

Abstract
Cardiovascular disease, until now, is still the leading cause of death in the United States. Due to the limited regenerative capacity of adult hearts, the damage caused by heart injury cannot be reversed and eventually progress into heart failure. In need of cardiovascular disease treatment, many therapies aimed at either cell transplantation or cell regeneration have been proposed. Direct reprogramming of somatic cells into induced cardiomyocytes (iCMs) is considered to be a promising strategy for regenerative medicine. The induction of cardiomyocytes from non-myocytes can be achieved efficiently via ectopic expression of reprogramming factors both in vitro and in vivo in the mouse model, however, the generation of human induced cardiomyocyte-like cells (hiCMs) remains challenging. The inefficiency of hiCMs production called for the identification of the additional epigenetic memories in non-myocytes which might be damping the hiCM reprogramming. Here, we conducted an unbiased loss-of-function screening focusing on epigenetic regulators and identified enhancer of zeste homolog 2 (EZH2) as an important epigenetic barrier during hiCM reprogramming. We found that the removal of EZH2 via genetic knockdown or treatment of EZH2 selective degrader significantly increased the hiCM reprogramming efficiency and led to profound activation of cardiac genes and repression of collagen and extracellular matrix genes. Furthermore, EZH2 inhibitors targeting its catalytic activity also promotes hiCM reprogramming, suggesting that EZH2 may restrain cardiac conversion through H3K27me3-mediated gene repression. Indeed, genomic profiling of H3K27me3 revealed a subset of cardiac genes that remain repressed with high levels of H3K27me3 despite of the delivery of the reprogramming factors. Inhibition of EZH2, however, leads to reduced H3K27me3 occupancy and robust activation of these cardiac genes. Taken together, our data suggested that EZH2 inhibition facilitates the activation of cardiac genes in fibroblasts and eases the production of hiCMs.
Keywords: Direct cardiac reprogramming, Epigenetic regulation, EZH2, H3K27me3
1. Introduction
As the leading cause of death in the United States, cardiovascular disease and its relevant therapies have attracted many researching attentions. During heart attack, approximately a billion of cardiomyocytes from left ventricle die in the matter of hours (Murry et al., 2006). Because adult mammalian hearts have very limited regenerative capacity, novel regenerative strategies to replenish damaged cardiomyocytes hold promise to be important therapeutic approaches (Cahill and Kharbanda, 2017; Go et al., 2013; Oyama et al., 2013; Porrello et al., 2011). Direct reprogramming of somatic cells into induced cardiomyocytes (iCMs) by overexpression of cardiac-lineageenriched transcriptional factors emerges as a promising strategy for regenerative medicine (Addis and Epstein, 2013; Fu et al., 2013; Ieda et al., 2010; Jayawardena et al., 2012; Nam et al., 2014; Qian and Srivastava, 2013; Song et al., 2012; Wada et al., 2013; Zhou et al., 2016). This strategy advantages in bypassing the unstable differentiation process, possessing the potential for in situ regeneration, lower chance of teratoma formation, minimization of alternative cell fates and potential to be induced into more specific subtypes of cardiomyocytes (Liu et al., 2017; Nam et al., 2014; Qian et al., 2012; Song et al., 2012; Zhou et al., 2017).
The induction of functional cardiomyocytes from non-myocytes has been achieved efficiently via ectopic expression of Mef2c (M), Gata4 (G), and Tbx5 (T) both in vitro and in vivo with mice models (Ieda et al., 2010; Ma et al., 2015; Muraoka et al., 2014; Qian et al., 2012; Wang et al., 2015). However, more complicated reprogramming cocktails including M, G, T, MESP1, MYOCD, ESRRG, HAND2, and microRNAs are required to generate human induced cardiomyocyte-like cells (hiCMs) in a less efficient way when compared with mouse cardiac reprogramming (Fu et al., 2013; Nam et al., 2013; Wada et al., 2013). We recently reported a simplified reprograming cocktail containing human polycistronic transgenes in the splicing order of M, G, and T together with an extra microRNA miR-133 (hMGT133 for short) and achieved cell fate conversion in two weeks with ~ 40% of transduced cells showing cardiac marker expression and sarcomere assembly (Zhou et al., 2019). However, our following single cell RNA-sequencing analysis of hMGT133-infected cells revealed a refractory reprogramming trajectory and immature molecular features during reprogramming, which suggested insufficient and incomplete conversion of fibroblasts into cardiomyocytes. Therefore, to improve hiCM production for potential therapeutic application, it is important to advance our understanding of the molecular basis underlying the process including removal of fibroblast fate features and acquisition of cardiomyocyte identity.
Epigenetic regulation, which stably alters gene expression or cellular phenotype without affecting DNA sequences (Bird, 2007; Goldberg et al., 2007), has emerged as a critical mechanism that can help understand how cells with identical DNA differentiate or transdifferentiate into different cell types and retain their cell identity through mitosis (Apostolou and Hochedlinger, 2013; Cantone and Fisher, 2013; Jaenisch and Young, 2008). Increasing studies on cardiac differentiation and heart development have demonstrated the critical roles of epigenetic regulation to activate cardiac genes during cardiomyocyte cell fate determination (Gilsbach et al., 2018; Paige et al., 2012). In the likely manner, direct cardiac reprogramming has also been found accompanied with epigenetic regulation and dynamic changes in the epigenetic landscape (Fu et al., 2013; Ieda et al., 2010; Liu et al., 2016a, 2016b; Stone et al., 2019; Zhou et al., 2016). To generate functional cardiomyocytes from fibroblasts, the epigenetic barriers to cardiac gene expression have to be removed and fibroblast signatures have to be silenced at epigenetic level.
In our study, we performed an shRNA-based loss-of-function screen and identified the enhancer of zeste homolog 2 (EZH2), which encodes a histone methyltransferase enzyme targeting histone H3 lysine 27 methylation, playing an inhibitory role on cardiac cell fate conversion. Genetic and pharmacological inhibition of EZH2 significantly increased reprogramming efficiency and the expression of a subset of cardiac-specific genes which were previously repressed by EZH2-mediated H3K27me3. Our work identified EZH2 as a critical epigenetic barrier of hiCM reprogramming and established EZH2 inhibition as an effective approach for improving hiCM production.
2. Materials and method
2.1. Fibroblast cell culture
The fibroblast cell line we used in this study was the H9 human ESC-derived fibroblast line (Fu et al., 2013). H9Fs were cultured in DMEM medium supplement with 20% FBS and 1% Pen-strep (HDF medium). For passaging, the medium was aspirated, and cells were washed with PBS once. Incubate the cells with 0.05% Trypsin at 37 °C for 5 min and neutralize the cell mixture with HDF medium. Centrifuge cells at 200 rpm for 5 min and seed the cells at desired concentration for further experiment.
2.2. Virus package
For reprogramming, pMXs-puro-hMGT and pBabe-miR-133 plasmids were used to produce retroviruses (Nam et al., 2013; Zhou et al., 2019). The gag/pol and VSV.G were used as package plasmids and purchased from Addgene. Desired plasmid and packaging plasmids were transfected with NanoFect (ALSTEM) into 293T cells. To generate lentiviruses expressing shRNAs, we transfected pLKO.1 constructs with package plasmids pMD2G and psPAX2 (purchased from Addgene) into 293T cells using NanoFect transfection reagent. Retrovirus or lentivirus particles were collected on 72 and 96 h after 293T transfection and precipitated with 8% PEG6000.
2.3. Direct cardiac reprogramming
H9Fs were seeded in 24-well plate pre-coated with SureCoat at a cell density of 4X104 per well. On Day 0, fibroblasts were transduced with hMGT133 cocktails 24 h after seeding in DMEM medium supplemented with 10% FBS and 20% Medium 199 (iCM medium). For the shRNA knockdown assay, lentiviral shRNAs targeting genes or control (Turbo or empty vector) were added 48 h after cell seeding (Day 1). For the pharmacological inhibition of EZH2, the addition of small molecules was performed one day after hMGT133 infection. On Day 2, cells were selected with puromycin at concentration of 2 μg/mL for 4 days. Puromycin containing medium was replaced with regular iCM medium after selection (Day 6). On Day 10 of reprogramming process, culture medium was changed to RPMI-1640 medium supplemented with 2% B27 supplement, 2% FBS, 0,05% bovine serum albumin (BSA), 50 μg/mL ascorbic acid, and 1X NEAA (hiCM Medium). The following reprogramming assessments were all performed on Day 14.
2.4. Immunofluorescence staining.
Cells were fixed with 4% PFA for 10 min at room temperature and permeabilized with 0.01% Triton X, washed with 0.1% TWEEN20 in PBS (PBST) three times, blocked with 10% donkey serum (Sigma-Aldrich; D9663) for 30 min at room temperature. The fixed cells were then incubated with desired primary antibodies (Anti-cTnT: Abcam #ab91605 at 1:200 dilution; Anti-αActinin: Sigma-Aldrich #A7811 at 1:200 dilution; Anti-cTnI: Abcam #ab47003 at 1:100 dilution; Anti-αMHC: DSHB #MF-20 at 1:100 dilution) overnight at 4 °C, washed three times with PBST, and incubated with corresponding secondary antibodies (FITC AffiniPure Donkey Anti-Mouse IgG: Jacksonimmuno #715–095-150; TRITC AffiniPure Donkey Anti-Rabbit IgG: Jacksonimmuno # 711–025-152) for 30 min at room temperature. After staining, cells were mounted with DAPI-containing mounting medium (Vector Laboratories; H-1200). Images were obtained using Olympus IX83 fluorescence microscope, and data analysis was performed with Image J software. Quantification was performed by counting the ratio of positively stained cells compared with DAPI positive total cells (~40 images were randomly taken under 20x magnification at the same exposure setting blindly). Differences between groups were examined for statistical significance using Student’s t test, or one-way ANOVA followed by Dunnett’s multiple comparisons test. Error bars indicated mean ± SEM, p < 0.05 will be considered as significant.
2.5. Flow cytometry
Cells were trypsinized with 0.05% trypsin and fixed using BD cytofix/cytoperm solution for 30 min at 4°C. The cells were then washed with 1X wash buffer and incubated in primary antibody at room temperature for 1 h. After washing the cells with 1X wash buffer, the cells were then incubated in secondary antibody at room temperature for 1 h. The secondary antibody was washed away with 1X wash buffer and the cells were resuspended in 1% PFA. The cells were analyzed with BD flow cytometer and data were processed with FlowJo. Differences between groups were examined for statistical significance using Student’s t test, or one-way ANOVA followed by Dunnett’s multiple comparisons test. Error bars indicated mean ± SEM, p < 0.05 will be considered as significant.
2.6. RT-qPCR and western blotting
Total RNA was isolated with TRIzol and reverse-transcribed into cDNA using Superscript IV Reverse Transcriptase. qPCR was performed with applied biology system and Power Up SYBR Green PCR Mix. All the primers used for qPCR were list in Table S1. Protein was collected using RIPA buffer with 1X protease inhibitor and concentration were determined using BCA assay. Samples were denatured at 99°C for 10 min and run through a 4–20% precast gel (BIORAD # 4561093). After transfer, the membrane was blocked with 5% dry milk (BIORAD # 1706404) at room temperature for 1 h. The membrane was then incubated with GAPDH (Abcam #ab22555 at 1:2000 dilution), EZH2 (BD Bioscences #612666 at 1:2000 dilution), and H3K27me3 (Active Motif #39155 at 1:1000 dilution) at 4°C overnight. After incubation, the membrane was washed with 1X TBST (1X TBS with Tween 20) three times 5 min each. Following, the membrane was incubated in corresponding secondary antibodies (Anti-Rabbit IgG: Cell Signaling Technology #7074 V at 1:2000 dilution; Anti-Mouse IgG: Cell Signaling Technology #7076 V at 1:2000 dilution) for 1 h at room temperature. After washed with 1X TBST three times 5 min each, the membrane was ready for imaging.
2.7. RNA sequencing and data analysis
RNA sequencing (RNA-seq) was performed in triplicate using independent biological samples. Total RNA was extracted from cells with TRIzol. Sequencing libraries were generated using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) following manufacturer’s manual and applied on a HiSeq 4000 sequencer (Illumina) for a pair-end 150 bp reads sequencing run. RNA-seq reads were aligned to the Ensemble reference genome and gene model annotation using Tophat program. The raw counts were then analyzed using iDEP.91 tool (http://bioinformatics.sdstate.edU/idep/#) (Ge et al., 2018). We ranked genes by their standard deviation across all samples and use the top 1000 genes in hierarchical clustering. With the DESeq2 package, we identified differentially expressed genes (DEGs) using a threshold of false discovery rate (FDR) < 0.1 and fold-change > 2. GSEA analysis was performed using GSEA_4.0.3 (https://www.gsea-msigdb.org/gsea/index.jsp) (Mootha et al., 2003; Subramanian et al., 2005).
2.8. CUT&RUN-sequencing and data analysis
CUT&RUN assay was performed as previously described (Zhang et al., 2020). In brief, 0.5 million cells were collected for each sample. The H3K27me3 antibody (9733S, Cell Signaling Technology) was used at a 1:100 dilution for binding of primary antibody. pAG-MNase was purchased from EpiCypher (15–1116) and used at 1:20 dilution for each 50 μL CUT&RUN reaction. Library construction was performed using the NEBNext UltraII DNA Library Prep Kit from NEB (E7645S). Indexed samples were run using the Illumina HiSeq 4000 platform with paired-end sequencing. CUT&RUN reads were processed with ChIP-seq pipeline as previously described with modifications (Lu et al., 2016). Briefly, raw reads were subjected to adapter removal (cutadapt 2.10) and mapped genome hg19 (bowtie2 2.4.1) using –end-to-end –very-sensitive –no-mixed –no-discordant –phred33 -I 10 -X 700 parameters. Duplicated reads were removed using picard MarkDuplicates tools (version 2.23.3). Bigwig files were generated by bamCoverage (version 3.3.0) and used for visualized in IGV (version 2.8.6). Heatmaps were generated by deeptools (version 3.3.0) averaged prolife plots across TSS regions were generate by sitepro within the cis-regulatory element annotation system (CEAS 0.9.9.7) (Shin et al., 2009). For the qPCR validation, the constructed library samples were diluted 1:90 with molecular degrade water before use as templates. For each qPCR reaction, 4 μL of templates were used with applied biology system and Power Up SYBR Green PCR Mix. The primers designed for CUT&RUN were listed in Table S1.
2.9. ChIP-qPCR and data analysis
~1 million cells reprogrammed with hMGT133 with shEV or shEZH2 were harvest on Day 14 of reprogramming. 200,000 cells were used for each IP reaction. The samples were then processed using MAGnify Chromatin Immunoprecipitation System kit (Invitrogen Life Technologies) according to manufacture guidance using EZH2 (BD Bioscences #612666), H3K27me3 (Active Motif #39155) and IgG (included in MAGnify Kit). The following qPCR was performed with applied biology system and Power Up SYBR Green PCR Mix. The obtained CT value was adjusted to the input controls. Averaged numbers from technical triplicates were used for statistics. Differences between groups were examined for statistical significance using Student’s t test. Error bars indicated mean ± SEM, p < 0.05 will be considered as significant. All the primers used for qPCR were list in Table S1.
3. Results
3.1. Loss-of-function screen identified EZH2 as a critical epigenetic barrier to human iCM reprogramming
We used a polycistronic vector controlling stoichiometry of human MGT expression and additional miR-133 (hMGT133 for short) as the basic cocktail to generate hiCMs from H9 human embryonic stem cell-derived fibroblast line (H9Fs). To identify potential epigenetic repressors or facilitators of hiCM reprogramming, we performed a loss-of-function screen of 50 selected components of epigenetic modulators. Each component was knocked down individually with pooled shRNAs (3–5 oligos for each gene) in hMGT133-infected H9Fs. Upon knockdown, hiCM reprogramming efficiency was determined by flow cytometry analysis of cardiac troponin T (cTnT) + cells (Fig. 1A). Among the 50 candidate epigenetic regulators, ablation of enhancer of zeste homolog 2 (EZH2), which encodes a member of polycomb group protein acting as a histone methyltransferase, resulted in the most significant increase in cTnT expression (Fig 1B and S1A). EZH2 knockdown led to a 3- to 4-fold increase in cTnT + cell percentages (8.03%±0.24 versus 31%± 1.6; Fig. 1C). In addition, three individual shRNA oligos were used to treat hMGT133-infected fibroblasts and all resulted in a significant increase in the percentage of cTnT + cells (Fig. 1D), excluding the off-target effects of shRNAs. Pooled EZH2 shRNA oligos were used for further experiment otherwise indicated.
Fig. 1. shRNA screen identified EZH2 as an epigenetic regulator of human iCM reprogramming.
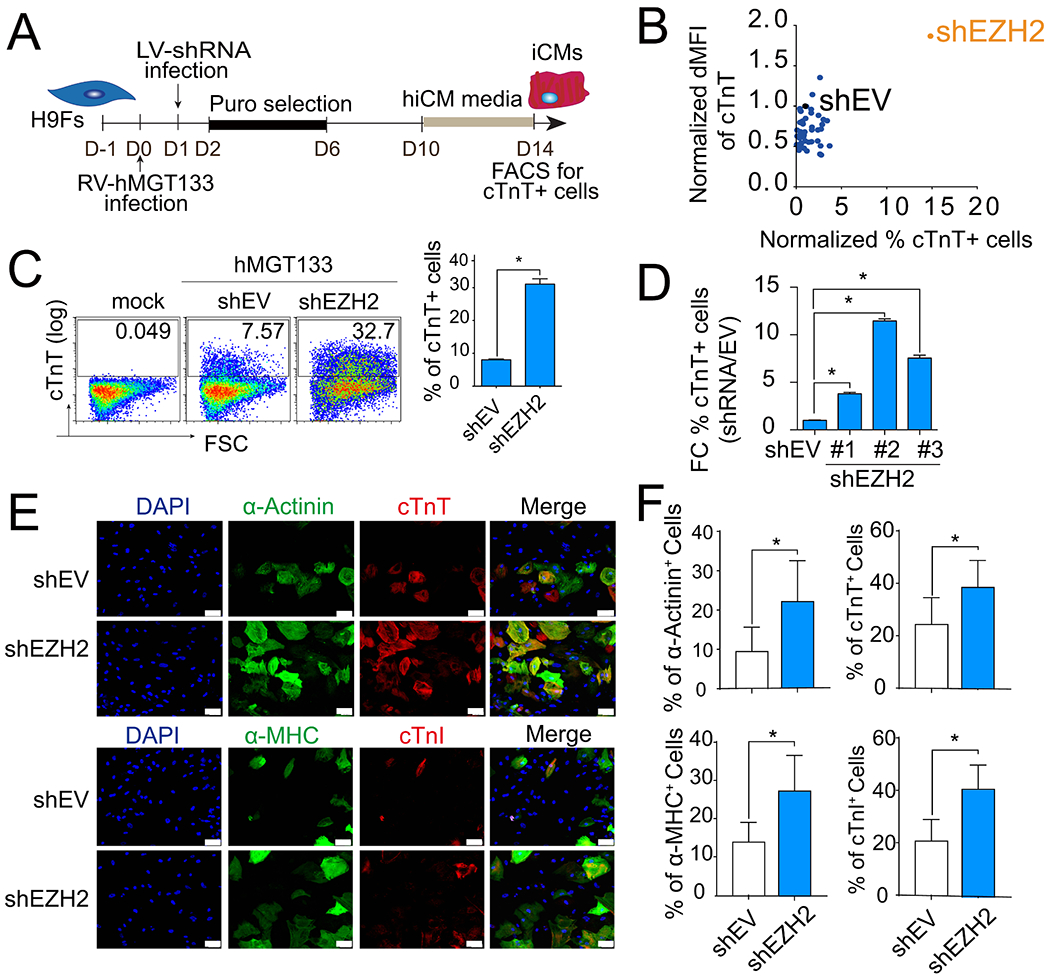
(A) Schematic of the shRNA-mediated loss of function screen. (B) FACS analysis of percentage and florescence intensity of cTnT + cells in hiCMs following RNAi screen. dMFI, delta median florescence intensity; shEV, empty vector control. (C) Representative flow plots and quantification of cTnT+ cells on H9F 2 weeks after transduction as indicated. (D) Quantification of cTnT+ cells after reprogramming with individual shRNAs targeting EZH2. (E-F) Representative images (E) and quantification (F) of immunofluorescence staining of α-Actinin+, cTnT+, α-MHC+, and cTnI + cells on H9F 2 weeks after transduction as indicated. Scale bars, 100 μm. Error bars indicate mean ± SEM; *P < 0.05.
Furthermore, expression of cTnT and additional cardiac sarcomeric proteins, including cardiac alpha-Actinin (α-Actinin), alpha myosin heavy chain (α-MHC), and cardiac troponin I (cTnI), were determined by immunocytochemistry staining (ICC) on shEZH2 and control shRNA (shEV) treated cultures 14 days after viral infection (Fig. 1E). The quantification results showed significant increases of all sarcomeric protein expressions after EZH2 knockdown (Fig. 1F), but merely changes in sarcomere formation (Figs .S1B–1C). Taken together, our data demonstrate that knockdown of EZH2 remarkably enhanced reprogramming efficiency of hiCMs defined by sarcomeric protein expression.
3.2. EZH2 knockdown results in activation of CM genes
To better understand the effects of EZH2 during hiCM reprogramming process, we tested cardiac genes and fibroblast genes expressions in EZH2 knocked down cells and control cells. The knockdown of EZH2 was confirmed with qPCR on reprogramming day 6 and day 14 (Fig. S2A). Consistent with previous results, the relative mRNA expressions of cardiac genes were significantly increased in EZH2 knockdown cells while the expressions of collagen genes were dramatically decreased (Fig. 2A and 2B). Furthermore, we performed a time-course analysis to assess mRNA expression levels of extracellular matrix (ECM) genes during the reprogramming process after EZH2 knockdown (Fig. 2C). The results showed that the matrix metalloproteinase-1/2 (MMP1 & MMP2) and metallopeptidase inhibitor 1/2 (TIMP1 & TIMP2) were downregulated by EZH2 depletion to some levels during the reprogramming process (day 6) and completely repressed to an undetectable level at the later stage of reprogramming (day 14), indicating the further repression of ECM and collagen deposit genes associated with fibroblast functions after EZH2 knockdown. Moreover, the time-course analysis of cardiac structure and contractility genes (MYL2, RYR2 and GJA5) showed no significant activation at day 6 but robustly boosted at day 12 of reprogramming (Fig. 2D), suggesting a sequential cardiac gene activation followed by the early repression of fibroblast genes. The similar two-phase gene changes were also found in EZH2-ablated hiCMs derived from human cardiac fibroblasts (Zhou et al., 2019) (Fig. S2B).
Fig. 2. Knockdown of EZH2 transcriptionally activated cardiac gene expression.
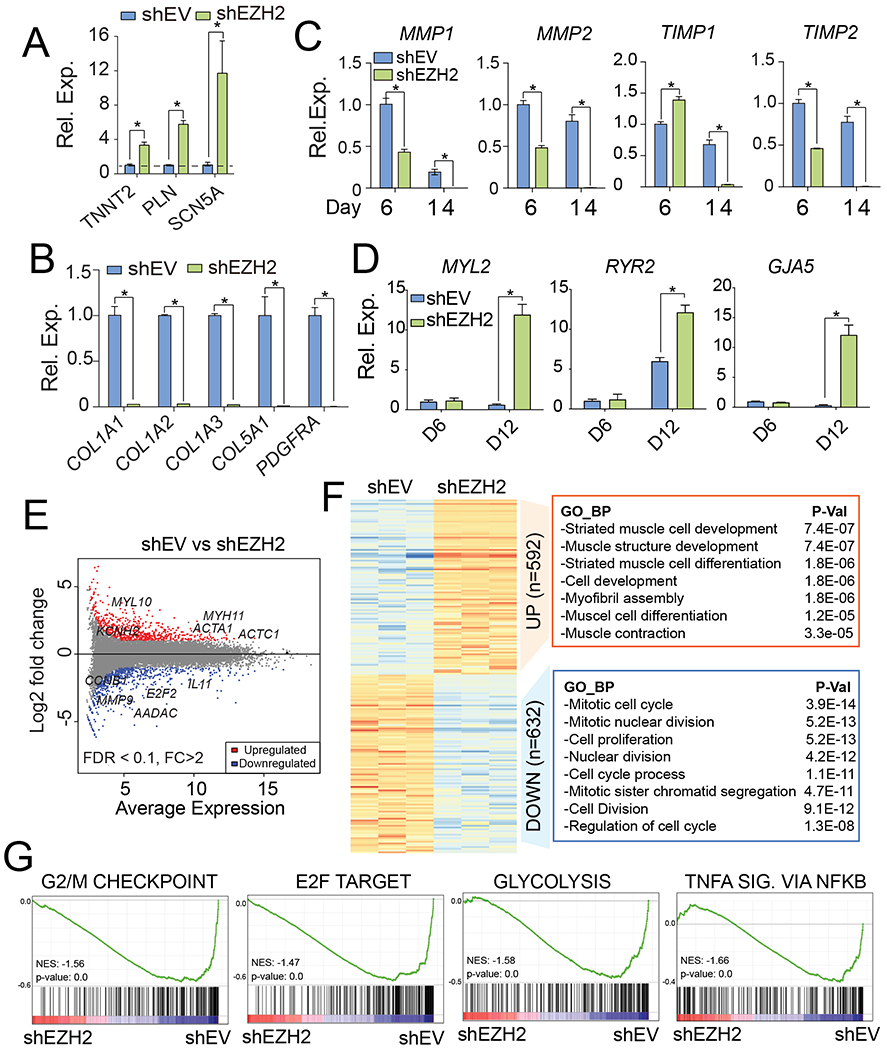
(A-B) Relative expression changes of cardiac (A) and fibroblast (B) genes in knockdown hiCMs. Dash line indicates normalized value 1 in shEV control cells. (C) Time course analysis for mRNA levels of ECM genes after EZH2 knockdown. (D) Time course analysis for mRNA levels of cardiac genes after EZH2 knockdown. (E) MA plot showing global transcriptomic changes in hiCMs upon EZH2 inhibition. Red and blue dots represent the significant up- or down-regulated DEGs (FDR < 0.1, FC > 2). (F) GO terms enriched in down- or up-regulated genes after EZH2 knockdown. (G) GSEA analysis showing the enriched gene sets in control cells versus EZH2 knockdown cells. NES, normalized enrichment score. SIG. stands for signaling. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. The removal of EZH2 leads to the transcriptomic changes promoting hiCM reprogramming
Through transcriptomic comparison between hiCMs infected with shEZH2 or shEV, we were able to visualize the global changes of gene expression after EZH2 knockdown in hiCMs (Fig. S2C). We identified 1224 differentially expressed genes (DEGs) by using DESeq2 tool (FDR < 0.1, fold change > 2). The identified DEGs were presented as scatter plots of log2-fold change (y-axis) versus mean of normalized counts in control cells (x-axis) (Fig. 2E). Among which, we identified 592 upregulated genes and 632 downregulated genes. To further determine what gene programs were associated with EZH2’s inhibitory function to hiCM reprogramming, we performed gene ontology (GO) analysis for both up- and down-regulated DEGs. GO results showed that downregulated genes in EZH2 knocked down cells were highly associated with “mitotic cell cycle” and “cell proliferation” (Fig. 2F). Through gene set enrichment analysis (GSEA), we also found the enrichment score (ES) for gene sets including “G2/M checkpoint” and “E2F target”, which are known to be involved with cell cycle regulation and DNA synthesis in mammalian cells, were higher in control cells compared to EZH2 knocked down cells (Fig. 2G). Interestingly, additional gene sets of “glycolysis” and “TNF-α signaling via NF-kB” were also enriched in control cells, suggesting that EZH2 knockdown might facilitate the metabolic switch and suppression of viral induced immune response occurring during direct cardiac reprogramming (Muraoka et al., 2019; Zhou et al., 2019, 2017). On the other hand, the upregulated genes after EZH2 knockdown were significantly associated with “striated muscle cell development”, “muscle cell development”, “muscle cell differentiation” and “muscle contraction” (Fig. 2F), supporting the notion that removal of EZH2 promotes the acquisition of cardiac cell fate. Thus, the transcriptomic data further demonstrated an inhibitory role of EZH2 on hiCM generation.
3.4. The function of EZH2 during hiCM reprogramming is dependent on its enzyme catalytic activity
EZH2 encodes a histone methyltransferase and serves as the catalytic subunit of Polycomb Repressive Complex 2 (PRC2) which mediates methylation of lysine 27 on histone H3 (H3K27me). We performed western blot analysis and found that knockdown of EZH2 reduced the global level of H3K27me3 by 42.3% (Fig. 3A). In order to test whether the function of EZH2 during hiCM reprogramming process is dependent on its enzyme catalytic activities, we applied two small molecules, UNC1999 and GSK343, to hiCM reprogramming. These two pharmacological inhibitors potently impede the enzymatic activity of EZH2 by binding to its catalytic SET domain (Kim et al., 2013; Konze et al., 2013; Tan et al., 2007). Western blot analysis showed that >50% of H3K27me3 level was reduced with treatment of small molecules at 0.3 μM or 1 μM in hMGT133-infected cells (Fig. 3B). Meanwhile, the reprogramming efficiency of cTnT+ and α-MHC+ cells was significantly increased compared to DMSO-treated group (Fig. 3C–3D). Of note, higher dosage of EZH2 inhibitors did not give rise to higher reprogramming efficiency. Additionally, a panel of cardiac gene markers including MYH6, myosin light chain 2 (MYL2), sodium voltage-gated channel alpha subunit 5 (SCN5A), connexin 40 (GJA5), alpha-cardiac actin (ACTC1), and ryanodine receptor 2 (RYR2) were transcriptionally upregulated in the cells treated with 0.3 μΜ or 1 μM EZH2 inhibitors during reprogramming (Fig 3E and S3A). The lower dosage at 0.1 μM failed to reduce global H3K27me3 level (data not shown) and could not activate any of the cardiac genes detected (Fig 3E and S3A). These results suggest that the EZH2 catalytic activity on H3K27me3 deposition is associated with the inhibitory role of EZH2 on hiCM reprogramming.
Fig. 3. Inhibition of global H3K27me3 using small molecules promoted hiCM generation.
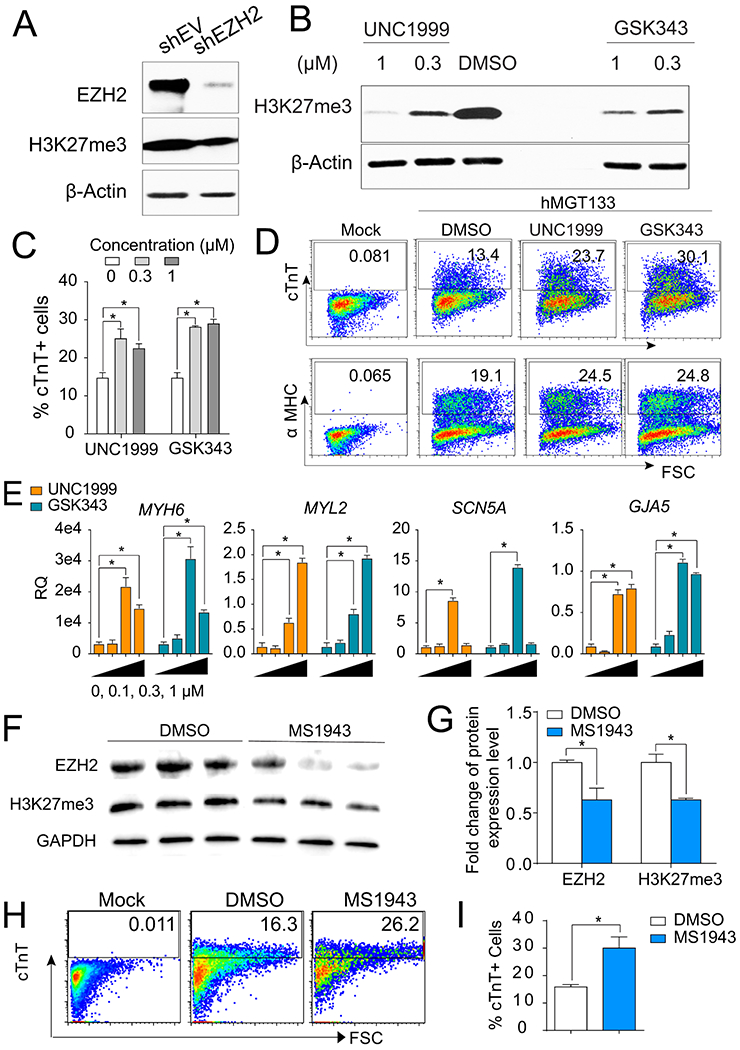
(A) WB of EZH2 and H3K27me3 in reprogrammed cells after EZH2 knockdown. β-Actin serves as a loading control. (B) WB for H3K27me3 after 14 days reprogramming under treatment of DMSO or EZH2 inhibitors at different concentration as indicated. DMSO was used as a vehicle control. (C) Quantification of FACS analysis for cTnT+ hiCMs at 14 days under treatment of EZH2 inhibitors at different dosage as indicated. (D) Flow plots of cTnT+ or α-MHC+ cells in hMGT133-infected cells treated with 0.3 μM of indicated small molecules. (E) RT-qPCR analysis of mRNA levels for cardiac genes MYH6, MYL2, SCN5A, and GJA5 with treatment of EZH2 inhibitors at 0 μM, 0.1 μM, 0.3 μM and 1 μM. (F) WB of EZH2 and H3K27me3 in reprogrammed cells after applying EZH2 degrader (MS1943 at 4 μM). (G) Quantification of WB with treatment of DMSO (control) or MS1943 (EZH2 degrader). (H) Flow plots of cTnT+ cells in hMGT133-infected cells treated with DMSO or 4 μM of MS1943. (I) Quantification of FACS analysis for cTnT+ hiCMs at 14 days post-transduction with treatment of DMSO or MS1943. Error bars indicate mean ± SEM; *P < 0.05; ns, not significant.
As reported, UNC1999 and GSK343 are enzymatic inhibitors for both EZH2 and its paralog EZH1; moreover, the catalytically independent functions of EHZ2 have also been reported in cancer studies (Kim et al., 2018; Zhao et al., 2019). To exclude the off-target effect of EZH1 and understand whether EZH2’s methylation-independent role also contributes to direct cardiac reprogramming, we applied an EZH2 selective degrader (MS1943), which is highly selective for EZH2 over a wide range of methyltransferases including EZH1 (Ma et al., 2020), to the reprogramming cultures. hMGT133-infected cells treated with 4 μm of MS1943 showed significant degradation of EZH2 proteins, ~50% reduction of H3K27me3 (Fig. 3F–3G) and a 2-fold increase in the percentage of cTnT + cells when compared to DMSO-treated group (Fig. 3H–3I). We also found that EZH2 methyltransferase inhibitors and degrader generated similar percentage of cTnT + cells (25.1%±2.5 for UNC1999, 29.5%±1.05% for GSK343, and 30.3%±4.08 for MS1943), suggesting that EZH2 blocks hiCM reprogramming mainly through its methyltransferase activity. Furthermore, the gene signature evaluation of MS1943-treated and control hiCMs by RT-qPCR analyses of a panel of cardiac genes (TNNT2, MYL2, and SCN5A) and a panel of fibroblast genes (MMP2, COL1A1, and PDGFRA) revealed higher cardiac gene expression and reduced fibroblasts signature in the EZH2 degrader-supplemented culture (Figs. S3C–S3D). Therefore, the pharmacological inhibition of EZH2 could also promote hiCM reprogramming and the underlying mechanism is mainly dependent on its methyltransferase activity.
3.5. Reduced H3K27me3 primes the reactivation of cardiac genes by EZH2 knockdown
To further dissect whether EZH2 directly targets those genes occupied with H3K27me3 to repress cardiac cell fate conversion, we first profiled H3K27me3’s chromatin occupancy in hMGT133-induced hiCMs through cleavage under targets and release using nuclease followed by deep sequencing (CUT&RUN-seq), which is a robust and efficient method for genomic profiling when using limited cell numbers (Skene and Henikoff, 2017). Biological duplicates of H3K27me3 CUT&RUN-seq showed robust, specific, and reproducible signals, where no peaks were detected from the IgG control (Fig 4A and S4A). At the global level, the genes derepressed by EZH2 inhibition harbored more abundant H3K27me3 marks comparing to the signals at downregulated genes or randomly selected genes (Fig. 4B). Interestingly, despite of the delivery of MGT reprogramming factors, many of the cardiac lineage-specific genes that were up-regulated by EZH2 inhibition, including sarcomere genes MYH6, MYH7, and MYL2, ion channel gene SCN5A and gap junction gene GJA5 (Fig. 3), were broadly marked by the H3K27me3 as demonstrated by sequencing results (Fig. 4C) and qPCR validation (Fig. 4D). Meanwhile, those cardiac genes moderately activated after EZH2 knockdown, such as ACTC1, SLC8A1 and ATP2A2 (Fig. S3), showed low H3K27me3 signals on their genome loci (Fig. 4E and S4B). To further determine the EZH2-regulated H3K27me3 changes on these cardiac loci, we performed chromatin immunoprecipitation (ChIP) analysis of H3K27me3 in EZH2 knockdown and control hiCMs. The qPCR analysis using the primers matching the binding sites of MYH6, MYH7, SCN5A, MYL2 and SCN10A confirmed the significant loss of H3K27me3 signals upon EZH2 knockdown during reprogramming (Fig. 4F and Fig. S4C). Furthermore, the EZH2 ChIP-qPCR for MYH6, MYH7 and SCN5A showed direct binding of EZH2 on these selected cardiac loci (Fig. 4C, 4G and S4D). Taken together, our data support that EZH2 maintained the repressive status of important cardiac genes and restricted them from fully activation by the reprogramming factors.
Fig. 4. Knockdown of EZH2 derepressed the cardiac genes silenced with high level of H3K27me3.
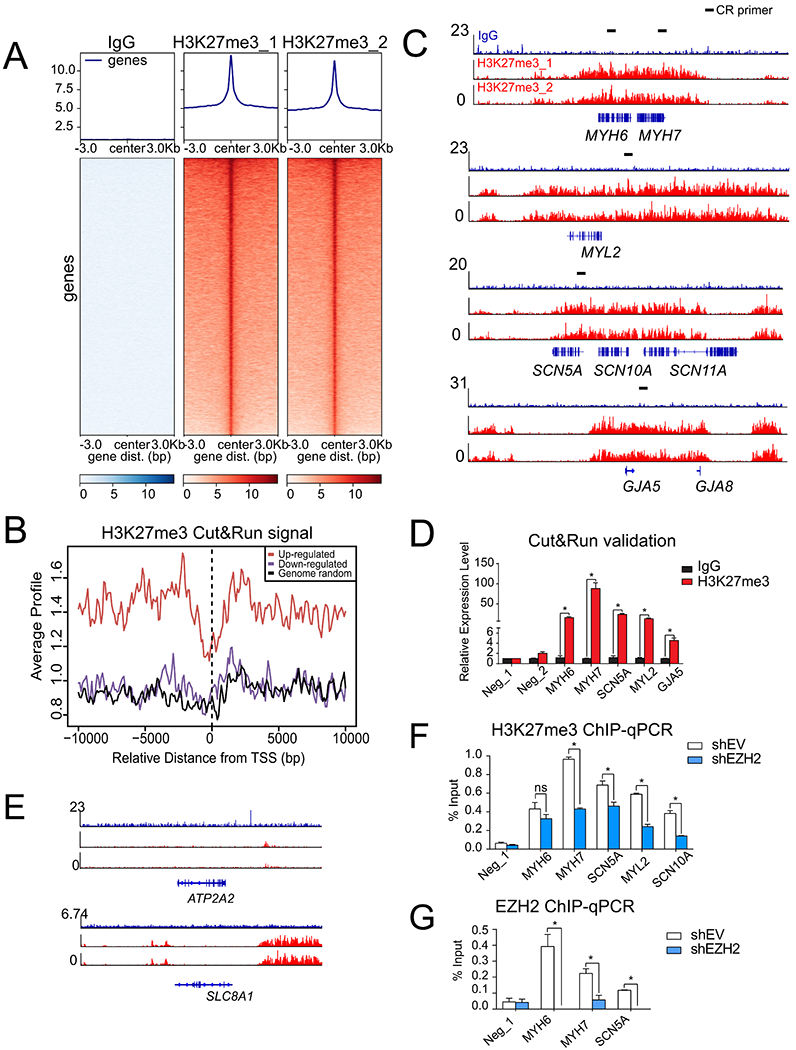
(A) Overall CUT&RUN-seq signals for negative control IgG and H3K27me3 in hMGT133-induced hiCMs. (B) Average CUT&RUN signals across transcription start site (TSS) of upregulated or downregulated genes after EZH2 knockdown. The signals of randomly picked genes severe as a negative control. (C) IGV browser tracks showing enriched CUT&RUN-seq peaks of H3K27me3 (red) at selected cardiac genes MYH6, MYH7, MYL2, SCN5A and GJA5. IgG signals were shown in blue. Primers used for qPCR validation were illustrated by black dashes. (D) qPCR validation for selected gene loci in IgG and H3K27me3 CUT&RUN samples. (E) IGV browser tracks showing no H3K27me3 (red) binding at selected ATP2A2 and SLC8A1 loci. (F) H3K27me3 ChIP-qPCR evaluation for selected cardiac genes at their H3K27me3 binding loci after EZH2 knockdown. (G) EZH2 ChIP-qPCR evaluation of EZH2 occupancy on the selected cardiac gene loci after EZH2 knockdown. Error bars indicate mean ± SEM; *P < 0.05; ns, not significant. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
4.1. Conclusion
Although studies in mouse iCM reprogramming have advance our understanding of cardiac reprogramming, generation of human iCMs has been challenging due to its low efficiency and quality. In this study, we performed a functional screen and identified EZH2 as a major epigenetic barrier during the hiCM reprogramming process. We found that the reprogramming efficiency could be significantly increased upon silencing of EZH2. Furthermore, knocking down EZH2 led to a profound repression of collagen and extracellular matrix genes, which are related to formation of fibrosis. In an effort to explore the potential underlying mechanism of EZH2, we demonstrated that EZH2 inhibition-mediated decrease of H3K27me3 was involved in the enhancement of hiCM reprogramming, accompanied with the decrease of fibrotic gene expression and increase of cardiac sarcomere genes. Therefore, we identified EZH2 as a critical epigenetic barrier, and removing of which could achieve more efficient hiCM generation.
4.2. Discussion
During normal development, cell fate decisions are made at the transcriptional level in response to environmental cues. To achieve the cell fate conversion, it is important to remove the transcriptional “memory” maintained by epigenetic mechanisms (Francis and Kingston, 2001). The polycomb group (PcG) proteins are one of the key proteins have been implicated in maintaining chromatin structure as well as regulating cell proliferation and differentiation. The reduction of H3K27me3 levels has been reported along mouse iCM reprogramming (Dal-Pra et al., 2017; Liu et al., 2017). However, H3K27me3 level is not simply correlated with reprogramming efficiency as we found further reduction of H3K27me3 could no longer increase the percentage of cTnT+ cells (Fig. 3B–3E). Intriguingly, our chromatin occupancy profiling of H3K27me3 showed that the removal of EZH2 preferentially primes the activation of cardiac genes in the genome loci occupied with supper clusters of H3K27me3 on the gene body rather than cis-regulatory region (Fig. 4C and 4E).
From our results, EZH2 knockdown using shRNAs seems to generate slightly higher percentages of cTnT positive cells than EZH2 pharmacological inhibition. It is likely due to EZH2’s paralog EZH1, which is highly homologous to EZH2 with 76% sequence identity overall and 96% sequence identity within the catalytic SET domain. The shRNA screening showed that EZH1 is required for human cardiac reprogramming (Fig. S1A). Knockdown of EZH1 reduced the percentage of cTnT+ cells, suggesting opposite roles of EZH1 and EZH2 during direct cardiac reprogramming. Since two pharmacological inhibitors (UNC1999 and GSK343) used in this study are dual inhibitors of EZH1 and EZH2, the reprogramming efficiency upon pharmacological inhibition, compared with shRNA treatment, might be impeded by EZH1 inhibition. We thus further tested an EZH2 selective degrader (MS1943). MS1943 maintained high potency for inhibiting EZH2 methyltransferase activity and was highly selective for EZH2 over a wide range of methyltransferases including EZH1 (Ma et al., 2020). Consistently, cTnT+ cell percentages after MS1943 treatment (30.03%±4.08) are comparable with shEZH2 knockdown, further supporting EZH2’s inhibitory role during reprogramming.
We found that the epigenetic barriers identified in mouse and human are different. In our previous screening in mouse using the similar MGT cocktail showed limited increase of reprogramming efficiency after Ezh2 knockdown in mouse fibroblasts (Zhou et al., 2016). Of note, one study reported different findings that the treatment of another Ezh2 inhibitor GSK126 at the early stage promotes the mouse iCM reprogramming (Hirai and Kikyo, 2014). However, they used Gata4, Hand2, Tbx5, and the fusion gene MM3 between Mef2c and the transactivation domain of MyoD to perform reprogramming, which might lead to inconsistent results. Additional evidence is that the previously identified epigenetic factors in mouse cardiac reprogramming, such as Zrsr2, Bcor and Stag2 (Zhou et al., 2018), showed moderate effects in the current study using human cells (Fig. S1), further supporting our notion that not like the conserved reprogramming factors used for reprogramming, unique epigenetic regulations may need to be removed for more efficient cell fate conversion.
To gain a complete transcriptomic understanding of EZH2’s effect on human cardiac reprogramming, we thoroughly analyzed RNA sequencing results. Interestingly, knocking down EZH2 inhibited many cell cycle genes compared to control groups, which is consistent with its suppressive effect on regulation of cell cycle and proliferation (Gonzalez et al., 2009). The inhibition of cell cycle has been known as a critical event for the maturation of endogenous cardiomyocytes (Karbassi et al., 2020), and also recently reported essential for iCM reprogramming (Bektik et al., 2018; Liu et al., 2017; Zhou et al., 2019). Moreover, it has been reported that EZH2 knockdown in human stem cell-derived cardiomyocytes resulted in an increased expression of cardiac maturation genes and improved mitochondrial function, suggesting a potential effect of EZH2 on cardiomyocyte maturation (Kuppusamy et al., 2015). As such, further assessment of sarcomere ultrastructure and mitochondrial function in EZH2 depleted hiCMs might help to fully determine the EZH2’s role on hiCM maturation. Other than that, repressing the expression of EZH2 reduced repressive histone modification H3K27me3 occupancy on those genes related to muscle striation and contraction and re-activated their expression. Our integrative analysis of transcriptomic and epigenomic data provide new mechanistic understanding of hiCM reprogramming.
Supplementary Material
Acknowledgments
J.Z is supported by NIH R01s, HL95077, HL114120, HL131017, HL149137, and NIH UO1 HL134764. L.Q. is supported by NHLBI 1R35HL155656 and AHA 18TPA34180058, 20EIA35310348. J.J. is supported by R01CA218600 and R01CA230854. R.L. is supported by Concern Foundation, Leukemia Research Foundation and Institutional Research Grant number IRG15-59-IRG from the American Cancer Society. Y.Z. is supported by NIH NHLBI R01 HL153220. We would like to thank Dr. Marion Spell from UAB Flow Cytometry Core for all the technical support.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Addis RC, Epstein JA, 2013. Induced regeneration-the progress and promise of direct reprogramming for heart repair. Nat. Med 19, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E, Hochedlinger K, 2013. Chromatin dynamics during cellular reprogramming. Nature 502, 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektik E, Dennis A, Pawlowski G, Zhou C, Maleski D, Takahashi S, Laurita KR, Deschenes I, and Fu JD (2018). S-phase Synchronization Facilitates the Early Progression of Induced-Cardiomyocyte Reprogramming through Enhanced Cell-Cycle Exit. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A, 2007. Perceptions of epigenetics. Nature 447, 396–398. [DOI] [PubMed] [Google Scholar]
- Cahill TJ, Kharbanda RK, 2017. Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: Mechanisms, incidence and identification of patients at risk. World J. Cardiol 9, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantone I, Fisher AG, 2013. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol 20, 282–289. [DOI] [PubMed] [Google Scholar]
- Dal-Pra S, Hodgkinson CP, Mirotsou M, Kirste I, Dzau VJ, 2017. Demethylation of H3K27 Is Essential for the Induction of Direct Cardiac Reprogramming by miR Combo. Circ. Res 120, 1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE, 2001. Mechanisms of transcriptional memory. Nat. Rev. Mol. Cell Biol 2, 409–421. [DOI] [PubMed] [Google Scholar]
- Fu JD, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, Srivastava D, 2013. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 1, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Son EW, Yao R, 2018. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinf. 19, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach R, Schwaderer M, Preissl S, Gruning BA, Kranzhofer D, Schneider P, Nuhrenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, et al. , 2018. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun 9, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. , 2013. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127, e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E, 2007. Epigenetics: a landscape takes shape. Cell 127, 635–638. [DOI] [PubMed] [Google Scholar]
- Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, Ljungman M, Merajver SD, Kleer CG, 2009. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 28, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Kikyo N, 2014. Inhibitors of suppressive histone modification promote direct reprogramming of fibroblasts to cardiomyocyte-like cells. Cardiovasc. Res 102, 188–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D, 2010. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Young R, 2008. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ, 2012. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res 110, 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X, Murry CE, 2020. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol 17, 341–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, Licht JD, Zhao JC, Yu J, 2018. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep 25 (2808–2820), e2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH, 2013. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol 9, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F Gao C, Huang XP, Kuznetsova E, et al. , 2013. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol 8, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, Pabon L, Zhu WZ, Tulloch NL, Yang X, et al. , 2015. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A 112, E2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lei I, Karatas H, Li Y, Wang L, Gnatovskiy L, Dou Y, Wang S, Qian L, Wang Z, 2016a. Targeting Mll1 H3K4 methyltransferase activity to guide cardiac lineage specific reprogramming of fibroblasts. Cell Discov. 2, 16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Zheng M, Wang L, Zhou Y, Yin C, Liu J, Qian L, 2016b. Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 16, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. , 2017. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, Chen WY, Abdel-Wahab O, Wade PA, Zheng D, et al. , 2016. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 30, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X, Schwarz M, Leshchenko V, Rialdi A, Dale B, et al. , 2020. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol 16, 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wang L, Yin C, Liu J, Qian L, 2015. In vivo cardiac reprogramming using an optimal single polycistronic construct. Cardiovasc. Res 108, 217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. , 2003. PGC-1alpharesponsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Muraoka N, Nara K, Tamura F, Kojima H, Yamakawa H, Sadahiro T, Miyamoto K, Isomi M, Haginiwa S, Tani H, et al. , 2019. Role of cyclooxygenase-2-mediated prostaglandin E2-prostaglandin E receptor 4 signaling in cardiac reprogramming. Nat. Commun 10, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, et al. , 2014. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 33, 1565–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Reinecke H, Pabon LM, 2006. Regeneration gaps: observations on stem cells and cardiac repair. J. Am. Coll. Cardiol 47, 1777–1785. [DOI] [PubMed] [Google Scholar]
- Nam YJ, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, Bassel-Duby R, Olson EN, Munshi NV, 2014. Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development 141, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, et al. , 2013. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. U.S.A 110, 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama K, El-Nachef D, MacLellan WR, 2013. Regeneration potential of adult cardiac myocytes. Cell Res. 23, 978–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, et al. , 2012. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA, 2011. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, 2012. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Srivastava D, 2013. Direct cardiac reprogramming: from developmental biology to cardiac regeneration. Circ. Res 113, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, Liu XS, 2009. CEAS: cis-regulatory element annotation system. Bioinformatics 25, 2605–2606. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S, 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. , 2012. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone NR, Gifford CA, Thomas R, Pratt KJB, Samse-Knapp K, Mohamed TMA, Radzinsky EM, Schricker A, Ye L, Yu P, et al. , 2019. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 25 (87–102), el09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. , 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q, 2007. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21, 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, et al. , 2013. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. U.S.A 110, 12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursae N, Liu J, Qian L, 2015. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res 116, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang Y, Zhou X, Wright S, Hyle J, Zhao L, An J, Zhao X, Shao Y, Xu B, et al. , 2020. Functional interrogation of HOXA9 regulome in MLLr leukemia via reporter-based CRISPR/Cas9 screen. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ding L, Wang D, Ye Z, He Y, Ma L, Zhu R, Pan Y, Wu Q, Pang K, et al. , 2019. EZH2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. EMBO J. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Alimohamadi S, Wang L, Liu Z, Wall JB, Yin C, Liu J, Qian L, 2018. A Loss of Function Screen of Epigenetic Modifiers and Splicing Factors during Early Stage of Cardiac Reprogramming. Stem Cells Int. 2018, 3814747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu Z, Welch JD, Gao X, Wang L, Garbutt T, Keepers B, Ma H, Prins JF, Shen W, et al. , 2019. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 25 (149–164), el49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang L, Liu Z, Alimohamadi S, Yin C, Liu J, Qian L, 2017. Comparative Gene Expression Analyses Reveal Distinct Molecular Signatures between Differentially Reprogrammed Cardiomyocytes. Cell Rep. 20, 3014–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu JD, Wang GG, Liu J, et al. , 2016. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.