

Abstract
Testing multiple biomarkers, as opposed to one, has become a preferred approach for diagnosing many heterogeneous diseases, such as cancer and infectious diseases. However, numerous technologies, including gold standard ELISA and PCR, can detect only one type of biomarker, either protein or nucleic acid (NA), respectively. In this work, we report for the first time simultaneous detection of proteins and NAs in the same solution, using solely functional NA (FNA) molecules. In particular, we combined the thrombin binding aptamer (TBA) and the 10-23 RNA-cleaving DNA enzyme (DNAzyme) in a single aptazyme molecule (Aptazyme1.15-3′), followed by extensive optimization of buffer composition, sequences and component ratios, to establish a competitive bioassay. Subsequently, to establish a multiplex bioassay, we designed a new aptazyme (Aptazyme2.20-5′) by replacing the target recognition and substrate sequences within Aptazyme1.15-3′. This designing process included an in silico study, revealing the impact of the target recognition sequence on the aptazyme secondary structure and its catalytic activity. After proving the functionality of the new aptazyme in a singleplex bioassay, we demonstrated the capability of the two aptazymes to simultaneously detect thrombin and NA target, or two NA targets in a multiplex bioassay. High specificity in target detection was achieved with the limits of detection in the low nanomolar range, comparable to the singleplex bioassays. The presented results deepen the barely explored features of FNA for diagnosing multiple targets of different origins, adding an extra functionality to their catalogue.
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00216-021-03458-6.
Keywords: DNAzyme, Aptazyme, Competitive bioassay, Multiplexing, Simultaneous detection
Introduction
The best therapeutic strategy for all diseases is an early and accurate diagnosis [1, 2], which, however, relies on the availability of biomarkers. Discovering a unique one for each disease can be fairly complex, especially when working with heterogeneous diseases, such as infectious diseases or cancer. To further advance their diagnosis, simultaneous evaluation of a panel of biomarkers, including both proteins and nucleic acids (NAs), has been shown as a promising strategy [2, 3]. For example, it has been proven that integrating the host response factors can significantly improve the diagnosis of infectious diseases [4]. Moreover, targeting multiple regions of a viral genome provides specific information about the type of virus next to circumventing problems with high viral mutation rates [1, 5]. All these aspects are essential to successfully control infections during outbreaks, as we are currently witnessing during the COVID-19 pandemic. Also, it has been demonstrated that the detection of colorectal cancer by the widely used carcinoembryonic antigen reaches 30–40% of sensitivity, which remarkably increases to 88% when combined with the detection of free circulating DNA [6]. Moreover, the combination of biomarkers can also provide information about the stage of cancer as well as about the response to a therapy [7]. According to a recent review, one of the next goals in the detection of cancer biomarkers is to develop NA-based techniques for personalized and point-of-care (POC) diagnostics that can detect both NA and non-NA targets [8]. This, however, poses a considerable challenge as multiple read-out platforms are still needed to accomplish this, which significantly complexifies diagnosis. For instance, despite being of high value as the gold standards, neither ELISA nor PCR can detect other types of targets but solely proteins or NAs, respectively.
Functional nucleic acids (FNA) are a group of DNA and RNA molecules, holding functionalities beyond the storage and transfer of genetic information [9]. Among numerous FNA types, the two exploited in this work are aptamers, capable of specifically binding to a target, and DNA enzymes (DNAzymes), capable of catalyzing a reaction. Both FNAs have become increasingly popular over the years, thanks to the sizeable possibilities for their design and modifications [10]. Aptamers are single-stranded (ss) DNA or RNA molecules, which are selected from a substantial pool of random sequences through an in vitro iterative process, known as SELEX (systematic evolution of ligands by exponential enrichment), leading to the enrichment of those sequences showing the highest affinity for the target [11]. The use of aptamers as recognition elements offers several advantages over their protein analogues, such as high synthesis reproducibility and high stability in a wide range of conditions [12]. Moreover, the ease of labeling DNA sequences with functional molecules, or their immobilization on a surface makes aptamers suitable to developing bioassays in combination with different types of read-outs, such as optical, electrochemical, or mass-sensitive [13]. Similar to aptamers, DNAzymes are also selected through an in vitro iterative procedure for their capability of performing catalytic activities [14]. They can catalyze many different reactions, such as redox [15], phosphorylation [16], ligation [17], or RNA-cleavage [18], the latter forming the base of this work. RNA-cleaving DNAzymes are composed of a catalytic core and two substrate-binding arms, responsible for enzymatic activity and target recognition, respectively. Similar to protein enzymes, their catalytic activity is dependent on specific cofactors, namely metal ions [19]. The option to integrate DNAzymes with several read-out methods expands their range of applications as well [20]. Interestingly, aptamers and DNAzymes have been also merged into a single molecule, known as aptazyme [21], which has unlocked a myriad of opportunities for applications in biosensing [22]. In this context, approaches using the properties of DNA beyond base-pairing have been studied, including aptamers for target recognition and DNAzymes for signal generation [23–25]. Although these approaches made an important contribution by demonstrating the potential of FNAs, to the best of our knowledge, they have never been used for multiplex detection of different types of targets in the same bioassay.
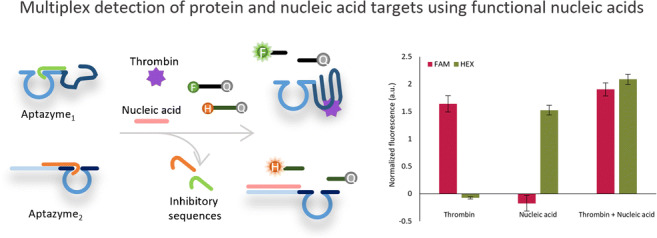
In this context, here we present an aptazyme-based DNA-only competitive bioassay capable of detecting both protein and NA targets or two different NA targets in the same reaction (Fig. 1). To accomplish this, we design two different aptazymes: (1) Aptazyme1.15-3′, for detecting a protein target as well as an NA1 target, and (2) Aptazyme2.20-5′ for detecting an NA2 target. Aptazyme1.15-3′ is constructed by combining a thrombin binding aptamer (TBA) as the target recognition element and a recently reported redesigned version of 10-23 core RNA-cleaving DNAzyme [26] for signal generation, referred to here as DNAzyme1. TBA has been selected as an attractive model system due to its extensive characterization and applicability in numerous bioassays [27]. The DNAzyme1 was recently made in our group for optimal catalysis at the standard room temperature (RT, i.e., 20–25 °C) [26], starting from the traditional DNAzyme that cleaves at elevated temperatures (ranging between 37 and 55 °C) [28, 29]. Aptazyme2.20-5′ is designed by joining a random NA2 target recognition sequence with DNAzyme2. In the latter, the substrate binding arms of DNAzyme1 are modified to allow binding of a different substrate, thus enabling multiplexing. In both cases, to prevent the enzymatic activity of the DNAzyme in the absence of a target, the aptazyme is blocked with a complementary inhibitory sequence, which hinders the binding and the cleavage of the DNAzyme substrate. Adding thrombin or NA1 target to the solution competitively removes the inhibitory sequence, allowing the substrate to bind. Subsequently, the DNAzyme can cleave the RNA bases in the substrate, resulting in its dissociation. Since the substrate sequence is labelled with a fluorophore (FAM or HEX, respectively referred to as F and H) on one end and a quencher (Iowa Black® FQ referred to as Q) on the other, the distance increases between them upon the cleavage, resulting in a fluorescence signal increase. Because one aptazyme can catalyze the cleavage of multiple substrates, this approach provides an amplification of the signal.
Fig. 1.

Schematic illustration of the proposed competitive bioassay for the simultaneous detection of A protein and NA2 target, and B two NA targets together (NA1 and NA2). The detection of a NA1 target by Aptazyme1.15-3′ was simply achieved by using the complementary sequence of TBA as the target. DNAzyme1 is always part of Aptazyme1.15-3′ and correspondingly cleaves Substrate1, whereas DNAzyme2 is part of Aptazyme2.20-5′, thereby cleaving Substrate2. TBA stands for thrombin binding aptamer
To establish such a competitive (multiplex) bioassay, in this paper we first optimize conditions permitting uncompromised functionality of both aptazyme elements (i.e., target recognition part and DNAzyme) in the same buffer. Next, different aspects of the competitive bioassay are scrutinized, including the concentrations of the different reaction elements and the incubation times to inhibit aptazyme activity. Finally, the specificity and sensitivity of the multiplexed bioassays are evaluated. As such, we report here the first ever study on using aptazymes for multiplex detection of different types of targets in the same bioassay, which offers massive advantage for implementing such bioassays on POC platforms and/or when having limited patients’ sample volume (both highly relevant for instance in the context of infectious diseases diagnostics). Although demonstrated using simple model systems, the flexibility and ease of designing NA receptors demonstrate the huge potential of this approach to be implemented for a variety of different targets and is aligned with the expectations in the field to establish NA-based techniques for detecting both NA and non-NA targets.
Materials and methods
Reagents and buffers
The oligonucleotides presented in this paper were purchased from Integrated DNA Technologies (IDT, Leuven, Belgium) and used without further purification. All the sequences are summarized in Table S1 in Supplementary Information (ESM). Potassium chloride (KCl) and ethylenediaminetetraacetic acid (EDTA) were purchased from Acros Organics (Geel, Belgium). Tris-ethylenediaminetetraacetic acid (TE, 100×), and glycine were obtained from Sigma-Aldrich (St. Louis, USA). Magnesium chloride solution (MgCl2) was acquired from Fisher Scientific (Leicestershire, England) and Tris-HCl (pH 8.8) was obtained from Genetex Inc. (Irvine, USA). Human α-thrombin was purchased from Haematologic Technologies (Essex Junction, USA). TE was diluted 100× and used as a buffer solution for the preparation of intermediate solutions of the DNA sequences. The pH of the buffers was adjusted using hydrochloric acid (HCl) from Sigma-Aldrich (St. Louis, USA). All dilutions were prepared using UltraPure distilled water (DNAse-RNAse free, Invitrogen, Carlsbad, USA) unless stated otherwise. All reaction mixes were prepared in DNA or Protein LoBind tubes (Eppendorf, Hamburg, Germany) and the fluorescence was measured in 384-well black-clear bottom microplates (Glasatelier Saillart, Meerhout, Belgium) using the SpectraMax ID3 microplate reader (Molecular Devices LLC, San Jose, USA). The composition of all the buffers used in this paper is summarized in Table 1. All the concentrations of different bioassay components refer to the concentration in the final volume, unless stated otherwise.
Table 1.
Composition of the buffers used in this work
| Name | Composition |
|---|---|
| BufferDNAzyme | 50 mM KCl and 20 mM MgCl2 in 10 mM Tris-HCl, pH 8.3 |
| BufferA | 5 mM KCl and 1 mM MgCl2 in 20 mM Tris-HCl, pH 8.3 (used in all the measurements, unless stated otherwise) |
| BufferB | 5 mM KCl, 1 mM MgCl2, and 0.1 mM EDTA in 10 mM Tris-HCl, pH 8.0 |
| BufferC | 5 mM KCl and 1 mM MgCl2 in 10 mM Tris-HCl, pH 8.0 |
Development of the competitive assay
Buffer screening
The thrombin binding of the two aptamers used (i.e., TBA15 and TBA28, where 15 and 28 correspond to their respective lengths in nucleotides (nts) and are represented as sequences nr 1 and 2 respectively in Table S1, ESM) was evaluated in four different buffers: BufferDNAzyme, BufferA, BufferB, and BufferC (for composition, see Table 1). This was done using the fiber optic surface plasmon resonance (FO-SPR) biosensor from FOx Biosystems (Diepenbeek, Belgium) (Fig. S1, ESM). The FO-SPR probes were manufactured and functionalized as described in Daems et al. [30]. Briefly, FO-SPR probes, sputtered with a gold layer, were functionalized with the aptamer TBA15 or TBA28 through thiol binding (Fig. S2, ESM). Subsequently, the functionalized FO-SPR probes were immersed in 150 μL of the four different buffers containing a range of thrombin concentrations (0, 62, 124, and 248 nM). The resonance wavelength was measured for 20 min at RT and the shift in the resonance wavelength was calculated per tested concentration.
In parallel, the cleavage activity of the DNAzyme1 (nr 3 in Table S1, ESM), used for establishing the competitive bioassay, was tested in 25 μL of the four buffers (BufferDNAzyme, BufferA, BufferB, and BufferC) containing 250 nM of DNAzyme1 and 250 nM of Substrate1, the latter dually labelled with F on the 5′ end and Q on the 3′ end (nr 5 in Table S1, ESM). The activity of DNAzyme1 in this and all subsequent experiments with Aptazyme1.15-3′ was monitored on a microplate reader, by measuring the fluorescence signal for 10 min at RT using 485 and 535 nm as excitation and emission wavelengths, respectively, whereas all measurements were performed in triplicates.
Validation of Aptazyme1.15-3′
The binding activity of the TBA15 within the constructed aptazyme was evaluated in BufferA (selected as the most optimal based on buffer screening as described above) using the same FO-SPR technique. The FO-SPR probes, functionalized with Aptazyme1.15-3′Thiol through thiol binding (nr 7 in Table S1, ESM), were immersed in 150 μL of BufferA containing different concentrations of thrombin (0, 62, 124, and 248 nM). The FO-SPR probes functionalized with TBA15 were used as a reference.
The cleavage activity of the DNAzyme1 within the Aptazyme1.15-3′ and Aptazyme1.PolyT (nr 8 and 9, respectively in Table S1, ESM) was tested in 25 μL of BufferA containing 250 nM of one of the respective aptazymes and 250 nM of Substrate1. The same reaction performed with DNAzyme1 served as a positive control.
Optimization of Aptazyme1.15-3’blocking
The blocking of the Aptazyme1.15-3’ activity was tested using two different inhibitory sequences (Inhibitor1A and Inhibitor1B at 2500 nM, nr 16 and 17, respectively in Table S1, ESM) at two different incubation times (15 and 30 min). After selecting Inhibitor1B and 15 min of reaction as the most optimal conditions, we next tested the potential of thrombin to unblock the Aptazyme1.15-3’ by using different concentrations of Inhibitor1B (250, 500, 1250, and 2500 nM) and one thrombin concentration (248 nM). In all these experiments, 250 nM of Substrate1 was added to generate fluorescence signal, and monitored for 10 min on a microplate reader as previously described. The signal generated upon the addition of thrombin was calculated by subtracting the signal in the absence of thrombin from the signal in its presence. All measurements were performed in 35 μL with 250 nM of Aptazyme1.15-3’.
Competitive bioassay
The possibility to detect either protein or NA target using the same approach was assessed by incubating 250 nM of Aptazyme1.15-3’ with 500 nM of Inhibitor1B (1:2 ratio). Next, 0, 15.5, 31, 62, 124, and 248 nM of the target, being either thrombin or NA1 (nr 19 in Table S1, ESM), and 750 nM of Substrate1 were added, in a total volume of 35 μL. The optimization of the concentrations of the components in the competitive bioassays has been described in Optimization of Substrate1 concentration (ESM).
Development of the multiplex bioassay
Aptazyme2design
For establishing the multiplex bioassay, DNAzyme2, Aptazyme2.28-3’ (nr 4 and 10, respectively in Table S1, ESM), and the variations of Aptazyme2.28-3’ (nr 11-15 in Table S1, ESM) were designed. The secondary structures of newly designed aptazymes were studied using the OligoAnalyzer 3.1 tool of IDT Technologies (https://eu.idtdna.com/calc/analyzer). Their cleavage activity was tested in 25 μL of BufferA, containing one of these sequences at 250 nM, together with 250 nM of Substrate2, which is dually labelled with H and Q on the 5’ and 3’ end, respectively (nr 6 in Table S1, ESM). The kinetic activity was monitored by measuring the fluorescence signal for 10 min at RT using 536 and 606 nm as excitation and emission wavelengths, respectively. All measurements involving multiplex bioassay were performed in triplicates.
Validation of the second competitive bioassay using Aptazyme2.20-5’
The second competitive bioassay was developed with Aptazyme2.20-5’, selected as the best performing sequence, under the same conditions as described above: 250 nM of Aptazyme2.20-5’ was incubated together with Inhibitor2 (nr 18 in Table S1, ESM) in a 1:2 ratio for 15 min at RT, followed by adding several concentrations of the NA2 target (nr 20 in Table S1, ESM) — 0, 15.5, 31, 62, 124, and 248 nM and 750 nM of Substrate2. All measurements were performed in a total volume of 35 μL.
Evaluation of competitive bioassay specificity in multiplex conditions
The compatibility of the two substrates was tested in a volume of 35 μL containing Substrate1 or Substrate2, or both together, each at 250 nM concentration. The fluorescent signal was measured for 10 min using 485–535 and 536–606 nm as excitation-emission wavelengths pair for the F and H fluorescent labels, respectively. Similarly, the specificity of the aptazymes for their substrate was evaluated by combining 250 nM of each aptazyme with 250 nM of each substrate, separately or in combination. Next, the specificity of the bioreceptors for their targets was evaluated in a solution of 35 μL containing Aptazyme1.15-3’ and Aptazyme2.20-5’ (each at 125 nM), previously inhibited with 250 nM of their respective inhibitors (thus keeping 1:2 ratio). Then, 248 nM of one or both targets was added to the solution together with 375 nM of Substrate1 and 375 nM of Substrate2.
Multiplex bioassay
The performance of the multiplex bioassay was tested by combining, in a 35 μL solution, 125 nM of Aptazyme1.15-3’ inhibited with 250 nM of Inhibitor1B and 125 nM of Aptazyme2.20-5’ inhibited with 250 nM of Inhibitor2. Then, several concentrations (0, 15.5, 31, 62, 124, and 248 nM) of either thrombin and NA2, or NA1 and NA2 were added in the solution. After adding 375 nM of each substrate, the fluorescence was measured as previously explained.
Building calibration curves and data analysis
For all experiments involving the measurement of the DNAzyme or the aptazyme activity, a solution containing only the corresponding substrate, or combination of substrates, was used to normalize the measured fluorescence intensity. The difference between the conditions tested was analyzed with a one-way ANOVA followed by a Tukey test for multiple comparisons.
Calibration curves were composed using the normalized fluorescence intensity at 7 min after the start of the reaction and subtracting the background intensity (in the absence of target) for each repetition. Limit of detection (LOD) values for different competitive bioassays were calculated based on the signal of the background plus three standard deviations. Finally, the difference between the singleplex and multiplex LOD was evaluated with a two-sided T-test with significance level alpha equal to 5%. The statistical analysis was performed in Python 3.7 using the scipy and statsmodels packages.
Results and discussion
Screening of buffers for the optimal Aptazyme1.15-3’performance
In order to have a functional Aptazyme1.15-3’ for developing a competitive bioassay, we first needed to select the most optimal buffer conditions, permitting both efficient TBA interaction with its thrombin target and efficient DNAzyme catalytic activity. Several studies have reported the presence of K+ ions as stabilizers of the TBA guanosine (G)-quadruplex structure [31, 32]. Moreover, the DNAzyme requires the presence of the Mg2+ as a cofactor to express cleavage activity [18]. Therefore, to meet the needs of both aptazyme elements, we selected three candidate buffers from literature containing both K+ and Mg2+ ions, but with different components and pH values [33–35] (referred to in this paper as BufferA, BufferB, and BufferC; for composition, see Table 1).
Although several TBAs have been discovered to date, only two are frequently used, TBA15 and TBA28 [36, 37], both of which undergo a similar secondary conformation based on an intramolecular G-quadruplex despite differing in length. To select one for establishing the competitive bioassay, we first tested their interaction with several thrombin concentrations (0, 62, 124, and 248 nM) in BufferA, BufferB, and BufferC. This was done on a FO-SPR sensor, commercialized by FOx Biosystems (a platform based on an in-house developed technology [29, 30]), by immobilizing thiol-modified aptamers on the FO-SPR probes (Fig. S2). As depicted in Fig. 2A (TBA15) and Fig. S3 in ESM (TBA28), TBA15 showed overall more consistent interaction with thrombin across different concentrations and buffers and was selected as the preferred aptamer in this context. In addition, we observed no statistically significant differences for TBA15-thrombin interaction among BufferA, BufferB, and BufferC (based on a one-way ANOVA followed by a Tukey test). Interestingly, we have also tested in the same context the DNAzyme buffer, which seemed not to be suitable for the interaction of TBA15 with thrombin, despite having the same components as the other buffer candidates. These results could be explained by the previously reported decrease in performance of TBA at high Mg2+ concentrations [38, 39].
Fig. 2.
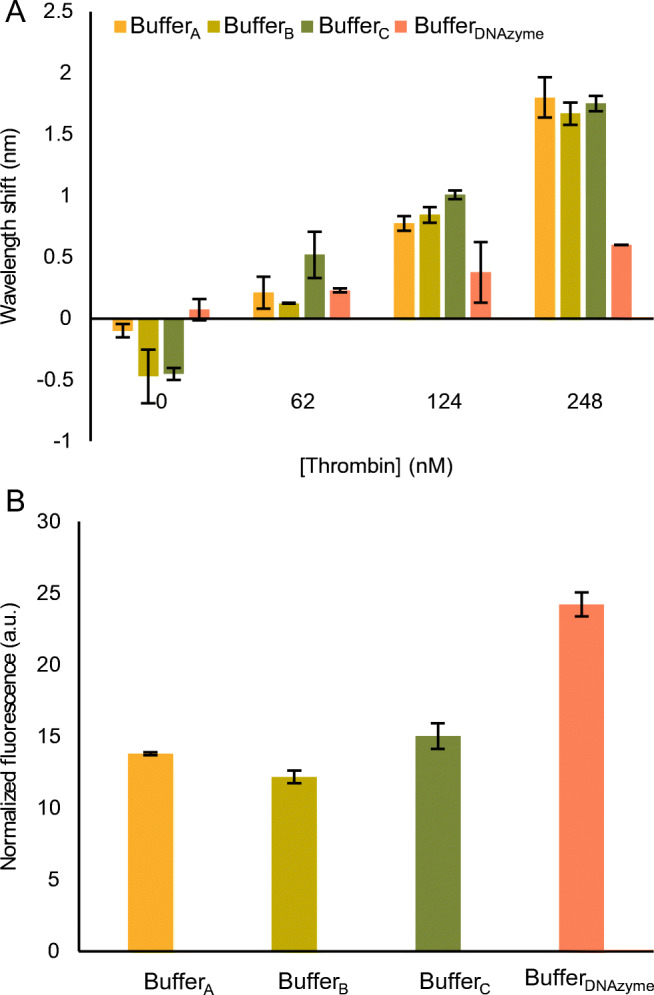
Performance of the aptazyme components in various buffers. A Performance of TBA15, immobilized on the FO-SPR sensor, obtained by measuring the wavelength shift in the presence of a dilution series of thrombin. B Fluorescence signal generated by DNAzyme1 (250 nM) in the presence of Substrate1 (250 nM) as measured on the microplate reader. The error bars in A and B represent the standard deviations of two and three repetitions, respectively
For assessing DNAzyme1 activity in BufferA, BufferB, and BufferC, we used a microplate reader as explained in the “Materials and methods.” Among the three candidate buffers, we only observed a statistical difference between BufferB and BufferC, based on a one-way ANOVA with Tukey test (Fig. 2B). BufferDNAzyme was used here as a positive reference and expectedly showed the highest signal, due to the highest concentration of Mg2+ in this buffer. However, because of the complete absence of TBA interaction with thrombin in this buffer (Fig. 2A), it could not be taken into account for developing competitive bioassays with aptazymes. Therefore, BufferA was selected for all the subsequent experiments, based on (1) Fig. 2A and Fig. 2B, (2) the fact that BufferA has the same pH (8.3) like the reference buffer for DNAzyme and (3) the same pH like TGK buffer used for optimal binding of TBA aptamer [40] (50 mM Tris, 192 mM glycine and 5 mM K2HPO4, 0.1% v/v Tween20 and 0.15% w/v BSA, pH 8.3).
Next, we independently verified the functionality of TBA15 and DNAzyme1 when combined in Aptazyme1.15-3’. To test the aptamer functionality, Aptazyme1.15-3’ was immobilized on FO-SPR probes and its binding to thrombin (0, 62, 124, and 248 nM) was monitored using BufferA (Fig. 3A). The results showed a superior binding of TBA15 within Aptazyme1.15-3’ compared to the control TBA15 immobilized on the FO-SPR probes, especially pronounced at higher thrombin concentrations. This improvement could be due to a reduction of the steric hindrance, and an increased spatial freedom for TBA15 to adopt its secondary conformation within Aptazyme1.15-3 because DNAzyme1 acts as a sort of a linker (Fig. S2, ESM).
Fig. 3.
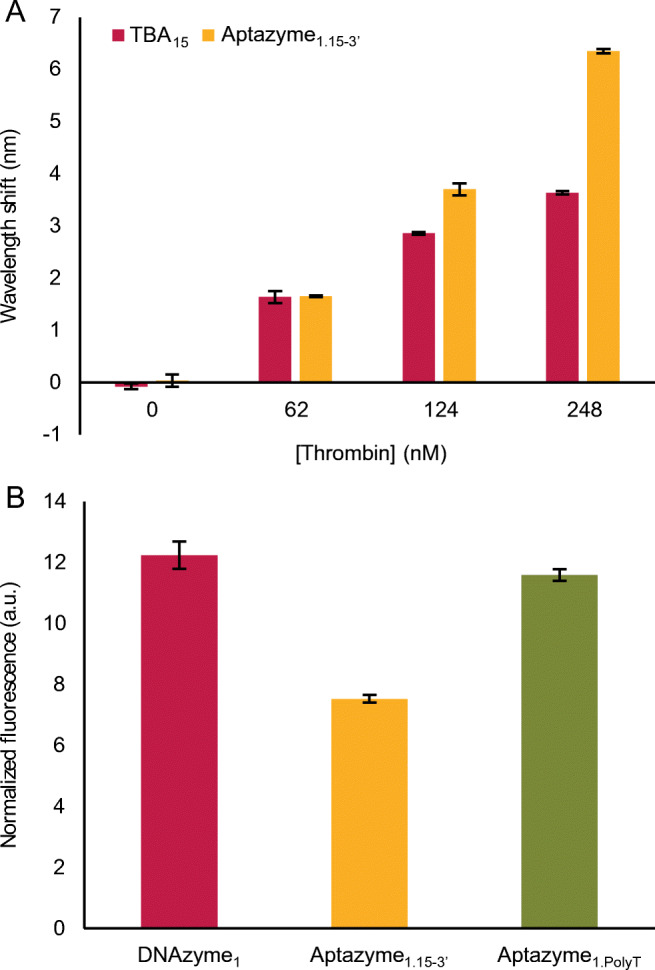
Validation of the functionality of TBA15 (A) and DNAzyme1 (B) within the hybrid Aptazyme1.15-3’. A FO-SPR probes were functionalized with either TBA15 (positive control) or Aptazyme1.15-3’ and their interaction with thrombin at different concentrations was monitored by a shift in the resonance wavelength. B The performance of DNAzyme1 within the hybrid Aptazyme1.15- 3’ and Aptazyme1.PolyT, the latter having TBA15 replaced with a PolyT of the same length, was validated by measuring the fluorescence intensity of the cleaved F/Q — labelled Substrate1 and compared to the original DNAzyme1 activity. Aptazyme1.15-3’ or DNAzyme1 and Substrate1 were used in these experiments at 250 nM concentration each. The error bars in A and B represent the standard deviation of two and three repetitions, respectively
In parallel, we compared the cleaving activity of Aptazyme1.15-3’ to that of DNAzyme1 by measuring the fluorescence signal increase after the addition of Substrate1 (Fig. 3B and ESM Fig. S4 for the signal increase over time). In this case, the results showed an inferior fluorescence intensity for Aptazyme1.15-3’. According to the literature, the quantum yield of a fluorescence reporter can be affected by the contiguous nucleobases, with G having the greatest impact [41, 42]. Given that TBA15 contains 60% of G, we replaced its sequence within Aptazyme1.15-3’ with a PolyT of the same length (Aptazyme1.PolyT, nr 7 in Table S1, ESM) and measured its cleaving activity. Here, the same signal was obtained for both the DNAzyme1 and the Aptazyme1.PolyT (Fig. 3B), suggesting that the decrease in the signal of Aptazyme1.15-3’ was probably due to G-mediated quenching of fluorescence. Therefore, we decided to proceed with Aptazyme1.15-3’ without any further optimization of its DNAzyme1 activity.
Development of DNA-only competitive bioassay for thrombin and NA target detection
To establish a competitive bioassay as depicted in Fig. 1, we first had to determine the optimal length of the inhibitory sequence. Here, we considered the percentage of the DNAzyme1 and TBA15 sequence within Aptazyme1.15-3’ that had to be blocked in order to, on one hand, maintain DNAzyme1 inhibition as stable as possible in the absence of thrombin, but on the other hand allow removal of the inhibitor upon addition of thrombin. In this context, we evaluated the blocking efficiency of two inhibitory sequences covering either the substrate binding arm of DNAzyme1 alone or in conjunction with three bases of the core as well (respectively, Inhibitor1A and Inhibitor1B, corresponding to nr 14 and 15 in Table S1, ESM) (Fig. 4A). It is important to note that both sequences blocked four nts of TBA15, which was based on the previous work demonstrating that blocking 26% of the aptamer results in a low background and a significant detection of thrombin at low temperatures [43].
Fig. 4.
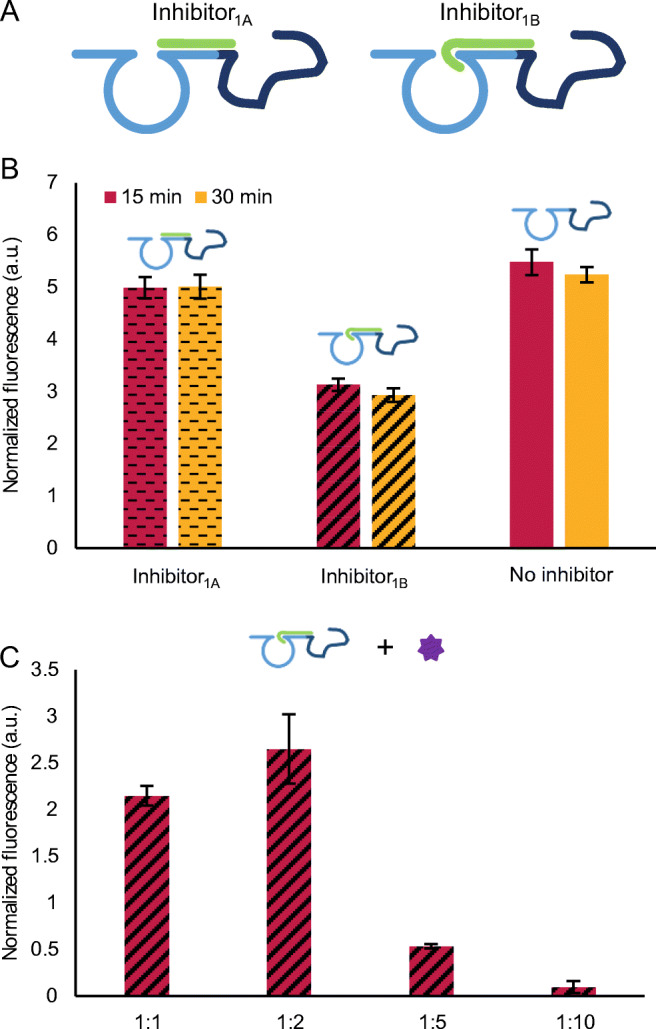
Optimization of the inhibition of Aptazyme1.15-3’. A Schematic representation of the two inhibitory sequences, Inhibitor1A and Inhibitor1B. B Testing two inhibitory sequences at two hybridization times (15 and 30 min) at 250 nM of Aptazyme15- 3’ and 2500 nM of Inhibitor1A or Inhibitor1B. The generated fluorescence signal was recorded upon addition of 250 nM of Substrate1. C Testing the capacity of thrombin (248 nM) to reactivate Aptazyme1.15-3’ inhibited with Inhibitor1B in different ratios (1:1, 1:2, 1:5, and 1:10, which correspond to 250 nM of Aptazyme1.15-3’: 250, 500, 1250, and 2500 nM of Inhibitor1B, respectively). In this case, the signal in the absence of thrombin was subtracted from the signal in the presence of thrombin. In both graphs, the error bars represent the standard deviation of three repetitions. The description of the fluorescence normalization process can be found in “Materials and methods” section
In a first attempt, 250 nM of Aptazyme1.15-3’ was incubated with each inhibitory sequence in a 1:10 ratio (i.e., 2500 nM) for 15 and 30 min and the generated fluorescence signal was measured after adding 250 nM of Substrate1 (Fig. 4B). Regardless of the incubation time, the obtained fluorescence signal when using Inhibitor1A was comparable to the non-inhibited Aptazyme1.15-3’, meaning that covering only the substrate binding arm of DNAzyme1 was not sufficient to prevent the cleavage of substrate. Contrary to this, covering part of the catalytic core in addition to the substrate binding arm resulted in half of the signal when compared with the non-inhibited Aptazyme1.15-3’, without significant difference between two tested times. Consequently, we selected Inhibitor1B and shorter incubation (15 min) for further optimization in the following experiments.
In a next step, we tested the ability of thrombin to remove the inhibitor and as such reactivate DNAzyme1 within Aptazyme1.15- 3’ (Fig. 4C). Therefore, 248 nM of thrombin (the highest concentration tested on the FO-SPR device, see Figs. 2 and 3) and the same concentration of Substrate1 as in the previous experiment (250 nM) were added to the inhibited Aptazyme1.15- 3’. Here, in parallel, we also investigated other ratios between the aptazyme and the inhibitor, rather than just 1:10 tested in Fig. 4B, being 1:1, 1:2, and 1:5. Although thrombin was capable of reactivating inhibited Aptazyme1.15-3’ for all the ratios, this success was clearly different among tested conditions. The one-way ANOVA with Tukey test showed a statistical difference between 1:2 and the other ratios, except for 1:1. However, based on the higher average value obtained for the 1:2 ratio compared to 1:1, the former was selected for all the subsequent experiments. After additional optimization of Aptazyme1.15-3’ and Substrate1 concentrations, we selected 250 nM of Aptazyme1.15-3’ and 750 nM of Substrate1 to proceed (Fig. S5, ESM), while keeping 1:2 ratio between aptazyme and inhibitor.
Using the optimized conditions, we tested the capacity of competitive bioassay to detect not only thrombin but an NA target as well (0 – 248 nM) (Fig. 5). The latter was achieved by simply using the complementary sequence of TBA15 as a target (NA1, nr 17 in Table S1, ESM). The competitive bioassay was performed as explained above and the generated fluorescence was measured after the addition of 750 nM of Substrate1. For both targets, we obtained a similar concentration-dependent response, although for NA1 the overall signal intensity was higher than that for thrombin. The calculated LOD was 45.6 ± 0.8 nM and 14.1 ± 0.9 nM for thrombin and NA1, respectively.
Fig. 5.
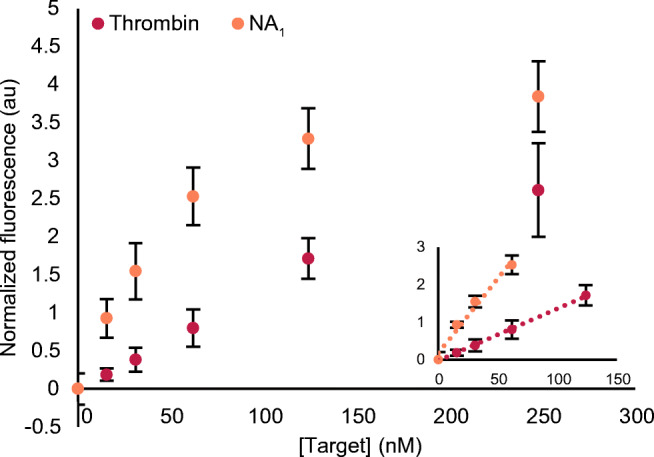
Concentration-dependent response of the competitive bioassay in the presence of thrombin or NA1. The inset shows the linear range for both targets, being 0–124 nM for thrombin (R2 = 0.99) and 0–62 nM for NA1 (R2 = 0.99). The error bars represent the standard deviation of three repetitions, whereas the LOD values have been calculated as described in the “Materials and methods” section
Design of Aptazyme2for establishing multiplex DNA-only bioassay
After demonstrating that the same competitive bioassay concept can be used to detect both protein and NA targets separately, we attempted to detect them simultaneously in the same reaction. However, because both targets were detected in singlepex reactions using the same Aptazyme1.15-3’, we first needed to construct a new aptazyme. Here, the original Aptazyme1.15-3’ was kept for thrombin detection, while a new aptazyme was designed for detecting a new NA target (i.e., NA2), motivated by the much easier design of a new target recognition sequence for the NA target. To achieve this, we made two changes to the original Aptazyme1.15-3’. First, to fluorescently differentiate between the two targets, we modified the substrate-binding arms of DNAzyme1 for binding Substrate2, labelled with a different fluorophore (nr 20 in Table S1, ESM), while keeping the same catalytic core. This substrate sequence was previously reported by Mokany et al. [44] and redesigned by Ven et al. for optimal performance at RT [26]. Second, we swapped the TBA15 for a random 28-mer NA sequence (nr 8 in Table S1, ESM the same bioreceptor). This resulted in generation of Aptazyme2.28-3’.
Contrary to the expectations, Aptazyme2.28-3’ was able to generate only 20% of the fluorescence signal generated by the corresponding DNAzyme2 (Fig. 6). To have a better understanding of the observed difference, we studied their secondary structures using the OligoAnalyzer 3.1 tool of IDT Technologies (https://eu.idtdna.com/calc/analyzer) (Fig. S6A and Fig. S6B, ESM). Aptazyme1.15-3’ was used here as a reference considering that the successful competitive bioassay was already accomplished with it. Based on the analysis of the most stable secondary structures, we observed that the addition of TBA15 to DNAzyme1 did not affect its secondary structure. However, Aptazyme2.28-3’ had an extra hairpin compared to DNAzyme2, which was composed by part of the (1) core, (2) substrate-binding arm, and (3) target recognition sequence (highlighted as yellow, red, and blue respectively in Fig. S6B, ESM). The fact that the target recognition sequence induced a change in the secondary structure of DNAzyme2 could explain the decrease in the enzymatic activity of Aptazyme2.28-3’.
Fig. 6.
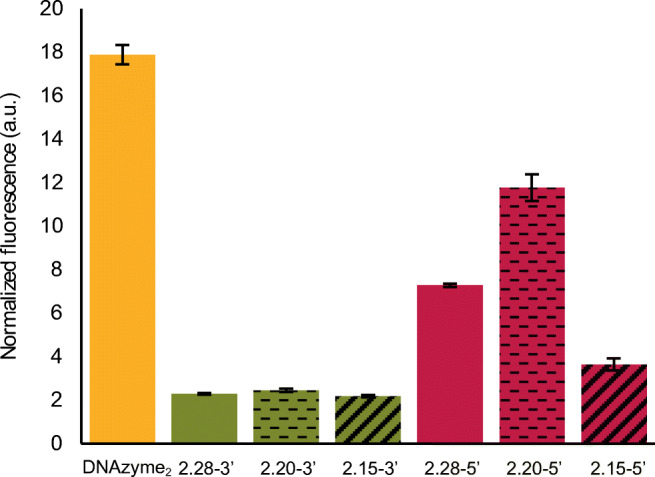
Screening of different Aptazyme2 designs based on their catalytic activity in the presence of Substrate2. The influence of different lengths (28, 20, and 15 nts) and positions (3’ or 5’ end of the DNAzyme2) of the target recognition sequences were studied. The error bars represent the standard deviation of three repetitions. Additionally, the fluorescence normalization process is described in the “Materials and methods” section
In order to develop an aptazyme of similar catalytic activity as DNAzyme2, we designed five additional aptazymes: two with target recognition sequences (20 and 15 nts) positioned on the 3’ side of DNAzyme2 and three with target recognition sequences (28, 20, and 15 nts) on the 5’ side (nr 8–13 in Table S1, ESM). Their secondary structures were then analyzed using the same simulation tool as above, followed by measuring their enzymatic activity. Aptazyme2.20-3’ and Aptazyme2.15-3’ revealed similar secondary structures as Aptazyme2.28-3’, suggesting that the target recognition sequences positioned on the 3’ side of DNAzyme2 affected its intrinsic conformation (Fig. S6C, ESM). This was further confirmed with the similar (low) level of enzymatic activity among these three aptazymes (Fig. 6). Contrary to this, the target recognition sequences introduced on the 5’ side of DNAzyme2 gave noticeable increase in the activity, which was different for all three aptazymes, while being the highest for Aptazyme2.20-5’ that was capable of generating 66% of the signal generated by the corresponding DNAzyme2. This observed disparity in their activities could be due to their secondary structures. To understand the differences, we considered two different aspects of the secondary structure: its stability and the involvement of DNAzyme2 in this secondary structure. For instance, while the target recognition sequence in Aptazyme2.28-5’ created an additional hairpin involving the substrate binding arm, it did not affect the intrinsic conformation of DNAzyme2 as in the case of Aptazyme2.15-5’. This could be the explanation why this aptazyme showed higher activity than Aptazyme2.15-5’ despite having a more stable secondary structure. In a similar manner, the target recognition sequence in Aptazyme2.20-5’ generated an additional hairpin, but with a shorter stem than Aptazyme2.28-5’, which resulted in a less stable conformation and thus higher activity. Taking all this into account, we can conclude that the degree to which the target recognition sequence affects the secondary structure of the DNAzyme is crucial for the aptazyme performance. This was further confirmed by the observation that, for all tested aptazymes, signal generation increased upon adding the target (data not shown), which can be explained by the fact that this target interacted with the part of the aptazyme that actually interfered with the DNAzyme activity. Importantly, however, even under these conditions, Aptazyme2.20-5’ remained the construct with the highest activity, therefore being selected for developing the multiplex bioassay.
Before using Aptazyme2.20-5’ directly in the multiplex bioassay, we also evaluated its performance in the competitive bioassay. Here, 250 nM of Aptazyme2.20-5’ was inhibited using Inhibitor2 as previously described. Subsequently, several concentrations of NA2 target were added in the range from 0 to 248 nM together with 750 nM of Substrate2, followed by measuring the generated fluorescence signal for each target concentration (Fig. S7, ESM). The fitting in the linear range resulted in a LOD of 5.1 ± 0.1 nM, similar to what was previously obtained for NA1.
DNA-only multiplex bioassay for detection of thrombin and NA target or two NA targets
To develop a multiplex bioassay, we first ensured the possibility of independently detecting both fluorescence reporters in the same solution without any interference. Therefore, we measured the intensity of Substrate1 and Substrate2 separately and in the same solution, in both F and H channels (Fig. S8A, ESM). The fluorescence intensities observed in both channels when both substrates were present together were comparable to their independent measurements, indicating no interference between the fluorescent labels. In the next step, to corroborate the specificity of each aptazyme for its substrate, we measured the fluorescence signal generated by each aptazyme separately and in the same solution, in the presence of both substrates (Fig. S8B, ESM, observing no non-specific signal generation). Following this, we evaluated the specificity of the multiplex approach in terms of the bioreceptors. Hereto, we measured the fluorescence signal generated by the 125 nM of Aptazyme1.15-3’ or Aptazyme2.20-5’, respectively inhibited with 250 nM of Inhibitor1B or Inhibitor2 prior to adding each individual target (248 nM of thrombin or NA2) or both targets together in the same reaction (Fig. 7A). In parallel, we also performed a similar experiment to determine the bioreceptor specificity when detecting two NA targets (Fig. 7B). In all these experiments, both Substrate1 and Substrate2 were always added together, each at 375 nM concentration. The results revealed high specificity of the bioreceptors towards their respective targets in both tested bioassays. All this also pointed out that the observed interactions are not promoted by the buffer selection, but are a reflection of the specific interactions between the aptazymes and their respective NA targets.
Fig. 7.

DNA-only multiplex bioassay for simultaneous detection of different targets. Evaluation of the bioreceptor specificity in multiplex conditions using A 248 nM of thrombin and NA2 as targets, and B 248 nM of NA1 and NA2 as targets. C Concentration-dependent response for the detection of thrombin and NA2. Inset figure represents the linear range of thrombin between 0 and 124 nM (R2 = 0.99) and of NA2 between 0 and 62 nM (R2 = 0.99). D Concentration-dependent response for the detection of NA1 and NA2. Inset figure represents the linear range between 0 and 124 nM for NA1 (R2 = 0.99) and 0 and 62 nM for NA2 (R2 = 0.99). For all graphs, the error bars represent the standard deviation of three repetitions and the LOD values have been calculated as described in the “Materials and methods” section
Finally, we evaluated the multiplex detection of thrombin and NA2 over the concentration range from 0 to 248 nM using Aptazyme1.15-3’ and Aptazyme2.20-5’ (each at 125 nM), inhibited with 250 nM of their respective inhibitors prior to adding the rest of the bioassay components. In the same manner, we attempted the simultaneous detection of NA1 and NA2. In both cases, we monitored the fluorescence intensity after the addition of Substrate1 and Substrate2, each at 375 nM concentration (Fig. 7C and Fig. 7D). The performed multiplex bioassays revealed LOD values in the nM range like the singleplex versions of the same bioassay (Table 2). The former, however, showed slightly lower LOD values for all targets, except for NA2 when detected together with NA1 (statistically significant as evaluated with a T-test). This improvement in LOD for the multiplex measurements could be attributed to the lower variability observed for the blank measurements, which was not the case for NA2 in the NA1 and NA2 multiplexing. Another possible explanation could be because of the reduced concentrations of each reaction component to half in the multiplexing bioassay in order to maintain the total amount of reaction elements in the solution. Nevertheless, taken altogether, these results revealed that the developed bioassay can be successfully performed both in singleplex and multiplex formats with sensitivities in the nM range. This is in accordance with similar reported LOD values for other aptazymes (including those with a different catalytic cores), ranging from 6.9 pM to 18 μM when using fluorescence-based read-out [25, 45, 46].
Table 2.
Overview of the obtained LOD values in the different bioassays established in this work, as depicted in Fig. 1
| Singleplex | Multiplex | |
|---|---|---|
| Thrombin | 45.6 ± 0.8 nM | 15.6 ± 0.5 nM |
| NA1 | 14.1 ± 0.9 nM | 7.5 ± 0.3 nM |
| NA2 | 5.1 ± 0.1 nM | 3.1 ± 0.1* / 6.4 ± 0.3 nM** |
In the case of NA2, two LODs were calculated: one from the multiplex bioassay for thrombin and NA2 (*) and the other one from NA1 and NA2 (**)
Conclusions
In the present work, we have shown for the first time the simultaneous detection of proteins and NAs in a competitive bioassay by using exclusively FNA molecules, i.e., aptazymes, which combine target recognition and signal generation in a single molecule. To attain this, we first screened several buffer compositions for preserving the optimal performance of both the aptamer and the DNAzyme within Aptazyme1.15-3’, followed by establishing the optimal concentrations for all the bioassay components. Subsequently, we successfully proved the feasibility to detect both thrombin and NA1 target in a singleplex competitive bioassay with calculated LOD values of 45.6 ± 0.8 nM and 14.1 ± 0.9 nM, respectively. Next, for achieving multiplexing, we modified the Aptazyme1.15-3’ by replacing the substrate binding arms of the DNAzyme and target recognition sequence. Here, we successfully designed aptazymes without affecting their catalytic activity based on the in silico analysis of their secondary structures. From the study of several aptazyme sequences, we concluded that the activity of the DNAzyme within an aptazyme can be strongly impacted by the interference of the target recognition sequence with the DNAzyme secondary structure. By using this prediction tool, we designed a new aptazyme, Aptazyme2.20-5’, for detecting NA2 target with an LOD of 5.1 ± 0.1 nM in a singleplex bioassay. Finally, we combined both Aptazyme1.15-3’ and Aptazyme2.20-5’ in the same solution for the simultaneous detection of thrombin and NA2 target or NA1 and NA2. In these multiplex bioassays, we demonstrated the high specificity of each aptazyme with no decrease in the bioassay efficiency compared to their performance in the singleplex bioassay. Although previous studies have reported the detection of thrombin [46–48] or various NA targets [49–51] with picomolar or even femtomolar sensitivities, many of those approaches required elevated temperatures and/or longer assay time (30 min – 5 h). Contrary to this, the approach we presented in this work not only is capable of detecting the targets at RT within 10 min, but is also unique in its capacity to detect multiple targets, regardless of their origin or chemical nature, using the same bioassay components and the same read-out method. Considering this premise, such a multiplex bioassay could be conveniently modified for different targets of interest, by replacing the aptamer or target recognition sequence. Similarly, the number of targets could be increased by incorporating new substrate-binding arms of the DNAzyme. Furthermore, due to the advantages associated to the NA nature of the bioassay, the possibility to work at RT, and the short assay time, this approach could be a good candidate for further implementation in POC platforms.
Supplementary Information
(DOCX 2.18 mb)
Acknowledgements
This work has received funding from European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 675412 (H2020-MSCA-ITN-ND4ID), the KU Leuven (postdoctoral mandate Saba Safdar PDM/20/080, research project C3/20/057), and the Research Foundation−Flanders (FWO SB/1S30116N, Research Project G042918N).
Biographies
Aida Montserrat Pagès
obtained her Master in Advanced Nanoscience and Nanotechnology (Nanobiotechnology) from Universitat Autònoma de Barcelona (UAB) in 2016. She is now a PhD student in the Biosensors group in the division Mechatronics, Biostatistics and Sensors (MeBioS) of the Biosystems department at KU Leuven, Belgium. She is currently working on studying the kinetics and reaction mechanism of RNA-cleaving DNAzymes for new strategies in biosensor development.

Saba Safdar
obtained her Master’s in Biomedical Engineering from Arizona State University, USA, in 2016 and her PhD in Bioscience Engineering from KU Leuven, Belgium, in 2020. She is currently a postdoctoral researcher in the Biosensors group in the division Mechatronics, Biostatistics and Sensors (MeBioS) of the Biosystems department at KU Leuven, Belgium. Her current work focusses on the development of innovative CRISPR-based assays for various in vitro and in vivo applications.

Karen Ven
received her Bachelor and Master degrees in Bioscience Engineering from KU Leuven in 2013 and 2015, respectively. In 2020, she received a PhD degree in Bioscience Engineering from KU Leuven for her work on novel concepts for digital bioassays, in the Biosensors group in the division Mechatronics, Biostatistics and Sensors (MeBioS) of the Biosystems department. She focused on novel fabrication strategies for the platform, as well as NAzyme-based approaches for signal generation and amplification. Currently, she is working in the same group on microfluidic solutions for single-cell analysis and isolation.

Jeroen Lammertyn
is Full Professor in Bionanotechnology at the KU Leuven. He founded the Biosensors group in 2005. The group closely follows the emerging field of food and medical diagnostics and is active in bio-assay development, DNA-nanotechnology, optical biosensors, and microfluidics. He is (co-)author of >300 peer-reviewed research papers, conference papers, and book chapters. He acts as reviewer for many international peer-reviewed journals, is co-inventor of 12 patent applications, and founder of the spin-off company FOx Biosystems. He is also chairman of the DIATECH 2014 and 2020 International Conferences on Novel technologies for in vitro Diagnostics in Leuven.

Dragana Spasic
received her MSc and PhD degrees in Medical Sciences from KU Leuven in 2004 and 2009, respectively. After working as a post-doctoral researcher at the Faculty of Medicine and Bioscience Engineering (KU Leuven), since 2015 she has held a position of Research Manager in the Biosensors group (MeBioS division, KU Leuven). She is (co-)author of >100 peer-reviewed research papers, conference papers, and book chapters, and she is involved in research and projects related to development of bioassays and their implementation on various life science research tools and diagnostics platforms.

Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Souf S. Recent advances in diagnostic testing for viral infections. Biosci Horiz. 2016. 10.1093/biohorizons/hzw010.
- 2.Etzioni R, Urban N, Ramsey S, McIntosh M, Schwartz S, Reid B, et al. The case for early detection. Nat Rev Cancer. 2003. 10.1038/nrc1041. [DOI] [PubMed]
- 3.Shipp G. Ultrasensitive measurement of protein and nucleic acid biomarkers for earlier disease detection and more effective therapies clinical decision making stands to benefit from ultrasensitive molecular diagnostic. Biotechnol Healthc. 2006. [PMC free article] [PubMed]
- 4.Langelier C, Kalantar KL, Moazed F, Wilson MR, Crawford ED, Deiss T, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci. 2018. 10.1073/pnas.1809700115. [DOI] [PMC free article] [PubMed]
- 5.Vemula SV, Zhao J, Liu J, Xue XW, Biswas S, Hewlett I. Current approaches for diagnosis of influenza virus infections in humans. Viruses. 2016. 10.3390/v8040096. [DOI] [PMC free article] [PubMed]
- 6.Flamini E, Mercatali L, Nanni O, Calistri D, Nunziatini R, Zoli W, et al. Free DNA and carcinoembryonic antigen serum levels: an important combination for diagnosis of colorectal cancer. Clin Cancer Res. 2006. 10.1158/1078-0432.CCR-06-1931. [DOI] [PubMed]
- 7.Khan K, Aslam MA, Zahra FT, Bashir H, Bilal M, Sumrin A. Potential biomarkers for the diagnosis of respiratory tract infection and lungs cancer. Cell Mol Biol. 2017. 10.14715/cmb/2017.63.11.9. [DOI] [PubMed]
- 8.Yan Y, Ma C, Tang Z, Chen M, Zhao H. A novel fluorescent assay based on DNAzyme-assisted detection of prostate specific antigen for signal amplification. Anal Chim Acta. 2020. 10.1016/j.aca.2020.01.014. [DOI] [PubMed]
- 9.Li Y, Lu Y. Functional nucleic acids for analytical applications. Springer New York. 2009. 10.1021/ja905562z.
- 10.Xiao X, Zhu L, He W, Luo Y, Xu W. Functional nucleic acids tailoring and its application. TrAC-Trends Anal Chem. 2019. 10.1016/j.trac.2019.05.027.
- 11.Gold L, Tuerk C. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990. 10.1126/science.2200121. [DOI] [PubMed]
- 12.Chen A, Yang S. Replacing antibodies with aptamers in lateral flow immunoassay. Biosens Bioelectron. 2015. 10.1016/j.bios.2015.04.041. [DOI] [PubMed]
- 13.Song S, Wang L, Li J, Fan C, Zhao J. Aptamer-based biosensors. TrAC - Trends Anal Chem. 2008. 10.1016/j.trac.2007.12.004.
- 14.Breaker RR, Joyce GF. A DNA enzyme that cleaves RNA. Chem Biol. 1994. 10.1016/1074-5521(94)90014-0. [DOI] [PubMed]
- 15.Zhu X, Gao X, Liu Q, Lin Z, Qiu B, Chen G. Pb2+-introduced activation of horseradish peroxidase (HRP)-mimicking DNAzyme. Chem Commun. 2011. 10.1039/c1cc11349f. [DOI] [PubMed]
- 16.Camden AJ, Walsh SM, Suk SH, Silverman SK. DNA oligonucleotide 3′-phosphorylation by a DNA enzyme. Biochemistry. 2016. 10.1021/acs.biochem.6b00151. [DOI] [PMC free article] [PubMed]
- 17.Yin H, Kuang H, Liu L, Xu L, Ma W, Wang L, et al. A ligation Dnazyme-induced magnetic nanoparticles assembly for ultrasensitive detection of copper ions. ACS Appl Mater Interfaces. 2014. 10.1021/am405482a. [DOI] [PubMed]
- 18.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Biochemistry. 1997. 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed]
- 19.McGhee CE, Loh KY, Lu Y. DNAzyme sensors for detection of metal ions in the environment and imaging them in living cells. Curr Opin Biotechnol. 2017. 10.1016/j.copbio.2017.03.002. [DOI] [PMC free article] [PubMed]
- 20.Gong L, Zhao Z, Lv YF, Huan SY, Fu T, Zhang XB, et al. DNAzyme-based biosensors and nanodevices. Chem Commun. 2015. 10.1039/c4cc06855f. [DOI] [PubMed]
- 21.Achenbach JC, Nutiu R, Li Y. Structure-switching allosteric deoxyribozymes. Anal Chim Acta. 2005. 10.1016/j.aca.2004.03.080.
- 22.Rossetti M, Porchetta A. Allosterically regulated DNA-based switches: from design to bioanalytical applications. Anal Chim Acta. 2018. 10.1016/j.aca.2017.12.046. [DOI] [PubMed]
- 23.Liu X, Wang F, Aizen R, Yehezkeli O, Willner I. Graphene oxide/nucleic-acid-stabilized silver nanoclusters: functional hybrid materials for optical aptamer sensing and multiplexed analysis of pathogenic DNAs. J Am Chem Soc. 2013. 10.1021/ja403485r. [DOI] [PubMed]
- 24.Zhang L, Guo S, Zhu J, Zhou Z, Li T, Li J, et al. Engineering DNA three-way junction with multifunctional moieties: sensing platform for bioanalysis. Anal Chem. 2015. 10.1021/acs.analchem.5b02468. [DOI] [PubMed]
- 25.Huang J, He Y, Yang XH, Quan K, Wang KM. Inhibited aptazyme-based catalytic molecular beacon for amplified detection of adenosine. Chin Chem Lett. 2014. 10.1016/j.cclet.2014.05.039. [DOI] [PubMed]
- 26.Ven K, Safdar S, Dillen A, Spasic D, Lammertyn J. Re-engineering 10–23 core DNA- and MNAzymes for applications at standard room temperature. Anal Bioanal Chem. 2018. 10.1007/s00216-018-1429-4. [DOI] [PubMed]
- 27.Deng B, Lin Y, Wang C, Li F, Wang Z, Zhang H, et al. Aptamer binding assays for proteins: the thrombin example-a review. Anal Chim Acta. 2014. 10.1016/j.aca.2014.04.055. [DOI] [PubMed]
- 28.Yin HS, Li BC, Zhou YL, Wang HY, Wang MH, Ai SY. Signal-on fluorescence biosensor for microRNA-21 detection based on DNA strand displacement reaction and Mg2+-dependent DNAzyme cleavage. Biosens Bioelectron. 2017. 10.1016/j.bios.2017.04.049. [DOI] [PubMed]
- 29.Peeters B, Daems D, Van Der Donck T, Delport F, Lammertyn J. Real-time FO-SPR monitoring of solid-phase DNAzyme cleavage activity for cutting-edge biosensing. ACS Appl Mater Interfaces. 2019. 10.1021/acsami.8b18756. [DOI] [PubMed]
- 30.Daems D, Peeters B, Delport F, Remans T, Lammertyn J, Spasic D. Identification and quantification of celery allergens using fiber optic surface plasmon resonance PCR. Sensors. 2017. 10.3390/s17081754. [DOI] [PMC free article] [PubMed]
- 31.Marathias VM, Bolton PH. Determinants of DNA quadruplex structural type: sequence and potassium binding. Biochemistry. 1999. 10.1021/bi982604+. [DOI] [PubMed]
- 32.Tan SY, Acquah C, Tan SY, Ongkudon CM, Danquah MK. Characterisation of charge distribution and stability of aptamer-thrombin binding interaction. Process Biochem. 2017. 10.1016/j.procbio.2017.06.003.
- 33.Nutiu R, Li Y. Structure-switching signaling aptamers. J Am Chem Soc. 2003. 10.1021/ja028962o. [DOI] [PubMed]
- 34.Li N, Ho CM. Aptamer-based optical probes with separated molecular recognition and signal transduction modules. J Am Chem Soc. 2008. 10.1021/ja076787b. [DOI] [PubMed]
- 35.Tang Z, Mallikaratchy P, Yang R, Kim Y, Zhu Z, Wang H, et al. Aptamer switch probe based on intramolecular displacement. J Am Chem Soc. 2008. 10.1021/ja804119s. [DOI] [PubMed]
- 36.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992. [DOI] [PubMed]
- 37.Tasset DM, Kubik MF, Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J Mol Biol. 1997. 10.1006/jmbi.1997.1275. [DOI] [PubMed]
- 38.Kang Y, Feng KJ, Chen JW, Jiang JH, Shen GL, Yu RQ. Electrochemical detection of thrombin by sandwich approach using antibody and aptamer. Bioelectrochemistry. 2008. 10.1016/j.bioelechem.2008.04.024. [DOI] [PubMed]
- 39.Baldrich E, Restrepo A, O’Sullivan CK. Aptasensor development: elucidation of critical parameters for optimal aptamer performance. Anal Chem. 2005. 10.1021/ac049258o. [DOI] [PubMed]
- 40.Buchanan DD, Jameson EE, Perlette J, Malik A, Kennedy RT. Effect of buffer, electric field, and separation time on detection of aptamer-ligand complexes for affinity probe capillary electrophoresis. Electrophoresis. 2003. 10.1002/elps.200390176. [DOI] [PubMed]
- 41.Behlke MA, Huang L, Bogh L, Rose S, Devor EJ. Fluorescence quenching by proximal G-bases. Integr DNA Technol. 2005.
- 42.Mao H, Luo G, Zhan Y, Zhang J, Yao S, Yu Y. The mechanism and regularity of quenching the effect of bases on fluorophores: the base-quenched probe method. Analyst. 2018. 10.1039/c8an00116b. [DOI] [PubMed]
- 43.Nutiu R, Li Y. Structure-switching signaling aptamers: transducing molecular recognition into fluorescence signaling. Chem - A Eur J. 2004. 10.1002/chem.200305470. [DOI] [PubMed]
- 44.Mokany E, Bone SM, Young PE, Doan TB, Todd AV. MNAzymes, a versatile new class of nucleic acid enzymes that can function as biosensors and molecular switches. J Am Chem Soc. 2010. 10.1021/ja9076777. [DOI] [PMC free article] [PubMed]
- 45.Zhou Z, Xiao L, Xiang Y, Zhou J, Tong A. A general approach for rational design of fluorescent DNA aptazyme sensors based on target-induced unfolding of DNA hairpins. Anal Chim Acta. 2015. 10.1016/j.aca.2015.06.036. [DOI] [PubMed]
- 46.Li J, Wang S, Jiang B, Xiang Y, Yuan R. Target-induced structure switching of aptamers facilitates strand displacement for DNAzyme recycling amplification detection of thrombin in human serum. Analyst. 2019. 10.1039/c9an00030e. [DOI] [PubMed]
- 47.Chen J, Zuehlke A, Deng B, Peng H, Hou X, Zhang H. A Target-triggered DNAzyme motor enabling homogeneous, amplified detection of proteins. Anal Chem. 2017. 10.1021/acs.analchem.7b03529. [DOI] [PubMed]
- 48.Jie G, Tan L, Zhao Y, Wang X. A novel silver nanocluster in situ synthesized as versatile probe for electrochemiluminescence and electrochemical detection of thrombin by multiple signal amplification strategy. Biosens Bioelectron. 2017. 10.1016/j.bios.2017.03.015. [DOI] [PubMed]
- 49.Liu Y, Shen T, Li J, Gong H, Chen C, Chen X, et al. Ratiometric fluorescence sensor for the MicroRNA determination by catalyzed hairpin assembly. ACS Sensors. 2017. 10.1021/acssensors.7b00313. [DOI] [PubMed]
- 50.Wang H, Li C, Liu X, Zhou X, Wang F. Construction of an enzyme-free concatenated DNA circuit for signal amplification and intracellular imaging. Chem Sci. 2018. 10.1039/c8sc01981a. [DOI] [PMC free article] [PubMed]
- 51.Li C, Li H, Ge J, Jie G. Versatile fluorescence detection of microRNA based on novel DNA hydrogel-amplified signal probes coupled with DNA walker amplification. Chem Commun. 2019. 10.1039/c9cc00565j. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2.18 mb)