

Abstract
Epstein-Barr virus nuclear antigens 2 (EBNA2) mediated super-enhancers, defined by in silico data, localize near genes associated with B cell transcription factors including RUNX3. However, the biological function of super-enhancer for RUNX3 gene (seR3) remains unclear. Here, we show that two seR3s, tandemly-located at 59- and 70-kb upstream of RUNX3 transcription start site, named seR3 –59h and seR3 –70h, are required for RUNX3 expression and cell proliferation in Epstein-Barr virus (EBV)-positive malignant B cells. A BET bromodomain inhibitor, JQ1, potently suppressed EBV-positive B cell growth through the reduction of RUNX3 and MYC expression. Excision of either or both seR3s by employing CRISPR/Cas9 system resulted in the decrease in RUNX3 expression and the subsequent suppression of cell proliferation and colony forming capability. The expression of MYC was also reduced when seR3s were deleted, probably due to the loss of trans effect of seR3s on the super-enhancers for MYC. These findings suggest that seR3s play a pivotal role in expression and biological function of both RUNX3 and MYC. seR3s would serve as a potential therapeutic target in EBV-related widespread tumors.
Keywords: RUNX3, Super-enhancer, Epstein-Barr virus, JQ1, MYC
1. Introduction
Epstein-Barr virus (EBV) is implicated in the development of human cancers including Burkitt lymphoma, NK/T cell lymphoma, nasopharyngeal cancer and gastric cancer (Young et al., 2016). EBV transforms resting B cells to continuously proliferating lymphoblastoid cells, which gain immortalized property in vitro. Although extensive studies have been performed on EBV-mediated tumorigenesis, the molecular mechanisms for this EBV-associated immortalization remain not fully elucidated. EBV produces viral proteins after infection into human cells. These viral proteins are shown to underlie the tumorigenesis. Among the EBV-proteins, Epstein-Barr virus nuclear antigens 2 (EBNA2) is the firstly synthesized viral protein after infection. EBNA2 plays important roles in the B cell transcriptional reprograming (Cohen et al., 1989; Alfieri et al., 1991). Although EBNA2 functions as a transcriptional factor, they do not bind DNA directly. Instead, EBNA2 facilitates DNA-binding of RBP-J, a final mediator of Notch signaling, resulting in activation of various gene expression (Grossman et al., 1994; Henkel et al., 1994; Kaiser et al., 1999). Recently, EBNA2 is reported to promote the formation of super-enhancers, which drive abundant expression of target genes including oncogenes. EBNA2 likely affects enhancer-promoter architecture to induce EBV-related cell proliferation (Jiang et al., 2017).
Super-enhancers are recently defined as transcriptional regulatory elements exhibiting extraordinarily high and broad signals for active histone modification mark by H3K27ac, or Bromodomain and Extra-terminal protein (BRD4), in chromatin immunoprecipitation with high throughput sequencing (ChIP-seq) (Pott and Lieb, 2015). BRD4 inhibition by a bromodomain inhibitor, JQ1, has larger effects on super-enhancer than on typical enhancer in gene expression. Previous reports described that super-enhancers regulate expression of critical oncogenic driver genes. BRD4 inhibition leads to preferential disruption of these super-enhancers in cancer cells such as multiple myeloma (Loven et al., 2013). Another report demonstrated that EBNA2 mediated super-enhancers are essential for MYC expression in an EBV-positive lymphoblastoid cell line (LCL) (Liang et al., 2016). EBNA2 is also shown to promote MYC super-enhancer looping to MYC transcription start site (TSS) for activating MYC transcription. In this way, EBNA2 promotes the formation of super-enhancers and interaction of enhancer-promotor, resulting in cell proliferation. A previous report showed that EBNA2 mediated super-enhancers were formed in the vicinity of genes encoding B cell transcription factors such as MYC, MAX, EBF, and RUNX3 (Zhou et al., 2015). However, the biological effect of super-enhancer for these genes in cell proliferation remains uncovered.
The RUNX family transcription factor, RUNX3, is expressed in a variety of tissues and functions as developmental regulators. RUNX3 is reported to act as tumor suppressor of gastric, colon, and many other solid tumors (Ito et al., 2015). Recent studies, however, demonstrated that RUNX3 also plays an oncogenic role in a certain subset of cancers, therefore called “conditional” oncogene, including head and neck cancer, pancreatic cancers, myelodysplastic syndrome and EBV-positive NK/T cell lymphoma (Tsunematsu et al., 2009; Whittle et al., 2015; Neil et al., 2017; Selvarajan et al., 2017; Yokomizo-Nakano et al., 2020). Multiple earlier studies reported RUNX3 induction by EBNA2 in human B cells (Spender et al., 2002, 2005). Subsequently, ChIP-seq data implied that EBNA2 activates RUNX3 through a specific cis-regulatory element (Gunnell et al., 2016). Another report showed that two EBNA2-mediated super-enhancer sites are localized in the vicinity of RUNX3 (Zhou et al., 2015). These reports clearly demonstrated the existence of super-enhancers for RUNX3, henceforth referred to as seR3s, which likely causes elevated RUNX3 expression. However, the biological function of seR3s remains unknown. We therefore elucidate the biological effects of EBNA2-associated seR3s on EBV-mediated cell proliferation, employing a BET bromodomain inhibitor and CRISPR/Cas9 system.
2. Materials and methods
2.1. Cell culture
Four hematopoietic cell lines, GM12878, Raji, Reh and K562 cells, were used in this study. GM12878 and Raji are EBV-positive B cell LCL and Burkitt lymphoma cell line, respectively. Reh is an EBV-negative precursor B cell acute lymphoblastic leukemia cell line bearing t (12;21) in which the ETV6(TEL)-RUNX1(AML1) fusion gene is generated. K562 is an EBV-negative chronic myeloid leukemia cell line, which is shown to be resistant to treatment of a BET bromodomain inhibitor JQ1 (Dawson et al., 2014). All hematopoietic cell lines were maintained in RPMI-1640 medium (Nacalai Tesque, Japan) supplemented with 10% fetal bovine serum (FBS) (BioWest, France). A non-hematopoietic human embryonic kidney cells that express a mutant version of the SV40 large T antigen, HEK293T cell line, was also used as a packaging cell line for a lentiviral vector. HEK293T cells were maintained in Dulbecco’s modified Eagle medium (Nacalai Tesque, Japan) with 10% FBS.
2.2. Cell proliferation, cell cycle, and apoptosis assays
Cell proliferation following treatment with the BET bromodomain inhibitor, JQ1 (Sigma, St. Louis, MO), was assessed using the Cell Counting kit-8 (Dojindo, Japan) according to the manufacturer’s recommendation. 15,000 cells/well were plated in a 96-well plate with 1 μM of JQ1 or vehicle DMSO (Sigma, St. Louis, MO) in 100 μl of growth media. Cell viability was measured by addition of Cell Counting kit-8 reagent and luminescence measurement on a Tecan infinite M200 plate reader (Tecan, Switzerland).
Cell cycle analysis was performed 72 h after JQ1 treatment using BrdU flow kit (BD Pharmingen, San Diego, CA) according to the manufacture’s instructions. The cells were resuspended in ice cold 70% ethanol and fixed for 30 min. The cells were washed twice with PBS, then incubated for 30 min at room temperature with 0.5% RNase A and stained with 25 μg/ml propidium iodide (PI; BioLegend, San Diego, CA). Cell cycle data was obtained by using LSRII (BD Biosciences, San Diego, CA).
Apoptosis was analyzed with Annexin V and PI. Cells were incubated with Annexin V binding Buffer (BD Pharmingen, San Diego, CA), 2 μl of Annexin V-APC (BD Pharmingen, San Diego, CA) and 5 μg/ml PI. After 15 min incubation at room temperature, the cells were analyzed.
2.3. Expression of RUNX3 and MYC
For quantitative reverse transcription-polymerase chain reaction (qRT-PCR), three days after the cell seeding, total RNA was extracted and complementary DNA was synthesized with SuperScript VILO kit (Invitrogen, Waltham, MA). qRT-PCR was conducted by QuantStudio 3 Real-Time PCR system using Power Up SYBR Green Kit (Applied Biosystems, Waltham, MA) and normalized against β-actin. Primer sequences are listed in Table S1.
For western blot analysis, harvested cells were resuspended in ice-cold suspension buffer supplemented with protease inhibitor cocktail (Sigma) and incubated on ice for 5 min. An equal volume of lysis buffer (4% SDS, 100 mM Tris, pH 6.8, 20% glycerol) was added and samples were boiled for 10 min at 95 °C and spin down at 13,500 g for 15 min at room temperature. Supernatant was used as the total cell extract. Protein concentration was determined using the BCA protein assay reagent (Pierce, Rockford, IL). To the lysates, 4x Laemmli buffer (Bio-Rad, Hercules, CA) was added and incubated at 95 °C for 10 min (Wang et al., 2014), and subsequently subjected to sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE). Antibodies against RUNX3 (1:500, R3-5G4, sc-101553, Santa Cruz, Dallas, TX), c-Myc (1:1000, D84C12, # 5605, Cell Signaling Technology, Danvers, MA), and Beta-actin (1:10000, AC-15, ab6276, Abcam, Cambridge, UK) were used for immunoblotting. Enhanced chemiluminescence (ECL) activated by horseradish peroxidase (HRP) conjugated with secondary antibodies (1:20000, ab205719 for mouse ab97051 for rabbit, Abcam) was analyzed using LAS3000 imaging system.
2.4. CRISPR/Cas- mediated excision of seR3s
Single guide RNAs (sgRNAs) were designed using CRISPRdirect, a publicly available webtool (https://crispr.dbcls.jp/). Off target effect was also checked using COSMID webtool (https://crispr.bme.gatech.edu/). Selected sgRNAs listed in Table S2 were individually cloned into lentiCRISPR vectors. These constructs were transfected into HEK293T cells with pMDLg/pRRE, pRSV-Rev and pMD2.G using FuGENE 6 reagent (Promega, Madison, WI). The cells were infected with virus in the presence of polybrene (Millipore, Burlington, MA) and HEPES (BioWest, France) and selected by 2.5 μg/l puromycin (Gibco, Waltham, MA). After drug selection by puromycin, transfected cells were subjected to cell proliferation assay and colony formation assay, using Cell Counting kit-8 and MethoCult H4034 Optimum (STEMCELL technologies, Canada) respectively. Genomic DNAs (gDNAs) and RNAs were extracted using DNeasy Blood and Tissue kit (Qiagen, Germany) and NcleoSpin RNA kit (Macherey-Nagel, Germany) respectively, according to the manufacturer’s instructions.
2.5. Inhibition of RUNX protein function
The small molecule Ro5-3335 (EMD Millipore, Billerica, MA) is reported to be an inhibitor for RUNX/CBFB protein function (Cunningham et al., 2012). Ro5-3335 stock solution (5 mM) was prepared by dissolving it in DMSO. 15,000 cells/well were plated with 1 μM, 5 μM and 10 μM of Ro5-3335 or DMSO as control. Cell viability in the presence of various concentrations of Ro5-3335 was assessed by Cell Counting kit-8 reagent.
2.6. Short interfering RNA (siRNA) targeting RUNX3
The short interfering RNAs (siRNAs) for control and Runx3 were purchased from GE healthcare Dharmacon, Inc (Lafayette, CO). Cells were transfected with siRNA using NEPA21 Transfection System (Nepa Gene, Japan) and cultured for 72 h.
2.7. Statistical analysis
Statistical significance was determined with t-tests and two-way ANOVA with multiple testing in GraphPad Prism software Version 7.05 (GraphPad Software, San Diego, CA).
3. Results
3.1. BET bromodomain inhibition suppresses cell proliferation via G1 arrest
EBNA2 binding sequence data were obtained from publicly available ChIP-seq database, ChIP-Atlas (https://chip-atlas.org/) and the results revealed the presence of a large enhancer region upstream of the RUNX3 (Fig. 1A). This region shows high-level H3K27ac ChIP-seq signals, which are compatible with characteristics of super-enhancers, specifically in EBV-positive B cell lines, GM12878 and Raji, but not in EBV-negative K562 and Reh. Two seR3s are tandemly-located at 59- and 70-kb upstream of RUNX3 TSS, and named seR3 ‒59h and seR3 ‒70h, respectively (Gunnell et al., 2016).
Fig. 1.
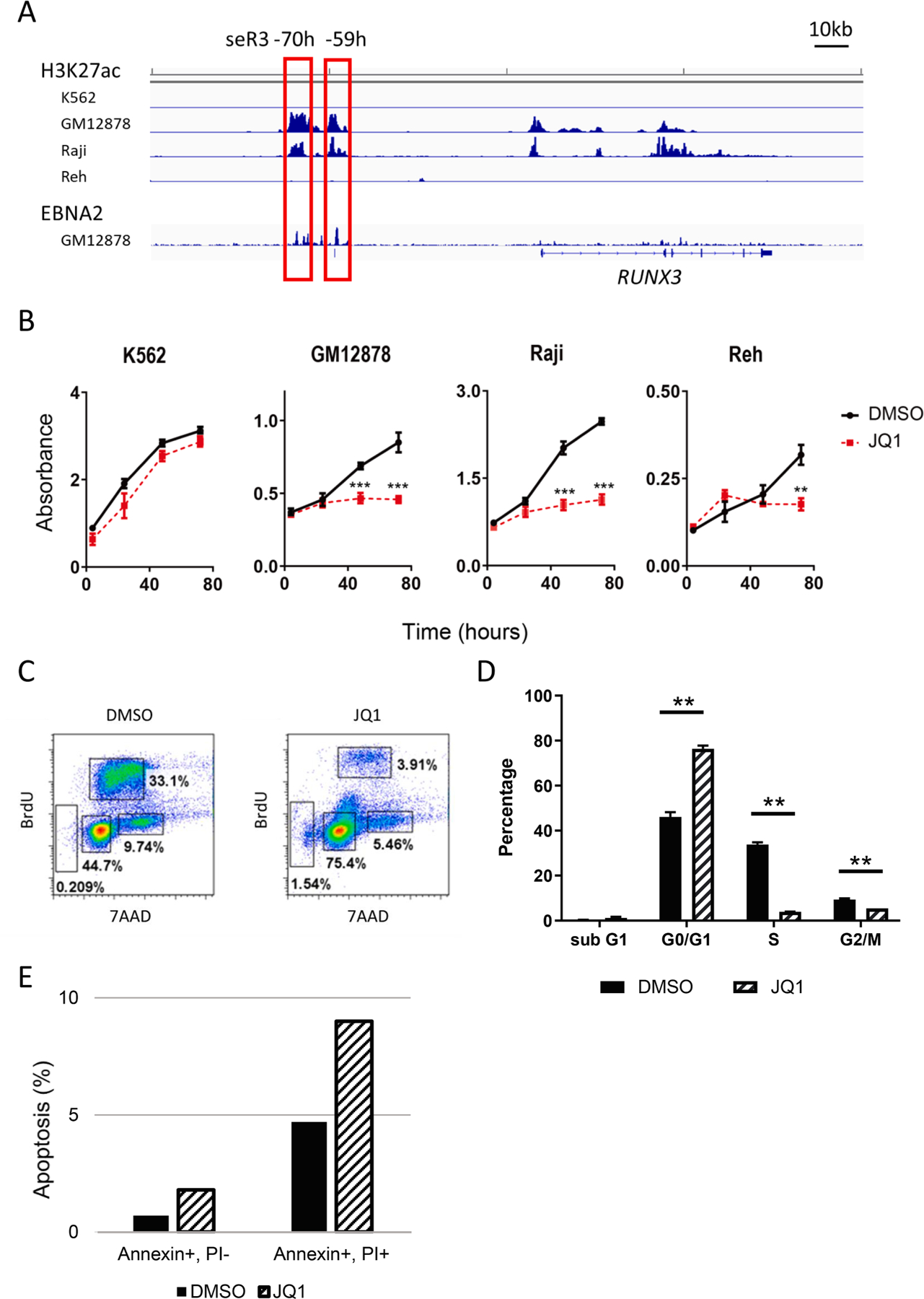
BET bromodomain inhibition suppresses the proliferation of EBV-positive B cells via G1 arrest. (A) ChIP-seq reads for H3K27ac signals in K562, GM12878, Raji and Reh cells and for EBNA2 protein in GM12878. RUNX3 runs left to right in the human genome. The red boxes indicate the super-enhancer regions. Numbers indicate the distance from RUNX3 transcription start site. (B) Cell proliferation of K562, GM12878, Raji and Reh cells treated with DMSO or 1 μM JQ1 for 1–3 days. Error bars represent SEM. Asterisks indicate p value using t-tests on 48 h and 72 h after treatment. **p < 0.01. ***p < 0.001. (C) Flowcytometric analysis of cell cycle in Raji cells after treatment of DMSO or JQ1. Data are representative of two independent experiments. (D) Assessment of cell cycle in Raji cells following 72 h of treatment with either DMSO or 1 μM JQ1. The percentages of each phase of the cell cycle are shown. (E) Apoptosis assessment by Annexin V/PI staining in Raji cell following 72 h of treatment with either DMSO or 1 μM JQ1.
To assess the role of super-enhancers in cell proliferation, the sensitivity to a super-enhancer specific inhibitor JQ1 in various cell lines was examined. JQ1 is frequently employed to suppress cell proliferation in which super-enhancers play a critical role. K562, GM12878 LCL, Raji and Reh were treated with 1 μM JQ1 or DMSO for 1–3 days. K562 was resistant to JQ1 as previously described, while EBV-positive GM12878 and Raji, and Reh were highly or modestly sensitive to JQ1 respectively (Fig. 1B). Cell cycle profile of Raji cell line showed a drastic decrease in the percentage of cells in S-phase and increase in G0/G1 peak by JQ1 treatment, suggesting that JQ1 causes the G1 arrest (Fig. 1C, D). JQ1 treatment also increased the percentage of sub-G1 cells in cell cycle analysis, and induced an increase in the number of Annexin V positive apoptotic cells (Fig. 1E). These results indicate that BET bromodomain inhibition induces cell cycle arrest and apoptosis, resulting in reduced cell proliferation in EBV-positive cell lines and, to a lesser extent, in Reh cells.
3.2. BET bromodomain inhibition suppresses RUNX3 and MYC expression
To assess the JQ1 effect on transcriptional level, expression of RUNX3 and MYC were evaluated by qRT-PCR. As already reported, JQ1 suppressed MYC expression, probably through the inhibition of its super-enhancers, seMYC (Mertz et al., 2011). In addition, JQ1 treatment reduced RUNX3 expression (Fig. 2A, B). JQ1 also suppressed the expression of MYC and RUNX3 proteins (Fig. 2C). This result suggests that increased RUNX3 expression in EBV-positive cells is likely attributable to the formation of super-enhancers.
Fig. 2.
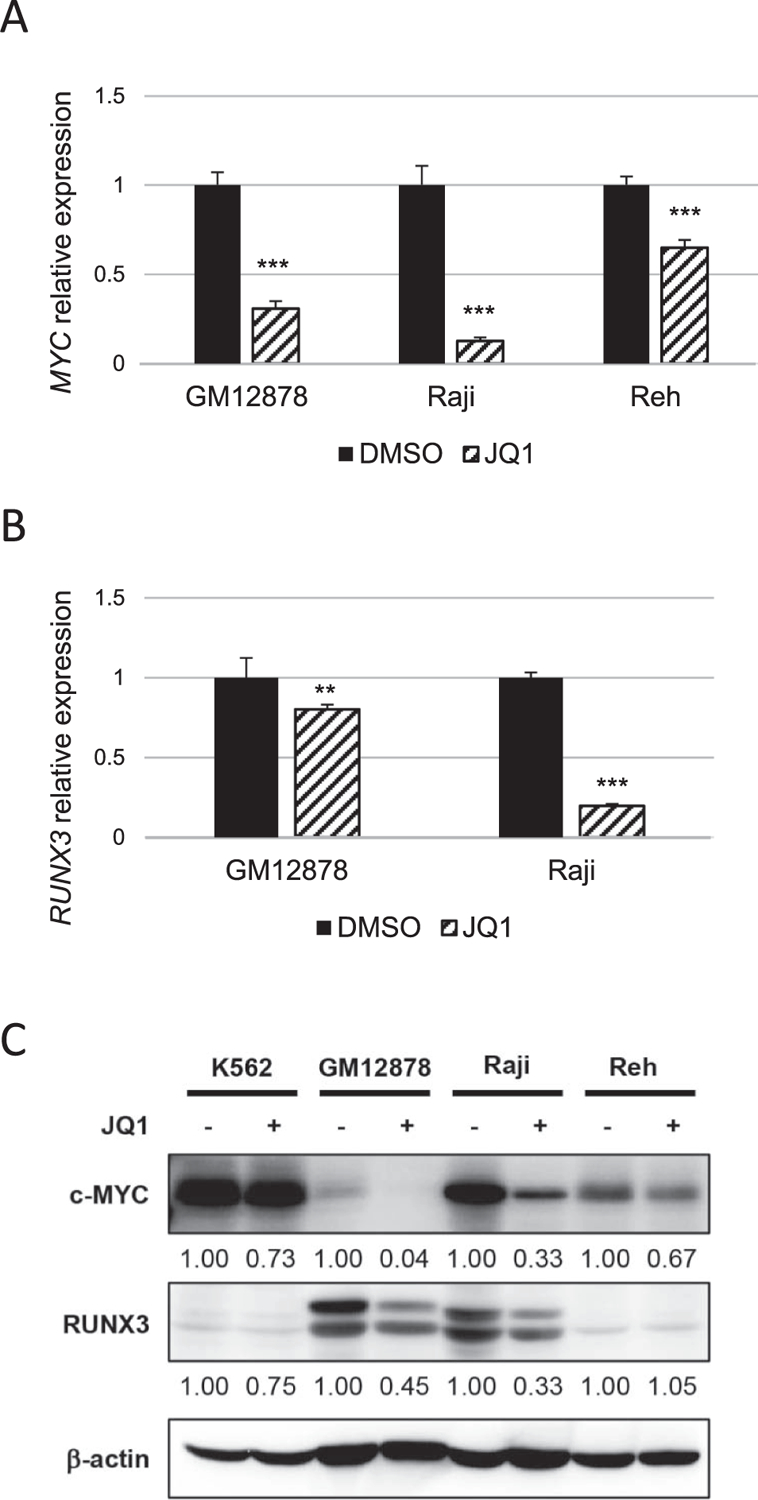
BET bromodomain inhibition suppresses RUNX3 and MYC expression. (A) qRT-PCR of MYC expression in GM12878, Raji and Reh cells treated with DMSO or JQ1. mRNA levels were first normalized to β -actin. MYC expression levels after JQ1 treatment were shown relative to DMSO control, which was set to 1. Error bars indicate SD of triplicate data. Asterisks indicate p < 0.001 using t-tests. (B) qRT-PCR of RUNX3 expression in indicated cells treated with DMSO or JQ1. RUNX3 expression levels after JQ1 treatment were shown relative to DMSO control. Asterisks represent significant differences (** p < 0.01, *** p < 0.001, Student’s t-test). (C) Western blot of MYC and RUNX3 expression in K562, GM12878, Raji and Reh. RUNX3 expression levels in the presence of JQ1 treatment were shown relative to those with DMSO controls.
3.3. Excision of seR3 suppresses the expression of RUNX3 and MYC
It is plausible that JQ1 suppresses the cell proliferation via inhibiting thousands of super-enhancers throughout the genome. In order to examine to what extent the particular seR3s are critical for cell proliferation in EBV-positive cell lines, excision of seR3s by employing CRISPR/Cas9 system was conducted. Pairs of single guide RNAs were designed for seR3 –59h and –70h regions (Fig. 3A). GM12878 and Raji have ChIP-seq peaks in seR3 –59h and –70h regions, while Reh does not (Fig. 1A). The lenti CRISPR vectors carrying both sgRNA and Cas9 were transfected into the EBV-positive B cell lines. After transfection and selection of infected cells by puromycin, gDNA was extracted for excision check. The targeted regions were indeed deleted, and the expression of RUNX3 was reduced in seR3 excised cells (Fig. 3B, C). Intriguingly, the expression of MYC was also reduced when seR3s were excised in GM12878 cells, suggesting that the expression of MYC is associated with the presence of seR3.
Fig. 3.
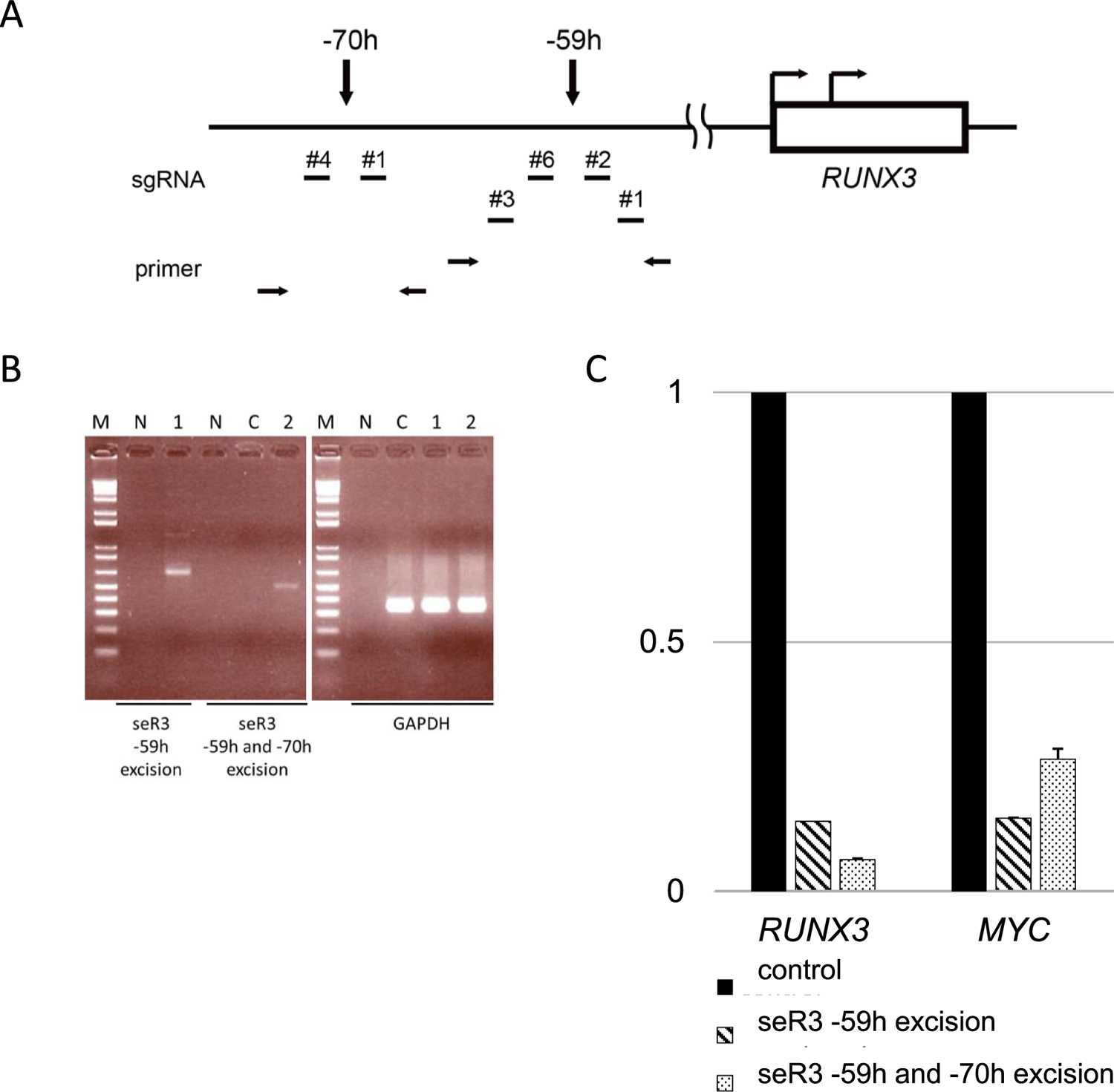
Excision of seR3 suppresses the expression of RUNX3 and MYC. (A) The location of single guide RNAs (sgRNAs) upstream of RUNX3 was shown. The arrows on the box indicate P1 and P2 promotors of RUNX3. The arrows over the line show the location of seR3 –59h and –70h. Two independent pairs of sgRNAs targeting seR3 –59h were designed, numbered as 1, 2, 3 and 6. The arrows at the bottom of this schema indicate the primers for excision check of seR3 regions. (B) seR3 excision was validated. Total genomic DNA was prepared from GM12878 cells transduced with dual sgRNAs. The targeted region was amplified by PCR. The bands indicate the deletion of seR3 regions and the presence of GAPDH regions. M: 1 kb DNA ladder marker. N: negative control. C: GM12878 cells were transduced with Cas9 only as control. The presence of seR3 was not detected by PCR because the size of deleting region is around 10 kb. Lane 1: GM12878 cells were transduced with sgRNA –59h #1 and –59h #3. The deletion of seR3 –59h were shown with 624 bp band after PCR using –59h excision check primers. Lane 2: GM12878 cells were transduced with sgRNA –59h #1 and –70h #4. The deletion of seR3 –59h and –70h regions were shown with 484 bp band after PCR using –59h forward and –70h reverse excision check primers. Results are representative of several independent assays. (C) seR3 excision reduced the expression of RUNX3 and MYC in GM12878 cells. The expression was reduced both by excised –59h region and by –59h and –70h regions. RNA levels were first normalized to β -actin. RUNX3 and MYC expression levels after seR3 deletion were shown relative to control, which was set to 1.
3.4. Excision of seR3s leads to reduction in proliferation and colony forming capability of EBV-positive B cells
The biological effect by the seR3 excision was next examined by performing two distinct assays, namely cell proliferation and colony forming assays. The result in cell proliferation assay clearly demonstrated that the simultaneous excision of seR3 –59h and seR3 –70h regions led to potent inhibition of cell propagation in EBV-positive cell lines, GM12878 and Raji, but only modest suppression in EBV-negative cell line, Reh (Fig. 4A). The colony forming capability was also reduced in the Raji cells when seR3 –59h and/or –70h region were deleted (Fig. 4B, C). To investigate which particular super-enhancer out of the two seR3s is more critical than the other in biological behavior, the excision of individual seR3 was performed and compared amongst three distinct excision patterns indicated in Fig. 4C. The result exhibited that the contribution of seR3 –70h to colony forming capability was slightly higher than that of seR3 –59h. However, the simultaneous excision of two seR3 caused further decrease as compared to the single seR3 excision. Therefore, seR3 –59h or seR3 –70h regions are both indispensable for the proliferation and colony forming capability in EBV-positive B cells. In addition, these results suggest that the above-described JQ1-induced suppression is at least in part due to its inhibition against seR3s, and the biological behavior of EBV-positive cells heavily relies on the super-enhancer function of seR3s.
Fig. 4.
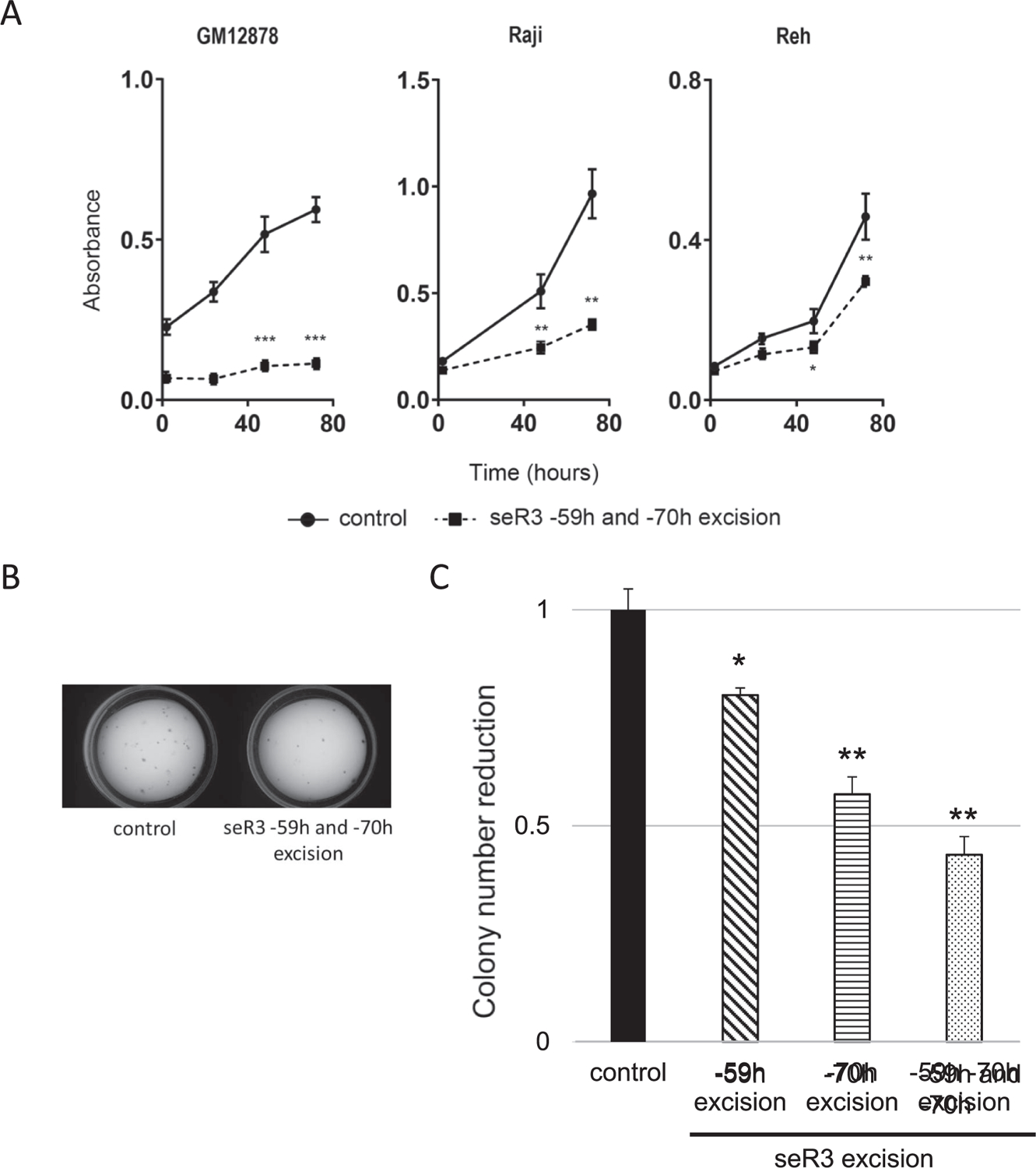
Excision of seR3 leads to reduction of EBV-positive cell proliferation and colony forming capability. (A) Cell proliferation of seR3 deleted GM12878, Raji and Reh cells. Cells were transduced with sgRNA –59h #1 and –70h #4 for excision of seR3 –59h and –70h simultaneously. After puromycin selection, proliferation of cell lines was assessed using Cell Counting kit-8. Error bars represent SEM. Asterisks indicate p value using t-tests on 48 h and 72 h after treatment. *p < 0.05. **p < 0.01. ***p < 0.001. (B) Colony formation assays of parental/control and seR3-deleted Raji cells. Image is a representative after excision of seR3 –59h and –70h. (C) Relative reduction against DMSO control in colony numbers of Raji cells with indicated seR3 excision. Asterisks indicate p value using t-tests. *p < 0.05. **p < 0.01.
3.5. seR3s might function in trans on seMYC
The formation of seR3s initiated by EBNA2 is expected to drive constitutive expression of RUNX3 and the elevated expression of protooncogenic RUNX3 would in turn facilitate cell proliferation in EBV-positive B cells. To examine this straightforward biological consequence by the seR3 formation, we next carried out an experiment for the indispensable role of RUNX3 by using Ro5-3335, a pharmacological inhibitor against pan-RUNX function. As expected, Ro5-3335 strongly suppressed the cell proliferation of GM12878 in a dose dependent manner (Fig. 5A). siRNA against RUNX3 (siRUNX3), which decreased RUNX3 expression in Western blot analysis, also reduced the proliferation of GM12878 cells (Fig. 5B and C). In addition, suppression in Reh and, to a lesser extent, in K562 was observed in the presence of Ro5-3335. This suppression in EBV-negative cell lines suggests the essential role of RUNX family proteins in cell viability in various types of cells, which was recently unveiled and has since been extensively investigated (Young et al., 2007; Morita et al., 2017). Indeed, the suppression of Reh proliferation by Ro5-3335 was previously reported by others (Cunningham et al., 2012). Most surprisingly, EBV-positive B cell line, Raji, did not show any suppression by Ro5-3335 and siRUNX3. Considering the above-described two consequences by the excision of seR3s in Raji cells, namely the suppression of MYC expression and significant cell death, seR3s are considered to have a hitherto unknown trans-function on seMYC and MYC expression, besides the expected cis-function for RUNX3 expression.
Fig. 5.
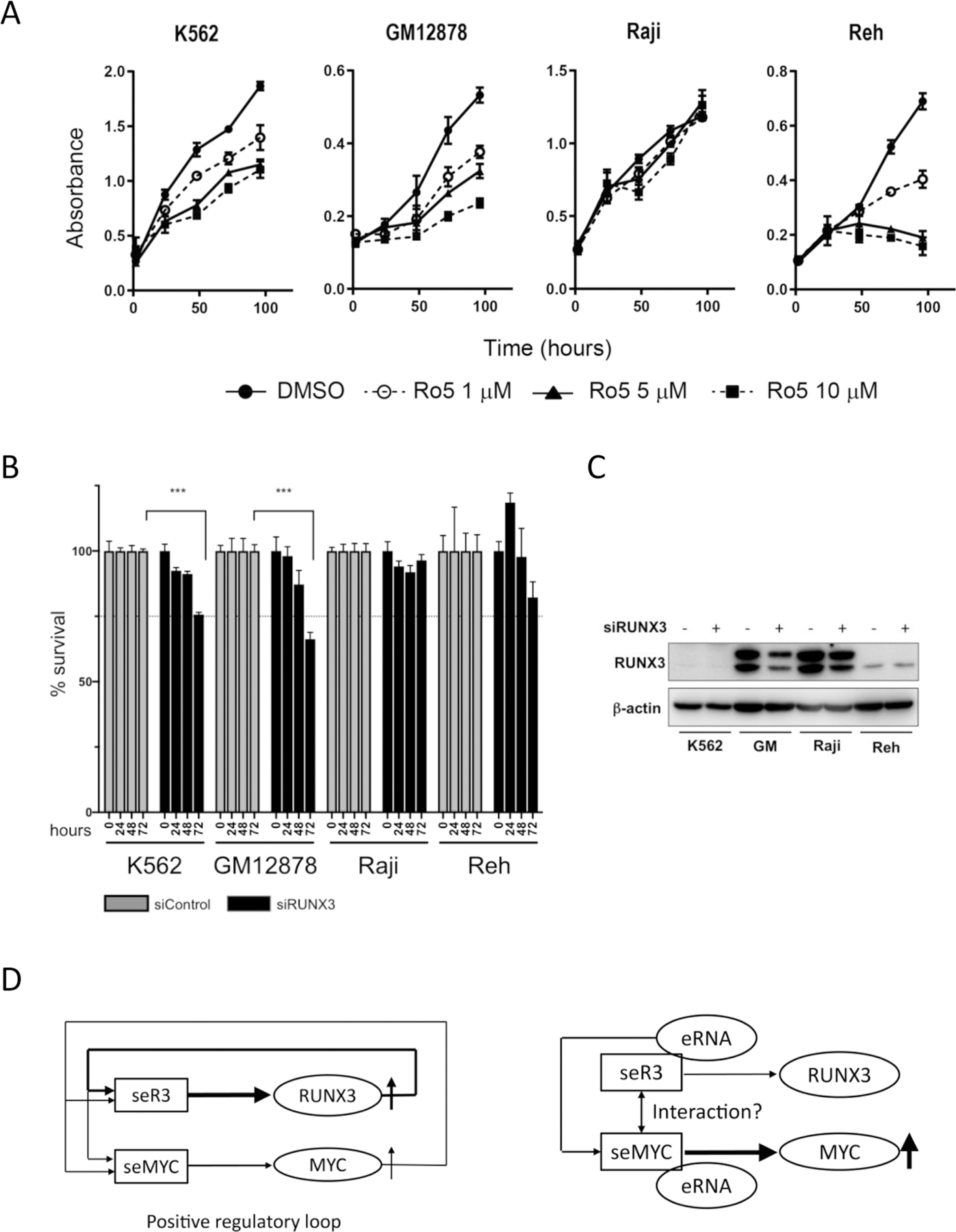
seR3s might function in trans on seMYC. (A) Cell proliferation of K562, GM12878, Raji and Reh cells treated with DMSO or indicated concentration of Ro5-3335 for 1–3 days is shown. Error bars represent SEM. (B) % cell viabilities of indicated cells transfected with siRUNX3, relative to the viabilities of cells transfected with siControl at indicated corresponding time points after transfection are shown. Asterisks indicate p value calculated by two-way ANOVA tests. ***p < 0.001. (C) Western blot of RUNX3 expression in indicated cells with siControl or siRUNX3. (D) The schematic diagrams represent the two distinct mode of seR3 function. The left diagram indicates the cis-function of seR3 and seMYC. seR3 and seMYC drive expression of their canonical target genes, RUNX3 and MYC, individually. seR3 driven RUNX3 protein in turn binds to both seR3 and seMYC, and seMYC driven MYC protein does the same, thereby forming a positive regulatory loop that ensures the constitutive expression of oncogenes RUNX3 and MYC. In GM12878 cells, this mechanism works dominantly. On the other hand, in Raji cells, seR3s likely function in trans on seMYC (right diagram), probably through the abundant production of enhancer RNA (eRNA) generated from seR3s. The direct physical interaction between seR3 and seMYC is also hypothetically proposed although not confirmed.
4. Discussion
In this study, we elucidated the role of EBNA2-bound super-enhancers, seR3s, tandemly located at 59- and 70-kb upstream of the RUNX3 gene in the proliferation of EBV-associated malignant cells. RUNX3 is well documented to be upregulated and play a critical role in EBV-positive malignant B cell behavior; however, the molecular basis for its elevation remains unknown. In the previous studies, ChIP-seq data identified seR3s which may underlie upregulation of RUNX3 expression in EBV-positive B cells. To examine whether seR3s indeed drive abundant expression of RUNX3 and facilitate cell proliferation, a super-enhancer inhibitor, JQ1, was first employed. The result clearly demonstrated the suppression of RUNX3 expression and cell proliferation by JQ1. To examine whether this suppression on cell proliferation is specifically due to seR3 amongst a number of super-enhancers formed throughout the genome, excision of seR3s by CRISPR/Cas9 method was subsequently conducted. Pronounced reduction in both proliferation and colony forming capability accompanied by the decrease in RUNX3 expression was confirmed. These results indicate that seR3s formation facilitates the elevation of RUNX3 expression and cell proliferation in EBV-positive B cells.
The super-enhancer-driven constitutive oncogene expression is well characterized in T lymphoblastic leukemia cells (T-ALL), where super-enhancers are formed at the loci of the key oncogene TAL1 and its regulatory partner genes, such as GATA3, RUNX1 and MYB (Kwiatkowski et al., 2014; Mansour et al., 2014). TAL1, GATA3, and RUNX1 form transcriptional complexes on the individual super-enhancer regions, thereby reinforcing and stabilizing increased expression of these genes, and eventually establish the solid interconnected positive regulatory loop of multiple oncogenes (Sanda et al., 2012). Similarly, RUNX3 and MYC, together with EBNA2, appear to form the potent auto-regulatory loop in EBV-positive B cells (Fig. 5D, left diagram). As for MYC, two EBNA2-bound super-enhancers are indeed identified at 428- and 525-Kb upstream of MYC TSS. The cross-bindings of RUNX3 and MYC with seMYCs and seR3s were in fact clearly observed in the publicly available ChIP-seq data in ChIP-Atlas.
Super-enhancers are also shown to generate enhancer RNAs (eRNAs) very abundantly. These eRNAs contribute to the enhancer-promoter looping and allow for elevated expression of its target gene (Liang et al., 2016; Alvarez-Dominguez et al., 2017; Tan et al., 2019). Intriguingly, eRNAs are also documented to be frequently included in the super-enhancer complexes in distally located gene loci, some of which are even located on the different chromosomes. This finding suggests that eRNAs transcribed from a certain super-enhancer function not only in cis but also in trans, by facilitating the formation of super-enhancer for non-canonical target gene(s), particularly in the case of super-enhancer complex composed of the similar constituting molecules. RUNX and MYC overexpression through the super-enhancer formation is well documented to underlie the development of various cancers (Kwiatkowski et al., 2014; Kubota et al., 2019). In EBV-positive B cells, super-enhancers for RUNX3 and MYC may functionally interact each other, through their eRNAs (Fig. 5D, right diagram).
The cis-effect of super-enhancer on its own target gene, as shown in Fig. 5D left diagram, has been widely believed to be the only mechanism which accounts for the super-enhancer mediated tumorigenesis; however, the result of pan-RUNX inhibitor Ro5-3335 carried out in this study raises the possibility that the trans-effect of super-enhancers, schematically summarized in Fig. 5D right diagram, also underlie the super-enhancer driven tumorigenesis. Ro5-3335 is a benzodiazepine which binds to RUNX proteins and inhibits its function (Illendula et al., 2016). If the cis-function of seR3s is the only scenario to induce EBV-mediated B cell transformation, RUNX3 inhibition by Ro5-3335 should result in the same outcome by seR3 excision by CRISPR-Cas9 method. As expected, potent suppression by Ro5-3335 was in fact observed in LCL GM12878 (Fig. 5A). In clear contrast, little or no suppression was induced in EBV-positive B cell line, Raji, although seR3 excision led to decrease in MYC expression and cell death. Therefore, we speculate that seR3s may function in trans on seMYC, probably through eRNAs transcribed from seR3.
The difference in the effect of Ro5-3335 on GM12878 versus Raji cells may be due to the distinct stage in the malignant transformation of B cells by EB virus. GM12878 is one of the LCLs which still retain the characteristic features of original primary B cells. On the other hand, Raji represents the more advanced EBV-related malignant cells. The cell proliferation may be reliant on the cis-function of seR3s, namely RUNX3 expression, at the early stage of EBV-mediated transformation, whereas on the trans-effect of seR3s at the late stage. This hypothetical mechanism warrants further investigation.
EBV oncoproteins remodel the host genome organization to form super-enhancers for key oncogenes, MYC and RUNX3, during the malignant transformation in B cells. These super-enhancers primarily make cis-effect on RUNX3 and MYC expression, and may have trans-effect on super-enhancers for other genes in a cross-regulatory manner. We unveiled the possibility of trans-effect of super-enhancers in this study. Further dissection of its molecular mechanism would provide us with deeper insights into EBV-related B cell transformation and other tumorigenesis, and may lead to a novel therapeutic avenue.
Supplementary Material
Acknowledgments
The authors thank members of MD1 Vivarium, NUS, for mouse husbandry.
This work was supported by research funding from 2016 Wakayama Medical Award for Young Researchers and Kaigai Kyoin Haken Jigyo by Wakayama Medical University Internal Grant, Japan, to H.H., JSPS Kakenhi (18KK0440), Japan, to A.N., the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award, Singapore, and the National Institutes of Health (R35CA197697 and P01HL131477), USA, to D.G.T., JSPS Kakenhi (16K14613 and 19K07668), Japanese Society of Hematology, Leukemia Research Foundation, Japan, and NCIS Centre Grant Seed Funding Program, Singapore, to M.O., and National Medical Research Council, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative, Singapore, to T.S., A.P.K., D.G.T., Y.I., and M.O.
Abbreviations:
- ChIP-seq
chromatin immunoprecipitation assays with sequencing
- CRISPR/Cas9
Clustered regularly interspaced short palindromic repeats/CRISPR-associated (Cas) gene 9
- eRNA
enhancer RNA
- EBNA2
Epstein-Barr virus nuclear antigens 2
- EBV
Epstein-Barr virus
- ECL
Enhanced chemiluminescence
- gDNA
genomic DNA
- LCL
lymphoblastoid cell line
- NK/T cell
natural killer T cell
- PI
propidium iodide
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- seMyc
super-enhancers for MYC
- seR3
super-enhancer for RUNX3
- SDS-PAGE
sodium dodecyl sulfate- polyacrylamide gel electrophoresis
- sgRNA
single guide RNA
- siRNA
short interfering RNA
- T-ALL
T lymphoblastic leukemia
- TSS
transcription start site
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gene.2021.145421.
References
- Alfieri C, Birkenbach M, Kieff E, 1991. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 181, 595–608. [DOI] [PubMed] [Google Scholar]
- Alvarez-Dominguez JR, Knoll M, Gromatzky AA, Lodish HF, 2017. The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell Rep 19, 2503–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CQ, Krishnan V, Tay LS, Chin DW, Koh CP, Chooi JY, Nah GS, Du L, Jacob B, Yamashita N, Lai SK, Tan TZ, Mori S, Tanuichi I, Tergaonkar V, Ito Y, Osato M, 2014. Disruption of Runx1 and Runx3 leads to bone marrow failure and leukemia predisposition due to transcriptional and DNA repair defects. Cell Rep 8, 767–782. [DOI] [PubMed] [Google Scholar]
- Cohen JI, Wang F, Mannick J, Kieff E, 1989. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. U.S.A 86, 9558–9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, Dehdashti SJ, Reinhold WC, Alemu L, Zhao L, Yeh JR, Sood R, Pommier Y, Austin CP, Jeang KT, Zheng W, Liu P, 2012. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc. Natl. Acad. Sci. U.S.A 109, 14592–14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, Cannizzaro E, Osaki H, Wiese M, Putwain S, Fong CY, Grove C, Craig J, Dittmann A, Lugo D, Jeffrey P, Drewes G, Lee K, Bullinger L, Prinjha RK, Kouzarides T, Vassiliou GS, Huntly BJ, 2014. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 28, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E, 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. U.S.A 91, 7568–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnell A, Webb HM, Wood CD, McClellan MJ, Wichaidit B, Kempkes B, Jenner RG, Osborne C, Farrell PJ, West MJ, 2016. RUNX super-enhancer control through the Notch pathway by Epstein-Barr virus transcription factors regulates B cell growth. Nucleic Acids Res 44, 4636–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG, 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265, 92–95. [DOI] [PubMed] [Google Scholar]
- Illendula A, Gilmour J, Grembecka J, Tirumala VSS, Boulton A, Kuntimaddi A, Schmidt C, Wang L, Pulikkan JA, Zong H, Parlak M, Kuscu C, Pickin A, Zhou Y, Gao Y, Mishra L, Adli M, Castilla LH, Rajewski RA, Janes KA, Guzman ML, Bonifer C, Bushweller JH, 2016. Small molecule inhibitor of CBFbeta-RUNX binding for RUNX transcription factor driven cancers. EBioMedicine 8, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Bae SC, Chuang LS, 2015. The RUNX family: developmental regulators in cancer. Nat. Rev. Cancer 15, 81–95. [DOI] [PubMed] [Google Scholar]
- Jiang S, Zhou H, Liang J, Gerdt C, Wang C, Ke L, Schmidt SCS, Narita Y, Ma Y, Wang S, Colson T, Gewurz B, Li G, Kieff E, Zhao B, 2017. The epsteinbarr virus regulome in lymphoblastoid cells. Cell Host Microbe 22 (561–573), e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B, 1999. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol 73, 4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Tokunaga K, Umezu T, Yokomizo-Nakano T, Sun Y, Oshima M, Tan KT, Yang H, Kanai A, Iwanaga E, Asou N, Maeda T, Nakagata N, Iwama A, Ohyashiki K, Osato M, Sashida G, 2019. Lineage-specific RUNX2 super-enhancer activates MYC and promotes the development of blastic plasmacytoid dendritic cell neoplasm. Nat. Commun 10, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS, 2014. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhou H, Gerdt C, Tan M, Colson T, Kaye KM, Kieff E, Zhao B, 2016. Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. U.S.A 113, 14121–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA, 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, Loh ML, Hunger SP, Sanda T, Young RA, Look AT, 2014. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd, 2011. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Suzuki K, Maeda S, Matsuo A, Mitsuda Y, Tokushige C, Kashiwazaki G, Taniguchi J, Maeda R, Noura M, Hirata M, Kataoka T, Yano A, Yamada Y, Kiyose H, Tokumasu M, Matsuo H, Tanaka S, Okuno Y, Muto M, Naka K, Ito K, Kitamura T, Kaneda Y, Liu PP, Bando T, Adachi S, Sugiyama H, Kamikubo Y, 2017. Genetic regulation of the RUNX transcription factor family has antitumor effects. J. Clin. Invest 127, 2815–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil JC, Gilroy K, Borland G, Hay J, Terry A, Kilbey A, 2017. The RUNX genes as conditional oncogenes: insights from retroviral targeting and mouse models. Adv. Exp. Med. Biol 962, 247–264. [DOI] [PubMed] [Google Scholar]
- Pott S, Lieb JD, 2015. What are super-enhancers? Nat. Genet 47, 8–12. [DOI] [PubMed] [Google Scholar]
- Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, Ma W, Tatarek J, Ahn Y, Kelliher MA, Jamieson CH, Staudt LM, Young RA, Look AT, 2012. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 22, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajan V, Osato M, Nah GSS, Yan J, Chung TH, Voon DC, Ito Y, Ham MF, Salto-Tellez M, Shimizu N, Choo SN, Fan S, Chng WJ, Ng SB, 2017. RUNX3 is oncogenic in natural killer/T-cell lymphoma and is transcriptionally regulated by MYC. Leukemia 31, 2219–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spender LC, Cornish GH, Sullivan A, Farrell PJ, 2002. Expression of transcription factor AML-2 (RUNX3, CBF(alpha)-3) is induced by Epstein-Barr virus EBNA-2 and correlates with the B-cell activation phenotype. J. Virol 76, 4919–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spender LC, Whiteman HJ, Karstegl CE, Farrell PJ, 2005. Transcriptional cross-regulation of RUNX1 by RUNX3 in human B cells. Oncogene 24, 1873–1881. [DOI] [PubMed] [Google Scholar]
- Tan SH, Leong WZ, Ngoc PCT, Tan TK, Bertulfo FC, Lim MC, An O, Li Z, Yeoh AEJ, Fullwood MJ, Tenen DG, Sanda T, 2019. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 134, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T, Kudo Y, Iizuka S, Ogawa I, Fujita T, Kurihara H, Abiko Y, Takata T, 2009. RUNX3 has an oncogenic role in head and neck cancer. PLoS ONE 4, e5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, Goggins M, Hruban RH, Chang AE, Calses P, Thorsen SM, Hingorani SR, 2015. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell 161, 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo-Nakano T, Kubota S, Bai J, Hamashima A, Morii M, Sun Y, Katagiri S, Iimori M, Kanai A, Tanaka D, Oshima M, Harada Y, Ohyashiki K, Iwama A, Harada H, Osato M, Sashida G, 2020. Overexpression of RUNX3 represses RUNX1 to drive transformation of myelodysplastic syndrome. Cancer Res 80, 2523–2536. [DOI] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, Nickerson JA, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS, 2007. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature 445, 442–446. [DOI] [PubMed] [Google Scholar]
- Young LS, Yap LF, Murray PG, 2016. Epstein-barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer 16, 789–802. [DOI] [PubMed] [Google Scholar]
- Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, Johannsen EC, Kharchenko P, Gewurz BE, Kieff E, Zhao B, 2015. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 17, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.