

Abstract
‘Oxidative and Nitrative Stress in Toxicology and Disease’ was the subject of a symposium held at the EUROTOX meeting in Dresden 15th September 2009. Reactive oxygen (ROS) and reactive nitrogen species (RNS) produced during tissue pathogenesis and in response to viral or chemical toxicants, induce a complex series of downstream adaptive and reparative events driven by the associated oxidative and nitrative stress. As highlighted by all the speakers, ROS and RNS can promote diverse biological responses associated with a spectrum of disorders including neurodegenerative/neuropsychiatric and cardiovascular diseases. Similar pathways are implicated during the process of liver and skin carcinogenesis. Mechanistically, reactive oxygen and nitrogen species drive sustained cell proliferation, cell death including both apoptosis and necrosis, formation of nuclear and mitochondrial DNA mutations, and in some cases stimulation of a pro-angiogenic environment. Here we illustrate the pivotal role played by oxidative and nitrative stress in cell death, inflammation and pain and its consequences for toxicology and disease pathogenesis. Examples are presented from 5 different perspectives ranging from in vitro model systems through to in vivo animal model systems and clinical outcomes.
1. INTRODUCTION
A symposium was held at the EUROTOX meeting in Dresden September 2009 entitled ‘Oxidative and Nitrative Stress in Toxicology and Disease’. In this lively and well-attended session, inflammation and the role of reactive oxygen and reactive nitrogen species (ROS and RNS) were discussed initially followed by illustrative examples from diverse tissue and model systems. The concept behind the session was to seek cross-fertilisation between the different illustrative examples, revealing common themes.
Inflammation begins when tissues react to a local irritation usually caused by a physical injury, by infection or by exposure to a toxicant. Fluid and accompanying white blood cells traverse the vascular barrier leading to swelling, erythema, further inflammation and attraction of further white blood cells (Figure 1). The resolution phase of inflammation involves repair and cell proliferation ultimately leading to tissue regeneration. During the inflammatory phase, there is a burst of respiration leading to the creation and release of free radicals, including production of both reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS utilize three main pathways of signalling which results in damage to DNA, proteins and lipids. Lipid peroxidation triggers the arachidonic-acid cascade, with the production of cell-proliferation-stimulating eicosanoids. While free radicals induce DNA damage directly, byproducts of the arachidonic acid pathway such as malondialdehyde (MDA) and 4-hydroxynonenol (4-HNE) also are DNA-damaging agents. Free radicals can also modify enzyme systems involved in DNA repair and alter cell death signalling by modification of caspases and modulation of cell survival pathways regulated by key molecules such as JNK and p38 MAP kinase (reviewed in Aslan et al 2008, Roberts et al, 2009).
Figure 1: Inflammation and the role of ROS and RNS in tissue damage.

Inflammation begins with a reaction to an irritant or infection that is characterized by movement of fluid and white blood cells into extravascular tissue. This is followed by tissue repair and regeneration and involves cell proliferation. Associated with these processes are the release of free radicals, such as reactive oxide species (ROS) and reactive nitrogen species (RNS). This can activate a process called lipid peroxidation and the arachidonic-acid cascade, with the production of cell-proliferation-stimulating eicosanoids. Also, DNA-damaging agents, such as malondialdehyde (MDA) and 4-hydroxynonenol (4-HNE), are byproducts of the arachidonic-acid cascade. The free radicals can also damage DNA and modify the structure and function of cancer-related proteins. OH•, hydroxyl radical; O2-, superoxide; NO, nitric oxide; ONOO-, peroxynitrite; N2O3, nitrous anhydride.
Although inflammation and the associated generation of free radicals can have a deleterious effect on the host, these processes also play a normal physiological role, especially in the elimination of invading pathogenic organisms. Since free radicals are ubiquitous in our body and are generated during normal physiological processes, mutiple defense systems have evolved to protect our cells from these potentially damaging free radicals. Antioxidant enzymes and scavenger systems serve to remove free radicals, whereas DNA repair pathways are triggered to repair damage to DNA after it has occurred.
It is clear that in certain situations of toxicant exposure or disease, the generation of ROS and RNS and the associated oxidative and nitrative stress are key mediators of inflammation, cell proliferation and cell death. In this review, evidence is presented in diverse tissues including the liver, brain, heart and skin in order to highlight common themes.
2. Oxidative and Nitrative stress: Role in the Response to Liver Toxicants (Ruth A Roberts)
The liver is the front line of defence and is therefore a target organ for many toxicants. Persistent inflammation plays a pivotal role in the response of liver to both chemical and viral damage, ranging from transient liver injury and repair through to hepatocarcinogenesis (Figure 2).
Fig 2: Liver damage, inflammation and cancer: lessons from PPARα.

A schematic depicting the pathway from toxicant exposure to inflammation and ultimately to tumor development. Inflammation can be triggered by toxicant exposure or by viral infection; if this inflammation persists it can progress to the development of tumors. Similalry, exposure of rodent liver to peroxisome proliferators causes activation of PPARα and hepatic growth changes ultimately leading to hepatocellular carcinoma. Cytokines such as TNFα and survival signalling molecules such as NFκB are implicated in both processes.
There is a major drive to understand the mechanisms of toxicant and viral-induced liver disease to facilitate prevention and treatment and also to understand the molecular basis of individual susceptibility. WHO mortality data for the Americas (WHO, 2003) show liver cirrhosis as responsible for 4% of deaths in the 45–64 age group, making it the 3rd most common cause of death. As illustrated by these figures, cirrhosis itself is a major health issue which is exacerbated by the association of chronic cirrhosis with quite frequent progression to hepatocellular adenoma and carcinoma.
There are multiple causes of cirrhosis and associated carcinogenesis, with the majority of cases attributed to alcohol, viral infection or a fatty diet (Ryder and Beckingham, 2001). The statistics surrounding Hepatitis B Infection are shocking; approximately third of the world’s population (~ 2 billion people) have been infected with hepatitis B, with 600 000 deaths each year due to the acute or chronic consequences of viral infection and 25% of adults who become chronically infected during childhood later die from liver cancer or cirrhosis (WHO 2003).
Accumulating evidence implicates the dedicated hepatic macrophage, the Kupffer cell, as a key mediator of the response to chemical and viral insult as well as other liver stresses such as transplantation (Krohn et al., 2009). Examination of the liver architecture (reviewed in Roberts et al., 2007) shows that the location of Kupffer cells, which are distributed around the key interface between the parenchyma, the sinudoidal space and the portal space allow these specialized macrophages to respond to incoming toxicants by stimulation of parenchyma.
The primary mechanism for the stimulation of parenchyma is via release of cytokines (Krohn et al., 2009; McVicker et al., 2007) and ROS (Rusyn, 2001; Tsuchiya et al., 2008) and RNS (Powell et al., 2006). The release of ROS by Kupffer cells was well illustrated recently by Lin et al (2009) who showed an increase in hepatic ROS during D-galactosamine – induced acute liver injury and apoptosis. Interestingly, staining for 4-HNE which identified oxidized protein revealed co-localization with lipid accumulation in these livers (Lin et al., 2009). Similarly, Powell et al (2006) reported that a subtoxic dose of acetaminophen significantly increased nitrotyrosine adducts in rat liver in vivo, especially in the centrilobular region, as a marker for peroxynitrite formed by the combination of the ROS, superoxide anion, and nitric oxide (NO). Thus, Kupffer cells orchestrate the hepatic response to chemical and viral insult via production and release of many mediators including ROS and RNS. So, how do these mediators impact on both normal liver biology and, more importantly, the adverse response to toxicants?
Toxicants cause tissue damage via diverse mechanisms, many of which involve activation of cell survival and apoptosis pathways. Two broad categories of apoptosis networks, referred to as the intrinsic and extrinsic pathways have been identified (reviewed in Aslan, et al. 2008). The extrinsic pathway is driven by signalling from death receptors such as the tumour necrosis factor a (TNFα) receptor and Fas through the death inducing signalling complex (DISC) and caspase 8. The intrinsic pathway is triggered by cellular stress and signals through survival kinases to the mitochondria which then engage caspase 9 through cytochrome c release. Both pathways converge on caspase 8 leading to cellular execution (reviewed in Aslan, et al. 2008). Oxidative and nitrative species can impact on these pathways at several key points; reactive nitrogen species (RNS) and reactive oxygen species (ROS) can modulate survival signalling molecules such as JNK and p38 MAP kinase (reviewed in Aslan et al 2008, Roberts et al., 2009). Similarly RNS such as nitric oxide (NO) can induce DNA damage, leading to p53 stabilisation and engagement of apoptosis pathways. Conversely, NO down regulates NFκB which protects from apoptosis often via perturbation of hepatocyte proliferation and death via apoptosis and necrosis (reviewed in Roberts et al 2007).
2.1. Carcinogenesis and Inflammation
As described, Kupffer cells detect and respond to viral and chemical stress by release of ROS, RNS and cytokines that in turn regulate cell survival and proliferation. So how would this perturbation of cell growth lead to carcinogenesis? These concepts are well described in the early publications on multistage carcinogenesis models (Weinstein et al., 1984; Cohen 1984), as summarised briefly here. First, in order to proliferate, a cell has to survive; thus any cell that should have died due to DNA damage but persists due to perturbation of signalling remains to be stimulated into proliferation possibly with an enhanced risk of mutation due to missense – mismatch repair (Radman and Wagner, 1993). These cells are then subjected to further mitotic stimulation and genetic instability progressing through to cancer. Overall, this explanation leads to the interesting question, ‘is cancer linked to unresolved inflammation?’
One of the most interesting papers to address this point looked at the immune pathogenesis of hepatocellular carcinoma (HCC) in response to HBV infection (Nakamoto et al., 1998). As mentioned earlier, HCC is a common complication of HBV infection and it has long been assumed that the clonal integration of viral DNA into hepatocytes would play a key role in the process. However, these authors showed that HCC occurred in the absence of viral transactivation and insertional mutagenesis. In contrast, the immune response to HBV was shown to be critical for tumour development. Overall, these data suggest that the HCC accompanying chronic hepatitis may result from the immune response to viral infection possibly mediated by Kupffer cells. In this respect, non-genotoxic and viral carcinogenesis may share common features.
2.2. Cross-talk with PPARα?
In addition to the regulation of hepatocyte survival and proliferation by ROS and RNS released by Kupffer cells, there is the possibility of complex interplay between ligand-activated receptors such as the peroxisome proliferators activated receptors (PPARs) which appear to play different roles in Kupffer cells versus the hepatocytes. PPARα is the receptor for the peroxisome proliferators (PPs), a diverse group of rodent nongenotoxic hepatocarcinogens of broad environmental and therapeutic significance (see Oliver and Roberts, 2002 for review). In rodents, PPs induce hepatocyte proliferation and suppress apoptosis mediated by PPARα, ultimately leading to clonal expansion and tumours. Both in vitro and in vivo experiments have shown that the hepatic response to PPs is absolutely dependent upon Kupffer cells (Hasmall et al., 2002, Parzefall et al., 2001) but interestingly, Kupffer cells do not have to express PPARα to support these changes in proliferation (Hasmall et al., 2002) Indeed, in a separate series of experiments, it was demonstrated that PPARα is absent in Kupffer cells that instead express PPARγ (Peters et al., 2000).
Overall, it’s clear that oxidative and nitrative stress play a pivotal role in the response to liver toxicants and viral injury. This is often orchestrated by complex interplay between the non-parenchymal Kupffer cells and hepatocytes with cross-talk between survival mechanisms and transcriptional regulation. Further development of our understanding of these mechanisms provides increased opportunities in the future for vital work on prevention, intervention and treatment.
3. Characterization of Oxidative Stress Using Neuronal Cell Culture Models (Robert A Smith)
The brain consumes more oxygen under physiological conditions than other organs, thereby increasing its susceptibility to oxidative stress since generation of higher levels of ROS can lead to pathological changes when these are in excess of the buffering capacity of endogenous antioxidant systems (Fatokun et al., 2008a). Excessive stimulation of glutamate receptors, especially N-methyl-D-aspartate (NMDA) receptors, also contributes significantly to the production of ROS and/or NO. Glutamate toxicity is known to underlie many neurological disorders including Alzheimer’s, Parkinson’s and Huntington’s diseases and stroke (Ischiropoulos and Beckman, 2003): with mitochondrial dysfunction triggering neuronal death pathways and subsequent neuronal loss in such conditions (Trushina and McMurray, 2007). Fully elucidating the mechanisms involved remains pivotal in the development of strategies to ameliorate these neurodegenerative disorders (Fatokun et al., 2008a).
Much data pertaining to oxidative and nitrative stress in the nervous system comes from postmortem findings and experimental mammalian in vivo studies. In view of the complexities of the nervous system however, mechanistic studies of oxidative stress have benefitted also from using reductive in vitro culture models. Effects on neuronal and glial components can be investigated in both combined mixed cultures or using individual cell types in culture to monitor specific responses. The merits and limitations of different cell systems appropriate to neurtoxicological approaches have recently been reviewed (see Smith, 2009).
Continuous cell lines are available for the elucidation of the death pathways activated by oxidative stress insults. The dopaminergic neuronal cell line, SH-SY5Y, derived from a human neuroblastoma, has been used in many studies since treatments can be applied both to proliferating undifferentiated cells or to cultures triggered to differentiate (Cheung et al., 2009). SH-SY5Y cells have also been used successfully in recent work demonstrating the protection afforded by the naturally occurring antioxidants, resveratrol and oxy-resveratrol, against oxidative stress induced by the parkinsonian mimetic 6-hydroxydopamine (Chao et al., 2008).
Primary cultures, maintained either as neuronal and glial cell co-cultures, or as neuronal- or astrocyte-enriched cultures, have the advantage of expressing a phentoype more closely resembling the in vivo state. They can be routinely prepared from most brain areas in which oxidative stress is of interest, including cortical regions (Tauskela et al., 2008). Postnatal cerebellar granule neurons (CGNs), maintained in culture for at least a week, undergo extensive regrowth of neurite processes and express functional glutamate receptors. As such, they remain as one of the most frequently used model systems for in vitro investigation of excitoxicity and oxidative stress. The cerebellum, comprises of over 95% of this neuronal type, and therefore represents a source of highly purified and homogenous neurons which are known to release glutamate as their principal neurotransmitter (Smith et al., 2003; Ward et al., 2005; Smith et al., 2008). Extensive data on oxidative stress, signalling pathways, cell death and neuroprotection have been generated in many studies using this culture model, including those illustrated in Figure 3 and described below.
Figure 3:

A summary diagram of the oxidative stress insults applied to primary cultures of postnatal cerebellar granule neurons (Smith et al., 2003), the mechanistic and signalling pathways investigated, and the neuroprotective strategies employed to attenuate neuronal damage and loss (see text for details of the individual studies).
Fatokun and co-workers (2007; 2008b) reported on the oxidative stress induced by hydrogen peroxide and glutamate. Means of attenuating the damage resulting from exposure of CGNs to the oxidative stressors were investigated by administrating the adenosine receptor A1 agonist, N6-cyclopentyladenosine, and the A2A antagonist, ZM 241385, 4-(2-[7-amino 2{furyl}{1,2,4}trichloro{2,3-a} {1,3,5}triazin-5-yl-amino] ethyl)phenol, which proved effective in protecting CGNs. The death pathways triggered were a mixed profile of both apoptosis and necrosis (Fatokun et al., 2008b). The free radical-generating system of xanthine and xanthine oxidase, frequently used as a source of the superoxide anion to cause oxidative stress leading to cellular death, was investigated in CGN cultures (Fatokun et al., 2007). Addition of superoxide dismutase failed to prevent damage by xanthine and xanthine oxidase, while catalase completely ameliorated it. When applied alone, xanthine oxidase significantly lowered cell viability, an effect that was blocked by allopurinol and catalase, but not by superoxide dismutase. These observations suggest that hydrogen peroxide, rather than the superoxide anion, mediates the main stress leading to cell death in this system (Fatokun et al., 2007).
The role of stimulation of NMDA receptors in the cell death induced by mitochondrial disruption following exposure to 3-nitroproprionic acid (3-NPA) and potassium cyanide (inhibitors of respiratory chain complexes II and IV respectively) was demonstrated using primary cultured CGNs: the NMDA channel blocker (MK-801), and the antagonist, 2-amino-5phosphopentanoic acid (D-AP5), proving effective, although resistance to blockade by kynurenic acid suggested NMDA receptor activation independent of the glycine site (Fatokun et al., 2008c). Protection of cultured CGNs against subsequent lethal challenges of 3-NPA and glutamate toxicity was also achieved by preconditioning with sublethal doses of NMDA (Smith et al., 2008). Less severe protocols, in which neurons were pre-conditioned by use of longer sublethal exposures with the potassium channel blocker, 4-aminopyridine (4-AP), and the GABAA receptor antagonist, bicuculline, have been shown to protect cortical neurons from oxidative stress by up-regulating bcl-2 expression and by stimulating the phosphorylation of the cyclic-AMP response element binding protein (CREB) (Tauskela et al., 2008). CREB phosphorylation increases in CGNs preconditioned with 4-AP, although activation of bcl-2 and caspase 3 levels did not differ significantly from untreated controls (Smith et al., 2009a).
Investigation of the signalling pathways activated by oxidative stress has been the focus of studies of a number of neurotoxic intermediates generated by the metabolism of tryptophan to nicotinamide via the kynurenine pathway (Smith et al., 2009b). Neuronal loss, resulting from treatments with 3-hydroxykynurenine, 3- or 5-Hydroxyanthranilic acids, appeared to be mediated by H2O2 since the effects were prevented by catalase, but not by superoxide dismutase. Activation of the p38 pathway was demonstrated, whereas caspase-3 levels were not increased, indicating the involvement of a caspase-3 independent mechanism (Smith et al., 2009b). Neuronal culture also permits the quantification of neurite outgrowth and its modification by oxidative stressors which offers a additional endpoint for assessing damage in in vitro studies (Smith, 2009).
Despite the acknowledged limitations implicit in the simplicity of neuronal culture models, primary cells such as CGNs have proved most valuable in generating data that extends our understanding of the mechanisms underlying the response of the nervous system to oxidative stress. When taken together with in vivo studies, it is clear that in vitro culture systems can play a vital role in the development of novel drug therapies targeting pathways associated with oxidative stress, with potential relevance to the management of a number of neurodegenerative conditions.
4. Nitrative Stress and Glial-Neuronal Interactions in the Pathogenesis of Parkinson’s disease (PD) (Ronald B Tjalkens and Stephen Safe)
Parkinson’s disease (PD) is a severely debilitating movement disorder resulting from progressive degeneration of dopaminergic neurons within the substantia nigra pars compacta of the midbrain. Unfortunately, pharmacologic treatment for PD has not progressed beyond the use of dopamine mimetics, such as L-dopa, that only transiently alleviate motor symptoms. Furthermore, chronic use of L-dopa is associated with its own array of resultant pathologies such as dyskinesia, cardiac arrhythmia and ischemic injury, and cerebral vascular dysfunction. Ultimately, individuals suffering from PD will progress to the end stage of the disease, which is characterized by significant gait abnormalities and frequent falls, as well as a deficit in non-motor functions resulting in dementia, psychosis, and other autonomic disturbances (Djaldetti et al., 2004).
Currently, a precise etiology explaining PD remains to be discovered but recent research has revealed features of the disease that represent realistic targets for neuroprotective chemotherapeutic intervention that could mitigate the progressive loss of dopaminergic neurons Tjalkens et al., 2008. Among these observations are the presence of chronic inflammation and sustained expression of inducible nitric oxide (NOS2), accompanied by activation of the surrounding astrocytes and microglia (Teismann and Schulz, 2004).
4.1. Neuroinflammation and PD
Neuroinflammation is now understood to play a critical role in the progression of PD. Unfortunately, current therapies do not address this problem, being focused on ameliorating the symptoms of dopamine loss rather than on targeting the underlying causes of injury to dopaminergic neurons. Thus, there remains no ‘disease modifying’ therapeutic that has been approved for the treatment of PD. Although much attention has been devoted to understanding the role that activated microglia have in neuroinflammation, it has become clear that a more complex interplay between microglia and astrocytes underlies the neuroinflammatory phenotype observed in PD (Tansey et al., 2008). Both microglia and astrocytes produce neurotoxic levels of inflammatory mediators such as TNFα and NO that are increased in PD patients and in animal models of the disease (Good et al., 1998). However, recent clinical trials aimed at suppressing microglial-mediated inflammation have, to date, yielded poor results. Thus, a better understanding of fundamental inflammatory pathways and cell-cell interactions between microglia and astrocytes is required to decipher the pathogenesis of neuroinflammation in degenerative CNS disorders such as PD. Although the pro-inflammatory role played by microglia in PD has been extensively studied, the neuroinflammatory phenotype of activated astrocytes is less well understood.
Astrocytes have diverse and critical functions in the CNS that include providing metabolic substrates to neurons, antioxidant protection, and the synthesis of trophic factors essential for the survival and function of neurons (Hermann et al., 2001; Hunot et al., 1996). However, many neurological disease states, including PD, Alzheimer’s disease, and ischemic injury are typically accompanied by varying degrees of astrocyte activation, or astrogliosis. While the exact cause of astrogliosis in PD is unknown, several reports have suggested that the activation of astrocytes is due to secretion of inflammatory cytokines, such as TNFα and Interferon gamma (IFNγ), by the surrounding microglial cells (Liberatore et al., 1999). While some degree of activation is likely beneficial, reactive astrogliosis results in neuronal injury. Astrogliosis results in increased production of various neurotoxic inflammatory mediators, including NO, that contribute to the progressive loss of nigro-striatal neurons. Supporting a deleterious role for excessive NO production in PD are postmortem observations of increased NOS2 expression in patients diagnosed with PD (Ebadi and Sharma, 2003), as well as reports that deletion of the Nos2 gene in mice confers protection against MPTP-mediated neurotoxicity (Liberatore et al., 1999).
4.2. Regulation of Neuroinflammatory Genes in Astrocytes
Expression of NOS2 in response to diverse neurotoxicants is highly dependent upon the NF-κB signaling pathway. Recent studies demonstrated that NF-κB is required for expression of NOS2 in activated astrocytes following stimulation with various inflammatory cytokines and neurotoxicants (Carbone et al., 2009; Moreno et al., 2008). Multiple signaling pathways activate NF-κB through the IκB kinase (IKK) complex, leading to phosphorylation and degradation of the inhibitory IκB subunit and nuclear translocation of the transcriptionally active p65 subunit. Ensuing induction of Nos2 mRNA then typically requires binding of p65 to enhancer sequences on the Nos2 promoter, acetylation of histones, and removal of constitutively bound nuclear co-repressor proteins such as NCoR2 by the nuclear proteosome (Ghisletti et al., 2007). Increasing attention is being given to NF-κB as a therapeutic target for blocking neuroinflammation in PD because of its central role in inflammatory signaling in glia. Recent data from transgenic animals containing microglial- and astrocyte-specific gene deletion of NF-κB support the neuroprotective efficacy of blocking this signaling pathway in models of inflammatory neurodegeneration (Brambilla et al., 2009). The importance of NF-κB-dependent transcriptional regulation of inflammatory genes in PD was highlighted in recent studies demonstrating that the orphan nuclear receptor, Nurr1, normally inhibits NF-κB-regulated neuroinflammatory genes in glia and its deficiency associates with loss of dopaminergic neurons (Saijo et al., 2009). Deficiency in expression of Nurr1 is also associated with a late-onset form of PD (Le et al., 2008), suggesting that nuclear regulators of NF-κB are linked to inflammatory activation of glia and loss of dopaminergic neurons.
4.3. Therapeutic Strategies to Interdict Neuroinflammation
Suppressing neuroinflammation has emerged as a potential strategy for treating disorders such as PD. Specifically, modulation of nuclear orphan receptors has been examined as a possible approach for suppressing inflammatory gene expression in astrocytes using traditional thiazoladinedione (TZD) ligands of PPARγ (Dehmer et al., 2004). The TZD ligand rosaglitazone appears to antagonize NF-κB by stabilizing NCoR2 at the proximal p65 enhancer element in RAW macrophages (Pascual et al., 2005) and another drug in this series, pioglitazone, confers partial neuroprotection in MPTP-induced parkinsonism, preserving dopaminergic cell bodies in the substantia nigra but not dopaminergic fibers in the striatum (Dehmer et al., 2004). The work presented here (Figure 4) demonstrates that a novel para-substituted diindolylmethane compound, 1,1-bis(3-’indolyl)-1-(p-t-butyl)methane (DIM-C-pPhtBu) (Qin et al., 2004), suppresses protein nitration in dopaminergic neurons in vivo caused by exposure to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). It was also recently reported that DIM-C-pPhtBu blocked NF-κB-dependent expression of NOS2 in astrocytes and protected co-cultured neurons from astrocyte-mediated apoptosis following treatment with MPTP and inflammatory cytokines (Carbone et al., 2009). Furthermore, the mechanism underlying DIM-C-pPhtBu-mediated inflammatory suppression differs from the trans-repressive mechanism described for rosiglitazone (Pascual et al., 2005), presenting a potentially novel avenue for suppression of inflammatory genes and neuroprotection using this class of compounds. The anti-neuroinflammatory efficacy of DIM-C-pPhtBu was further examined in astrocytes derived from a unique transgenic mouse expressing a reporter construct consisting of high affinity NF-κB consensus elements driving expression of enhanced green fluorescent protein (EGFP). DIM-C-pPhtBu potently suppressed MPTP-dependent NF-κB activation in transgenic astrocytes isolated from these transgenic mice that directly correlated with inhibition of NOS2 expression (Carbone et al., 2009). Thus, inhibition of key molecular signaling pathways responsible for induction of NOS2, such as NF-κB, may offer a strategy for preventing neuronal injury resulting from excessive production of NO.
Figure 4: Inhibition of NF-κB prevents protein nitration in dopaminergic neurons in vivo.
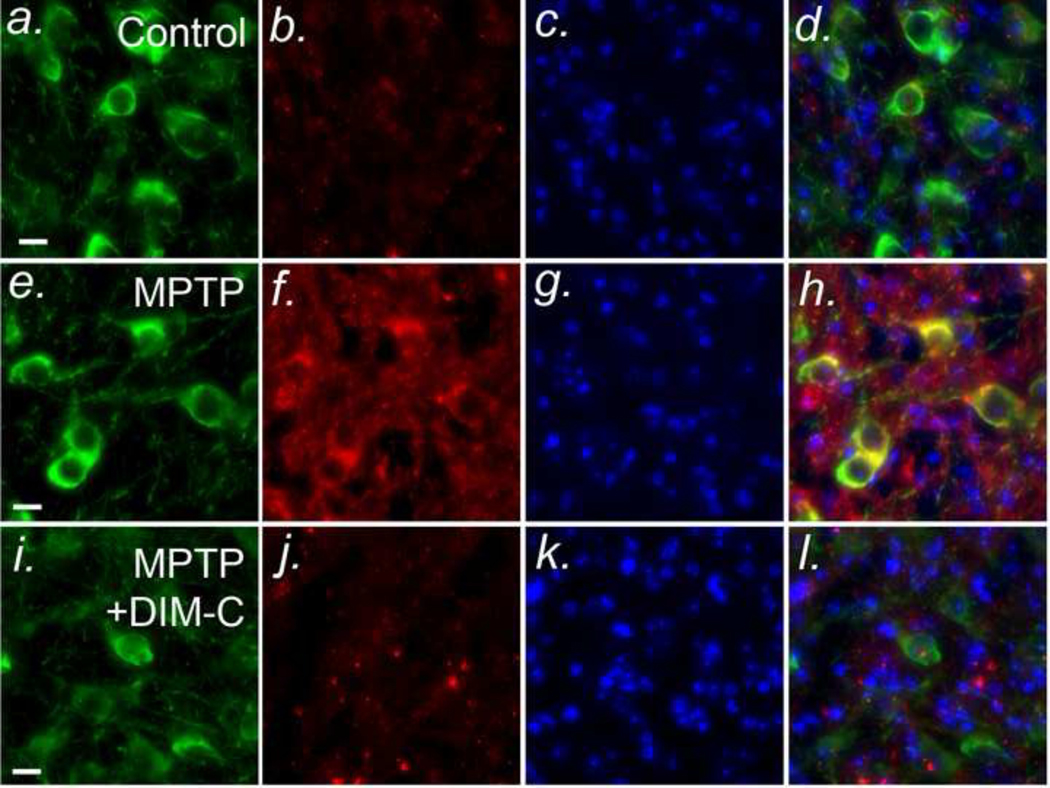
C57Bl/6 mice were treated with MPTP (4×10 mg/Kg, 2 hr intervals) in the presence or absence of DIM-C-pPhtBu (DIM-C) (4 daily doses by intragastric gavage, 50 mg/Kg) and sacrificed after 7 days. Frozen sections were stained by immunofluorescence for typrosine hydroxylase (green), 3-nitrotyrosine (red; an indicator of peroxynitrite formation), and nuclear morphology (DAPI; blue). (a - d) - Control; (e - h) MPTP; (i - l) MPTP + DIM-C-pPhtBu. Yellow staining the merged images in d, h, and l indicates co-localization.
5. Oxidative and Nitrative Stress in Multi-Stage Carcinogenesis (Fredika M Robertson)
The murine model of multistage skin carcinogenesis has been used to define the sequential genetic and epigenetic mechanisms responsible for development of human squamous cell carcinomas (Abel, et al., 2009). This classic model of skin inflammation and carcinogenesis has also been used to identify chemicals that have potential carcinogenic activity and is a primary screening tool to evaluate the toxicological profile of drugs under development. The process of murine multistage carcinogenesis is operationally defined as consisting of discreet steps of initiation, promotion, and malignant progression. Initiation is the first step in inducing growth of skin tumors and occurs following treatment of the dorsal skin of genetically susceptible mice with a chemical or physical carcinogen such as 7,12-dimethylbenz[a]anthracene (DMBA) or exposure to ultraviolet light in the 290–320 nm range, which is uvB light derived from solar irradiation. Exposure to DMBA induces stable, permanent and heritable genetic alterations that consist of specific point mutations within the celluar v-Ha-ras gene, while exposure to uvB light induces signature mutations in the p53 tumor suppressor gene. These mutations occur in “label retaining” epidermal keratinocytes located within both the basal epidermis and in the “bulge region” of the hair follicles of the dermis, which are now recognized to represent the stem cell compartments of adult skin (Affara et al., 2006).
The second stage of skin carcinogenesis is tumor promotion, which is induced by multiple applications of the tumor promoter 12–0-tetradecanolyphorbol-13-acetate (TPA) or by multiple exposures to uvB light. While initiation is characterized by genetic alterations, there are multiple epigenetic changes that are critical to the process of tumor promotion. As an example, tumor promoters stimulate inflammation characterized by the production of reactive intermediates and pro-inflammatory mediators with contributions by multiple cell types, including epidermal keratinocytes, leukocytes and vascular endothelial cells (Figure 5). Multiple types of pro-inflammatory cytokines including Interleukin-1 alpha, tumor necrosis factor and granulocyte macrophage stimulating factor are produced during tumor promotion (Oberyszyn et al., 1993; Robertson, et al., 1996; Robertson et al., 1994). To add to the pro-inflammatory microenvironment in the skin during growth of papillomas and conversion to malignant carcinomas, the process of tumor promotion is associated with the production of ROS including the cell permeant mediator hydrogen peroxide (Robertson et al,, 1990; Sirak et al, 1991; Ariza et al, 1996). ROS are produced in sufficient quantities during tumor promotion to result in formation of signatures of oxidant-mediated DNA damage, detectable by the presence of the sentinel DNA adduct, 8-oxo-deoxyguanosine (8-oxodG).
Figure 5:

Schematic of the roles of reactive oxygen species (ROS), reactive nitrogen species (RNS) oxidative DNA damage, pro-inflammatory cytokines, and vascular endothelial growth factor (VEGF) in multi-stage skin carcinogenesis.
In addition to production of ROS, NO and peroxynitrite (ONOO-) play a role in the process of tumor promotion in the skin (Robertson et al., 1996), and are collectively defined as RNS. Peroxynitrite, formed by the interaction of ROS with NO and detectable by the presence of nitrotyrosine, was produced by infiltrating leukocytes within the dermis during tumor promotion (Robertson FM et al, 1996). In addition, there was a notable absence of NOS2 in hyperplastic epidermis, consistent with the known anti-proliferative effect of NO.
In contrast to the localization of NOS2 to infiltrating leukocytes in the dermis, NOS, also known as endothelial NOS, was localized specifically to the vascular endothelium during tumor promotion. Additionally, the NOS inhibitor L-NG-monomethyl-arginine (L-NMMA) was found to block vascular permeability, suggesting that targeting NOS3 effectively inhibits the early changes in vessel permeability that are characteristic of the “angiogenic switch” during skin carcinogenesis. Data generated showed that VEGF regulates vascular permeability at early times during tumor promotion and at later times in this process, mediates the robust tumor-associated angiogenesis that is a requirement for formation of skin papillomas and their malignant progression to form carcinomas (Affara and Robertson, 2004).
Taken together, these studies suggest that the pro-inflammatory signatures of tumor promotion are regulated in part by pro-inflammatory cytokines, as well as by ROS and RNS. Furthermore, NO and VEGF are interlinked and are an integral part of the process of multistage skin carcinogenesis (Figure 5).
6. Role of Peroxynitrite in The Pathogenesis of Doxorubicin-Induced Cardiotoxicity (Csaba Szabo)
Doxorubicin is a broad-spectrum antitumor anthracycline antibiotic that is commonly used to treat a variety of cancers, including severe leukemias, lymphomas and solid tumors. It continues to be an effective and widely used broad-spectrum chemotherapeutic agent. However, its clinical use is limited because of its serious dose-limiting cardiotoxicity (Blum and Carter, 1974; Young et al., 1981; Singal et al., 1987; Singal and Iliskovic, 1988). Clinical and experimental investigations suggested that increased oxidative stress associated with an impaired antioxidant defense status plays a critical role in subcellular remodeling, calcium-handling abnormalities, alteration of cardiac energetics and subsequent cardiomyopathy and heart failure associated with doxorubicin treatment (Myers et al., 1977; Doroshov et al., 1986; Siveski-Iliskovic et al., 1994; Li and Singal, 2000; Weinstein et al., 2000; Mihm et al., 2002).
Increased expression of NOS2 and formation of nitrotyrosine, a marker of RNS, have been shown to be present in cardiomyocytes after doxorubicin exposure (Weinstein et al., 2000; Mihm et al., 2002; Pacher et al., 2002). The causative role of peroxynitrite in the cardiotoxic effects of doxorubicin is evidenced by the protective effects of peroxynitrite neutralizing agents in experimental models of doxorubicin cardiotoxicity. For instance, studies with FP15, an N-PEGylated-2-pyridyl iron porphyrin, a potent peroxynitrite decomposition catalyst, demonstrated that FP15 exerts cardioprotective effects in various rodent models of doxorubicin cardiotoxicity (Pacher et al., 2002). FP15 attenuated the development of cardiac dysfunction, increased survival and reduced the doxorubicin-induced increase in serum Lactate dehydrogenase (LDH) and creatine phosphokinase (CK) activities, consistent with protection against peroxynitrite-mediated necrosis of cardiac myocytes. FP15 also abolished tyrosine nitration in the hearts of doxorubicin-treated animals (Pacher et al., 2002a) Although tyrosine nitration can be induced by alternative pathways, not only by the reaction of peroxynitrite with protein tyrosine residues (Szabo et al., 2007), the increase in nitrotyrosine in myocytes of doxorubicin-treated mice, and its abolishment by FP15, suggests that a causative link exists between oxidative and nitrosative stress and cardiotoxicity of doxorubicin.
FP15 also prevented doxorubicin-induced cardiac lipid peroxidation and activation of matrix metalloproteases (MMPs). MMP are significant contributors to the development of various pathophysiological conditions including dilated cardiomyopathy and congestive heart failure. The activation of proMMPs is triggered by peroxynitrite generation, via an extensive S-glutathiolation reaction (Okamoto et al., 2001). By inhibiting this reaction, peroxynitrite decomposition catalysts may reduce MMP activation. In addition to direct oxidation, peroxidation and nitration reactions and MMP activation, likely additional downstream, cytotoxic mechanisms elicited by peroxynitrite during doxorubicin-induced cardiac injury include DNA damage and activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), as well as the inhibition of myofibrillar creatine kinase (Weinstein et al., 2002; Mihm et al., 2002; Pacher et al., 2002b). Consistent with these findings, direct, pharmacological inhibition of PARP activation, as well as genetic deletion of the PARP gene has been shown to be effective in protecting against doxorubicin-induced cardiotoxicity in rodent models (Pacher et al 2002b; Pacher et al., 2006). Peroxynitrite, via activation of PARP, can also induce myocardial calcium overload and contractile dysfunction in doxorubicin-treated hearts (Szenczi et al., 2005). Although peroxynitrite can also induce the activation of myocardial MMPs, this effect is independent of PARP activation (Bai et al., 2004; Mukhopadhyay et al., 2009).
6.1. Molecular Mechanisms of peroxynitrite formation
A recent series of in vitro and in vivo studies have evaluated in detail, the molecular mechanisms of peroxynitrite formation and cardiotoxicity in response to doxorubicin exposure, and tested the effect of various peroxynitrite-neutralizing molecules (Mukhopadhyay et al., 2009). Using a well-established mouse model of doxorubicin-induced heart failure, marked increases in myocardial apoptosis (caspase-3 and caspace 9 expression/activity, cytochrome-c release, TUNEL, etc.), NOS2 gene expression, generation of NO and mitochondrial superoxide generation, nitrotyrosine formation, MMP2 and MMP9 gene expression and PARP activation were demonstrated, without major changes in expression of the multiple other genes linked to ROS and RNS production including NOX1, NOX2, p22phox, p40phox, p47phox, p67phox, xanthine oxidase, NOS1 and NOS3, genes. These changes were associated with a decrease in myocardial contractility after doxorubicin treatment. Pre-treatment of mice with two different peroxynitrite decomposition catalysts (FeTMPyP and MnTMPyP) markedly attenuated doxorubicin-induced myocardial apoptosis and dysfunction (Mukhopadhyay et al., 2009). Doxorubicin also increased mitochondrial superoxide and nitrotyrosine generation in a dose dependent manner, and apoptosis/necrosis in cardiac-derived H9c2 and/or human coronary artery endothelial cells as measured by flow cytometry and fluorescence microscopy. The doxorubicin- and/or peroxynitrite-induced apoptosis/necrosis positively correlated with intracellular nitrotyrosine formation, and was abolished by FeTMPyP/MnTMPyP. Likewise, the doxorubicin-induced cell death was also attenuated by selective NOS2 inhibitors. Importantly, co-administration of various NO donors and doxorubicin, but not NO donors alone, dramatically enhanced doxorubicin-induced cell death with concomitant increased nitrotyrosine formation and decreased mitochondrial superoxide generation (Mukhopadhyay et al., 2009). These studies lend additional support to the hypothesis that peroxynitrite, and not NO or superoxide, is the major trigger of doxorubicin-induced cell death (Figure. 6). It is hoped that future experimental therapies will be designed around neutralization of peroxynitrite, or around modulation of the pathways leading to peroxynitrite generation.
Figure 6:
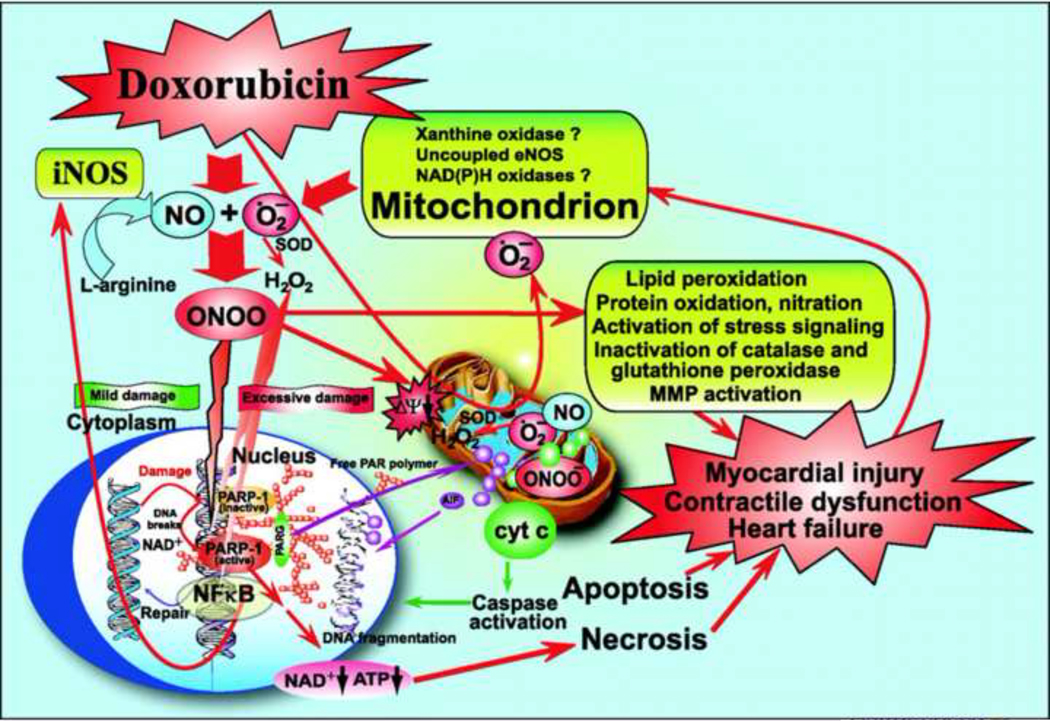
Schematic diagram of doxorubicin-induced cardiotoxicity: role of superoxide, NO, and peroxynitrite. Doxorubicin initially increases mitochondrial superoxide and, consequently, the generation of other ROS (e.g., H2O2) in cardiomyocytes and/or endothelial cells by redox cycling. Increased doxorubicin-induced ROS generation in cardiomyocytes triggers the activation of the transcription factor NF-kB, leading to enhanced NOS2 expression and NO generation. NO reacts with superoxide to form peroxynitrite both in the cytosol and mitochondria, which, in turn, induces cell damage via lipid peroxidation, inactivation of enzymes and other proteins by oxidation and nitration, and activation of stress signaling pathways (e.g., MAPK), MMPs, and PARP-1, among others. In the mitochondria, peroxynitrite, in concert with other ROS/RNS, impairs various key mitochondrial enzymes, leading to more sustained intracellular ROS generation (persistent even after doxorubicin already metabolized), triggering further activation of transcription factor(s) and NOS2 expression, resulting in the amplification of oxidative/nitrosative stress. In the mitochondria, peroxynitrite also triggers the release of proapoptotic factors (e.g., Cytochrome C and apoptosis-inducing factor) mediating caspase-dependent and -independent cell death pathways, which are also pivotal in doxorubicin-induced cardiotoxicity. Peroxynitrite, in concert with other oxidants, also causes strand breaks in DNA, activating the nuclear enzyme PARP-1. Once excessive oxidative and nitrosative stress-induced DNA damage occurs, overactivated PARP initiates an energy-consuming cycle by transferring ADP-ribose units from NAD+ to nuclear proteins, resulting in the rapid depletion of intracellular NAD+ and ATP pools, slowing the rate of glycolysis and mitochondrial respiration, eventually leading to cellular dysfunction and death, mostly by necrosis. Overactivated PARP may also facilitate the expression of a variety of inflammatory genes leading to increased inflammation (PARP-1 is a known co-activator of NF-kB) and associated oxidative stress, thus facilitating the progression of cardiovascular dysfunction and heart failure. PARG, poly(ADP-ribose) glycohydrolase (Reproduced with permission from Mukhopadhyay et al., 2009).
7. CONCLUSIONS
The examples presented by the different authors illustrate that oxidative and nitrative stress play a pivotal role in the tissue damage that occurs both in response to toxicants and in the pathogenesis of multiple human diseases. The pathways activated in cells by ROS and RNS include those that regulate cell survival or cell death and are a common feature of the response of tissues to chemical and viral toxicant exposure. ROS and RNS trigger a complex interplay of signalling pathways, with the potential central role of NFκB in transcriptional regulation of these responses. Additionally, there is evidence to support the differential role of peroxynitrite, formed from the interaction of superoxide anion and NO, in mediating tissue responses to toxicants.
Taken together, these studies suggest that further understanding of the multiple roles of ROS and RNS in the molecular responses to toxicants using model systems of pathological conditions is an interesting and fruitful route of investigation which may lead to identification of compounds to abrogate the deleterious effects of oxidative and nitrative stress.
Acknowledgements
Supported by a grant from the Michael J. Fox Foundation for Parkinson’s Disease Research (RBT) and by NIH/CA124998 (SS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production processerrorsmaybediscoveredwhichcouldaffectthecontent,andalllegaldisclaimers that apply to the journal pertain.
References
- Abel EL2009 Angel JM, Kiguchi K, J. and DiGiovanni J, Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 4(9) (2009), pp. 1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affara NI2004 and Robertson FM, Vascular endothelial growth factor as a survival factor in tumor-associated angiogenesis. In Vivo. 18(5) (2004), pp. 525–42. [PubMed] [Google Scholar]
- Affara NI2006 Trempus CS, Schanbacher BL, Pei P, Mallery SR, Bauer JA and Robertson FM, Activation of Akt and mTOR in CD34+/K15+ keratinocyte stem cells and skin tumors during multi-stage mouse skin carcinogenesis. Anticancer Res. 26(4B) (2006), pp. 2805–20. [PubMed] [Google Scholar]
- Ariza ME1996 Oberyszyn AS, Robertson FM and Williams MV, Mutagenic potential of peripheral blood leukocytes: in vivo exposure to the carcinogen 7,12 dimethylbenz[a]anthracene, and the tumor promoter 12-O-tetradecanoylphorbol acetate followed by in vitro co-culture with AS52 cells. Cancer Lett. 106(1) (1996) pp. 9–16. [DOI] [PubMed] [Google Scholar]
- Aslan M. 2008Cort A. and Yucel I, Oxidative and nitrative stress markers in glaucoma, Free Radical Biol. Med. 45 (2008), pp. 367–376. [DOI] [PubMed] [Google Scholar]
- Bai P.2004 Mabley JG, Liaudet L, Virág L, Szabó C. and Pacher P, Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep. 11 (2004), pp. 505–8. [PubMed] [Google Scholar]
- Blum RH 1974 and Carter SK, Adriamycin. A new anticancer drug with significant clinical activity. Ann Intern Med 180 (1974), pp. 249–259. [DOI] [PubMed] [Google Scholar]
- Brambilla R. 2009Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR and Bethea JR, Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol 182 (2009), pp. 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone DL 2009 Popichak KA, Moreno JA, Safe S, and Tjalkens RB, Suppression of 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-Induced Nitric-Oxide Synthase 2 Expression in Astrocytes by a Novel Diindolylmethane Analog Protects Striatal Neurons against Apoptosis. Mol Pharmacol 75 (2009), pp. 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J. 2008 Yu M-S. Y-S., Y-S, Wang M. and Chang RC-C, Dietary oxy-resveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity, Free Rad. Biol. Med 45 (2008), pp. 1019–1026. [DOI] [PubMed] [Google Scholar]
- Cheung Y-T 2009 Lau WK-W, Yu M-S, Lai CS-W, Yeung S-C, So K-F and Chang RC-C, ffects of all-trans retinoic acid on human SHSY5Y neuroblastoma as in in vitro model in neurotoxicity research NeuroToxicology 30 (2009) pp. 127–135. [DOI] [PubMed] [Google Scholar]
- Fatokun AA 2007 Stone TW and Smith RA, Hydrogen peroxide mediates damage by xanthine and xanthine oxidase in cerebellar granule neuronal cultures, Neurosci. Lett 416 (2007), pp. 34–38. [DOI] [PubMed] [Google Scholar]
- Cohen SM 1983 Promotion in urinary bladder carcinogenesis. Environmental Health Perspectives 50 (1983) pp. 51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehme T. 2004 Heneka MT, Sastre M, Dichgans J. and Schulz JB, Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88, (2004) pp. 494–501. [DOI] [PubMed] [Google Scholar]
- Djaldetti R. 2004 Hellmann M. and Melamed E, Bent knees and tiptoeing: late manifestations of end-stage Parkinson’s disease. Mov Disord 19, pp. 1325–1328. [DOI] [PubMed] [Google Scholar]
- Doroshow JH 1986 and Davies KJ, Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 261 (1986), pp. 3068–3074. [PubMed] [Google Scholar]
- Ebadi M. 2003 Sharma SK, Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid Redox Signal 5 (2003), pp. 319–335. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, 2008b Stone TW and Smith RA, Adenosine receptor ligands protect against a combination of apoptotic and necrotic cell death in cerebellar granule neurons. Exptl. Brain Res 186 (2008b), pp. 151–160. [DOI] [PubMed] [Google Scholar]
- Fatokun AA 2008a Stone TW and Smith RA, Oxidative stress in neurodegeneration and available means of protection. Front. Biosci 13 (2008a), pp.3288–3311. [DOI] [PubMed] [Google Scholar]
- Fatokun AA 2008c Stone TW and Smith RA, Resistance to kynurenic acid of the NMDA receptor-dependent toxicity of 3-nitropropionic acid and cyanide in cerebellar granular neurons. Brain Res. 1215 (2008c) pp., 200–207. [DOI] [PubMed] [Google Scholar]
- Ghisletti S. 2007 Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG and Glass CK, Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma, Mol Cell 25 (2007), pp. 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good PF 1998 Hsu A, Werner P, Perl DP and Olanow CW, Protein nitration in Parkinson’s disease, J Neuropathol Exp Neurol 57 (1998), pp. 338–342. [DOI] [PubMed] [Google Scholar]
- Hermann DM 2001 Kilic E, Kugler S, Isenmann S. and Bahr M, Adenovirus-mediated GDNF and CNTF pretreatment protects against striatal injury following transient middle cerebral artery occlusion in mice. Neurobiol Dis 8 (2001), pp. 655–666. [DOI] [PubMed] [Google Scholar]
- Hunot S. 1996 Bernard V, Faucheux B, Boissiere F, Leguern E, Brana C, Gautris PP, Guerin J, Bloch B and Agid Y, et al., Glial cell line-derived neurotrophic factor (GDNF) gene expression in the human brain: a post mortem in situ hybridization study with special reference to Parkinson’s disease. J Neural Transm 103 (1996), pp. 1043–1052. [DOI] [PubMed] [Google Scholar]
- Hunot S. 1999 Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, and Hirsch EC, FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells, J Neurosci 19 (1999), pp. 3440–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H. 2003 Beckman JS, Oxidative stress and nitration in neurodegeneration: Cause, effect or association. J.Clin. Invest 111 (2003), pp. 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krohn N, 2009 Kapoor S, Enami Y, Follenzi A, Bandi S, Joseph B. and Gupta S, Hepatocyte transplantation-induced liver inflammation is driven by cytokines-chemokines associated with neutrophils and Kupffer cells, Gastroenterology 136 (2009), pp. 1806–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W. 2008 Pan T, Huang M, Xu P, Xie W, Zhu W, Zhang X, Deng H, and Jankovic J, Decreased NURR1 gene expression in patients with Parkinson’s disease, J Neurol Sci 273 (2008), pp. 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. 2000 and Singal PK, Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation 102 (2000), pp. 2105–2110. [DOI] [PubMed] [Google Scholar]
- Liberatore GT 1999 Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM and Przedborski S, Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5 (1999), pp. 1403–1409. [DOI] [PubMed] [Google Scholar]
- Lin B-R 2009 Yu C-J, Chen W-C, Lee H-S, Chang H-M, Lee Y-C, Chien C-T, and Chen C-F, Green tea extract supplement reduces D-galactosamine-induced acute liver injury by inhibition of apoptotic and proinflammatory signalling, Journal of Biomedical Science 16 (2009), pp. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker BL 2007, Tuma DJ, Kharbanda KK, Kubik JL, and Casey CA, Effect of chronic ethanol administration on the in vitro production of proinflammatory cytokines by rat Kupffer cells in the presence of apoptotic cells, Alcoholism: Clinical & Experimental Research 31 (2007), pp.122–129. [DOI] [PubMed] [Google Scholar]
- Mih MJ. 2002 Yu F. and Weinstein DM et al. Intracellular distribution of peroxynitrite during doxorubicin cardiomyopathy: evidence for selective impairment of myofibrillar creatine kinase. Br J Pharmacol 135 (2002) pp. 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA 2008. Sullivan KA, Carbone DL, Hanneman WH and Tjalkens RB, Manganese potentiates nuclear factor-kappaB-dependent expression of nitric oxide synthase 2 in astrocytes by activating soluble guanylate cyclase and extracellular responsive kinase signaling pathways. J Neurosci Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P. 2009 Rajesh M, Bátkai S, Kashiwaya Y, Haskó G, Liaudet L, Szabó C. and Pacher P, Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 296 (2009) H1466–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE 1977 McGuire WP, and Liss RH et al. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science 197 (1977) pp.165–167. [DOI] [PubMed] [Google Scholar]
- Nakamoto Y. 1998 Guidotti LG, Kuhlen CV, Fowler P. and Chisari FV, Immune Pathogenesis of Hepatocellular Carcinoma, J.Exp.Med 188 (1998) pp. 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberyszyn TM 1993 Sabourin CL, Bijur GN GN, Oberyszyn AS LG Boros and F.M. Robertson, Interleukin-1 alpha gene expression and localization of interleukin-1 alpha protein during tumor promotion. Mol Carcinog. 7(4) (1993) pp. 238–48. [DOI] [PubMed] [Google Scholar]
- Okamoto T. 2001 Akaike T, and Sawa T, et al. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem 276 (2001) 29596–29602. [DOI] [PubMed] [Google Scholar]
- Oliver JD 2002. Roberts RA, Receptor-Mediated Hepatocarcinogenesis: Role of Hepatocyte Proliferation and Apoptosis. Pharmacology and Toxicology 91 pp. 1–7. [DOI] [PubMed] [Google Scholar]
- Pacher P. 2003 Liaudet L, Bai P, Mabley JG, Kaminski PM, Virág L, Deb A, Szabó E, Ungvári Z, Wolin MS, Groves JT and Szabó C, Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 107 (2003) pp. 896–904. [DOI] [PubMed] [Google Scholar]
- Pache P. 2002 Liaudet L, Bai P, Virag L, Mabley JG, Haskó G. and Szabó C, Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 300 (2002) pp. 862–7. [DOI] [PubMed] [Google Scholar]
- Pacher P. 2006 Liaudet L, Mabley JG, Cziráki A, Haskó G. and Szabó C, Beneficial effects of a novel ultrapotent poly(ADP-ribose) polymerase inhibitor in murine models of heart failure. Int J Mol Med. 17 (2006) pp. 369–75. [PMC free article] [PubMed] [Google Scholar]
- Pacher P. 2002b Liaudet L. and Bai P, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. JPET 300 (2002b) pp. 862–867. [DOI] [PubMed] [Google Scholar]
- Parzefall W. 2001 Berger W, Kainzbauer E, Teufelhofer O, Schulte-Hermann R. and Thurman RG, Peroxisome proliferators do not increase DNA synthesis in purified rat hepatocytes, Carcinogenesis 22. (2001), pp. 519–523. [DOI] [PubMed] [Google Scholar]
- Pascual G. 2005 Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, and Glass CK, A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437 (2005), pp. 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM 2000 Rusyn I, Rose ML, Gonzalez FJ, and R RG.G. Ghurman, Peroxisome proliferator-activated receptor alpha is restricted to hepatic parenchymal cells, not Kupffer cells: implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis, Carcinogenesis. 21 (2000), pp. 823–826. [DOI] [PubMed] [Google Scholar]
- Powell CL 2006 Kosyk O, Ross PK, Schoonhove and R, Boysen G, Phenotypic Anchoring of Acetaminophen-Induced Oxidative Stress with Gene Expression Profiles in Rat Liver, Toxicological Sciences 93 (2006), pp. 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman M. 1993 Wagner R, Carcinogenesis. Missing mismatch repair, Nature 366 pp. 722. [DOI] [PubMed] [Google Scholar]
- Roberts RA 2007 Ganey PE, Ju C, Kamendulis LM, Rusyn I, and Klaunig JE, Role of the Kupffer cell in mediating hepatic carcinogenesis and toxicity. Toxicological Sciences 96 (2007), pp. 2–15. [DOI] [PubMed] [Google Scholar]
- Roberts RA 2009. Smith R, Laskin D, Robertson F, Doorn J and Slikker W. Nitrative and Oxidative Stress in Toxicology and Disease. Toxicological Sciences, 112, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, Laskin DL, Smith CV, Robertson FM, Allen EMG, Doorn JA and Slikker W. (2009). Nitrative and Oxidative Stress in Toxicology and Disease. Tox Sci. 112, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson FM 1996Long BW, Tober KL, Ross MS and Oberyszyn TM, Gene expression and cellular sources of inducible nitric oxide synthase during tumor promotion. Carcinogenesis. 17(9) (1996), pp. 2053–9. [DOI] [PubMed] [Google Scholar]
- Robertson FM 1990 Beavis AJ, Oberyszyn TM, O’Connell SM, Dokidos A, Laskin DL, Laskin JD and Reiners JJ Jr. Production of hydrogen peroxide by murine epidermal keratinocytes following treatment with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 50 (1990) pp. 6062–7. [PubMed] [Google Scholar]
- Robertson FM 1994 Bijur GN, Oberyszyn AS, Pellegrini AE, Boros LG, Sabourin CL and Oberyszyn TM, Granulocyte-macrophage colony stimulating factor gene expression and function during tumor promotion. Carcinogenesis 15 (1994) pp.1017–29. [DOI] [PubMed] [Google Scholar]
- Robertson FM 1996 Ross MS, Tober KL, Long BW and Oberyszy TM, Inhibition of pro-inflammatory cytokine gene expression and papilloma growth during murine multistage carcinogenesis by pentoxifylline. Carcinogenesis. Aug; 17 (1996) pp. 1719–28. [DOI] [PubMed] [Google Scholar]
- Rusyn I. 2000 Rose ML, Bojes HK and Thurman RG, Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antioxidants & Redox Signaling 2 (2000), pp. 607–621. [DOI] [PubMed] [Google Scholar]
- Ryder S. 2001 Beckingham I, “ABC of diseases of liver, pancreas, and biliary system: Acute hepatitis”. BMJ 322 (2001), pp. 151–153. http://www.who.int/mediacentre/factsheets/fs204/en/index.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K. 2009 Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH and Glass CK, A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death, Cell 137 (2009), pp. 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singal PK 1987 Deally CM and Weinberg LE. Subcellular effects of adriamycin in the heart: a concise review. J Mol Cell Cardiol 19 (1987) pp. 817–828. [DOI] [PubMed] [Google Scholar]
- Singal PK 1998 and Iliskovic N, Doxorubicin-induced cardiomyopathy. N Engl J Med. 339 (1998) pp. 900–905. [DOI] [PubMed] [Google Scholar]
- Sirak AA. 1991 Beavis AJ and Robertson FM, Enhanced hydroperoxide production by peripheral blood leukocytes following exposure of murine epidermis to 12-O-tetradecanoylphorbol-13-acetate. Carcinogenesis. Jan 12 (1) (1991) pp. 91–5. [DOI] [PubMed] [Google Scholar]
- Siveski-Iliskovic N. 1994 Kaul N. and Singal PK, Probucol promotes endogenous antioxidants and provides protection against adriamycin-induced cardiomyopathy in rats. Circulation 89 (1994) pp. 2829–2835. [DOI] [PubMed] [Google Scholar]
- Smith AJ 2008 Stone TW, and Smith RA, Preconditioning with NMDA protects against toxicity of 3-nitropropionic acid or glutamate in cultured cerebellar granule neurons. Neurosci. Lett 440 (2008), pp. 294–298. [DOI] [PubMed] [Google Scholar]
- Smith AJ 2009a Tauskela JS, Stone TW, and Smith RA, Preconditioning with 4-aminopyridine protects cerebellar granule neurons against excitotoxicity, Brain Res. 1294 (2009a), pp.165–175. [DOI] [PubMed] [Google Scholar]
- Smith AJ 2009b Smith RA, and Stone TW, 5-Hydroxyanthranilic acid, a tryptophan metabolite, generates oxidative stress and neuronal death via p38 activation in cultured cerebellar granule neurons, Neurotoxicity Res.15 (2009b), pp. 303–310. [DOI] [PubMed] [Google Scholar]
- Smith RA 2003 Walker T, Xie X. and Hou ST, ST Involvement of the transcription factor E2F1/Rb in kainic acid-induced death of murine cerebellar granule cells, Mol. Brain Res 116 (2003), pp. 70–79. [DOI] [PubMed] [Google Scholar]
- Smith RA 2009 Twenty first century challenges for in vitro neurotoxicity. ATLA 37 (2009), pp. 367–375. [DOI] [PubMed] [Google Scholar]
- Szabó C 2007 Ischiropoulos H. and Radi R, Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 6 (2007) pp. 662–80. [DOI] [PubMed] [Google Scholar]
- Szenczi O 2005 Kemecsei P, Holthuijsen MF, van Riel NA, van der Vusse GJ, Pacher P, Szabó C, Kollai M, Ligeti L. and Ivanics T, Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem Pharmacol. 69 (2005) pp. 725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG 2008 Frank-Cannon TC, McCoy MK, Lee JK, Martinez TN, McAlpine FE, Ruhn KA and Tran TA, Neuroinflammation in Parkinson’s disease: is there sufficient evidence for mechanism-based interventional therapy, Front Biosci 13 (2008), pp. 709–717. [DOI] [PubMed] [Google Scholar]
- Tauskela JS 2008 Fang H, Hewitt M, Brunette E, Ahuja T, Thivierge JP, Comas T, and Mealing G, Elevated synaptic activity preconditions neurons against an in vitro model of ischemia. J. Biol. Chem (2008) 283 pp. 34667–34676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P. 2004 and Schulz JB, Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation, Cell Tissue Res 318 (2004), pp. 149–161. [DOI] [PubMed] [Google Scholar]
- Tjalkens RB. 2008 Liu X. Mohl B. Wright T. Moreno JA. Carbone DL. Safe S. The peroxisome proliferator-activated receptor-gamma agonist 1,1-bis(3’-indolyl)-1-(p-trifluoromethylphenyl)methane suppresses manganese-induced production of nitric oxide in astrocytes and inhibits apoptosis in cocultured PC12 cells. J. Neuroscience Res 86:618–29, 2008. [DOI] [PubMed] [Google Scholar]
- Trushina E. 2007 McMurray CT, Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145 (2007), pp. 1233–1248. [DOI] [PubMed] [Google Scholar]
- Tsuchiya M. 2008 Kono H, Matsuda M, Fujii H, and Rusyn I, Protective effect of Juzentaiho-to on hepatocarcinogenesis is mediated through the inhibition of Kupffer cell-induced oxidative stress, International Journal of Cancer 123 (2008), pp. 2503–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW 2005 Kushnareva Y, Greenwood S. and Connolly CN, Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J. Neurochem 92 (2005), 1081–1090. [DOI] [PubMed] [Google Scholar]
- Weinstein DM 2000 Mihm MJ, and Bauer JA, Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther 294 (2000) pp. 396–401. [PubMed] [Google Scholar]
- Weinstei IB. 1984 Gattoni-Celli S, Kirschmeier P, Lambert M, Hsiao W, Backer J. and Jeffrey A, Initial cellular targets and eventual genomic changes in multistage carcinogenesis, IARC Scientific Publications 56 (1984), pp. 277–97. [PubMed] [Google Scholar]
- WHO 2003. Health Statistics from the Amercicas. www.paho.org/english/dd/pub/SP_591.htm
- Young RC 1981 Ozols RF and Myers CE, The anthracycline antineoplastic drugs. N Engl J Med 305 (1981) pp. 139–153. [DOI] [PubMed] [Google Scholar]