

Abstract
Ventilator-induced lung injury is associated with an increase in mortality in patients with respiratory dysfunction, although mechanical ventilation is an essential intervention implemented in the intensive care unit. Intrinsic molecular mechanisms for minimizing lung inflammatory injury during mechanical ventilation remain poorly defined. We hypothesize that Yes-associated protein (YAP) expression in endothelial cells protects the lung against ventilator-induced injury. Wild-type and endothelial-specific YAP-deficient mice were subjected to a low (7 mL/kg) or high (21 mL/kg) tidal volume (VT) ventilation for 4 h. Infiltration of inflammatory cells into the lung, vascular permeability, lung histopathology, and the levels of inflammatory cytokines were measured. Here, we showed that mechanical ventilation with high VT upregulated YAP protein expression in pulmonary endothelial cells. Endothelial-specific YAP knockout mice following high VT ventilation exhibited increased neutrophil counts and protein content in bronchoalveolar lavage fluid, Evans blue leakage, and histological lung injury compared with wild-type littermate controls. Deletion of YAP in endothelial cells exaggerated vascular endothelial (VE)-cadherin phosphorylation, downregulation of vascular endothelial protein tyrosine phosphatase (VE-PTP), and dissociation of VE-cadherin and catenins following mechanical ventilation. Importantly, exogenous expression of wild-type VE-PTP in the pulmonary vasculature rescued YAP ablation-induced increases in neutrophil counts and protein content in bronchoalveolar lavage fluid, vascular leakage, and histological lung injury as well as VE-cadherin phosphorylation and dissociation from catenins following ventilation. These data demonstrate that YAP expression in endothelial cells suppresses lung inflammatory response and edema formation by modulating VE-PTP-mediated VE-cadherin phosphorylation and thus plays a protective role in ventilator-induced lung injury.
Keywords: inflammation, vascular endothelial-cadherin, vascular endothelial protein tyrosine phosphatase, ventilator-induced lung injury, Yes-associated protein
INTRODUCTION
Mechanical ventilation is an indispensable intervention in the management of patients with respiratory failure. However, in patients with acute lung injury and acute respiratory distress syndrome (ARDS), mechanical ventilation with conventional tidal volumes (VT) contributes to ∼9% increase in mortality (1). This so-called ventilator-induced lung injury was also seen in patients without respiratory dysfunction (2–4). Ventilator-induced lung injury is characterized by inflammatory cell infiltration in the lung and alveolar capillary barrier dysfunction with leakage of proteinaceous fluid into the airspace (5). Despite widespread investigation of this serious clinical problem, the intrinsic molecular mechanism for minimizing lung injury during mechanical ventilation remains enigmatic.
Alveolar epithelial cells, pulmonary endothelial cells, macrophages, and neutrophils all contribute to the pathogenesis of ventilator-induced lung injury (6). Among these types of cells, endothelial cells are exposed to both hemodynamic forces from flowing blood and mechanical forces resulting from respiratory cycles (7) and are the most sensitive cell type to mechanical stretch (8). The endothelium forms a semipermeable barrier between the blood and interstitium, which regulates the transport of macromolecules, leucocytes, and fluid across the vessel wall. The integrity of this barrier is predominantly determined by homophilic interactions of vascular endothelial (VE)-cadherin between neighboring endothelial cells via adherens junctions. The stabilization of VE-cadherin expression in endothelial junctions is mainly dependent on its interaction with p120-catenin and α-, β-, γ-catenins, which bind to the actin cytoskeleton (9). Disassembly of the VE‐cadherin–catenins complex is an essential step for breaching the endothelial barrier, leading to leukocyte extravasation and vascular hyperpermeability (10). Tyrosine phosphorylation of VE-cadherin serves as an important mechanism for preventing VE-cadherin binding to p120- and β-catenins and subsequently disrupting endothelial cell contacts (11–13). VE-cadherin phosphorylation at Tyr685 residue selectively induced vascular leakage, whereas phosphorylation of Tyr731 exclusively facilitated leukocyte extravasation in vivo (14). Vascular endothelial protein tyrosine phosphatase (VE-PTP) inhibits the tyrosine phosphorylation of VE-cadherin via direct interaction between these two proteins, which stabilizes endothelial barrier function (15).
Yes-associated protein (YAP) is a transcriptional regulator downstream of Hippo signaling and plays fundamental roles in cell survival, growth, and proliferation. In mammals, mammalian STE20-like 1/2 (orthologs of Drosophila Hippo) phosphorylate and activate large tumor suppressor 1/2 kinases, which in turn directly phosphorylate YAP, thereby resulting in its cytoplasmic sequestration and degradation. When dephosphorylated, YAP is translocated to nuclei and modulates gene transcription (16, 17). YAP acts as an important mechanosensor, which transfers mechanical signals to regulate gene expression profiles and functional consequences (18, 19). In response to disturbed flow, YAP induced atheroprone phenotypes and atherosclerotic lesion formation (20). Interestingly, our recent study showed that YAP expression in endothelial cells regulates nuclear factor-κB signaling and suppresses vascular inflammation during sepsis (21). Whether and how endothelial YAP modulates ventilator-induced lung injury remains unknown.
In the present study, using endothelial cell-specific YAP knockout mice, we identified a protective role of YAP in ventilator-induced lung injury. YAP stabilized the expression of VE-PTP, which suppressed tyrosine phosphorylation of VE-cadherin, leading to stabilization of endothelial barrier function and blockade of lung vascular leakage and neutrophil infiltration following mechanical ventilation at a high tidal volume.
METHODS AND MATERIALS
Generation of Endothelial Cell-Specific YAP Knockout Mice
Endothelial cell-specific YAP knockout mice on a C57BL/6 background were generated by crossing mice carrying the floxed yap gene with Tie2-Cre transgenic mice as described previously (21). Mice of genotype of yapf/f with Cre recombinase served as endothelial cell-specific YAP knockout mice while littermate mice homozygous for the floxed yap allele but not expressing Cre recombinase were used as wild-type controls. Age-matched (8–10 wk old) male and female mice (ratio 1:1) used in the studies were backcrossed for at least five generations. Mice had free access to food and water and were subjected to a 12-h day and night cycle. All animal procedures were strictly performed in accordance with NIH guidelines and approved by the University of Illinois Institutional Animal Care and Use Committee. Experimental groups were assigned using a simple randomization procedure by means of drawing lots.
Mouse Genotype Analysis
Mouse genomic DNA was prepared from the tail tips (3–5 mm) and amplified by using REDExtract-N-AMP Tissue PCR kit (Sigma-Aldrich, XNAT-100RXN) following manufacturer’s protocols (21). Briefly, the samples were incubated in a mixture of Tissue Preparation Solution and Extraction Solution at room temperature. After adding Neutralization Solution B, an aliquot of the DNA extract was then combined with REDExtract-N-Amp PCR Reaction Mix and PCR primers to amplify target DNA. The PCR primer sequences used in the studies are as follows: mouse Yap flox 5′- CACCAAACCTGGCATAGACATGTGTTC-3′ and 5′- CAGTCTGTAACAACCAGTCAGGG-ATAC-3′, mouse Tie2 Cre 5′- TTCCATGAGTGA- ACGAACCTGGTC-3′ and 5′- AGTGATGA- GGTTCGCAAGAACCTG-3′.
DNA Constructs
Cyan fluorescent protein-tagged murine full-length wild-type VE-PTP aa 1-1,998 was cloned from murine VE-PTP cDNA, into pAmCyan1-C1 (catalog number PT 3478-5 or 632441; Clontech) at flanking 59/39-SalI restriction sites. Cyan fluorescent protein-tagged deletion mutant VE-PTP aa 1,611-1,998 (ΔN) were generated from wild type VE-PTP and inserted into pAmCyan1-C1 at 59-BspEl and 39-Xhol sites. These VE-PTP cDNAs are generously provided by Dr. Dietmar Vestweber (Max Planck Institute for Molecular Biomedicine, Münster, Germany).
Isolation of Murine Lung Endothelial Cell
Primary pulmonary endothelial cells were isolated from adult mouse lung as previously described (22). Briefly, lungs were harvested, finely minced, and then digested with Collagenase/Dispase (Roche) before incubation with magnetic microbeads (Dynabeads Sheep anti-Rat IgG, Invitrogen) coupled to a rat anti-mouse platelet endothelial cell adhesion molecule-1 specific antibody (BD PharMingen). Primary purified endothelial cells and nonendothelial cells were lysed with radioimmunoprecipitation assay buffer (Boston BioProducts) for further Western blot analyses.
Isolation of Neutrophils
Neutrophils were isolated from bone marrow-derived cells by density gradient centrifugation (22). Briefly, mice were euthanized, and bone marrow was flushed from tibiae and femurs using PBS. The cell suspension was passed through a 0.70-μm filter, resuspended in HBSS without Ca2+, and then centrifuged at 400 g, 4°C for 10 min. Erythrocytes in the pellet were lysed with sterile distilled water and subsequent addition of 0.6 M KCl. Then cells were centrifuged on a Ficoll-Paque Plus gradient (GE Healthcare, Sweden) for 20 min at 800 g. The purity of isolated PMNs was >98%, and viability was >95% as evaluated by Trypan blue exclusion.
Real-Time PCR Analysis
RNA was extracted from mouse lung endothelial cell using Trizol (Invitrogen Life Technologies). The single-stranded cDNA was synthesized from RNA samples with high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR analysis was performed using ABI 7500 system with Brilliant II SYBR Green QPCR Master Mix (Agilent). The following gene-specific primers were used to amplify transcripts: mouse YAP, 5′- GTCCTCCTTTGAGATCCCTGA-3′ and 5′- TGTTGTTGTCTGATCGTTGTGAT-3′; mouse GAPDH, 5′- CCCAATGTGTCCGTCGTGGAT-3′ and 5′- TGTAGCCCAAGATGCCCT-TCAG-3′. The data output is expressed as a fold-change of control gene expression levels (21).
In Vivo Experimental Protocols
For induction of ventilator-induced lung injury (23), mice were anesthetized with ketamine/xylazine (75/5 mg/kg) intraperitoneally, intubated orally with a 20-gauge IV catheter, and then ventilated with a Harvard Apparatus ventilator (MiniVent, Harvard Biosciences). Mice were randomly assigned to the different treatment groups with blinding. Ventilated animals received either high VT (14, 21, or 28 mL/kg, injurious ventilation) with a respiratory rate of 60 breaths/min or a low VT (7 mL/kg) with a respiratory rate of 120 breath/min. All experiments were conducted with room air and zero end-expiratory pressure. Paco2 was maintained between 35 and 45 Torr (4.7–6.0 kPa) by adding ∼5 mL of dead space to the ventilator circuit. Mice were ventilated for 4 h, unless otherwise specified. During ventilation, a heating pad was used to maintain body temperature between 37 and 38°C. Immediately after the onset of mechanical ventilation, 0.25 mL of warm 0.9% normal saline was administered intraperitoneally every 2 h to ensure adequate blood volume. Additional doses of ketamine 0.5 mg/xylazine 0.025 mg were injected intraperitoneally for maintenance of adequate anesthesia throughout the experiment. At the end of the mechanical ventilation period, lung inflammation and injury were evaluated. Animals were given a lethal dose of the anesthetic agent at the end of 4-h mechanical ventilation before harvesting samples. The primary outcome was lung inflammatory injury; the secondary outcome was molecular signaling for regulating endothelial barrier function.
Neutrophil Depletion and Repletion
Mice were injected intraperitoneally with 350 μg of monoclonal anti-Ly6G antibody or isotype control Immunoglobulin (Ig) G2a antibody contained in 200 μL of PBS for 3 consecutive days (24). Neutrophils from anti-Ly6G antibody-treated mice were efficiently depleted (99%). To determine the role of YAP expression in neutrophils in the mechanism of ventilator-induced lung injury, neutrophil repletion experiments were performed in the neutropenic mice with neutrophils (2 × 106 cells) isolated from multiple wild-type or endothelial cell-specific YAP knockout mice.
Overexpression of VE-PTP in Mouse Pulmonary Endothelium In Vivo
VE-PTP was overexpressed in mouse pulmonary vasculature by tail vein injection of cationic liposome–cDNA complex as described previously (21). The liposome–cDNA complex was prepared by addition of 2 μg/g cDNA into 200 μL of liposome suspensions. An empty vector was employed as control. Successful overexpression of VE-PTP was verified by Western blot analysis of isolated pulmonary endothelial cells from transfected mice. All animal experiments were performed at 48 h posttransfection.
Western Blotting and Immunoprecipitation
Cells or lung tissues were lysed by radioimmunoprecipitation assay buffer supplemented with 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Sigma), and 1 mM Na3VO4. Lysates were sonicated and cleared of insoluble material by centrifugation at 4°C with 13,200 rpm for 15 min. Protein concentration was measured with DC Protein Assay Reagent (Bio-Rad). Samples were equally loaded for 8%–14% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, 0.45 µm, IPVH00010). Membranes were blocked with 5% BSA, incubated with primary antibody overnight at 4°C, and then probed with horseradish peroxidase-conjugated secondary antibody (1:3,000–5,000) for 1 h at room temperature. Protein bands were visualized using ECL SuperSignal reagent (Pierce), detected with Odyssey Fc Imager (LI-COR Biosciences), and quantified with ImageJ Software (NIH).
Immunoprecipitation was performed using protein A/G PLUS agarose beads as described previously (21). Briefly, cells were lysed in buffer containing 1% NP-40, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 150 mM NaCl, 50 mM Tris HCl, 1 mM EDTA, 1 mM NaF, and protease inhibitor cocktail. Lysates were precleared with 1–2 µg normal IgG together with protein A/G PLUS agarose beads (Santa Cruz) and then incubated with primary antibody at 4°C overnight, followed by further incubation with 25 µL beads at 4°C for 2 h. Immunoprecipitated proteins were washed, eluted, and analyzed by SDS-polyacrylamide gel electrophoresis and Western blotting analysis.
Plasma Membrane Extraction
The plasma membrane and cytosolic fractions were separately collected using a membrane protein extraction kit (Thermo Fisher) according to the manufacturer’s instructions. In brief, endothelial cells isolated from mouse lungs were washed and incubated with 0.75 mL of permeabilization buffer for 10 min at 4°C. The samples were then centrifuged for 15 min at 16,000 g, and the resulting supernatants (cytosol) were collected. The pellet was incubated with 0.5 mL of solubilization buffer for 30 min at 4°C and centrifuged at 16,000 g for 15 min at 4°C. The supernatant containing solubilized membrane and membrane-associated proteins was collected.
Analysis of Bronchoalveolar Lavage Fluid Content
Changes in neutrophils and protein contents of the bronchoalveolar lavage fluid were analyzed using conventional procedures (28). Briefly, at the end of ventilation, the airways were rinsed three times with intratracheal injection of 1-mL PBS. Collected bronchoalveolar lavage fluid was centrifuged at 500 g for 5 min at 4°C. Total cell and neutrophil counts were performed manually using a hemocytometer. Two-hundred microliter of cells (1 × 105 cells/mL) were cytospun onto slides at 300 g for 5 min with a cytocentrifuge (Shandon). Slides were immediately fixed, stained with Diff-Quick dye (Siemens), and examined by light microscopy. A differential count of 300 cells was quantified by their characteristic morphologies. The remaining bronchoalveolar lavage fluid was centrifuged, and the supernatant was collected. Total protein concentrations in bronchoalveolar lavage fluid were measured using Bradford protein assay (Bio-Rad) and calculated from a standard curve generated with serial dilutions of BSA.
Cytokine Measurements
Proinflammatory cytokines TNF-α and IL-6 in the bronchoalveolar lavage fluid were measured using ELISA kits (BioLegend) following the manufacturers’ instructions and protocols. The cytokine concentration is displayed in pg/mL.
Myeloperoxidase Activity of Lung
Myeloperoxidase (MPO) activity in the lung tissue as a marker of neutrophil infiltration was measured by spectrophotometric assay (22). In brief, the lung tissue was minced, homogenized, and then centrifuged at 16,000 g for 30 min at 4°C. MPO activity was assessed in 100 μL of supernatants in triplicate by using development reagent at 450 nm. Results are expressed as a change in absorbance per milligram of protein.
Vascular Permeability Assay In Vivo
Lung microvascular permeability was determined via Evans blue-albumin dye technique as previously described (21). Briefly, Evans blue (30 mg/kg) was injected into the tail vein 35 min before lung collection. Pulmonary circulation was flushed with PBS. Lung was excised, homogenized in ice-cold PBS, incubated with formamide for 18 h at 60°C, and centrifuged at 13,000 g for 20 min. Absorbance (A620 and A740) of the supernatant was measured and tissue Evans blue content (µg Evans blue/g lung/min) calculated as follows: A620 (corrected) = A620 − (1.1649 × A740 + 0.004). The Evans blue index was expressed as the amount of dye in the lung relative to the weight of lung tissue.
Lung Histology and Lung Injury Scoring
Lung samples were fixed immediately at the time of collection in 10% formalin solution and submitted for paraffin blocking and dehydrating in 70% ethanol. Multiple 5-µm sections of lung were stained with hematoxylin and eosin and observed by light microscopy. Lung injury scores were calculated by an investigator blinded to the treatment groups according to the pulmonary injury scoring system previously used (25). Briefly, four pathological processes were chosen to be scored on a scale of 0–4: 1) alveolar congestion, 2) hemorrhage, 3) infiltration of inflammatory cells or aggregation of neutrophils in air space or the vessel wall, and 4) alveolar wall thickness. The following criteria were used: 0 = normal lungs; 1 = mild, <25% lung involvement; 2 = moderate, 25%–50% lung involvement; 3 = severe, 50%–75% lung involvement; and 4 = very severe, >75% lung involvement. An overall score of ventilator-induced lung injury was obtained based on the summation of all the scores.
Immunohistochemistry
Immunohistochemistry was carried out on serial lung sections (26). Briefly, the sections were incubated with 0.5% hydrogen peroxide in methanol (Sigma-Aldrich) to quench endogenous peroxidase activity. This was followed by exposure to pepsin (2 mg/mL pepsin in 0.01 N HCl, 60 min at room temperature) (Sigma-Aldrich) to unmask antigens and incubation with BAS (1% in PBS) for 30 min. Incubation with primary antibodies was performed for 12 h at 4°C before treatment with horseradish peroxidase-conjugated secondary antibodies at room temperature for 30 min. The negative controls included omission of either the primary or secondary antibody. Each primary antibody was initially titrated to determine the optimal concentration for use in our experiments. The stained sections were observed under microscope, and quantitative analysis of positive staining areas (%) in five images of nonoverlapping fields from each section was performed by using Image J software (NIH).
Antibody Validation
The specificity of all antibodies used in this study has been tested and reported in previous studies. Anti-YAP (No. 4912) (21), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, No. 5174) (21), and anti-β-catenin (No. 8480) (27) antibodies were purchased from Cell Signaling. Antiplatelet endothelial cell adhesion molecule-1 (PECAM-1, sc-376764) (28), anti-VE-cadherin (sc-9989) (29), anti-p120 catenin (sc-23873) (30), and anti-VE-PTP (sc-1114) (31) antibodies were obtained from Santa Cruz. Anti-VE-cadherin-Tyr685 (ab119785) (31) antibody was from Abcam. Monoclonal anti-Ly6G antibody (1A8) and an isotype control Immunoglobulin (Ig) G2a (2A3) antibody (32) were purchased from Bio X Cell. Anti-green fluorescent protein (GF28R No. 2956), anti-VE-cadherin-Tyr658 (44–1144 G), and anti-VE-cadherin-Tyr731 (44–1145 G) antibodies (31) were ordered from Thermo Fisher.
Statistics
All data are expressed as means ± SD and analyzed using SPSS statistics software version 23.0 (IBM, USA). A preplanned, independent, two-sample t test was used for comparisons between two groups, and one-way ANOVA with post hoc test (Bonferroni test) for three or more groups. One-way ANOVA with Bonferroni post hoc test was performed for experiments with one single time point analysis. An ordinary two-way ANOVA (no repeated measures) with Bonferroni post hoc test was used with VT and mice as factors to be evaluated. All tests were two-tailed, and P < 0.05 was considered as the level of significance.
RESULTS
Mechanical Ventilation Upregulates YAP Expression
To determine the potential role of YAP in ventilator-induced lung injury, we first examined total YAP expression in the lung. Mechanical ventilation upregulated YAP protein expression in a VT-dependent manner (P < 0.05, n = 6/group) (Fig. 1A). Mechanical ventilation at a VT of 21 mL/kg also induced YAP protein expression in a time-dependent manner (P < 0.05, n = 6/group) (Fig. 1B). Four hours of mechanical ventilation at a normal VT (7 mL/kg) did not alter YAP protein expression. To identify the cell types in the lung responsible for the change of YAP expression, we isolated endothelial cells from the lung following ventilation. The expression of YAP protein in both endothelial (P < 0.01, n = 6/group) and nonendothelial cell fractions (P < 0.001, n = 6/group) statistically increased in response to mechanical ventilation (Fig. 1C). Furthermore, the changes in YAP expression in lung endothelial cells were time-dependent (P < 0.01, n = 6/group) (Fig. 1D). Increased YAP mRNA expression was also seen in lung endothelial cells after mechanical ventilation by real-time quantitative reverse transcription PCR (P < 0.05, n = 6/group) (Fig. 1E). These results indicate that mechanical ventilation regulates YAP expression in lung endothelial cells.
Figure 1.

Mechanical ventilation (MV) induces YAP expression in mouse lungs and endothelial cells (ECs). A: effects of different tidal volumes on expression of YAP in the lung. Mice were ventilated at indicated tidal volumes for 4 h. Top: representative Western blots for YAP expression; bottom: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from three male and three female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. B: time course of YAP expression in the lung following ventilation. Mice were ventilated at a tidal volume of 21 mL/kg for the indicated times. Top: representative Western blots for YAP expression; bottom: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from three male and three female mice). *P < 0.05, **P < 0.01 versus 14 mL/kg group, one-way ANOVA with Bonferroni post hoc test. C: YAP expression in mouse lung EC following ventilation. Lung endothelial cells (ECs) and nonendothelial cells (non-ECs) were isolated from mice ventilated at a tidal volume of 21 mL/kg for 4 h. Top: representative Western blots for YAP expression; middle and bottom: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from 3 male and 3 female mice). **P < 0.01, independent two-sample t test. D: time course of YAP expression in lung endothelial cells following ventilation. Lung endothelial cells were isolated from mice ventilated at a tidal volume of 21 mL/kg for the indicated times. Top: representative Western blots for total YAP expression; bottom: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. E: quantitative analysis of YAP mRNA levels in lung endothelial cells by real-time PCR and normalized to control. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.01, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. YAP, Yes-associated protein.
Endothelial Cell-Specific Deletion of YAP Exaggerates Mechanical Ventilation-Induced Lung Inflammation and Injury
To explore the involvement of endothelial YAP in mouse ventilator-induced lung injury, we utilized a double transgenic mouse model in which the yap gene is selectively deleted from endothelial cells (Tie2-Cre+/yapflox/flox mice). Previous studies demonstrated in this mouse line that yap was efficiently deleted in endothelial cells without affecting epithelial cell YAP expression in the lung (21). Following noninjurious ventilation (7 mL/kg), there was no difference between wild-type and endothelial cell-specific YAP knockout mice in albumin-bound Evans blue dye extravasation (4.6 ± 0.9 vs. 5.9 ± 1.4 μg/g, P = 0.861) and total protein concentration (128 ± 17 vs. 145 ± 14 μg/mL, P = 0.639) in the bronchoalveolar lavage fluid (Fig. 2, A and B). Mechanical ventilation with a high VT (21 mL/kg), as opposed to ventilation with low VT, induced a drastic increase in albumin-bound Evans blue dye extravasation (81 ± 14 vs. 5 ± 1 μg/g, P < 0.001) and total protein concentration (449 ± 62 vs. 128 ± 17 μg/mL, P < 0.001) in the bronchoalveolar lavage fluid in control mice. Deletion of YAP in endothelial cells exaggerated vascular leakage and lung edema during ventilator-induced lung injury, as indicated by the marked increase in albumin-bound Evans blue dye extravasation (145 ± 23 vs. 81 ± 14 μg/g, n = 6, P < 0.001) (Fig. 2A) and total protein concentrations (755 ± 109 vs. 449 ± 62 μg/g, n = 6, P < 0.001) in bronchoalveolar lavage fluid (Fig. 2B) after exposure to mechanical stress associated with high VT mechanical ventilation. Microscopic images of lung tissue exhibited increased levels of alveolar congestion, exudate, interstitial and alveolar cellular infiltrates, and intra-alveolar capillary hemorrhage in endothelial cell-specific YAP knockout mice as compared with control mice following mechanical ventilation (Fig. 2C, top). These histological changes were reflected in a statistically significant increase in lung injury score (6.4 ± 0.8 vs. 3.7 ± 0.6, n = 6, P < 0.001) in ventilated endothelial cell-specific YAP knockout mice (Fig. 2C, bottom). Neutrophil counts in bronchoalveolar lavage fluid (1.8 ± 0.4 vs. 0.9 ± 0.1 × 105 cells/mL, n = 6, P < 0.001) (Fig. 2D) and lung myeloperoxidase level (1.9 ± 0.3 vs. 1.0 ± 0.1 OD/g, n = 6, P < 0.001) (Fig. 2E) were also statistically increased in endothelial cell-specific YAP knockout mice ventilated with high VT compared with those in ventilated wild-type mice (Fig. 2, D and E). Furthermore, endothelial cell-specific deletion of YAP augmented mechanical ventilation-induced increases in inflammatory markers, such as TNF-α (156 ± 30 vs. 98 ± 14 pg/mL, n = 6, P < 0.001) and IL-6 (790 ± 48 vs. 547 ± 39 pg/mL, n = 6, P < 0.001) in bronchoalveolar lavage fluid (Fig. 2, F and G). These results suggest that endothelial YAP plays a protective role in ventilator-induced lung inflammation and injury.
Figure 2.
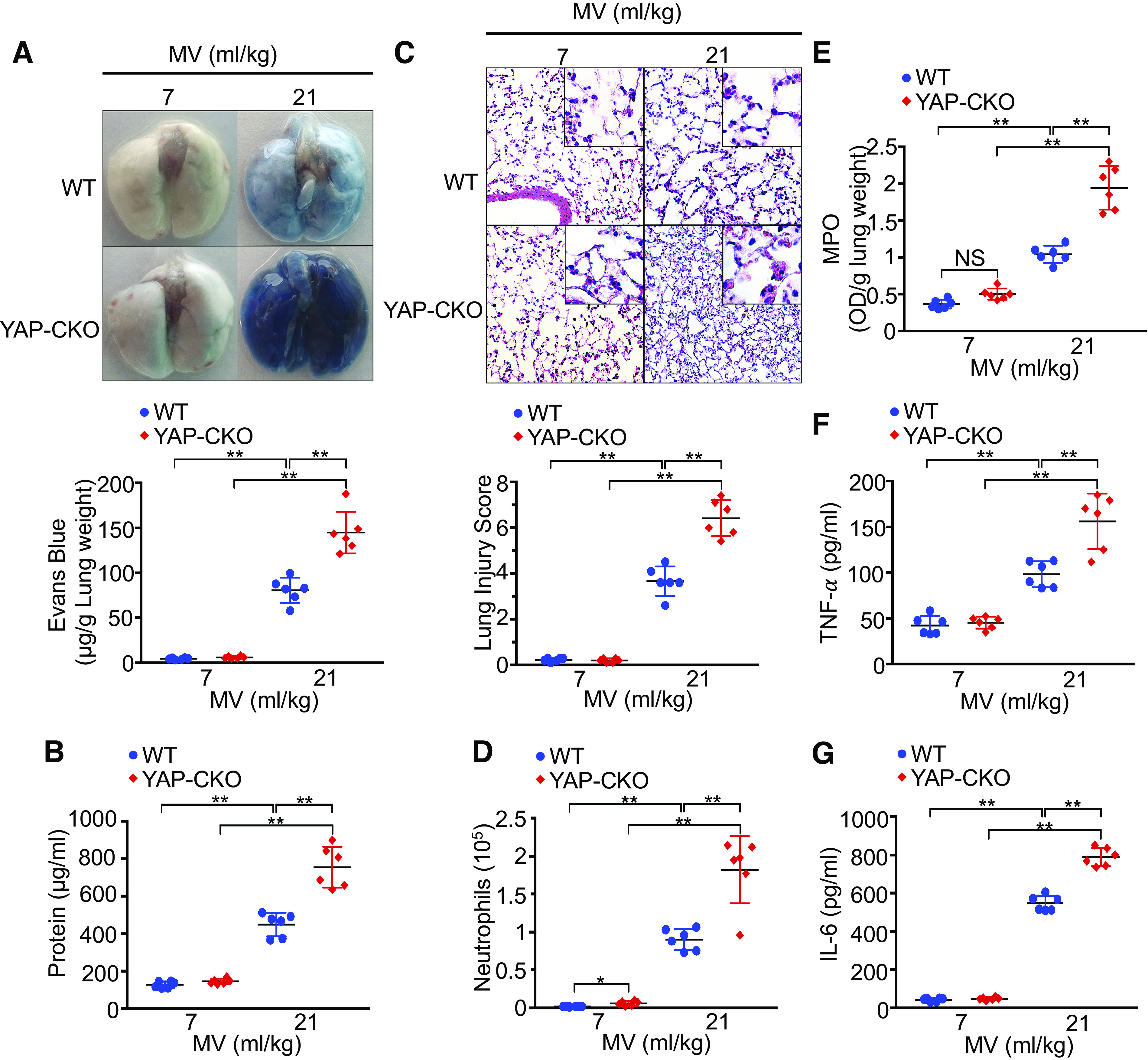
YAP deletion in endothelial cells increases lung inflammatory injury following mechanical ventilation (MV). Wild-type (WT) and endothelial cell-specific YAP knockout mice (YAP-CKO) were ventilated at indicated tidal volumes for 4 h. A: pulmonary vascular permeability measured by Evans blue dye extravasation. Top: representative lung appearance after Evans blue dye administration. Bottom: quantitative analysis of Evans blue-labeled albumin extravasation. n = 6/group. **P < 0.001, two-way ANOVA with Bonferroni post hoc test. B: protein concentrations in bronchoalveolar lavage fluid. C: hematoxylin and eosin staining of sections of lungs. Top: representative lung histology. Magnification, ×20; inset, ×40. Bottom: quantification of histopathological lung injury scores. n = 6/group (3 male and 3 female mice). **P < 0.001, two-way ANOVA with Bonferroni post hoc test. All images for control and treatment groups were collected at the same time under the same conditions. D: neutrophil count in bronchoalveolar lavage fluid by cytospin analysis. n = 6/group (3 male and 3 female mice). *P < 0.05, **P < 0.001, two-way ANOVA with Bonferroni post hoc test. E: MPO activity in the lung tissues. **P < 0.001, two-way ANOVA with Bonferroni post hoc test. F and G: levels of TNF-α (F) and IL-6 (G) in the bronchoalveolar lavage fluid of ventilated WT and endothelial cell-specific YAP knockout mice (YAP-CKO). n = 6/group (three male and three female mice). **P < 0.001, two-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. YAP, Yes-associated protein.
Since Tie2-Cre alleles also show Cre recombinase reporter gene expression in hematopoietic cells (21), Cre recombinase driven by the Tie2 promoter may also cause YAP deletion in hematopoietic cells. Using neutrophil depletion and repletion, we next investigated if YAP deletion in myeloid cells contributes to the enhanced lung inflammation and injury. As expected, neutrophil depletion markedly reduced neutrophil counts (Fig. 3A), total protein concentrations (Fig. 3B), and tumor necrosis factor-α (Fig. 3C) in bronchoalveolar lavage fluid and albumin-bound Evans blue dye extravasation (Fig. 3D) in ventilated endothelial cell-specific YAP knockout mice. These responses were similarly reversed by repletion of neutrophils from either wild-type or endothelial cell-specific YAP knockout mice. These results indicate that YAP ablation in hematopoietic cells per se did not contribute to augmented lung inflammatory injury in ventilated endothelial cell-specific YAP knockout mice.
Figure 3.

Effects of neutrophil depletion and repletion on lung inflammatory injury. Endothelial cell-specific YAP knockout mice (YAP-CKO) were intraperitonially injected with isotype control immunoglobulin (Ig) G2a antibody or monoclonal anti-Ly6G antibody for 3 consecutive days. Neutrophil repletion was performed in neutropenic mice with neutrophils (2 × 106 cells/mouse, iv) isolated from wild-type (WT) or endothelial cell-specific YAP-CKO mice. All mice were ventilated with a tidal volume of 21 mL/kg for 4 h. A: neutrophil count in bronchoalveolar lavage fluid. B: protein concentrations in bronchoalveolar lavage fluid. C: levels of TNF-α in the bronchoalveolar lavage fluid. D: pulmonary vascular permeability measured by Evans blue dye extravasation. n = 6/group (three male and three female mice). **P < 0.001, two-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. YAP, Yes-associated protein.
YAP Ablation Enhances Tyrosine Phosphorylation of VE-Cadherin following Mechanical Ventilation with a High Tidal Volume
Phosphorylation of VE-cadherin on tyrosine residues following an inflammatory insult has been shown to induce adherens junction disassembly and subsequent vascular hyperpermeability and leukocyte extravasation (14). We therefore investigated the role of YAP deletion in tyrosine phosphorylation of VE-cadherin in lung endothelial cells isolated from ventilated mice. As shown in Fig. 4, mechanical ventilation at a high VT caused an increase in VE-cadherin phosphorylation at Tyr685 (n = 6, P < 0.001), Tyr731 (n = 6, P < 0.001), and Tyr658 (n = 6, P < 0.001) compared with ventilation at a low VT. The phosphorylation of VE-cadherin at these three residues was dramatically enhanced in YAP-depleted lung endothelial cells isolated from endothelial cell-specific YAP knockout mice (n = 6, P < 0.001) (Fig. 4, A and B). YAP deletion alone did not alter tyrosine phosphorylation of VE-cadherin. These results suggest that YAP may regulate lung vascular leakage and neutrophil infiltration into the lung during mechanical ventilation by modulating the status of VE-cadherin phosphorylation at tyrosine residues.
Figure 4.

YAP deletion in endothelial cells increases tyrosine phosphorylation of VE-cadherin (VE-cad) following mechanical ventilation (MV). Wild-type (WT) and endothelial cell-specific YAP knockout mice (YAP-CKO) were ventilated at indicated tidal volumes for 4 h and endothelial cells were isolated from the lung. A: representative Western blots for expression of total VE-cadherin (VE-cad), phospho-Tyr685-VE-cad (VE-cad-Tyr685), VE-cad-Tyr731, and VE-cad-Tyr658 in endothelial cells. B: protein quantification (normalized to VE-cad) by densitometry. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.001, two-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. YAP, Yes-associated protein.
YAP Deletion Facilitates the Dissociation of VE-Cadherin from p120-Catenin and β-Catenin as well as VE-Cadherin Internalization in Lung Endothelial Cells from Ventilated Lungs
VE-cadherin interaction with p120-catenin and β-catenin stabilizes VE-cadherin at the junctions by preventing clathrin-mediated endocytosis of VE-cadherin (33). Tyrosine phosphorylation of VE-cadherin blocks binding of p120-catenin and β-catenin and induces endothelial barrier dysfunction (12, 13). As expected, mechanical ventilation at a high VT reduced the association of VE-cadherin with p120-catenin (n = 6, P < 0.001) and β-catenin (n = 6, P < 0.001) in endothelial cells from wild-type lungs compared with low VT ventilation, whereas YAP deletion completely abolished the association of VE-cadherin with p120-catenin (n = 6, P < 0.001) and β-catenin (n = 6, P < 0.001) in endothelial cells (Fig. 5A). YAP deletion alone had no effect on VE-cadherin association with p120-catenin (n = 6, P = 0.708) and β-catenin (n = 6, P = 0.396). To further determine whether YAP regulates VE-cadherin internalization, we analyzed cell surface and cytoplasmic expression of VE-cadherin following ventilation. Immunoblot analysis indicated that mechanical ventilation decreased cell surface VE-cadherin (n = 6, P < 0.05) but increased cytoplasmic VE-cadherin (n = 6, P < 0.05) in lung endothelial cells from wild-type mice. These changes were significantly enhanced in lung endothelial cells from ventilated endothelial cell-specific YAP knockout mice (n = 6, P < 0.001) (Fig. 5B). Overall, these data suggest that YAP stabilizes VE-cadherin expression in endothelial cell-cell junction by maintaining assembly of the VE-cadherin–p120-catenin/β-catenin complex and inhibiting VE-cadherin endocytosis.
Figure 5.

YAP ablation enhances the disassociation of VE-cadherin (VE-cad) from catenins and VE-cadherin internalization during mechanical ventilation (MV). Wild-type (WT) and endothelial cell-specific YAP knockout mice (YAP-CKO) littermates were ventilated at indicated tidal volumes for 4 h and endothelial cells were isolated from the lung. A: effect of YAP deletion on the association of VE-cadherin (VE-cad) with catenins. Top: the association between VE-cadherin and p120-catenin (p120) or between VE-cadherin and β-catenin was detected by immunoprecipitation with anti-VE-cadherin antibody followed by immunoblotting for anti-p120- and anti-β-catenin antibodies. Bottom: protein quantification (normalized to VE-cad) by densitometry. n = 6 lungs (from 3 male and 3 female mice). **P < 0.001, two-way ANOVA with Bonferroni post hoc test. B: effects of YAP depletion on subcellular localization of VE-cadherin. Left: immunoblot showing the levels of VE-cadherin expression in membrane and cytosolic fractions; right: protein quantification by densitometry. Bar graph shows the relative abundance of VE-cadherin protein (normalized to that of loading controls) from six independent experiments (lungs) (from 3 male and 3 female mice). *P < 0.05, **P < 0.001, two-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. YAP, Yes-associated protein.
YAP Deletion Downregulates VE-PTP Expression in Lung Endothelial Cells from Ventilated and Nonventilated Lungs
Because VE-PTP controls the status of VE-cadherin phosphorylation at tyrosine residues and regulates the induction of vascular permeability and leukocyte extravasation (34), we next investigated the effect of YAP deletion on VE-PTP expression following ventilation. Western blot analysis indicated that mechanical ventilation at a high VT induced a statistically significant decrease in VE-PTP expression in endothelial cells from wild-type mouse lungs compared to ventilation at a normal volume (n = 6, P < 0.01) (Fig. 6A). Interestingly, YAP ablation alone also reduced VE-PTP protein expression in endothelial cells from endothelial cell-specific YAP knockout mouse lungs. Following mechanical ventilation at a high VT, YAP deletion almost completely abolished VE-PTP expression in endothelial cells (n = 6, P < 0.01) (Fig. 6A). Mechanical ventilation at low tidal volume (7 mL/kg) did not change VE-PTP protein expression in both WT and YAP-CKO mice compared with spontaneous breathing (data not shown). To further verify these findings, immunohistochemical staining of lung tissue sections with antibody against VE-PTP was performed. The changes in VE-PTP expression in wild-type and endothelial cell-specific YAP knockout mouse lungs after ventilation are consistent with the results obtained by Western blotting (n = 6, P < 0.01) (Fig. 6B). Taken together, these results demonstrated that YAP modulates VE-PTP expression in endothelial cells.
Figure 6.

Effects of YAP deletion on VE-PTP expression in lung endothelial cells. A: VE-PTP expression in lung endothelial cells analyzed by Western blot. Endothelial cells were isolated from the lungs of wild-type (WT) and endothelial cell-specific YAP knockout mice (YAP-CKO) littermates mechanically ventilated with at indicated tidal volumes (MV) for 4 h. Top: levels of VE-PTP in endothelial cells were determined by Western blot analysis. Bottom: protein quantification (normalized to GAPDH) by densitometry. B: immunohistochemical staining of VE-PTP in lung sections. Top: low-power (×20) and inset high-power (×40) magnified images of immunohistochemical staining for VE-PTP in the lungs of WT and YAP-CKO mice. Bottom: quantification of VE-PTP. n = 6 lungs (5 slices/lungs) (from 3 male and 3 female mice). All images for control and treatment groups were collected at the same time under the same conditions. *P < 0.05, **P < 0.01, two-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. VE-PTP, vascular endothelial protein tyrosine phosphatase; YAP, Yes-associated protein.
Exogenous Expression of VE-PTP in Pulmonary Vasculature Rescues the Phenotype of Increased Vascular Permeability and Neutrophil Infiltration in Ventilated Endothelial Cell-Specific YAP Knockout Mice
To elucidate the role of VE-PTP in YAP deficiency-induced vascular leakage and neutrophil extravasation in vivo, we used liposome-mediated VE-PTP cDNA delivery to manipulate VE-PTP expression in endothelial cell-specific YAP knockout mouse lungs (21, 35). VE-PTP physically interacts with VE-cadherin via its extracellular domain (17 fibronectin-like domains) (15, 31). We utilized full-length VE-PTP and ΔN VE-PTP (lacking entire extracellular VE-PTP domain) (Fig. 7A) to determine whether and how VE-PTP regulates YAP deletion-mediated lung edema and inflammation. Figure 7B shows successful expression of wild-type VE-PTP and ΔN VE-PTP protein in endothelial cells isolated from endothelial cell-specific YAP knockout mouse lungs transfected with VE-PTP cDNAs. Ectopic expression of wild-type VE-PTP cDNA in lung vascular endothelial cells rescued the phenotype of increased albumin-bound Evans blue dye extravasation (n = 6, P < 0.001) (Fig. 7C), lung histopathological lesions (n = 6, P < 0.001) (Fig. 7D), protein content (n = 6, P < 0.001) (Fig. 7E), and neutrophil counts (n = 6, P < 0.001) (Fig. 7F) in bronchoalveolar lavage fluid after mechanical ventilation with a high VT. However, exogenous expression of ΔN VE-PTP protein failed to reverse YAP deletion-induced lung edema, histopathological damage, and inflammation. Our results suggest that YAP maintains pulmonary vascular endothelial barrier integrity during mechanical ventilation by facilitating interaction between VE-PTP and VE-cadherin.
Figure 7.

Exogenous expression of VE-PTP in pulmonary vasculature rescues YAP deficiency-mediated lung injury and inflammation. Endothelial cell-specific YAP knockout mice (YAP-CKO) overexpressing full-length VE-PTP and VE-PTP mutant (△N) were ventilated with a tidal volume of 21 mL/kg for 4 h. A: schematic representation of full length VE-PTP and △N VE-PTP (lacking extracellular domain) mutant. B: verification of overexpression of VE-PTP and VE-PTP mutant (△N) in lung endothelial cells of endothelial cell-specific YAP knockout mice (YAP-CKO) by Western blot. Green fluorescent protein (GFP) for detection of cyan fluorescent protein-tagged VE-PTP and VE-PTP mutant (△N). C: pulmonary vascular permeability measured by Evans blue dye extravasation. Top: representative lung appearance after Evans blue dye administration. Bottom: quantitative analysis of Evans blue-labeled albumin extravasation. n = 6/group (3 male and 3 female mice). **P < 0.001, one-way ANOVA with Bonferroni post hoc test. D: hematoxylin and eosin staining of lung sections. Top: representative lung histology. Magnification, ×20; inset, ×40. Bottom: quantification of histopathological lung injury scores. n = 6/group (3 male and 3 female mice). **P < 0.001, one-way ANOVA with Bonferroni post hoc test. All images for control and treatment groups were collected at the same time under the same conditions. E: protein concentrations in bronchoalveolar lavage fluid. F: neutrophil count in bronchoalveolar lavage fluid by cytospin analysis. n = 6/group (3 male and 3 female mice). **P < 0.001, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. VE-PTP, vascular endothelial protein tyrosine phosphatase; YAP, Yes-associated protein.
Exogenous Expression of VE-PTP in Pulmonary Vasculature Prevents VE-Cadherin Phosphorylation and Restores VE-Cadherin Association with Catenins in Ventilated Endothelial Cell-Specific YAP Knockout Mice
Expression of wild-type VE-PTP in lung endothelial cells prevented the phosphorylation of VE-cadherin at Tyr658 (n = 6, P < 0.001), Tyr731 (n = 6, P < 0.05), and Tyr685 (n = 6, P < 0.05) elicited by YAP depletion, whereas expression of ΔN VE-PTP had no effect on tyrosine phosphorylation of VE-cadherin induced by YAP deletion in endothelial cells from ventilated lungs (Fig. 8, A and B). Coimmunoprecipitation studies demonstrated that expression of wild-type VE-PTP but not ΔN VE-PTP increased the association between VE-cadherin and p120-catenin (n = 6, P < 0.001) and β-catenin (n = 6, P < 0.001) in pulmonary endothelial cells from ventilated endothelial cell-specific YAP knockout mice (Fig. 8, C and D). These data suggest that YAP inhibits VE-cadherin phosphorylation at tyrosine residues and stabilizes the VE-cadherin/catenin complex by enhancing interactions between VE-PTP and VE-cadherin.
Figure 8.

Exogenous expression of VE-PTP in pulmonary vasculature prevents VE-cadherin (VE-cad) phosphorylation and dissociation of VE-cadherin with catenins. Full-length VE-PTP and VE-PTP mutant (△N) cDNAs were delivered to YAP-CKO mice via cationic liposomes. Lung microvascular endothelial cells were isolated from these transfected mice. A: effects of overexpressed VE-PTP and VE-PTP mutant on tyrosine phosphorylation of VE-cadherin (VE-cad) in the endothelial cells of YAP-CKO mice. B: protein quantification (normalized to GAPDH) by densitometry. n = 6 lungs (from 3 male and 3 female mice). *P < 0.05, **P < 0.001, one-way ANOVA with Bonferroni post hoc test. C: association of VE-cadherin with p120- or β-catenin detected by immunoprecipitation after overexpression of VE-PTP and VE-PTP mutant (△N) in the YAP-CKO mice. D: protein quantification (normalized to VE-cadherin, VE-cad) by densitometry. n = 6 lung (from 3 male and 3 female mice). **P < 0.001, one-way ANOVA with Bonferroni post hoc test. Data represent means ± SD. VE-PTP, vascular endothelial protein tyrosine phosphatase; YAP, Yes-associated protein.
DISCUSSION
The functions of YAP expression in endothelial cells have primarily been explored in relation to vessel maintenance (53), angiogenesis (36–38), and vascular inflammation (21). Here, we demonstrate that endothelial YAP works as a molecular brake during mechanical ventilation to prevent uncontrolled impairment of endothelial barrier function, thereby restraining massive vascular leakage and lung injury. YAP expression is upregulated in pulmonary endothelial cells within hours after mechanical ventilation at a high VT and that YAP-deficiency specifically in endothelial cells leads to enhanced vascular hyperpermeability and neutrophil infiltration. We showed that YAP controls VE-PTP expression, which inhibits tyrosine phosphorylation of VE-cadherin, preventing dissociation of VE-cadherin from p120-catenin and β-catenin thus stabilizing endothelial barrier.
YAP senses mechanical signaling which activates its ability to regulate cell fate decisions including proliferation, survival, and differentiation (39–41). Our study showed that YAP protein expression was upregulated in pulmonary endothelial cells following mechanical ventilation. Similar results have been reported in mammary epithelial cells exposed to cyclic stretch (42). Because YAP messenger RNA expression was also increased in lung endothelial cells, mechanical stimulation may promote YAP protein synthesis and subsequently increased YAP protein expression. It has been shown that c-Jun N-terminal kinase is activated by mechanical strain, which inhibits Hippo signaling and increases YAP expression (42). Thus, c-Jun N-terminal kinase activation may also contribute to mechanical ventilation-induced YAP expression.
Our study reveals a protective role for YAP expression in endothelial cells in a murine model of ventilator-induced lung injury. No significant differences were detected between wild-type and endothelial cell-specific YAP knockout mice under low VT ventilation by all measures for lung vascular permeability and inflammation. YAP ablation specifically in all vascular endothelial cells dramatically enhanced lung inflammatory injury following high VT ventilation as evident by higher vascular permeability and neutrophil infiltration, severe lung histopathological changes, and increased proinflammatory cytokine production. Volume controlled ventilation may have some limitations in murine ARDS as compared with pressure-controlled ventilation (43). However, in our model of high VT ventilation, YAP was shown to be required for a negative feedback regulation of ventilator-induced lung injury. Mechanical ventilation causes an increase in YAP expression in endothelial cells, which in turn limits the lung inflammatory response by stabilizing endothelial barrier integrity. The clinical ARDS scenario often involves infection that is present before the onset of mechanical ventilation. We have recently reported that mice harboring an endothelial cell-specific deletion of YAP displayed increased lung edema and inflammation following endotoxemia and polymicrobial sepsis (21). Our studies clearly demonstrate that endothelial YAP not only respond to inflammatory stimuli but is also an important molecule of cellular responses to mechanical stress. We acknowledge that a slight increase in basal neutrophil count in bronchoalveolar lavage fluid may affect the development of ventilator-induced lung injury in endothelial cell-specific YAP knockout mice. Despite this limitation, we believe that this study strongly supports the notion that YAP signaling may play a pivotal role in restricting uncontrolled lung inflammation and injury during mechanical ventilation.
Inflammatory cells, including neutrophils (6, 44), macrophages (6), T cells (45), and natural killer cells (46), play important roles in the development of lung injury. Neutrophil infiltration and vascular hyperpermeability due to loss of endothelial barrier integrity are crucial mechanisms in the pathogenesis of ventilator-induced lung injury (8). Stable VE-cadherin expression in cell junctions is an important determinant of maintenance of endothelial barrier function. Tyrosine phosphorylation of VE-cadherin induces endothelial junction disassembly and vascular barrier dysfunction, leading to vascular leakage and neutrophil infiltration into tissues (47). Mechanical stimuli such as shear stress have also been shown to stimulate Src-dependent tyrosine phosphorylation of VE-cadherin in vivo, which promotes internalization of VE-cadherin via disruption of VE-cadherin association with p120-catenin and β-catenin (48). In the present study, we observed that mechanical ventilation increased VE-cadherin phosphorylation at Tyr685, Try731, and Tyr658. YAP depletion in endothelial cells enhanced mechanical ventilation-induced VE-cadherin phosphorylation at these residues. Coimmunoprecipitation studies showed that YAP deletion exaggerated mechanical ventilation-induced dissociation of VE-cadherin from p120-catenin and β-catenin. Further study demonstrated that mechanical ventilation-induced VE-cadherin internalization, which was enhanced by YAP deletion. Phosphorylation of VE-cadherin at residues Tyr658 and Tyr685 mediates endothelial hyperpermeability, whereas phosphorylation at Tyr731 causes leukocyte extravasation across the endothelial barrier (34). In our study, YAP ablation enhanced lung vascular barrier leakiness and inflammation in ventilated mice. Thus, these results strongly support the hypothesis that YAP protects against ventilator-induced lung injury by controlling VE-cadherin phosphorylation and internalization.
The next question addressed was how YAP regulates VE-cadherin phosphorylation-mediated inflammation and vascular leakage in the lung during mechanical ventilation. The status of tyrosine phosphorylation of VE-cadherin is dependent on Src family kinases that phosphorylate VE-cadherin, and protein tyrosine phosphatases including VE-PTP, SHP2, and Dep-1, which dephosphorylate VE-cadherin in response to noxious stimuli (48). VE-PTP physically interacts with VE-cadherin at adherens junctions and stabilizes the endothelial barrier via a VE-cadherin phosphorylation-independent pathway in the quiescent endothelium (31) and a VE-cadherin phosphorylation-dependent pathway in the activated endothelium (48). VE-PTP expression can be upregulated by hypoxia (49) and hypoxia-inducible factor 2α (50), while the redistribution of VE-PTP is induced by shear stress (51). In a mouse model of ventilator-induced lung injury, we observed that mechanical ventilation downregulated VE-PTP protein expression in pulmonary endothelial cells. Endothelial cell-specific YAP knockout mice exhibited a marked decrease in endothelial VE-PTP expression following ventilation. To further elucidate the role of VE-PTP in the YAP-mediated protective effect in ventilator-induced injury, we selectively overexpressed VE-PTP in the pulmonary vasculature of endothelial cell-specific YAP knockout mice by injection of liposome–VE-PTP cDNA complexes. We observed that overexpression of full-length VE-PTP completely reversed YAP deletion-mediated endothelial hyperpermeability and leukocyte extravasation in lungs following mechanical ventilation. Consistently, overexpression of VE-PTP also prevented YAP-deletion-mediated VE-cadherin phosphorylation at the three tyrosine residues and subsequent dissociation of VE-cadherin from p120 catenin and β-catenin. Because expression of ΔN VE-PTP mutant was unable to alter the phenotype of ventilated endothelial cell-specific YAP knockout mice, our data demonstrated that the extracellular VE-PTP domain is essential for stabilizing endothelial barrier. Taken together, these findings support the hypothesis that YAP expression strengthens the endothelial barrier and subsequently blocks vascular leakage and neutrophil extravasation during mechanical ventilation through VE-PTP-mediated VE-cadherin dephosphorylation. YAP deletion reduced VE-PTP expression following noninjurious ventilation and further exaggerated high tidal volume mechanical ventilation-induced decrease in VE-PTP expression. The exact mechanism by which YAP regulates VE-PTP expression remains unclear. It will be interesting to investigate how YAP maintains VE-PTP expression in future studies.
In summary, we identified the role of YAP in strengthening endothelial barrier by modulating the VE-cadherin-associated VE-PTP. YAP stabilizes VE-PTP expression, which prevents the VE-cadherin phosphorylation at Tyr658, Tyr685, and Tyr731, and thereby abrogates phosphorylation-dependent VE-cadherin internalization from adherens junctions (Fig. 9). Although gene expression profiles of a variety of murine models of acute lung injury including ventilator-induced lung injury have a high degree of correlation with human data (52), whether translation of insights gained from this study to human lung injury remains to be determined.
Figure 9.

Model of Yes-associated protein (YAP)-mediated protection against ventilator-induced lung injury. Yes-associated protein (YAP) expression is increased in lung endothelial cells after mechanical ventilation. YAP controls vascular endothelial protein tyrosine phosphatase (VE-PTP) expression, which inhibits tyrosine phosphorylation (p) of VE-cadherin (VE-cad), preventing dissociation of VE-cadherin from p120-catenin (p120) and β-catenin (β-cat), thus stabilizing vascular endothelial barrier function. YAP deletion in endothelial cells causes a decrease in VE-PTP expression, which increases phosphorylation of VE-cadherin, dissociation of p120-catenin and β-catenin from VE-cadherin, and subsequent VE-cadherin internalization from adherens junctions, which leads to enhanced vascular hyperpermeability and neutrophil infiltration.
GRANTS
This work was supported by NIH National Heart, Lung, and Blood Institute (NHLBI) Grant HL104092 (to G. Hu), NIH National Institute of Allergy and Infectious Diseases Grant R21AI152249 (to G. Hu), and NIH NHLBI Grant HL152696 (to G. Hu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.S., J.W., and G.H. conceived and designed research; K.S., J.W., and Y.L. performed experiments; G.H. analyzed data; Y.L., M.T., Y.-Y.Z., R.D.M., and G.H. interpreted results of experiments; K.S. prepared figures; K.S., J.W., and G.H. drafted manuscript; M.T., Y.-Y.Z., R.D.M., and G.H. edited and revised manuscript; K.S., J.W., Y.L., M.T., Y.-Y.Z., R.D.M., and G.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Yulia Komarova, PhD (Department of Pharmacology, University of Illinois College of Medicine) for providing the constructs.
REFERENCES
- 1.Acute Respiratory Distress Syndrome Network, Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301–1308, 2000. doi: 10.1056/nejm200005043421801. [DOI] [PubMed] [Google Scholar]
- 2.Futier E, Constantin J-M, Paugam-Burtz C, Pascal J, Eurin M, Neuschwander A, Marret E, Beaussier M, Gutton C, Lefrant J-Y, Allaouchiche B, Verzilli D, Leone M, De Jong A, Bazin JE, Pereira B, Jaber S, IMPROVE Study Group. A trial of intraoperative low-tidal-volume ventilation in abdominal surgery. N Engl J Med 369: 428–437, 2013. doi: 10.1056/NEJMoa1301082. [DOI] [PubMed] [Google Scholar]
- 3.Guay J, Ochroch EA. Intraoperative use of low volume ventilation to decrease postoperative mortality, mechanical ventilation, lengths of stay and lung injury in patients without acute lung injury. Cochrane Database Syst Rev, 7: CD011151, 2015. doi: 10.1002/14651858.cd011151.pub2. [DOI] [PubMed] [Google Scholar]
- 4.Neto AS, Cardoso SO, Manetta JA, Pereira VG, Espósito DC, Pasqualucci MO, Damasceno MC, Schultz MJ. Association between use of lung-protective ventilation with lower tidal volumes and clinical outcomes among patients without acute respiratory distress syndrome: a meta-analysis. JAMA 308: 1651–1659, 2012. doi: 10.1001/jama.2012.13730. [DOI] [PubMed] [Google Scholar]
- 5.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 369: 2126–2136, 2013. [Erratum in N Engl J Med 370: 1668–1669, 2014]. doi: 10.1056/NEJMra1208707. [DOI] [PubMed] [Google Scholar]
- 6.Dos Santos CC, Slutsky AS. Invited review: mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol (1985) 89: 1645–1655, 2000. doi: 10.1152/jappl.2000.89.4.1645. [DOI] [PubMed] [Google Scholar]
- 7.Woods SJ, Waite AA, O'Dea KP, Halford P, Takata M, Wilson MR. Kinetic profiling of in vivo lung cellular inflammatory responses to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 308: L912–L921, 2015. doi: 10.1152/ajplung.00048.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T, Gross C, Desai AA, Zemskov E, Wu X, Garcia AN, Jacobson JR, Yuan JX, Garcia JG, Black SM. Endothelial cell signaling and ventilator-induced lung injury: molecular mechanisms, genomic analyses, and therapeutic targets. Am J Physiol Lung Cell Mol Physiol 312: L452–L476, 2017. doi: 10.1152/ajplung.00231.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 10.Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30: 4157–4170, 2011. doi: 10.1038/emboj.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112: 2770–2779, 2008. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 179: 4053–4064, 2007. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 13.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem 280: 31906–31912, 2005. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 14.Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, Nottebaum AF, Vestweber D. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol 15: 223–230, 2014. doi: 10.1038/ni.2824. [DOI] [PubMed] [Google Scholar]
- 15.Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J 21: 4885–4895, 2002. [Erratum in EMBO J 24: 3158, 2005]. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94: 1287–1312, 2014. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 17.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev 27: 355–371, 2013. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15: 637–646, 2013. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183, 2011. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 20.Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA 113: 11525–11530, 2016. doi: 10.1073/pnas.1613121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao YY, Hu G, Pan D, Malik AB, Hu G. YAP controls endothelial activation and vascular inflammation through TRAF6. Circ Res 123: 43–56, 2018. doi: 10.1161/CIRCRESAHA.118.313143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu G, Ye RD, Dinauer MC, Malik AB, Minshall RD. Neutrophil caveolin-1 expression contributes to mechanism of lung inflammation and injury. Am J Physiol Lung Cell Mol Physiol. 294: L178–L186, 2008. doi: 10.1152/ajplung.00263.2007. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 307: L173–L185, 2014. doi: 10.1152/ajplung.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis RW 4th, Snyder E, Miller J, Carter S, Houser C, Klampatsa A, Albelda SM, Cengel KA, Busch TM. Luminol chemiluminescence reports photodynamic therapy-generated neutrophil activity in vivo and serves as a biomarker of therapeutic efficacy. Photochem Photobiol 95: 430–438, 2019. doi: 10.1111/php.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sue RD, Belperio JA, Burdick MD, Murray LA, Xue YY, Dy MC, Kwon JJ, Keane MP, Strieter RM. CXCR2 is critical to hyperoxia-induced lung injury. J Immunol 172: 3860–3868, 2004. doi: 10.4049/jimmunol.172.6.3860. [DOI] [PubMed] [Google Scholar]
- 26.Nishina K, Mikawa K, Takao Y, Shiga M, Maekawa N, Obara H. Intravenous lidocaine attenuates acute lung injury induced by hydrochloric acid aspiration in rabbits. Anesthesiology 88: 1300–1309, 1998. doi: 10.1097/00000542-199805000-00022. [DOI] [PubMed] [Google Scholar]
- 27.Andersson-Rolf A, Mustata RC, Merenda A, Kim J, Perera S, Grego T, Andrews K, Tremble K, Silva JC, Fink J, Skarnes WC, Koo BK. One-step generation of conditional and reversible gene knockouts. Nat Methods 14: 287–289, 2017. doi: 10.1038/nmeth.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruggisser J, Tarek B, Wyder M, Müller P, von Ballmoos C, Witz G, Enzmann G, Deutsch U, Engelhardt B, Posthaus H. CD31 (PECAM-1) serves as the endothelial cell-specific receptor of Clostridium perfringens β-toxin. Cell Host Microbe 28: 69–78, 2020. doi: 10.1016/j.chom.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Gong H, Gao X, Feng S, Siddiqui MR, Garcia A, Bonini MG, Komarova Y, Vogel SM, Mehta D, Malik AB. Evidence of a common mechanism of disassembly of adherens junctions through Gα13 targeting of VE-cadherin. J Exp Med 211: 579–591, 2014. doi: 10.1084/jem.20131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Sun D, Yan Z, Reynolds AB, Christman JW, Minshall RD, Malik AB, Zhang Y, Hu G. Differential role for p120-catenin in regulation of TLR4 signaling in macrophages. J Immunol 193: 1931–1941, 2014. doi: 10.4049/jimmunol.1302863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juettner VV, Kruse K, Dan A, Vu VH, Khan Y, Le J, Leckband D, Komarova Y, Malik AB. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J Cell Biol 218: 1725–1742, 2019. doi: 10.1083/jcb.201807210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boivin G, Faget J, Ancey PB, Gkasti A, Mussard J, Engblom C, Pfirschke C, Contat C, Pascual J, Vazquez J, Bendriss-Vermare N, Caux C, Vozenin MC, Pittet MJ, Gunzer M, Meylan E. Durable and controlled depletion of neutrophils in mice. Nat Commun 11: 2762, 2020. doi: 10.1038/s41467-020-16596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol 163: 535–545, 2003. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, Vestweber D. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med 208: 2393–2401, 2011. doi: 10.1084/jem.20110525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res 105: 676–685, 2009. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azad T, Ghahremani M, Yang X. The role of YAP and TAZ in angiogenesis and vascular mimicry. Cells 8: 407, 2019. doi: 10.3390/cells8050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boopathy GTK, Hong W. Role of hippo pathway-YAP/TAZ signaling in angiogenesis. Front Cell Dev Biol 7: 49, 2019. doi: 10.3389/fcell.2019.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L, Jain RK, Mazzone M, Schmidt T, Fischer T, Halder G, Ruiz de Almodóvar C. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev Cell 42: 462–478, 2017. doi: 10.1016/j.devcel.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154: 1047–1059, 2013. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 40.Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem 294: 17693–17706, 2019. doi: 10.1074/jbc.REV119.007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elbediwy A, Thompson BJ. Evolution of mechanotransduction via YAP/TAZ in animal epithelia. Curr Opin Cell Biol 51: 117–123, 2018. doi: 10.1016/j.ceb.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Codelia VA, Sun G, Irvine KD. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol 24: 2012–2017, 2014. doi: 10.1016/j.cub.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckle T, Füllbier L, Grenz A, Eltzschig HK. Usefulness of pressure-controlled ventilation at high inspiratory pressures to induce acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol 295: L718–L724, 2008. doi: 10.1152/ajplung.90298.2008. [DOI] [PubMed] [Google Scholar]
- 44.Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, Standiford TJ, Weng T, Fletcher AA, Barthel L, Masterson JC, Furuta GT, Cai C, Blackburn MR, Ginde AA, Graner MW, Janssen WJ, Zemans RL, Evans CM, Burnham EL, Homann D, Moss M, Kreth S, Zacharowski K, Henson PM, Eltzschig HK. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med 9: eaah5360, 2017. doi: 10.1126/scitranslmed.aah5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, Riegel AK, Westrich JA, Colgan SP, Eltzschig HK. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J 27: 2207–2219, 2013. doi: 10.1096/fj.12-225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoegl S, Ehrentraut H, Brodsky KS, Victorino F, Golden-Mason L, Eltzschig HK, McNamee EN. NK cells regulate CXCR2+ neutrophil recruitment during acute lung injury. J Leukoc Biol 101: 471–480, 2017. doi: 10.1189/jlb.3A0516-227R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121: 2115–2122, 2008. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 48.Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M, Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3: 1208, 2012. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, Kojima M, Xia H, Seidel C, Lima e Silva R, Dong A, Hackett SF, Wang J, Howard BW, Vestweber D, Kontos CD, Peters KG, Campochiaro PA. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest 124: 4564–4576, 2014. doi: 10.1172/JCI74527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, Zhao YY, Chatturvedi P, Zhao Y, Komarova YA, Komorova YA, Vogel SM, Malik AB. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125: 652–664, 2015. [Erratum in J Clin Invest 125: 1364, 2015]. doi: 10.1172/JCI77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mantilidewi KI, Murata Y, Mori M, Otsubo C, Kotani T, Kusakari S, Ohnishi H, Matozaki T. Shear stress-induced redistribution of vascular endothelial-protein-tyrosine phosphatase (VE-PTP) in endothelial cells and its role in cell elongation. J Biol Chem 289: 6451–6461, 2014. doi: 10.1074/jbc.M113.529503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sweeney TE, Lofgren S, Khatri P, Rogers AJ. Gene expression analysis to assess the relevance of rodent models to human lung injury. Am J Respir Cell Mol Biol 57: 184–192, 2017. doi: 10.1165/rcmb.2016-0395OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, Ando K, Miyazaki T, Yokota Y, Schmelzer E, Belting HG, Affolter M, Lecaudey V, Mochizuki N. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell 40: 523–536, 2017. doi: 10.1016/j.devcel.2017.02.019. [DOI] [PubMed] [Google Scholar]