

Abstract
Obesity increases incidence and severity of asthma but the molecular mechanisms are not completely understood. Hyperinsulinemia potentiates vagally induced bronchoconstriction in obese rats. Since bronchoconstriction results from airway smooth muscle contraction, we tested whether insulin changed agonist-induced airway smooth muscle contraction. Obesity-prone and resistant rats were fed a low-fat diet for 5 wk and treated with insulin (Lantus, 3 units/rat sc) 16 h before vagally induced bronchoconstriction was measured. Ex vivo, contractile responses to methacholine were measured in isolated rat tracheal rings and human airway smooth muscle strips before and after incubation (0.5–2 h) with 100 nM insulin or 13.1 nM insulin like growth factor-1 (IGF-1). M2 and M3 muscarinic receptor mRNA expression was quantified by qRT-PCR and changes in intracellular calcium were measured in response to methacholine or serotonin in isolated rat tracheal smooth muscle cells treated with 1 µM insulin. Insulin, administered to animals 16 h prior, potentiated vagally induced bronchoconstriction in both obese-prone and resistant rats. Insulin, not IGF-1, significantly increased methacholine-induced contraction of rat and human isolated airway smooth muscle. In cultured rat tracheal smooth muscle cells, insulin significantly increased M2, not M3, mRNA expression and enhanced methacholine- and serotonin-induced increase in intracellular calcium. Insulin alone did not cause an immediate increase in intracellular calcium. Thus, insulin acutely potentiated agonist-induced increase in intracellular calcium and airway smooth muscle contraction. These findings may explain why obese individuals with hyperinsulinemia are prone to airway hyperreactivity and give insights into future targets for asthma treatment.
Keywords: airway hyperreactivity, asthma, hyperinsulinemia, intracellular calcium, obesity
INTRODUCTION
The prevalence of obesity among adults in the United States is 42.4% and 8% of those individuals suffer from asthma (1). Obesity increases the incidence and severity of asthma. This is well established in multiple epidemiological studies involving thousands of subjects (2–9) and in metaanalyses of epidemiologic literature (10, 11). Several large clinical trials measuring airway responsiveness demonstrate that obese patients with asthma are more symptomatic, have greater severity of illness and are less responsiveness to antiinflammatory therapies compared with nonobese patients with asthma (12, 13). However, the mechanisms by which obese individuals are prone to have severe asthma are not completely understood, making it difficult to appropriately prevent and treat. Given the scale of the problem and the paucity of effective intervention, there is an urgent need to identify the molecular mechanisms of obesity-related asthma and associated airway hyperreactivity.
Insulin resistance is common in obesity (14). One third of children and adolescents with obesity have insulin resistance (15) and a strong dose-response relationship between obesity and insulin resistance has been reported in a large-scale cross-sectional study (16). Insulin resistance is tissue specific (17). Kleemann et al. (18) showed that as obesity developed in mice on a high-fat diet, liver and white adipose tissue became insulin resistant within 12 wk, whereas skeletal muscle remained insulin sensitive. Similarly, insulin administration still activated inflammatory cells in insulin-resistant patients (19). Thus, despite being insulin resistant in adipocytes and hepatocytes, insulin may still affect cellular function in other tissues, such as neurons and smooth muscle. Insulin resistance mediates a relationship between obesity and asthma, independent of other variables (20), but the molecular mechanism of how insulin resistance increases the incidence of asthma is still unknown.
Obesity-induced insulin resistance and compensatory hyperinsulinemia are common in obese individuals (21), and high levels of insulin may play a role in obesity-related asthma. Two cross-sectional studies have shown that high insulin is associated with moderate persistent asthma in adolescents (22) and with decreased lung function in men (23). In our previous study, diet-induced obese rats had increased body weight, body fat, levels of circulating insulin, and bronchoconstriction in response to electrical stimulation of the vagus nerve compared to lean rats (24). When insulin was reduced in diet-induced obese rats by treating them with streptozotocin, vagus nerve-meditated bronchoconstriction was similar to lean animals. Furthermore, when a subacute dose of insulin was administered to streptozotocin-treated rats, vagus nerve-mediated bronchoconstriction was potentiated (24). Combined, these data indicate that hyperinsulinemia potentiated bronchoconstriction induced by vagus nerve stimulation.
Airway vagus nerves contain parasympathetic nerves, which provide dominant autonomic control of the airway tone. Parasympathetic nerves release acetylcholine (ACh) that activates M3 muscarinic receptors on airway smooth muscle, resulting in smooth muscle contraction and bronchoconstriction. ACh also feeds back onto inhibitory M2 muscarinic receptors located on the parasympathetic nerves to limit further release of ACh (25).
To distinguish the effects of insulin, specifically, on lung function, separate from the effects of other metabolic disorders and separate from the systemic effects associated with chronic hyperinsulinemia, we treated nonobese rats with a single dose of insulin to directly test whether insulin potentiates bronchoconstriction induced by stimulation of the vagus nerves, or by iv ACh when the vagus nerves were cut. In addition, we tested whether insulin alone could directly contract or enhance airway smooth muscle contraction in tracheas isolated from nonobese rats in response to agonist stimulation. We performed these experiments ex vivo, since muscarinic agonists can induce cardiac death in rats with high levels of circulating insulin (24), truncating the dose-response curve. In addition, since insulin and IGF-1 are closely related hormones with homologous structures and can bind to and activate each other’s receptor, although with reduced affinity (26), we also tested whether IGF-1 behaves similar to insulin on the airway smooth muscle.
METHODS
Animals
Obese-prone, obese-resistant, and wild-type Sprague–Dawley male rats at 5 wk of age were purchased from Charles River Labs (Wilmington, MA). Obese prone and obese resistant rats were developed by selectively breeding rat pairs who gained the most weight or least weight, respectively (27). Wild-type Sprague–Dawley rats were used for ex vivo experiments. All animals were shipped in filtered crates and kept in a facility with high-efficiency particulate-filtered air. Animals were handled in accordance with the standards established by the United States Animal Welfare Acts set forth in NIH guidelines and approved by the Institutional Animal Care and Use Committee at Oregon Health & Science University.
Both obese prone and obese resistant rats were fed a low-fat diet (10% of calories come from fat, Test Diet 58124) for 5 wk (Fig. 1). After 5 wk, some animals were treated with 3 units insulin in 100 µL PBS s.c. (Lantus; (28)), during which they had free access to food to avoid hypoglycemia. Control animals were treated with PBS alone (100 µL s.c.) and fasted overnight. After a 16-h treatment with either insulin or PBS, airway function, blood glucose, body fat were measured as previously described (24).
Figure 1.
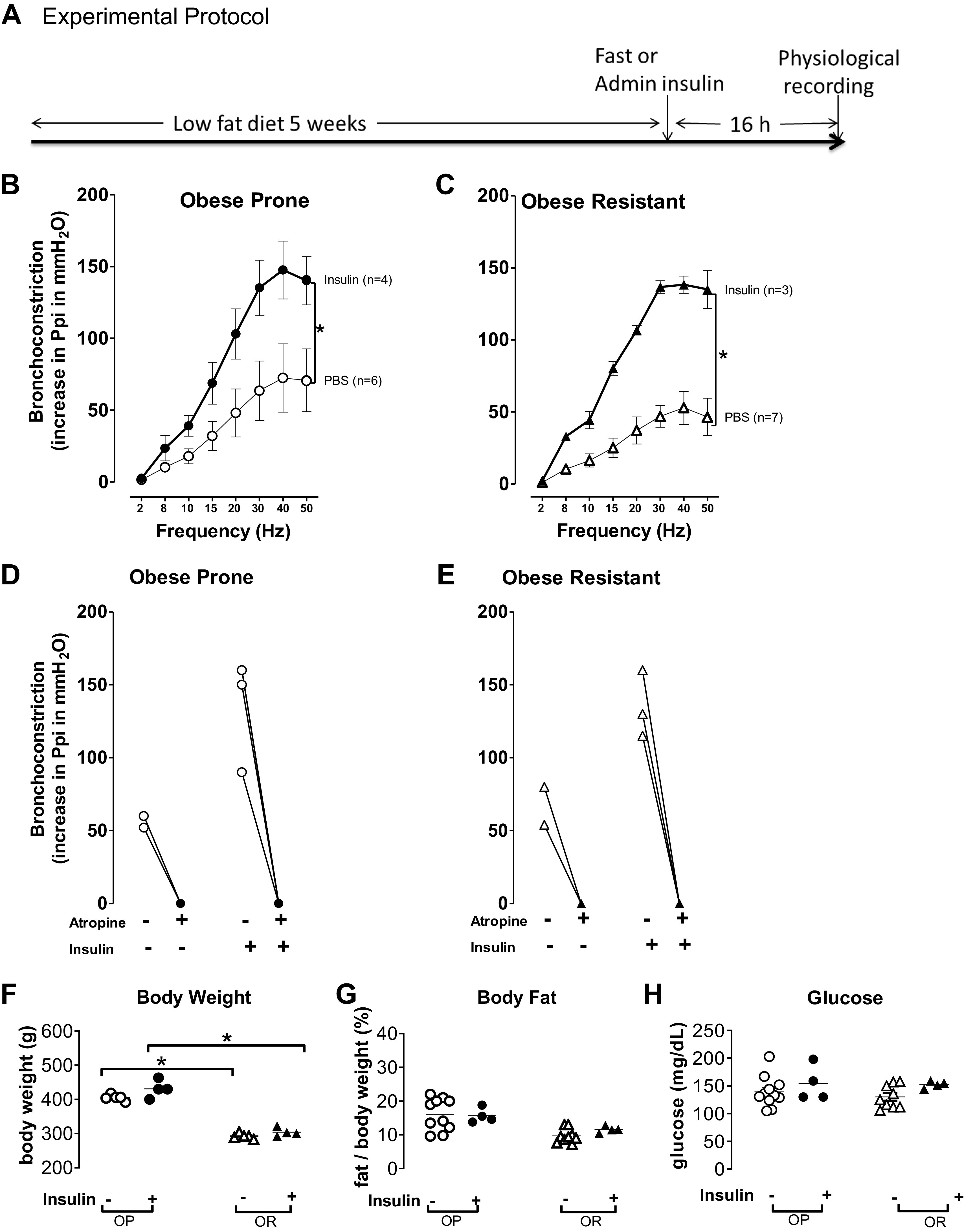
Insulin-potentiated vagally induced bronchoconstriction. A: obese-prone (OP) and obese-resistant (OR) rats were fed a low-fat diet for 5 wk. Sixteen hours before measuring airway function, rats were either treated with 100 μL of phosphate-buffered saline (PBS) subcutaneously and fasted overnight or treated with supplemental insulin (Lantus, 3 units/rat subcutaneously) with free access to food. B–C: electrical stimulation of both vagus nerves (2–50 Hz, 20 V, 0.4 ms pulse duration, for 6 s at 40 s intervals) caused frequency-dependent bronchoconstriction that was not significantly different between obese-prone rats (B, open circles) and obese-resistant rats (C, open triangles) treated with PBS. After a subcutaneous injection of insulin 16 h before measuring airway physiology, insulin significantly potentiated bronchoconstriction in both obese-prone (B, filled circles) and obese-resistant rats (C, filled triangles). D and E: atropine blocked vagally induced bronchoconstriction at 50 Hz (open symbols, bronchoconstriction before atropine; filled symbols, bronchoconstriction after atropine). F: obese-prone rats (OP, circles) on a high-fat diet for 5 wk weighed significantly more than obese-resistant (OR, triangles) rats on the same diet, regardless of insulin treatment. G: body fat, as a % of weight, was not significantly different among any groups. H: in rats not treated with insulin, fasting glucose was not different between obese-prone (open circles) or obese-resistant (open triangles) rats. Glucose was also not different between obese-prone and -resistant rats treated with insulin and with unlimited access to food, (filled symbols). Data are represented as means ± SE. *P < 0.05.
Measurement of Vagally Induced Bronchoconstriction
Bronchoconstriction in response to electrical stimulation of the vagus nerves was measured as previously described (24). Briefly, rats were anesthetized, ventilated, paralyzed, chemically sympathectomized, and vagotomized. Heart rate, blood pressure, and pulmonary inflation pressure (Table 1) were recorded by Lab Chart software (ADInstruments, Colorado Springs, CO). Bronchoconstriction was measured as the increase in pulmonary inflation pressure above baseline inflation pressure produced by the ventilator. Electrical stimulation of both vagus nerves (2–50 Hz, 20 V, 0.4 ms pulse duration, for 6 s at 40 s intervals) produced frequency-dependent bronchoconstriction that recovered upon cessation of electrical stimulation. After stimulating the nerves at 50 Hz, atropine (1 mg/kg; iv) was administered to some of the animals and bronchoconstriction induced by 50 Hz electrical stimulation was measured again to verify the role of muscarinic receptors in vagally induced bronchoconstriction.
Table 1.
Baseline pulmonary inflation pressure (Ppi) and cardiovascular parameters
| Treatment Group | Ppi, mmH2O | Heart Rate, beats/min | Systolic BP, mmHg | Diastolic BP, mmHg |
|---|---|---|---|---|
| OP/LFD | 101 ± 5 | 337 ± 28 | 74 ± 11 | 46 ± 7 |
| OR/LFD | 106 ± 3 | 355 ± 17 | 73 ± 5 | 51 ± 6 |
| OP/LFD/Insulin | 90.0 ± 5.8 | 350 ± 25.5 | 95.5 ± 15.5 | 48.5 ± 9.5 |
| OR/LFD/Insulin | 97.5 ± 2.5 | 342 ± 17.0 | 115.0 ± 5 | 71.5 ± 9.7 |
Data are shown as means ± SE of 4–6 animals/group. LFD, low-fat diet; OP, obese-prone; OR, obese-resistant.
Human Tracheal Smooth Muscle
De-identified human tracheas were obtained from nonasthmatic organ donors from the Pacific Northwest Transplant Bank (Portland, OR). Tracheal smooth muscle strips (1.5 cm × 0.5 cm) were used for ex vivo experiments 6–20 h after harvest.
Measurement of Tracheal Smooth Muscle Function Ex Vivo
Wild type Sprague–Dawley rats were euthanized with 30 mg/kg pentobarbital i.p. (Vortech Pharmaceuticals, Dearborn, MI) and tracheas were removed. Rat tracheal rings (spanning 4–5 cartilage rings) and human tracheal smooth muscle strips were placed in 5 mL organ baths (Radnoti, Colorado Springs, CO) containing Krebs buffer as previously described (24, 29). Rat tracheal epithelium was removed from some segments by inserting and rotating a plastic catheter inside the tracheal lumen before placing the segments in the organ bath. The removal of epithelium was confirmed by hematoxylin and eosin staining. After tissues equilibrated and stabilized, ascending doses of methacholine (0.1–100 µM, A2251 Sigma-Aldrich) were cumulatively added to the organ baths, followed by a series of washes, and then another ascending dose of methacholine (Fig. 2A). Changes in tension were measured as force (in grams) and recorded by Lab Chart software (ADInstruments). Tracheal smooth muscle was then treated with 1 µM insulin (I9278 Sigma-Aldrich) (30), 13.1 nM IGF-1 (100 ng/mL), or PBS for 10 min–2 h. Rat IGF-1 (4326-RG, R&D Systems, Minneapolis, MN) and human IGF-1 (I3769, Sigma-Aldrich) were used for rat and human tissue, respectively, and the PBS control was used to ensure tissue viability did not change over time. After treatment with insulin, IGF-1, or PBS, ascending doses of methacholine were again added to the organ bath and then repeated after washing the tissue (Fig. 2A). The average of the two methacholine dose-response curves before treatment was compared to the average of the two methacholine dose-response curves after treatment. For longer treatments with insulin (4 and 16 h), rat tracheal smooth muscle was treated with either 1 µM insulin or PBS for 4 h or 16 h. In all the organ bath experiments, each tissue was contracted with 0.1 M KCl at the end of each experiment to control for variations between samples due to tissue mass or contractibility, and this change in tension was used to normalize contractions induced by methacholine.
Figure 2.
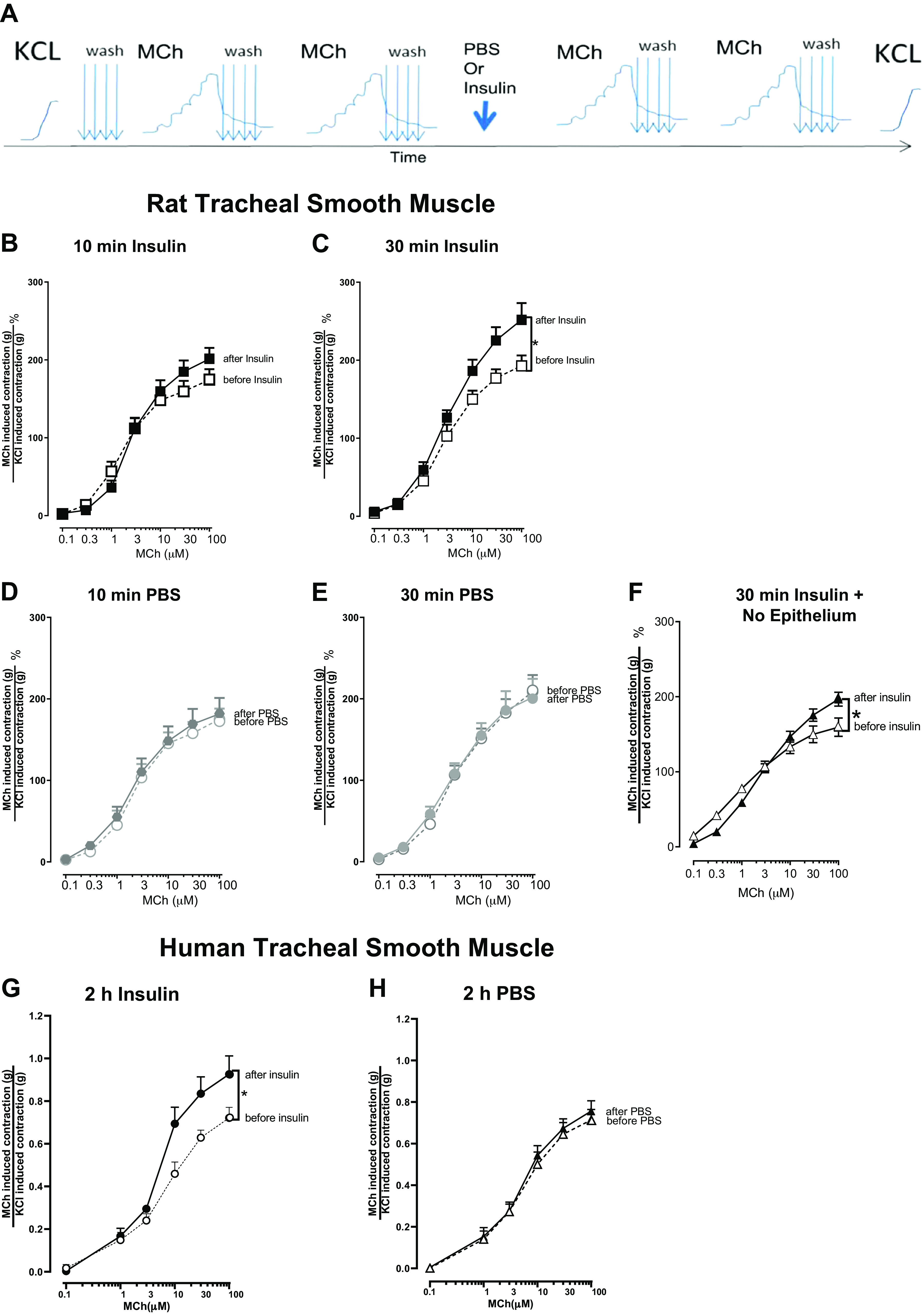
Methacholine (MCh)-induced contraction of tracheal smooth muscle is potentiated by insulin, in both rats and humans. A: the experimental protocol for organ baths experiments (see methods for more details). Methacholine-induced contraction was measured before and after treatment with 1 μM insulin. In intact rat tracheal rings, methacholine-induced contraction was measured before and after a 10- or 30-min treatment with 1 μM insulin (B and C) or PBS (D and E). In rat tracheal rings without airway epithelium, methacholine-induced contraction was measured before and after a 30-min treatment with 1 μM insulin (F). In intact human tracheal smooth muscle strips, methacholine-induced contraction was measured before and 2 h after treatment with 1 μM insulin (G) or PBS (H). Methacholine-induced contractions were normalized to contractions induced by 100 mM KCl. Data shown are means ± SE of two replicates each from 6–9 smooth muscle strips (n = 6–10). *P < 0.05. PBS, phosphate-buffered saline.
Rat Tracheal Smooth Muscle Cultures
Wild-type Sprague–Dawley rats were euthanized with 30 mg/kg pentobarbital intraperitoneally (Vortech Pharmaceuticals) and tracheas were removed antiseptically. After removing the epithelium and dissecting away extraneous tissue from the tracheal muscle, the muscle was minced and dissociated using the Papain Dissociation System followed by the addition of collagenase type IV (Worthington Biochemical Corp, Lakewood, NJ). After cells were strained through a 100-micron cell strainer, tracheal smooth muscle cells were cultured in media containing DMEM/F12, 10% fetal bovine serum, and antibiotic-antimycotic (Thermo Fisher Scientific, Waltham, MA).
Immunohistochemistry
Tracheal smooth muscle cells (passages 4–6) were plated on glass chamber slides (Thermo Scientific Nunc; Vernon Hills, IL). Cells were fixed with zinc buffered formalin (Anatech Ltd., Battlecreek, MI), rinsed, and blocked in 10% normal goat serum (Vector Laboratories, Burlingame, CA) in PBS containing 0.05% Tween-20 (USB Corporation, Cleveland, OH) for 1 h at room temperature. Actin expression was detected using a mouse anti-actin smooth muscle antibody (1:250; MAB 1522; EMD Millipore, Temecula, CA) and elucidated with a goat anti-mouse Alexa 555 secondary antibody (1:1,000; Invitrogen, Carlsbad, CA). Insulin receptor expression was detected using a rabbit anti-insulin receptor β antibody (1:250; SC-711; Santa Cruz Biotechnology, Dallas, TX) and elucidated with a goat anti-rabbit Alexa 488 secondary antibody (1:1,000; Invitrogen). Primary antibodies were incubated overnight at 4°C and secondary antibodies were incubated 1 h at room temperature. Control slides were treated similarly but did not receive the primary antibody. Slides were mounted with Vectashield containing DAPI (Vector Laboratories) to visualize nuclei. Images were obtained on a Zeiss ApoTome2 microscope (Thornwood, NY) at ×63 for both control and labeled slides using identical exposure times.
M2 and M3 Quantitative mRNA Expression
Media on rat tracheal smooth muscle cells (passages 4–6) was replaced with serum free media (DMEM/F12 with antibiotic-antimycotic) for 24 h, before cells were treated with 1 µM insulin in serum free media for 3 h. M2 and M3 mRNA expression was measured using real-time RT-PCR with the following primers: 18S 5′- GTAACCCGTTGAACCCCATT and 18S 3' CCATCCAATCGGTAGTAGCG; rat M2 5′- TACCCAGTTAAGCGGACCAC and M2 3' GCAGATAGAACGCTGCAATG; rat M3 5′- TCGGCAGACAAGACCACGGC and M3 3' GCGTCTGGGCGGCCTTCTTC. n = 5 experiments, each done in duplicate. Data were analyzed using the delta-delta CT (ddCT) method (31) and represented as a fold change over M2 or M3 mRNA expression in cells not treated with insulin.
Measuring Intracellular Calcium
Rat tracheal smooth muscle cells (passages 4–6) were plated in 35 mm glass bottom FluoroDishes (World Precision Instruments, Inc., Sarasota, FL) at 5 × 105 cells/dish, and the following day, media was replaced with serum-free media (DMEM/F12 with antibiotic-antimycotic). The next day, tracheal smooth muscle cells were loaded with 5 µM Fluo4 (Molecular Probes, Eugene, OR) for 30 min in DMEM (Thermo Fisher Scientific, Waltham, MA) containing 2 mM probenecid (Invitrogen, Carlsbad, CA). Cells were washed for 30 min in DMEM containing 2 mM probenecid and then placed in HBSS containing calcium and magnesium (Mediatech, Inc., Manassas, VA). Some cells were pretreated with 1 µM insulin (Sigma Aldrich, St. Louis, MO) for 3 h (which comprised of 2 h in serum free media, 30 min in DMEM with Fluo4, and 30 min in DMEM for the wash). Cells were recorded on a Nikon/Yokogawa CSU-W1 spinning disk confocal microscope (Tokyo, Japan) at 40× using the 488 nm laser and a 300 ms exposure. To obtain a baseline, 5 images were taken every 10 s, and then either 10 µM insulin, 10–1000 µM methacholine, or 0.1–10 µM serotonin was added to the cells. After this addition, images were recorded every second for a duration of 2 min. To ensure the cells loaded with Fluo4, 20 µM ionomycin (Sigma Aldrich, St. Louis, MO) was added and images were recorded every 3 s for another 30 s. Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Each cell within the image with clearly defined edges was manually circled and the average intensity of fluorescence was measured over time for each cell. For each experiment, 2–3 dishes of cells were used per treatment (HBSS control, insulin, methacholine, or serotonin) and 30–50 cells were analyzed per dish. The average of the fluorescence intensity for the first 5 images defined the baseline fluorescence for each cell and this baseline fluorescence was subtracted from all other fluorescence values recorded for that cell. After normalizing the baseline fluorescence for each cell, any fluorescence intensities less than zero (i.e., fluorescence intensity faded over time) were reported as a zero for that cell at that time point. After these adjustments, fluorescence intensity was graphed over time and area under the curve was calculated.
Statistical Analysis
Vagally induced bronchoconstriction and methacholine-induced tracheal smooth muscle contraction were analyzed using repeated-measures analysis of variance (ANOVA). Body weight, body fat, blood glucose, pulmonary inflation pressure (Ppi), blood pressure, and heart rate were analyzed by one-way ANOVA with Bonferroni correction. Differences in M3 and M2 mRNA expression and the effect of acute insulin (10 µM) on intracellular calcium in rat tracheal smooth muscle cells was analyzed by a two-tailed unpaired t test. The effect of insulin exposure on methacholine- and serotonin-induced changes in intracellular calcium was analyzed by a two-way ANOVA and corrected by the Sidak multiple comparison test (Prism 7). All data were analyzed with Prism (GraphPad, La Jolla, CA). P value < 0.05 was considered significant.
RESULTS
Vagally Mediated Bronchoconstriction Was Potentiated by Insulin Treatment Independent of Body Weight or Body Fat
After fed a low-fat diet for 5 wk, bronchoconstriction in response to vagus nerve stimulation was measured in obese-prone and obese-resistant rats, either fasted overnight or treated with insulin for 16 h with free access to food (Fig. 1A). Electrical stimulation of the vagus nerve produced frequency dependent bronchoconstriction (Fig. 1, B and C). Vagally mediated bronchoconstriction was not significantly different between obese-prone and obese-resistant rats (Fig. 1, B and C, respectively; open symbols). Insulin, administered 16 h before measuring airway physiology, significantly potentiated vagally mediated bronchoconstriction in both obese-prone and obese-resistant rats (Fig. 1, B and C, respectively, closed symbols) with a similar magnitude. At the end of the experiment, atropine (1 mg/kg, iv) completely blocked vagally mediated bronchoconstriction (Fig. 1, D and E), indicating that blocking muscarinic receptors prevented vagally induced bronchoconstriction.
Obese prone rats on a low-fat diet had higher body weight (Fig. 1F), but similar body fat (Fig. 1G), compared to obese resistant rats on a low-fat diet. Insulin treatment for 16 h did not significantly change body weight or body fat in either obese-prone or obese-resistant rats (Fig. 1, F and G).
Glucose levels were not significantly different between obese prone (Fig. 1H, open circles) and obese resistant rats (Fig. 1H, open triangles) treated with PBS and fasted for 16 h. In addition, both the obese prone and obese resistant rats were not hypoglycemic after treatment with insulin for 16 h and free access to food (Fig. 1H, closed symbols). Blood glucose levels were similar between rats treated with PBS and fasted overnight (open symbols) and rats treated with insulin but that had access to food (closed symbols; Fig. 1H).
Methacholine-Induced Rat Tracheal Smooth Muscle Contraction Was Potentiated by Insulin Ex Vivo
Methacholine-induced contractions of rat tracheal smooth muscle rings were measured before and after incubation with either PBS or insulin in organ baths (Fig. 2A). Methacholine causes dose-dependent contractions in tracheal smooth muscle from wild type rats (Fig. 2, B–F). When airway epithelium was intact, methacholine-induced contraction after a 10-min incubation with 1 µM insulin was slightly increased compared to methacholine-induced contraction before insulin incubation (Fig. 2B). Methacholine-induced contractions were significantly potentiated after a 30-min incubation with 1 µM insulin (Fig. 2C) but did not significantly change the EC50 (EC50 before insulin EC50 = 3.67 ± 0.82 µM, after insulin EC50 = 3.59 ± 0.65 µM; p = 0.9417 unpaired t test). Methacholine-induced contraction after incubation with PBS (control) for 10 min (Fig. 2D) or 30 min (Fig. 2E) was not significantly different from methacholine-induced contractions before incubation with PBS, indicating that smooth muscle contractibility was not changed over time. When airway epithelium was removed (Fig. 2F), methacholine-induced contractions were still significantly potentiated after a 30 min incubation with 1 µM insulin (after insulin EC50 = 3.02 ± 0.22 µM). Potentiation of methacholine-induced tracheal smooth muscle contractions persisted after a 4 h incubation with 1 µM insulin but was lost after a 16 h incubation (data not shown). Incubation with 0.1 µM or 0.5 µM insulin for 30 min did not significantly potentiate methacholine-induced tracheal smooth muscle contraction (data not shown).
Methacholine-Induced Human Tracheal Smooth Muscle Contraction Was Potentiated by Insulin
In organ baths, methacholine dose-dependently contracted human tracheal smooth muscle strips isolated from organ donors (Fig. 2, G and H). Methacholine-induced contraction after a 2 h incubation with 1 µM insulin was significantly potentiated compared the methacholine-induced contractions before incubation with insulin (Fig. 2G). Methacholine-induced contractions were similar before and after a 2-h incubation with PBS, the vehicle control for insulin (Fig. 2H), indicating that there was no change in smooth muscle contractibility over time.
IGF-1 Did Not Potentiate Methacholine-Induced Contraction of Rat or Human Tracheal Smooth Muscle Ex Vivo
Incubation with 13.1 nM (100 ng/mL) rat IGF-1 for 30 min to 2 h or 20 ng/mL human IGF-1 for 30 min did not potentiate methacholine-induced tracheal smooth muscle contraction in either rat tracheal smooth muscle (Fig. 3, A and B) or human tracheal smooth muscle strips (Fig. 3D), respectively. Again, methacholine-induced rat tracheal smooth muscle contractions were not different after administration PBS (the vehicle control for IGF-1), indicating that the tissue does not become fatigued over this interval (Fig. 3C).
Figure 3.

IGF-1 did not potentiate methacholine (MCh)-induced contraction of rat or human tracheal smooth muscle. Methacholine-induced smooth muscle contraction was measured in rat tracheal rings before and after a 30-min (A) or 2-h (B) exposure to 100 ng/mL rat IGF-1 and in human tracheal smooth muscle strips before and after a 30-min exposure to 20 ng/mL human IGF-1 (D). C: methacholine-induced smooth muscle contraction in rat tracheal rings was also measured before and after a 30-min treatment with PBS (vehicle for IGF-1). Methacholine-induced contractions were normalized to contraction induced by 100 mM KCl. Data shown are means ± SE of two replicates each from 3 rat tracheas (n = 3; A–C) and 2 replicates each from 5 human donors (n = 5; D). PBS, phosphate-buffered saline.
Insulin Increased M2, but Not M3, Muscarinic Receptor mRNA Expression in Rat Tracheal Smooth Muscle Cells
Tracheal smooth muscle cells were cultured from wild type rats. These cells expressed smooth muscle actin by immunohistochemistry, which confirms the identity of these cells (Fig. 4A). Immunohistochemistry also showed that rat tracheal smooth muscle cells express insulin receptors (Fig. 4A). Cells (passages 4–6) were treated with 1 µM insulin for 3 h. Insulin had no effect on M3 mRNA expression (Fig. 4D), however insulin caused a small, but significant, increase in M2 mRNA expression (Fig. 4C) compared to control, baseline expression in rat tracheal smooth muscle cells.
Figure 4.

Rat tracheal smooth muscle cells isolated from wild type rats. Cell phenotype was verified by staining with anti-actin antibody (A; red). Insulin receptor expression on these tracheal smooth muscle cells is shown by positive staining with an anti-insulin receptor β antibody (A; green). B: no primary control for both antibodies; (blue, DAPI nuclear stain). Insulin (1 μM for 3 h) increased M2 muscarinic receptor mRNA (C), but did not change M3 muscarinic receptor mRNA expression (D) in cultured rat tracheal smooth muscle cells. Data shown are means ± SE. *P ≤ 0.05 (n = 5 cultures from separate rats).
Methacholine- and Serotonin-Induced Increase in Intracellular Calcium Was Potentiated by Insulin
Rat tracheal smooth muscle cells (passages 4–6) were loaded with the fluorescent calcium indicator Fluo4. Fluorescence intensity was recorded before (to determine baseline fluorescence) and for 2 min after the addition of reagents to determine changes in intracellular calcium. When insulin (10 µM) was added to the cells, insulin did not cause an immediate increase in intracellular calcium within the 2 min of recording (Fig. 5A). Although 10 µM insulin did not cause an immediate increase in intracellular calcium (Fig. 5A), incubation with 1 µM insulin for 3 h significantly shifted the baseline fluorescence of the cells (Fig. 5B) before addition of methacholine or serotonin. Because of this, the baseline fluorescence was subtracted from all other fluorescence values before analyzing the methacholine and serotonin data (see methods). Methacholine (10 µM, 100 µM, and 1,000 µM added to separate dishes) caused a concentration-dependent increase in intracellular calcium (Fig. 5C, open circles). When cells were incubated with 1 µM insulin for 3 h, the methacholine-induced increase in intracellular calcium was significantly potentiated (Fig. 5C, closed circles). Serotonin (0.01 µM, 0.1 µM, 1 µM, and 10 µM added to separate dishes) also caused a concentration-dependent increase in intracellular calcium (Fig. 5D, open circles) that was also significantly potentiated after a 3-h exposure to 1 µM insulin (Fig. 5D, closed circles).
Figure 5.

Insulin potentiates agonist-induced increases in intracellular calcium in rat tracheal smooth muscle cells. Insulin (10 μM) did not cause an immediate, acute increase in intracellular calcium in cells preloaded with the calcium-sensitive indicator Fluo4 (A). However, pretreatment with 1 μM insulin for 3 h significantly increased baseline fluorescence (B). Methacholine (10, 1,000 μM, MCh) and serotonin (0.01–10 μM) each caused concentration-dependent increases in intracellular calcium (C and D, respectively, open circles), that were significantly potentiated in cells pretreated with 1 μM insulin for 3 h (C and D, filled circles). (n = 5). *P < 0.05.
DISCUSSION
In this study, bronchoconstriction induced by vagal stimulation was potentiated following a 16 h exposure to insulin. This potentiation was blocked by atropine, indicating muscarinic receptors mediated this effect. Ex vivo, a 30-min exposure to insulin was sufficient to significantly potentiate smooth muscle contraction induced by methacholine and a 3-h exposure to insulin similarly potentiated agonist-induced increase of intracellular calcium in tracheal smooth muscle cells. Our data identifies a potential mechanism of how insulin primes airway smooth muscle cells for a more robust contraction upon agonist stimulation, leading to a stronger bronchoconstriction in response to airway stimulation.
Because airway smooth muscle is the vital component for bronchoconstriction, many studies have investigated the effect of insulin or the role of obesity on airway smooth muscle function using different approaches. The differences and similarities between these studies highlights the complicated role of insulin and obesity has in mediating airway function. Orfanos et al. (32) showed that carbachol, a muscarinic and nicotinic acetylcholine receptor agonist, significantly increased intracellular calcium in airway smooth muscle cells isolated from obese patients compared to nonobese patients. It is known that obese patients usually have insulin resistance and compensatory hyperinsulinemia (21). We showed here that a 3-h exposure to 1 µM insulin in rat airway smooth muscle cells was sufficient to potentiate agonist-induced increase in intracellular calcium, which may explain why the carbachol-induced intracellular calcium was higher in airway smooth muscle cells isolated from obese patients. Orfanos et al. (32) also showed that when airway smooth muscle cells derived from either obese or nonobese patients were treated with 0.1 – 1 µM insulin for 24 h, there was no increase in intracellular calcium following stimulation with either histamine or carbachol. In our studies, the ability of insulin to potentiate methacholine-induced contractions in rat tracheal smooth muscle rings was lost after a 16-h exposure to insulin (data not shown) suggesting a transient effect of insulin or reduced calcium channel activity after 16 h in culture. Other studies by Schaafsma et al. (33) and Sharif et al. (34) showed guinea pig tracheal smooth muscle contracted in direct response to 1 µM insulin. Unlike these studies, we investigated the effect of insulin exposure on agonist induced contraction since this more closely models our in vivo studies. Our calcium data show that insulin primes the tissue for a stronger response to stimulatory agonist. Also unlike studies in guinea pigs (33, 34) we show that insulin potentiated agonist-induced contraction in rat tracheas independent of airway epithelium (Fig. 2F), and without changing the EC50. Furthermore, blocking prostaglandins in rat tracheal tissue with indomethacin did not alter insulin’s ability to potentiate agonist induced contraction effect (data not shown).
We have previously shown that hyperinsulinemia also causes dysfunction of neuronal M2 receptors on parasympathetic nerves in rodent models. Dysfunctional neuronal M2 receptors result in increased acetylcholine release onto airway smooth muscle and a significant increase in bronchoconstriction (24, 35). Results from the current study suggest that airway smooth muscle contraction is also potentiated by insulin. Therefore in an obese, hyperinsulemic animals, both parasympathetic nerve activity and smooth muscle function are enhanced. In these animals, parasympathetic nerve stimulation would lead to increased acetylcholine release, which activates primed airway smooth muscle, causing a more robust bronchoconstriction. Discovery of the multifaceted effects of insulin on potentiating vagally mediated bronchoconstriction may help direct the development of therapeutic targets for treatment of asthma in patients with hyperinsulinemia, type II diabetes, or obesity.
The potentiation effect on agonist-induced airway smooth muscle contraction is specific to insulin and not related to IGF-1, which is elevated in obese individuals (36) and may play a role in the development of asthma (37) via increasing proliferation of airway smooth muscle cells (38). IGF-1 and insulin have homologous structures and activate their own receptors, but also bind to and activate other receptors (26). Despite these similarities, our data showed no potentiation of methacholine-induced airway smooth muscle contraction by IGF-1.
Obesity and asthma have been shown to be associated with genetic background, body mass, and plasma glucose levels (39–41). In our study, the short exposure (16 h) to insulin was not expected to affect body weight or body fat, which indeed were not changed. Such subacute treatment of insulin nonetheless significantly potentiated vagally mediated bronchoconstriction, suggesting insulin’s capability to potentiate bronchoconstriction independent of metabolic changes. Organ bath results in the study corroborated this finding. Rat and human tracheal smooth muscle segments incubated with insulin and stimulated with methacholine showed significantly enhanced contractions. Together, these results suggest that insulin does not acutely cause smooth muscle contraction, but instead primes the tissue for a more robust contraction upon stimulation.
Mechanisms by which insulin may affect airway smooth muscle function include modulating muscarinic receptor expression or signaling pathways. Although a short incubation with insulin (3 h) did not change M3 muscarinic receptor mRNA expression, it significantly potentiated M3 muscarinic receptor agonist-induced increase of intracellular calcium, suggesting that insulin may amplify the effect of M3 muscarinic receptor stimulation. M2 receptors are also expressed on airway smooth muscle at a 4:1 ratio to M3 receptors (42). Our data showed that insulin modestly increased M2 mRNA expression on tracheal smooth muscle cells. Although smooth muscle M2 receptors also can increase airway contraction, their mechanism of action is via inhibition the relaxation of smooth muscle. M2 receptors may enhance acetylcholine-induced contraction (43) by inhibiting cAMP (44). Although, our previous study demonstrated that insulin reduces neuronal M2 receptors function (24), we have not directly tested smooth muscle M2 receptor function, especially as the observed increase in M2 receptor mRNA expression was small (less than 11%).
Smooth muscle contraction requires elevated intracellular calcium (45, 46), which can be induced by several factors. M3 muscarinic receptors on tracheal smooth muscle are Gq-linked receptors that are stimulated by acetylcholine, released from parasympathetic nerves or by exogenously applied methacholine. Gq-linked receptors activate phospholipase C which generates inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 binds to IP3 receptors on the sarcoplasmic reticulum and stimulates the release of calcium from this intracellular calcium store. Oscillations of released calcium are important for the activation of myosin light chain kinase leading to smooth muscle contraction. The sarcoplasmic reticulum calcium transport ATPase (SERCA) refills the internal calcium store by moving intracellular calcium back into the sarcoplasmic reticulum. DAG activates receptor operated calcium channels at the cell surface, and extracellular calcium entering the cell supports intracellular levels of calcium. In our study, direct administration of insulin did not stimulate an immediate increase in intracellular calcium in smooth muscle cells, although baseline calcium levels increased after a 3-h incubation with insulin, following which methacholine-induced increase in intracellular calcium was significantly potentiated. Under resting conditions, airway smooth muscle cells maintain a relatively low intracellular calcium concentration. An elevation of intracellular calcium may be the mechanism by which insulin significantly potentiated M3 muscarinic receptor agonist-induced airway smooth muscle contraction both in vivo and ex vivo. Our study also showed that serotonin-induced increase in intracellular calcium was also potentiated by 3 h incubation with insulin, suggesting that the increase of intracellular calcium is not due to a specific effect on M3 muscarinic receptors but instead an effect of insulin on smooth muscle cell function. The serotonin receptor 5HT2C, is also a Gq-linked receptor and is expressed on the airway smooth muscle (47). In summary, our data suggest that insulin primes tracheal smooth muscle, potentially by allowing a gradual increase in intracellular calcium. When a contractile agonist stimulates the cells, either via M3 muscarinic receptor or via serotonin receptor, increased baseline calcium allows for potentiation of calcium signaling and enhanced airway contraction.
The mechanism by which insulin primes tracheal smooth muscle, whether by allowing calcium entrance or by other mechanisms, will be the focus of future studies. There are several mechanisms by which insulin may increase intracellular calcium in trachea smooth muscle cells to prime the cells for contraction. Insulin receptors are tyrosine kinase receptors and, when activated, autophosphorylate to provide a docking site for insulin receptor substrates. Phosphorylated insulin receptor substrates then interact with other downstream signaling proteins like phosphoinositide 3-kinase. Akt (protein kinase B) is activated by phosphoinositide 3-kinase and has been shown to interact and activate L type-calcium channels (48), which would allow extracellular calcium to enter the cell. Insulin may have other mechanisms of increasing intracellular calcium. In other tissues, insulin receptor activation has been shown to modulate the activity of SERCA, thereby affecting the refilling of intracellular calcium store (49, 50).
Our data demonstrate that agonist-induced increases in intracellular calcium in airway smooth muscle cells can be potentiated by insulin, which primes tracheal smooth muscle to a stronger contraction upon agonist stimulation. This may explain why obese patients, who are usually accompanied by hyperinsulinemia (21), are prone to have stronger bronchoconstriction in response to airway stimulation. In addition, inhaled insulin has been found to affect lung function such as reduced FEV1 with unclear reasons in some clinical trials (51, 52). Our data indicate that acetylcholine induced airway smooth muscle constriction will be increased by treatment with high levels of insulin. Thus, inhaled insulin may prime the tracheal smooth muscle to a stronger response induced by acetylcholine released from parasympathetic nerves, increasing airway baseline tone and making it hard for patients to breathe. Therefore, caution should be assumed when giving insulin therapy to asthmatic diabetic patients because insulin may increase the risk of exacerbated asthma attacks. Calcium blockers have some benefits in improving lung function in patients with asthma (53) and have been used to treat hypertension and metabolic disorders in obese patients (54). However, their use in treating obesity-related asthma has not been adequately researched. Our data suggest that M3 muscarinic receptor antagonists and calcium blockers may be preferable in treating obesity-related asthma.
GRANTS
This research was supported by grants from NIH R01HL131525, R01 HL113023, R01 AR061567, and R01 HL124165.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.J.P., G.N.C., and Z.N. conceived and designed research; B.J.P., G.N.C., and Z.N. performed experiments; B.J.P., G.N.C., and Z.N. analyzed data; B.J.P., G.N.C., and Z.N. interpreted results of experiments; B.J.P., G.N.C., and Z.N. prepared figures; B.J.P., G.N.C., and Z.N. drafted manuscript; B.J.P., G.N.C., and Z.N. edited and revised manuscript; B.J.P., G.N.C., and Z.N. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Fryer and Dr. Jacoby for helping design the experiments, reviewing, and editing the manuscript.
REFERENCES
- 1.Centers for Disease Control and Prevention. FastStats! (Online). https://www.cdc.gov/nchs/fastats/default.htm.
- 2.Bibi H, Shoseyov D, Feigenbaum D, Genis M, Friger M, Peled R, Sharff S. The relationship between asthma and obesity in children: is it real or a case of over diagnosis? J Asthma 41: 403–410, 2004. doi: 10.1081/JAS-120026097. [DOI] [PubMed] [Google Scholar]
- 3.Brumpton B, Langhammer A, Romundstad P, Chen Y, Mai XM. General and abdominal obesity and incident asthma in adults: the HUNT study. Eur Respir J 41: 323–329, 2013. doi: 10.1183/09031936.00012112. [DOI] [PubMed] [Google Scholar]
- 4.Brumpton BM, Camargo CA , Jr, Romundstad PR, Langhammer A, Chen Y, Mai XM. Metabolic syndrome and incidence of asthma in adults: the HUNT study. Eur Respir J 42: 1495–1502, 2013. doi: 10.1183/09031936.00046013. [DOI] [PubMed] [Google Scholar]
- 5.Cottrell L, Neal WA, Ice C, Perez MK, Piedimonte G. Metabolic abnormalities in children with asthma. Am J Respir Crit Care Med 183: 441–448, 2011. doi: 10.1164/rccm.201004-0603OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosen DM, Schatz M, Magid DJ, Camargo CA. Jr.. The relationship between obesity and asthma severity and control in adults. J Allergy Clin Immunol 122: 507–511.e6, 2008. doi: 10.1016/j.jaci.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Nystad W, Meyer HE, Nafstad P, Tverdal A, Engeland A. Body mass index in relation to adult asthma among 135,000 Norwegian men and women. Am J Epidemiol 160: 969–976, 2004. doi: 10.1093/aje/kwh303. [DOI] [PubMed] [Google Scholar]
- 8.Shaheen SO, Sterne JA, Montgomery SM, Azima H. Birth weight, body mass index and asthma in young adults. Thorax 54: 396–402, 1999. doi: 10.1136/thx.54.5.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor B, Mannino D, Brown C, Crocker D, Twum-Baah N, Holguin F. Body mass index and asthma severity in the National Asthma Survey. Thorax 63: 14–20, 2008. doi: 10.1136/thx.2007.082784. [DOI] [PubMed] [Google Scholar]
- 10.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med 175: 661–666, 2007. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eneli IU, Skybo T, Camargo CA. Jr, Weight loss and asthma: a systematic review. Thorax 63: 671–676, 2008. doi: 10.1136/thx.2007.086470. [DOI] [PubMed] [Google Scholar]
- 12.Lessard A, Turcotte H, Cormier Y, Boulet LP. Obesity and asthma: a specific phenotype? Chest 134: 317–323, 2008. doi: 10.1378/chest.07-2959. [DOI] [PubMed] [Google Scholar]
- 13.Sutherland TJ, Cowan JO, Young S, Goulding A, Grant AM, Williamson A, Brassett K, Herbison GP, Taylor DR. The association between obesity and asthma: interactions between systemic and airway inflammation. Am J Respir Crit Care Med 178: 469–475, 2008. doi: 10.1164/rccm.200802-301OC. [DOI] [PubMed] [Google Scholar]
- 14.Toft I, Bonaa KH, Jenssen T. Insulin resistance in hypertension is associated with body fat rather than blood pressure. Hypertension 32: 115–122, 1998. doi: 10.1161/01.HYP.32.1.115. [DOI] [PubMed] [Google Scholar]
- 15.Viner RM, Segal TY, Lichtarowicz-Krynska E, Hindmarsh P. Prevalence of the insulin resistance syndrome in obesity. Arch Dis Child 90: 10–14, 2005. doi: 10.1136/adc.2003.036467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez KE, Tucker LA, Bailey BW, LeCheminant JD. Expanded normal weight obesity and insulin resistance in US adults of the National Health and Nutrition Examination Survey. J Diabetes Res 2017: 1–8, 2017. doi: 10.1155/2017/9502643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab 16: 144–152, 2012. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Kleemann R, van Erk M, Verschuren L, van den Hoek AM, Koek M, Wielinga PY, Jie A, Pellis L, Bobeldijk-Pastorova I, Kelder T, Toet K, Wopereis S, Cnubben N, Evelo C, van Ommen B, Kooistra T. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PloS one 5: e8817, 2010. doi: 10.1371/journal.pone.0008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghanim H, Green K, Abuaysheh S, Batra M, Kuhadiya ND, Patel R, Makdissi A, Dhindsa S, Chaudhuri A, Dandona P. Suppressive effect of insulin on the gene expression and plasma concentrations of mediators of asthmatic inflammation. J Diabetes Res 2015: 1–7, 2015. doi: 10.1155/2015/202406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardet JC, Ash S, Kusa T, Camargo CA , Jr, Israel E. Insulin resistance modifies the association between obesity and current asthma in adults. Eur Respir J 48: 403–410, 2016. doi: 10.1183/13993003.00246-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31: S262–S268, 2008. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- 22.Morishita R, Franco MD, Suano-Souza FI, Sole D, Puccini RF, Strufaldi MW. Body mass index, adipokines and insulin resistance in asthmatic children and adolescents. J Asthma 53: 478–484, 2016. doi: 10.3109/02770903.2015.1113544. [DOI] [PubMed] [Google Scholar]
- 23.Kim KM, Kim SS, Lee SH, Song WJ, Chang YS, Min KU, Cho SH. Association of insulin resistance with bronchial hyperreactivity. Asia Pac Allergy 4: 99–105, 2014. doi: 10.5415/apallergy.2014.4.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie Z, Jacoby DB, Fryer AD. Hyperinsulinemia potentiates airway responsiveness to parasympathetic nerve stimulation in obese rats. Am J Respir Cell Mol Biol 51: 251–261, 2014. doi: 10.1165/rcmb.2013-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caulfied MP, Birdsall N. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998; 50(2): 279–290, 1998. doi: 10.1111/j.2042-7158.1998.tb06861.x. [DOI] [PubMed] [Google Scholar]
- 26.Singh S, Prakash YS, Linneberg A, Agrawal A. Insulin and the lung: connecting asthma and metabolic syndrome. J Allergy (Cairo) 2013: 627384, 2013. doi: 10.1155/2013/627384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol 273: R725– R730, 1997. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- 28.Grant CW, Duclos SK, Moran-Paul CM, Yahalom B, Tirabassi RS, Arreaza-Rubin G, Spain LM, Guberski DL. Development of standardized insulin treatment protocols for spontaneous rodent models of type 1 diabetes. Comp Med 62: 381–390, 2012. [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufman EH, Fryer AD, Jacoby DB. Toll-like receptor 7 agonists are potent and rapid bronchodilators in guinea pigs. J Allergy Clin Immunol 127: 462–469, 2011. doi: 10.1016/j.jaci.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henagan TM, Stefanska B, Fang Z, Navard AM, Ye J, Lenard NR, Devarshi PP. Sodium butyrate epigenetically modulates high-fat diet-induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. Br J Pharmacol 172: 2782–2798, 2015. doi: 10.1111/bph.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Current protocols in molecular biology. 2006; Chapter 15:Unit 15.8. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 32.Orfanos S, Jude J, Deeney BT, Cao G, Rastogi D, van Zee M, Pushkarsky I, Munoz HE, Damoiseaux R, Di Carlo D, Panettieri R. Jr.. Obesity increases airway smooth muscle responses to contractile agonists. Am J Physiol Lung Cell Mol Physiol 315: L673–L681, 2018. doi: 10.1152/ajplung.00459.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaafsma D, Gosens R, Ris JM, Zaagsma J, Meurs H, Nelemans SA. Insulin induces airway smooth muscle contraction. Br J Pharmacol 150: 136–142, 2007. doi: 10.1038/sj.bjp.0706985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharif M, Khan BT, Ajmal K, Anwar MA. Acute effect of insulin on guinea pig airways and its amelioration by pre-treatment with salbutamol. J Pak Med Assoc 64: 932–935, 2014. [PubMed] [Google Scholar]
- 35.Coulson FR, Jacoby DB, Fryer AD. Insulin regulates neuronal M2 muscarinic receptor function in the ileum of diabetic rats. J Pharmacol Exp Ther 308: 760–766, 2004. doi: 10.1124/jpet.103.057570. [DOI] [PubMed] [Google Scholar]
- 36.Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer 19: F27–F45, 2012. doi: 10.1530/ERC-11-0374. [DOI] [PubMed] [Google Scholar]
- 37.Cohen P, Noveral JP, Bhala A, Nunn SE, Herrick DJ, Grunstein MM. Leukotriene D4 facilitates airway smooth muscle cell proliferation via modulation of the IGF axis. Am J Physiol Lung Cell Mol Physiol 269: L151–L157, 1995. doi: 10.1152/ajplung.1995.269.2.L151. [DOI] [PubMed] [Google Scholar]
- 38.Noveral JP, Bhala A, Hintz RL, Grunstein MM, Cohen P. Insulin-like growth factor axis in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 267: L761–L765, 1994. doi: 10.1152/ajplung.1994.267.6.L761. [DOI] [PubMed] [Google Scholar]
- 39.Choquet H, Meyre D. Genetics of obesity: what have we learned? Curr Genomics 12: 169–179, 2011. doi: 10.2174/138920211795677895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hjellvik V, Tverdal A, Furu K. Body mass index as predictor for asthma: a cohort study of 118,723 males and females. Eur Respir J 35: 1235–1242, 2010. doi: 10.1183/09031936.00192408. [DOI] [PubMed] [Google Scholar]
- 41.Lange P, Groth S, Kastrup J, Mortensen J, Appleyard M, Nyboe J, Jensen G, Schnohr P. Diabetes mellitus, plasma glucose and lung function in a cross-sectional population study. Eur Respir J 2: 14–19, 1989. [PubMed] [Google Scholar]
- 42.Eglen RM, Reddy H, Watson N, Challiss RA. Muscarinic acetylcholine receptor subtypes in smooth muscle. Trends Pharmacol Sci 15: 114–119, 1994. doi: 10.1016/0165-6147(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 43.White TA, Kannan MS, Walseth TF. Intracellular calcium signaling through the cADPR pathway is agonist specific in porcine airway smooth muscle. FASEB J 17: 482–484, 2003. doi: 10.1096/fj.02-0622fje. [DOI] [PubMed] [Google Scholar]
- 44.Fernandes LB, Fryer AD, Hirshman CA. M2 muscarinic receptors inhibit isoproterenol-induced relaxation of canine airway smooth muscle. J Pharmacol Exp Ther 262: 119–126, 1992. [PubMed] [Google Scholar]
- 45.Jude JA, Wylam ME, Walseth TF, Kannan MS. Calcium signaling in airway smooth muscle. Proc Am Thorac Soc 5: 15–22, 2008. doi: 10.1513/pats.200704-047VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol 7: a006023, 2015. doi: 10.1101/cshperspect.a006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tolloczko B, Jia YL, Martin JG. Serotonin-evoked calcium transients in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 269: L234–L240, 1995. doi: 10.1152/ajplung.1995.269.2.L234. [DOI] [PubMed] [Google Scholar]
- 48.Catalucci D, Zhang DH, DeSantiago J, Aimond F, Barbara G, Chemin J, Bonci D, Picht E, Rusconi F, Dalton ND, Peterson KL, Richard S, Bers DM, Brown JH, Condorelli G. Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability. J Cell Biol 184: 923–933, 2009. [Erratum in J Cell Biol 200: 851, 2013]. doi: 10.1083/jcb.200805063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Algenstaedt P, Antonetti DA, Yaffe MB, Kahn CR. Insulin receptor substrate proteins create a link between the tyrosine phosphorylation cascade and the Ca2+-ATPases in muscle and heart. J Biol Chem 272: 23696–23702, 1997. doi: 10.1074/jbc.272.38.23696. [DOI] [PubMed] [Google Scholar]
- 50.Borge PD , Jr, Wolf BA. Insulin receptor substrate 1 regulation of sarco-endoplasmic reticulum calcium ATPase 3 in insulin-secreting beta-cells. J Biol Chem 278: 11359–11368, 2003. doi: 10.1074/jbc.M209521200. [DOI] [PubMed] [Google Scholar]
- 51.Ceglia L, Lau J, Pittas AG. Meta-analysis: efficacy and safety of inhaled insulin therapy in adults with diabetes mellitus. Ann Intern Med 145: 665–675, 2006. doi: 10.7326/0003-4819-145-9-200611070-00009. [DOI] [PubMed] [Google Scholar]
- 52.Heinemann L. Inhaled insulin: dead horse or rising phoenix? J Diabetes Sci Technol 12: 239–242, 2018. doi: 10.1177/1932296817748231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiu KY, Li JG, Lin Y. Calcium channel blockers for lung function improvement in asthma: a systematic review and meta-analysis. Ann Allergy Asthma Immunol 119: 518–523.e3, 2017. doi: 10.1016/j.anai.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 54.Park HW, Park H, Semple IA, Jang I, Ro SH, Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS, Lee JH. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun 5: 4834, 2014. doi: 10.1038/ncomms5834. [DOI] [PMC free article] [PubMed] [Google Scholar]