry, which were selected for imaging.
(B) Representative immunoblots of actin, mCherry, and Aβ species (detected with antibody 6E10) from cortical neuronal lysates that were infected with CT84 or CT100 viruses. Neurons were incubated with a gamma secretase inhibitor (0.5 μM L685-458; GSI) where indicated.
(C) Top: representative FLIM images of neurons expressing indicated FRET probes for indicated conditions. Pseudocolor scale indicates GFP lifetime at each pixel for all figures. Scale bar, 5 μm. Bottom: bar graph of spine FRET efficiency (see STAR methods) of GluN1-GFP/GluN1-mCherry-expressing neurons for the indicated conditions. n > 20 neurons, >400 spines (for each condition). ***p < 0.0001, unpaired t test.
(D) Top: diagram of neurons expressing the FRET probes infected with CT100 + PSD-95 virus. Representative FLIM images of neurons expressing indicated FRET probes for indicated conditions. Scale bar, 5 μm. Bottom: Bar graph of spine FRET efficiency of GluN1-GFP/GluN1-mCherry-expressing neurons for the indicated conditions; n > 20 neurons; >400 spines (for each condition). ***p < 0.0001, unpaired t test.
To measure conformational movement of the NMDAR cytoplasmic domain, we used FRET-FLIM—FRET measured by fluorescence lifetime imaging microscopy (FLIM)—and imaged transfected hippocampal dissociated cultured neurons expressing GluN1 NMDAR subunits with C-terminus-tagged fluorophores (Figure 2A) (Dore et al., 2015). Incubation with the NMDAR glycine site blocker 7-chloro-kynurenic acid permitted measurement of ion-flux-independent NMDAR signaling (Aow et al., 2015). Neurons displaying green fluorescence in dendritic spines (indicating transfection of GluN1-GFP) and nuclear red fluorescence (indicating infection with the virus expressing nuc-mCherry and CT100) were selected for imaging. In neurons transfected with only GluN1-GFP, viral expression of CT84 or CT100 had no effect on GluN1-GFP lifetime (Figure 2C, top). As expected (Dore et al., 2015), neurons expressing GluN1-GFP and GluN1-mCherry displayed significantly reduced GFP lifetime in their spines, indicating FRET between GluN1-GFP and GluN1-mCherry (Figure 2C, top). Neurons also expressing CT84 displayed a similar FRET efficiency (uninfected: 5.8% ± 0.3%; CT84: 5.5% ± 0.3%; p = 0.55) (Figure 2C). However, CT100 expression reduced the FRET efficiency between GluN1-GFP and GluN1-mCherry by about 50% (CT100: 2.8% ± 0.3%; CT84: 5.5% ± 0.3%; p < 0.001). The effect on FRET efficiency by expression of CT100 was blocked by incubating cultures with the GSI L685-458 (CT100: 5.9% ± 0.3%; CT84: 5.4% ± 0.3%; p = 0.26), indicating that Aβ, and not CT100, affects the NMDAR conformation.
PSD-95 blocks Aβ-induced depression by restraining the NMDAR CTD conformational movement
How could PSD-95 block Aβ-induced depression? Because of its direct interaction with the NMDAR CTD (Kornau et al., 1995; Niethammer et al., 1996), PSD-95 could block the signaling by constraining NMDAR CTD conformational movements that can drive synaptic depression (Aow et al., 2015). To test this hypothesis, we measured the effect of PSD-95 overexpression on the NMDAR CTD conformational movement with the FRET-FLIM assay (Figure 2D). Elevated PSD-95 appeared not to change the NMDAR CTD conformation, as the FRET efficiency was similar between expression of CT84 and CT84 + PSD-95 (CT84: 5.5% ± 0.3%; CT84 + PSD-95: 5.3% ± 0.3%; p = 0.5; n > 400 spines) (Figure 2D). However, PSD-95 overexpression blocked the reduced FRET efficiency produced by expression of CT100 (CT100 + PSD-95: 6.0% ± 0.3%; CT100: 2.8% ± 0.3%; p < 0.001; n > 400 spines). Thus, our results suggest that interactions between PSD-95 and the NMDAR CTD blocked Aβ-induced conformational movement of the NMDAR CTD, thereby likely blocking the CTD signaling associated with such movement that produces synaptic depression. The GluN1 isoform used for these FRET experiments was GluN1-2a (formerly called NR1-C), the most commonly expressed GluN1 isoform in the brain (Laurie and Seeburg, 1994; Paoletti, 2011). This isoform does not contain a PDZ-binding motif that interacts with PSD-95, indicating that GluN2 subunits, which do bind PSD-95 PDZ domains (Doré et al., 2014; Kornau et al., 1995; Niethammer et al., 1996), are mediating NMDAR/PSD-95 interactions blocking Aβ-induced conformational movement of the NMDAR CTD.
For PSD-95 to restrain directly synaptic NDMAR CTD signaling, PSD-95 proximity to the post-synaptic membrane would be required. Palmitoylation of the PSD-95 N terminus is required for its association with the post-synaptic membrane (Topinka and Bredt, 1998). Therefore, we expressed a mutant form of PSD-95 (C3,5S-PSD-95), which is palmitoylation resistant (Topinka and Bredt, 1998) and does not potentiate transmission (Ehrlich and Malinow, 2004). Expression of C3,5S-PSD-95 failed to block the effect on FRET efficiency by CT100 (CT100: 2.8% ± 0.2%; CT100 + C3,5S-PSD-95: 2.9% ± 0.2%; p = 0.83; n > 700 spines) (Figure 2D). We also tested this mutant PSD-95 in the electrophysiology assay described earlier and found that it did not prevent Aβ-induced synaptic depression (CT100: 32% ± 6% depression, n = 17; CT100 + C3,5S-PSD-95: 45% ± 9% depression, n = 11; p = 0.3; Figure S2). These findings suggest that PSD-95 must be associated with the post-synaptic membrane to restrain the Aβ-driven NMDAR CTD conformational effects.
To rule out the possibility that overexpression of PSD-95 affected generation of Aβ from CT100, we infected cultures, previously transfected with NMDAR FRET probes, with a virus producing CT100 or a virus producing CT100 and PSD-95 and compared the FRET efficiency in infected and non-infected nearby cells closely surrounded by infected cells (Figure S3). We reasoned that, if PSD-95 did not reduce Aβ production from CT100, nearby non-infected cells (i.e., not expressing CT100) would still be exposed to secreted Aβ. FRET efficiency between GluN1-GFP and GluN1-mCherry was equally reduced in CT100-infected neurons compared to that in nearby non-infected cells also expressing the FRET probes (CT100 infected: 3.4% ± 0.3%; non-infected nearby neurons: 3.5% ± 0.2%; p = 0.7; n > 550 spines; Figure S3), indicating that extracellular Aβ can drive NMDAR CTD conformational movement. Importantly, in neurons expressing the FRET probes, CT100 and PSD-95, the reduced FRET signaling was blocked, but nearby cells expressing only FRET probes showed reduced FRET signaling, indicating that PSD-95 is not preventing production of Aβ, as it appears to be secreted and affect the NMDAR CTD of nearby cells (Figure S3). We note that, in hippocampal slice experiments (Figure 1), infection with CT100-expressing Sindbis virus was sparse; cell bodies of infected and non-infected cells were 50–100 μm apart. In these conditions, an infected neuron has little effect on a non-infected neuron (Kamenetz et al., 2003). In contrast, in primary neuronal cultures, CT100 expression was seen in about 80% of neurons. In these conditions, the dendrites of non-infected neurons were surrounded by CT100-expressing dendrites (usually within 5 μm; Figure S3C), which, our data indicate, produce effects on FRET in non-infected synapses. These results align with other studies demonstrating that the effects of Aβ are seen only within 20–50 μm of an Aβ source (Busche et al., 2008, 2012; Wei et al., 2010). Together, these experiments indicate that exogenously expressed PSD-95 blocks Aβ-driven NMDAR CTD conformational movement.
Aβ drives FRET changes between PP1 and the NMDAR cytoplasmic domain
How is movement in the NMDAR CTD producing the synaptic depression observed with elevated Aβ? PP1, which participates in synaptic depression (Morishita et al., 2001; Mulkey et al., 1993; Thiels et al., 1998), interacts with the GluN1 subunit through the scaffolding protein AKAP9 (Westphal et al., 1999) and could be affected by movement in the NMDAR CTD. Indeed, during chemically induced long-term depression (cLTD), NMDAR conformational movement induced changes in its interaction with PP1 (Aow et al., 2015). We reasoned that a similar effect may occur during Aβ-induced synaptic depression. To test this view, we measured the impact of elevated Aβ on FRET between GluN1-GFP and PP1-mCherry (Figures 3A and 3B). FRET between these proteins was unaffected by infection with the CT84-expressing control virus but was almost abolished in neurons expressing CT100 (uninfected: 3.6% ± 0.3%; CT84: 3.4% ± 0.3%; CT100, 0.5% ± 0.2%; CT84 versus CT100; p < 0.0001; n > 330 spines) (Figure 3B). Interestingly, PSD-95 blocked the effect of CT100 on the NMDAR-PP1 interaction as the FRET efficiency in neurons infected with CT84+PSD95 or CT100+PSD95 was restored to uninfected levels (Figure 3B). Aβ is, thus, inducing movement in the NMDAR CTD, which then decreases FRET between GluN1-GFP and PP1-mCherry. This Aβ-driven decreased interaction with NMDAR CTD could potentially drive PP1 activity.
Figure 3. Aβ-decreased FRET between NMDAR and PP1 and increased PP1 activity are blocked by PSD-95.

(A) Diagram of the NMDAR with its GluN1-GFP subunit acting as a FRET donor for PP1-mCherry, which interacts with the NMDAR complex through interactions with AKAP9.
(B) Bar graph of FRET efficiency for indicated conditions. n > 20 neurons, >400 spines (for each condition). ***p < 0.0001, unpaired t test.
(C) Representative images of P-Thr320-PP1 and total PP1 immunostaining in neurons overexpressing CT100+NLS-mCherry, as indicated. Scale bars: 10 μm (top image) and 5 μm (lower images).
(D) Bar graph of average immunostaining intensity for ratio of P-Thr320-PP1 to total PP1 (normalized to CT84 values) in spines of neurons overexpressing the indicated constructs. n = 7–18 neurons (>1,000 spines) per condition. ***p < 0.0001, unpaired t test.
To investigate this possibility, PP1 activity was assessed by measuring the relative amount of phosphorylation on PP1 at Thr-320, which inhibits phosphatase activity and has been used as a measure of PP1 activity (Hou et al., 2013). We found that CT100 expression significantly decreased the amount of P-Thr320-PP1, which suggests that PP1 activity increases in the presence of Aβ (P-Thr320-PP1/PP1 ratio, CT100: 0.65 ± 0.04; CT84: 1.00 ± 0.05; p < 0.0001; n = 7–18 neurons) (Figure 3D). Interestingly, synaptic NMDAR stimulation producing cLTD had a similar effect on this specific PP1 phosphorylation (Hou et al., 2013). Notably, overexpression of PSD-95 blocked the Aβ-induced change in P-Thr320-PP1. These data support the view that NMDAR CTD movement driven by elevated Aβ triggers PP1 activity and that this process is blocked by overexpressed PSD-95 constraining NMDAR CTD movement (Figure 3D).
Large spines are protected from Aβ
Since overexpression of PSD-95 protected spines from Aβ (Figures 1, 2, and 3), we tested whether endogenously expressed PSD-95 could also be protective. We used the previously established strongly positive correlation between spine size, post-synaptic density (PSD) area, and PSD-95 content (Harris and Stevens, 1989) (Aoki et al., 2001). Thus, if endogenous PSD-95 were protective, larger spines, by having more PSD-95, should be affected less by elevated Aβ than smaller spines with less PSD-95. We used neurons infected with CT84 or CT100 along with GFP (to measure spine size) and measured PSD-95 levels using immunohistochemistry. In agreement with previous reports (Almeida et al., 2005; Gylys et al., 2004), Aβ reduced PSD-95 levels significantly (CT84: 100% ± 1%; CT100: 77% ± 1%; p < 0.0001; n > 3,000 spines) (Figure S4). However, small spines were largely responsible for the decrease in PSD-95 levels, and bigger spines were resilient to Aβ (Figures 4A-4C). Thus, large spines, which (among other differences) contain more PSD-95, appear to be protected from this effect of Aβ. We note that CT100 reduced spine size while not affecting GluN1-GFP signal area or intensity (see Figure S5 for details). Moreover, we measured a similar reduction of PSD-95 levels by CT100 in the presence or absence of 7CK, suggesting that NMDAR metabotropic signaling is the main mechanism by which Aβ reduces PSD-95 (Figure S4). These results are consistent with the view that endogenous PSD-95 protects synapses from the damaging metabotropic NMDAR signaling driven by elevated Aβ.
Figure 4. Large spines are protected from Aβ.

(A) Representative examples of PSD-95 immunostaining in neurons expressing GFP and CT100 (left) or CT84 (right). Larger spines have bright PSD-95 puncta in both conditions (arrowheads), and smaller spines have lower PSD-95 intensity in CT100-expressing spines (arrows). Dendrites are masked for clarity. See Figure S5 for unmasked images. Scale bar, 5 μm.
(B) Graph of PSD-95 average intensity in spines expressing GFP and CT100 or CT84 (normalized to PSD-95 intensity values in all CT84-expressing spines). Spine contours were manually traced with the GFP channel, and spines were binned according to their size. (See the Immunohistochemistry and Quantification and statistical analysis” sections in STAR Methods for more information.) n > 3,000 spines per condition; 300–1,500 spines per size bin. CT100-expressing spines had lower PSD-95 intensity than spines expressing CT84 in most size bins; *p < 0.05; ***p < 0.001, unpaired t test. Both datasets are fitted with a second order polynomial curve.
(C) Mean PSD-95 intensity (same dataset as in B) divided into small (<0.6 μm2) and large (>0.6 μm2) dendritic spines of neurons expressing CT100 or CT84. CT100 only affects endogenous PSD-95 levels in small spines.
(D) Graph of NMDAR FRET efficiency in spines expressing CT100 or CT84 (data from Figure 2C). Spine size was assessed using the GluN1-GFP signal, and spines were binned according to their size (as in B). n > 2,500 spines per condition; 120–1,000 spines per size bin. CT100-expressing spines had lower FRET efficiency than spines expressing CT84 in most size bins. *p < 0.05; **p < 0.01; ***p < 0.001, unpaired t test. CT84 dataset is fitted with a linear regression, and CT100 dataset is fitted with a second order polynomial curve.
(E) Model of NMDAR conformation in the presence of Aβ. In small spines, Aβ is inducing a conformational change in the NMDAR CTD; in large spines, increased amounts of PSD-95 can effectively restrain NMDAR conformation and protect spines from Aβ.
Next, we tested whether higher endogenous PSD-95 prevented Aβ-induced NMDAR conformational movement. We found a strong positive correlation between spine size and FRET efficiency in neurons expressing CT100; in neurons expressing CT84, FRET efficiency was independent of spine size (Figure 4D). Although there are likely a number of molecular differences between large and small spines, these data support the following model: elevated Aβ modifies NMDAR conformation in small spines, which will trigger downstream signaling, leading to lower amounts of PSD-95 and synaptic depression; in larger spines, elevated amounts of endogenous PSD-95 restrains the NMDAR conformation protecting them from Aβ (Figure 4E).
Pharmacological blockade of PSD-95 depalmitoylation reverses synaptic damage caused by Aβ
Our results indicate that PSD-95 palmitoylation, which maintains PSD-95 at the PSD, is essential for the protection of synapses from Aβ. We reasoned that a drug increasing PSD-95 palmitoylation could enhance this protection and, potentially, be beneficial against AD. Interestingly, specific enzymes responsible for PSD-95 depalmitoylation, ABHD17A/B/C, were recently identified (Yokoi et al., 2016). Moreover, Palm B, an inhibitor of these enzymes, was shown to increase the amount of palmitoylated PSD-95 and the size of PSD-95 clusters in neurons (Jeyifous et al., 2016). To determine whether Palm B could rescue Aβ-induced depression by increasing PSD-95 palmitoylation, we performed dual-patch recordings (as in Figure 1) in hippocampal brain slices from WT and PSD-95 KO mice (Béïque et al., 2006). We sparsely infected these organotypic slices with a virus expressing CT100 for 15–18 h and applied Palm B for the last 3 h. We obtained dual-patch recordings from CT100-infected and control neurons 50–100 μm away. Slices incubated in Palm B displayed no depression in neurons expressing CT100 (CT100: 38% ± 10% depression, n = 20; CT100 + Palm B: 6% ± 8% potentiation, n = 21; p < 0.05) (Figure 5A). This was somewhat surprising, given that when Palm B was applied, CT100-expressing neurons would have already displayed depression; thus, Palm B actually reversed the depressed phenotype. We observed a similar rescue of Aβ-induced depression in hippocampal slices made from WT rats (Figure S7). In contrast to WT mice, in PSD-95 KO mice, similar amounts of synaptic depression were observed in slices treated with vehicle or Palm B (CT100: 34% ± 10% depression, n = 20; CT100 + Palm B: 30% ± 8% depression, n = 21; p > 0.05) (Figure 5B). Moreover, Palm B also reversed synaptic depression induced by expression of full-length APP (Figure S7). This result suggests that any effect of Palm B on Aβ production (a potential confound, since BACE1 cleavage of APP may be influenced by palmitoylation status [Andrew et al., 2017; Bhattacharyya et al., 2016], and both BACE1 and the α-secretase ADAM10 are located adjacent to PSD-95 [Lundgren et al., 2020]) does not prevent Palm B from reversing Aβ-induced synaptic depression. These results indicate that (1) while Palm B may block depalmitoylation of several molecules (Lin and Conibear, 2015), the effect of Palm B on the phenotypes measured as described earlier is through PSD-95; and (2) transient (3-h) Palm B treatment can reverse Aβ-induced depression.
Figure 5. Pharmacological blockade of PSD-95 depalmitoylation reverses synaptic damage caused by Aβ.

(A) Bar graph of dual-patch recordings obtained in slices made from WT mice; responses normalized to uninfected controls (light gray). Palm B-treated slices were incubated with 1 μM Palm B for 3 h. Dot plot with raw data for these recordings is shown in Figure S6A. *p < 0.05, unpaired t test; **p < 0.01, paired t test. n ≥ 20 for each group.
(B) Bar graph of dual-patch recordings obtained in slices made from PSD-95 KO mice; responses normalized to uninfected controls (light gray). Dot-plot for these recordings is shown in Figure S6B. *p < 0.05; **p < 0.01, paired t test; n.s., nonsignificant. n = 19 for each group.
(C) Top: FLIM images of neurons expressing CT100 before and 1 h after application of 3 μM Palm B (right) or vehicle (left). Scale bar, 2 μm. Dendrites masked for clarity. Bottom: FRET efficiency change in the same spines imaged before and after Palm B or vehicle treatment. n > 550 spines per condition. ***p < 0.001, unpaired t test.
(D) Top: representative images of PSD-95 immunostaining in neurons expressing GFP and treated or not treated with Palm B. Bottom: bar graph of PSD-95 intensity in spines of indicated size. Control neurons showed reduced PSD-95 intensity in spines smaller than 0.6 μm, Palm B treatment specifically increased PSD-95 levels in those small spines. n > 230 spines per condition. **p < 0.01, unpaired t test. Scale bar, 5 μm.
(E) Top: representative fluorescence images of GFP-expressing neurons along with CT100 or CT84 treated or not treated with 1 μM Palm B. Scale bar, 5 μm. Bottom: bar graph of spine density for indicated conditions. ***p < 0.001, unpaired t test.
See also Figure S6.
We next tested whether Palm B could reverse the Aβ-induced NMDAR conformation. One-hour treatment of CT100-expressing neurons with Palm B increased FRET between the NMDAR CTDs; the same spines were compared before and after Palm B treatment (Palm B: 1.7% ± 0.3% increase; vehicle: 0.4% ± 0.3% decrease; n > 550 spines; p < 0.0001) (Figure 5C; Figure S8). This suggests that Palm B increased PSD-95 levels in spines and reverted the NMDAR CTD conformation to that observed without Aβ. Also, Palm B did not affect FRET efficiency in control neurons in either small or big spines (Figure S8). This suggests that, in control cells, NMDARs are in a conformation not affected by increased synaptic PSD-95. Consistent with published results (Jeyifous et al., 2016), Palm B treatment also increased endogenous PSD-95 levels at spines. We found this effect to occur specifically in small dendritic spines (Figure 5D). Furthermore, we found that Palm B reversed the reduced spine density produced by Aβ (Figure 5E). These findings indicate that it is possible to reverse synaptic damage caused by Aβ with a pharmacological approach.
DISCUSSION
Previous studies indicate that elevated Aβ depresses synapses, reduces dendritic spine density, and decreases spine PSD-95 content (Almeida et al., 2005; Hsieh et al., 2006; Shankar et al., 2007). However, the molecular mechanisms underlying these effects are poorly understood. Interestingly, NMDA application (Aow et al., 2015; Nabavi et al., 2013; Stein et al., 2015) and Aβ elevation (Birnbaum et al., 2015; Kessels et al., 2013; Tamburri et al., 2013) can induce synaptic weakening that requires ligand binding to the NMDAR glutamate binding site but does not require ion flux through the NMDAR channel. This ion-flux-independent function of NMDARs upon agonist binding was associated with a FRET-detected movement of the NMDAR CTD (Dore et al., 2015) that drove signaling underlying LTD (Aow et al., 2015). Using the same approach, we show here that elevated Aβ produces a similar change in the NMDAR CTD. This supports the view that a common metabotropic action of the NMDAR underlies Aβ-induced synaptic depression and LTD.
We find that increased spine PSD-95 content, either endogenously higher levels in larger spines or exogenously overexpressed, effectively protected synapses from Aβ. This protection did not require PSD-95-induced enhanced transmission, as overexpressed PSD-95 failed to potentiate transmission in GluA1 KO animals but still protected synapses from elevated Aβ. Increased PSD-95 also blocks Aβ-driven NMDAR CTD movement and interaction with PP1, suggesting that PSD-95 constrains NMDAR CTD, blocking its metabotropic actions. Importantly, a drug that increases synaptic PSD-95 (by blocking its depalmitoylation) rescues the deleterious effects of Aβ on synapses. This effect is lost in tissue lacking PSD-95, indicating that the drug is acting through PSD-95. Notably, while many proteins are palmitoylated, most display little turnover (Martin and Cravatt, 2009; Yokoi et al., 2016), while PSD-95 has very fast turnover (Yokoi et al., 2016). Selectively blocking PSD-95 depalmitoylation may serve as a viable therapeutic option in developing treatment for AD.
We note that our studies were conducted on immature tissue, which may question the relevance of our finding to AD, a disease found in older individuals. Interestingly, increasing PSD-95 by epigenetic editing (genome modification allowing targeted overwriting of the epigenetic signature at endogenous loci) in aged or AD model mice enhanced cognitive function (Bustos et al., 2017). Furthermore, reduced levels of PSD-95 are seen in AD model mice (Hong et al., 2016; Shao et al., 2011) and brains of patients with AD (Perez-Nievas et al., 2013). These results suggest that metabotropic NMDAR mechanisms depressing transmission may be operative in AD and reversed by increasing PSD-95. Thus, our findings on tissue from young animals may be relevant to AD pathophysiology.
We also find that Aβ selectively targets smaller spines, producing larger NMDAR CTD movement and PSD-95 removal, compared to its actions on larger spines. While large and small spines likely have a number of molecular differences, the Aβ-protected status of large spines could be due to the higher levels of PSD-95 normally found in larger spines. It is tempting to extrapolate these findings to clinical observations regarding AD: (1) individuals with higher lifetime cognitive activity have reduced incidence of AD (Stern et al., 1994, 2012; Valenzuela and Sachdev, 2006); and (2) older memories are maintained better in patients with AD (Budson and Price, 2005; Gold and Budson, 2008; Orlovsky et al., 2017). In the first case, cognitive activity could drive the synaptic plasticity that increases spine size (Kopec et al., 2006; Matsuzaki et al., 2004); the higher PSD-95 content would be protective against AD. In the second case, older memories may be represented by larger spines, particularly if repeated remembering increases spine size and PSD-95 content, which would render them less sensitive to Aβ.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kim Dore (kdore@health.ucsd.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rats
Sprague-Dawley rats were used to prepare primary neuronal cultures and organotypic slices (see below for details). Pregnant dams were ordered from Envigo at E16-18 and housed in a UCSD School of Medicine facility and were maintained on a 12-hour light-dark cycle. All rats were given ad libitum access to food and water. Postnatal day (P) P0-P2 pups of both sexes were used for primary neuronal cultures. Similarly, P5-P7 pups were used for organotypic slice cultures. All procedures involving animals were approved by UCSD’s IACUC.
Mice
Mice were used to prepare organotypic slices (see below for details). For Figures 1D and 1E, we used GluA1-deficient mice (c57bl6/129 hybrid background, obtained from Dr. R. Huganir (Kim et al., 2005)). For experiments shown in Figure 5, PSD-95KO mice were used (c57bl6 hybrid background, obtained from Dr. R. Huganir (Béïque et al., 2006)). Genotype of each mice used was confirmed by PCR. All mice were housed at a UCSD School of Medicine facility, were maintained on a 12-hour light-dark cycle and given ad libitum access to food and water. P5-P7 mice pups of both sexes were used to prepare organotypic slice cultures. All procedures involving animals were approved by UCSD’s IACUC.
METHOD DETAILS
Primary neuronal cultures
Primary hippocampal or cortical (used only for biochemistry experiments shown in Figure 2B) neurons were made according to previously described protocols with minor modifications (Dore et al., 2015; Nabavi et al., 2013; Nault and De Koninck, 2010). Hippocampi from P0-P2 Sprague-Dawley rat pups were dissected in ice-cold dissection media (see Dore et al., 2015 for details) before being cut into fine pieces using a scalpel. The hippocampal tissue was resuspended in dissociation media (dissection media supplemented with 2mM L-cysteine hydrochloride, 10mg/mL papain, pH adjusted to ~7.4) and incubated at 37°C for 10min. DNaseI (24U/mL) was then added to digest precipitated DNA from dead cells and after mixing, cells were filtered through a 70 μm cell strainer to remove undissociated tissue and spun at 1000x g. Neurons were resuspended in plating media (Neurobasal-A, 10% FBS, 0.5% Pen/Strep and 0.25% Glutamax) at a concentration of 1-2 × 106 cells/mL and plated onto 18mm PDL-coated glass coverslips (Neuvitro) (≈0.5 × 106 neurons/12-well plate well). For cortical neuronal cultures, cells were plated onto PDL (Sigma P1024) coated 6-well plates (1-2 × 106 neurons/well). Additional media was added 2 hours later (Neurobasal-A, 5% FBS, 2% GS21, 0.5% Pen/Strep, 0.25% Glutamax). Thereafter, half the media was replaced every 2-4 days (Neurobasal-A, 2% GS21, 0.5% Pen/Strep, 0.25% Glutamax). When specified, neurons were incubated with 0.5μM of the gamma secretase inhibitor L685-458 for 1h prior to imaging or cell lysis for biochemical experiments. D-2-Amino-5-phosphonopentanoic acid (APV) was used to block NMDARs at a concentration of 100 μM when specified.
Organotypic slice cultures
Organotypic hippocampal slices were prepared from P5-P7 rat or mice pups as described (Stoppini et al., 1991). Slice cultures were maintained for 6–8 days, then infected using a Sindbis virus as indicated in the text. For experiments shown in Figures 1D and 1E, organotypic hippocampal slices were similarly prepared from GluA1-deficient mice (c57bl6/129 hybrid background, obtained from Dr. R. Huganir (Kim et al., 2005)). For experiments shown in Figures 5A and 5B, organotypic hippocampal slices were similarly prepared from PSD-95-deficient mice or their WT littermates.
Biochemistry
Western blotting experiments shown in Figure 1B were performed using primary cortical neurons. At 12-15 DIV, neurons were infected with Sindbis viruses expressing CT84/CT100 along with nuclear m-Cherry. 24h later, neurons were washed in HBSS solution for 5min and then lysed in ice-cold lysis buffer (in mM: 50 Tris-HCl pH 7.6, 150 NaCl, 2 EDTA, 1 PMSF; 1% Triton X-100, protease + phosphatase inhibitors (Sigma)). Cell lysates were very briefly vortexed and centrifuged at 2000x g for 10 minutes. The supernatant was collected and protein concentrations were measured using the BCA assay (Pierce) and normalized. Samples were separated on 10%–20% Tris-Tricine gels (Bio-Rad, #456-3115) and transferred onto 0.2 μm nitrocellulose membranes (Biorad). Samples were blotted in 1% milk for 48h at 4°C with gentle rocking against actin (1:1000 dilution), mCherry (1:1000 dilution) and Aβ species (6E10 antibody, 1:100 dilution). Appropriate secondary antibodies (HRP conjugates, Santa Cruz Biotechnologies) were used at a 1:5000-10000 dilution and blotted for one hour at room temperature with gentle rocking. ECL Prime (GE Healthcare Lifesciences) was used for chemiluminescence.
Transfection and infection
For experiments shown in Figures 1 and 5, CT100, CT100 + PSD-95 or CT84 + PSD-95 were expressed in CA1 neurons of organotypic hippocampal slices using Sindbis virus. Similarly, for Figure S7, human full-length APP was expressed using Sindbis. Virus was injected into CA1 of 6-8 days in vitro (DIV) slice cultures, and recombinant proteins were allowed to express for ~24h before recording. Primary hippocampal neurons were transfected at DIV 7-10 using Lipofectamine 2000 as previously reported (Dore et al., 2015). Briefly, ~2 μg of total DNA (GluN1-GFP, GluN2B and GluN1-mCherry for Figures 2 and 4; for Figure 3, PP1γ-mCherry was used instead of GluN1-mCherry) and 4 μL of Lipofectamine 2000 was used per well. GluN1-GFP and GluN1-mCherry were a kind gift of Paul De Koninck and are both derived from the GluN1-GFP described in (Rao and Craig, 1997); the GluN1 isoform in these constructs is GluN1-2a (formerly named NR1-C). 18-24h prior to imaging, neurons were infected with Sindbis viruses to express the APP derived peptides CT84 and CT100 (along with PSD-95 when specified) by adding 1-2uL of virus to 2mL of neuronal culture media. 7CK (100 μM final concentration) was added at the same time as infection. For experiments shown in Figure S5, 1 μM Palmostatin B (final concentration) was added to neurons infected with CT84 or CT100, 3 hours prior to imaging. 1 μM Palmostatin B was also added to the imaging solution.
Fluorescence lifetime imaging
Neurons expressing the desired constructs were imaged at 14-18DIV in a HBSS based solution containing: 0.87x HBSS, 5mM HEPES, 1mM Glucose, 2.5mM MgCl2, 0.5mM CaCl2, 0.1mM 7CK. To obtain data presented in Figure 5C, neurons were first imaged in the HBSS based solution, 3 μM Palmostatin B was added; then 1h later the same neurons were imaged a second time. For control neurons, the same volume of DMSO (vehicle) was added to the imaging solution. Fluorescence lifetime imaging was performed on a SliceScope two-photon microscope (Scientifica, UK) as previously described (Dore et al., 2015). Briefly, a Chameleon Ultra II IR laser (Coherent) tuned at 930 nm was used for the excitation (power was adjusted to 3mW after the microscope objective (LUMPLFLN 60XW, NA = 1.0, Olympus)). Fluorescence emission was detected with a hybrid PMT detector (HPM-100-40, Becker and Hickl, Germany) and synchronized by a TCSPC module (SPC-150, Becker and Hickl). The following parameters were kept constant for all acquired images: pixel size (80 nm; all 512 × 512 pixels), pixel dwell time (3.2 μs), FLIM acquisition time (~120 s/image), and number of time bins (256) in the fluorescence decay curves.
Electrophysiological recordings
Hippocampal organotypic slices were used for electrophysiological recordings shown in Figures 1, 5, S2, S6, S7, and S8 (see “Organotypic slice cultures” section for more details). For experiments shown in Figures 5A and 5B, 1 μM Palmostatin B (final concentration) was added to slices infected with CT100, 3 hours prior to recordings, 1 μM Palmostatin B was also added to external solution. Control slices, expressing CT100 but without Palmostatin B incubation, were interleaved. Simultaneous whole-cell recordings were obtained from two neurons, one infected and one neighboring control CA1 pyramidal neurons under visual guidance using differential interference contrast and fluorescence microscopy. Two stimulating electrodes (contact Pt/Ir cluster electrodes (Frederick Haer)), were placed between 100 and 300 μm down the apical dendrite, 100 μm apart, and 200 μm laterally in opposite directions. Whole-cell recordings were obtained with Axopatch-1D amplifiers (Molecular Devices) using 3 to 5 MΩ pipettes with an internal solution containing (in mM) 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na2ATP, 0.4 Na3GTP, 10 sodium phosphocreatine (Sigma), and 0.6 EGTA (Amresco), at pH 7.25. External perfusion consisted of artificial cerebrospinal fluid containing (in mM) 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, 0.004 2-chloroadenosine (Sigma), and 0.1 picrotoxin (Sigma) (pH 7.4), and gassed with 5% CO2/95% O2 at 27°C. The AMPAR-mediated excitatory postsynaptic current (EPSC) was measured as peak inward current at −60 mV. For experiments shown in Figure S1, a similar approach (using the same equipment, recording pipettes, and internal solution) was used to obtain whole-cell recordings of spontaneous miniature excitatory postsynaptic currents (sEPSCs) in 8-12DIV primary hippocampal neurons held at −60mV. External perfusion consisted of HBSS based solution containing: 0.87x HBSS, 5mM HEPES, 1mM Glucose, 1mM MgCl2, 1mM CaCl2, 0.4 μM 2-chloroadenosine, and 10 μM gabazine (Tocris) (pH 7.4). sEPSCs were recorded for 1-10min and events were analyzed manually using the MiniAnalysis program (Synapto-soft) blind to experimental conditions; event threshold was set at 6-8pA.
Immunohistochemistry
For experiments shown in Figures 3C, 3D, 4A, 4B, and 5D, live neurons were washed in HBSS based imaging solution (see Fluorescence lifetime imaging section above) for 5min, fixed in 4% paraformaldehyde (PFA) in HBSS for 10min. PFA reaction was then quenched by 5min incubation in 0.1M Glycine in PBS. Neurons were placed in a blocking/permeabilization solution containing 2% goat serum and 0.1% Triton X-100 in PBS for 30min, followed by immunostaining with primary antibodies (1/100 in the blocking/permeabilization solution for PSD-95 or P-Thr320-PP1 and PP1γ; overnight at 4°C). Samples were washed 3 times in PBS before secondary antibodies were applied (1/1000 of GAM-AF647 and/or GAR-AF488 for 1h at room temperature, from Life Technologies). After 3 finals washes in PBS, samples were mounted in Prolong Gold mounting media (Life Technologies). Neurons expressing the indicated constructs (identified with nuclear-mCherry expression) were imaged on an Olympus FV-1000 confocal microscope with a 60X oil immersion objective. Software recommended filters were used for each dye to acquire z stacks with a 0.5 μm separation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence lifetime images were analyzed with SPCImage (Becker and Hickl) using a binning factor between 6 and 10 pixels, minimum threshold of 10 photons at the peak time bin, a single exponential model and used the same calculated instrumental response function for each set of experiments (see (Dore et al., 2015) for more details). For further analysis, each FLIM image was exported as a matrix of lifetimes, photon counts, and goodness of fit values (chi-square) and analyzed blind to condition with a custom MATLAB script (see (Dore et al., 2015) for details). To calculate FRET efficiency the following formula was used: EFRET = 1-TDA/TD (TDA = lifetime of the donor in the presence of the acceptor (GluN1-GFP expressed with GluN1-mCherry (Figures 1, 2, 3, and 5) or with PP1γ-mCherry (Figure 4B)), TD = average lifetime of the donor alone; GluN1-GFP).
For immunohistochemistry experiment analysis, Fiji was used to generate maximum projections of the acquired z stacks. Then, all visible dendritic spines were hand traced on either the total PP1 (AF647, Figures 3C and 3D) or the GFP (Figures 4A, 4B, and 5D) channel. ROIs were pasted onto other channels and the mean intensity (after background subtraction) and area of each spine was measured. Data are presented as mean ± standard error of the mean (SEM) and t test (paired or unpaired, as indicated) was used to determine statistical significance. Please note that statistical details are found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-actin monoclonal | Santa Cruz Biotechnologies | sc-8432; RRID:AB_626630 |
| Mouse anti-beta-amyloid 1-16, Clone 6E10 | BioLegend | 803001; RRID:AB_2564653 |
| Rabbit anti-mCherry polyclonal | Abcam | ab167453; RRID:AB_2571870 |
| Mouse anti-PSD-95 monoclonal | Thermo | 6G6-1C9; RRID:AB_325399 |
| Mouse anti-PP1γ monoclonal | Santa Cruz Biotechnologies | sc-515943 |
| Rabbit anti-Phospho-PP1 (Thr320) | Cell Signaling | 2581; RRID:AB_330823 |
| Bacterial and virus strains | ||
| pSinRep5-CT84-T2A-NLS-mCherry | In house | – |
| pSinRep5-CT100-T2A-NLS-mCherry | In house | – |
| pSinRep5-dp-PSD95-CT84-T2A-NLS-mCherry | In house | – |
| pSinRep5-dp-PSD95-CT100-T2A-NLS-mCherry | In house | – |
| pSinRep5-dp-C3,5S-PSD95-CT100-T2A-NLS-mCherry | In house | – |
| pSinRep5-GFP | In house | Wei et al., 2010 |
| pSinRep5-APP695-IRES-GFP | In house | Hsieh et al., 2006 |
| Chemicals, peptides, and recombinant proteins | ||
| 7-Chlorokynurenic acid sodium salt | Tocris | 3697 |
| Gamma-secretase inhibitor L685-456 | Sigma | L1790 |
| Palmostatin B | EMD Millipore | 17-850 |
| D-APV | Tocris | 0106 |
| GS21 | MTI-GlobalStem | GSM-3100 |
| Papain | Sigma | P4762 |
| DNase 1 | Invitrogen | 18047-019 |
| Experimental models: Organisms/strains | ||
| Rat: Sprague-Dawley | Envigo | – |
| Mouse: GluA1 knock-out | From R. Huganir | Kim et al., 2005 |
| Mouse: PSD-95 knock-out | From R. Huganir | Béïque et al., 2006 |
| Recombinant DNA | ||
| GluN1-GFP | From P. De Koninck | Dore et al., 2015 |
| GluN1-mCherry | From P. De Koninck | Dore et al., 2015 |
| GluN2B | In house | Dore et al., 2015 |
| PP1γ-mCherry | Addgene | 45220 |
| Software and algorithms | ||
| MATLAB | Mathworks | R2011b |
| Fiji | ImageJ | – |
Highlights.
Elevated PSD-95 prevents Ab-induced synaptic depression
Aβ-triggered metabotropic NMDA receptor signaling is blocked by elevated PSD-95
Synapses with high amounts of endogenous PSD-95 are protected from Aβ
Drug inhibition of PSD-95 depalmitoylation reverses Aβ effects on synapses
ACKNOWLEDGMENTS
This work was supported by UCSD School of Medicine Microscopy Core grant P30 NS047101, NIH grant R01MH049159, and Alzheimer’s Disease Research Foundation grant 20192193 (to R.M.). We also would like to thank Nguyen Tran, Sacha DePoyen Brown, and Andrew Pham for help with data analysis.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109194.
REFERENCES
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, and Gouras GK (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Andrew RJ, Fernandez CG, Stanley M, Jiang H, Nguyen P, Rice RC, Buggia-Prévot V, De Rossi P, Vetrivel KS, Lamb R, et al. (2017). Lack of BACE1 S-palmitoylation reduces amyloid burden and mitigates memory deficits in transgenic mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 114, E9665–E9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Miko I, Oviedo H, Mikeladze-Dvali T, Alexandre L, Sweeney N, and Bredt DS (2001). Electron microscopic immunocytochemical detection of PSD-95, PSD-93, SAP-102, and SAP-97 at postsynaptic, presynaptic, and nonsynaptic sites of adult and neonatal rat visual cortex. Synapse 40, 239–257. [DOI] [PubMed] [Google Scholar]
- Aow J, Dore K, and Malinow R (2015). Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc. Natl. Acad. Sci. USA 112, 14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béïque JC, Lin DT, Kang MG, Aizawa H, Takamiya K, and Huganir RL (2006). Synapse-specific regulation of AMPA receptor function by PSD-95. Proc. Natl. Acad. Sci. USA 103, 19535–19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R, Fenn RH, Barren C, Tanzi RE, and Kovacs DM (2016). Palmitoylated APP Forms Dimers, Cleaved by BACE1. PLoS ONE 11, e0166400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum JH, Bali J, Rajendran L, Nitsch RM, and Tackenberg C (2015). Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 6, e1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budson AE, and Price BH (2005). Memory dysfunction. N. Engl. J. Med 352, 692–699. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, and Garaschuk O (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, and Konnerth A (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 109, 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos FJ, Ampuero E, Jury N, Aguilar R, Falahi F, Toledo J, Ahumada J, Lata J, Cubillos P, Henríquez B, et al. (2017). Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain 140, 3252–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, Wang C, Anwyl R, and Wang Q (2013). Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 253, 435–443. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, and Scheff SW (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. [DOI] [PubMed] [Google Scholar]
- Dore K, and Malinow R (2021). Elevated PSD-95 Blocks Ion-flux Independent LTD: A Potential New Role for PSD-95 in Synaptic Plasticity. Neuroscience 456, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré K, Labrecque S, Tardif C, and De Koninck P (2014). FRET-FLIM investigation of PSD95-NMDA receptor interaction in dendritic spines; control by calpain, CaMKII and Src family kinase. PLoS ONE 9, e112170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K, Aow J, and Malinow R (2015). Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA 112, 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, and Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J. Neurosci 24, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, and Malinow R (2007). PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. USA 104, 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, and Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368. [PubMed] [Google Scholar]
- El-Husseini AE-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, and Bredt DS (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863. [DOI] [PubMed] [Google Scholar]
- Gold CA, and Budson AE (2008). Memory loss in Alzheimer’s disease: implications for development of therapeutics. Expert Rev. Neurother 8, 1879–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, and Cole GM (2004). Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am. J. Pathol 165, 1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Harris KM, and Stevens JK (1989). Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci 9, 2982–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Sun L, Siddoway BA, Petralia RS, Yang H, Gu H, Nairn AC, and Xia H (2013). Synaptic NMDA receptor stimulation activates PP1 by inhibiting its phosphorylation by Cdk5. J. Cell Biol 203, 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, and Malinow R (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyifous O, Lin EI, Chen X, Antinone SE, Mastro R, Drisdel R, Reese TS, and Green WN (2016). Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc. Natl. Acad. Sci. USA 113, E8482–E8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Nabavi S, and Malinow R (2013). Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 110, 4033–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Takamiya K, Petralia RS, Sattler R, Yu S, Zhou W, Kalb R, Wenthold R, and Huganir R (2005). Persistent hippocampal CA1 LTP in mice lacking the C-terminal PDZ ligand of GluR1. Nat. Neurosci 8, 985–987. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, and Malinow R (2006). Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J. Neurosci 26, 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, and Seeburg PH (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, and Seeburg PH (1994). Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci 14, 3180–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, and Conibear E (2015). ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. eLife 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren JL, Vandermeulen L, Sandebring-Matton A, Ahmed S, Winblad B, Di Luca M, Tjernberg LO, Marcello E, and Frykman S (2020). Proximity ligation assay reveals both pre- and postsynaptic localization of the APP-processing enzymes ADAM10 and BACE1 in rat and human adult brain. BMC Neurosci. 21, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, and Cravatt BF (2009). Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW Jr., and Morris JC (2001). Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56, 127–129. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, and Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, and Malenka RC (2001). Regulation of synaptic strength by protein phosphatase 1. Neuron 32, 1133–1148. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, and Malenka RC (1993). An essential role for protein phosphatases in hippocampal long-term depression. Science 261, 1051–1055. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, and Malinow R (2013). Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 110, 4027–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault F, and De Koninck P (2010). Dissociated Hippocampal Neurons, Fourth Edition (Humana Press; ). [Google Scholar]
- Niethammer M, Kim E, and Sheng M (1996). Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci 16, 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KM, and Sheng M (2012). NMDA receptors and BAX are essential for Aβ impairment of LTP. Sci. Rep 2, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlovsky I, Huijbers W, Hanseeuw BJ, Mormino EC, Hedden T, Buckley RF, LaPoint M, Rabin JS, Rentz DM, Johnson KA, et al. (2017). The relationship between recall of recently versus remotely encoded famous faces and amyloidosis in clinically normal older adults. Alzheimers Dement. (Amst.) 10, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P (2011). Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci 33, 1351–1365. [DOI] [PubMed] [Google Scholar]
- Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, Fernandez-Carballo L, de Munain EL, Perez J, Marquie M, et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, and Craig AM (1997). Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19, 801–812. [DOI] [PubMed] [Google Scholar]
- Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, and Kessels HW (2016). Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 113, E6526–E6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, and Sabatini BL (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao CY, Mirra SS, Sait HB, Sacktor TC, and Sigurdsson EM (2011). Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 122, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, and Nicoll RA (2003). Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci 23, 5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Gray JA, and Zito K (2015). Non-Ionotropic NMDA Receptor Signaling Drives Activity-Induced Dendritic Spine Shrinkage. J. Neurosci 35, 12303–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y (2012). Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 11, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, and Mayeux R (1994). Influence of education and occupation on the incidence of Alzheimer’s disease. JAMA 271, 1004–1010. [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, and Muller D (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. [DOI] [PubMed] [Google Scholar]
- Tamburri A, Dudilot A, Licea S, Bourgeois C, and Boehm J (2013). NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS ONE 8, e65350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Norman ED, Barrionuevo G, and Klann E (1998). Transient and persistent increases in protein phosphatase activity during long-term depression in the adult hippocampus in vivo. Neuroscience 86, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Topinka JR, and Bredt DS (1998). N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20, 125–134. [DOI] [PubMed] [Google Scholar]
- Valenzuela MJ, and Sachdev P (2006). Brain reserve and dementia: a systematic review. Psychol. Med 36, 441–454. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, and Malinow R (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci 13, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, and Scott JD (1999). Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285, 93–96. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, et al. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci 30, 2636–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, and Malenka RC (2008). Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron 57, 248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, and Fukata M (2016). Identification of PSD-95 Depalmitoylating Enzymes. J. Neurosci 36, 6431–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.