

Abstract
Increasing evidence suggests an important role for deubiquitinating enzymes (DUBs) in modulating a variety of biological functions and diseases. We previously identified the upregulation of the DUB ubiquitin carboxyl terminal hydrolase 1 (UCHL1) in murine ventilator-induced lung injury (VILI). However, the role of UCHL1 in modulating vascular permeability, a cardinal feature of acute lung injury (ALI) in general, remains unclear. We investigated the role of UCHL1 in pulmonary endothelial cell (EC) barrier function in vitro and in vivo and examined the effects of UCHL1 on VE-cadherin and claudin-5 regulation, important adherens and tight junctional components, respectively. Measurements of transendothelial electrical resistance confirmed decreased barrier enhancement induced by hepatocyte growth factor (HGF) and increased thrombin-induced permeability in both UCHL1-silenced ECs and in ECs pretreated with LDN-57444 (LDN), a pharmacological UCHL1 inhibitor. In addition, UCHL1 knockdown (siRNA) was associated with decreased expression of VE-cadherin and claudin-5, whereas silencing of the transcription factor FoxO1 restored claudin-5 levels. Finally, UCHL1 inhibition in vivo via LDN was associated with increased VILI in a murine model. These findings support a prominent functional role of UCHL1 in regulating lung vascular permeability via alterations in adherens and tight junctions and implicate UCHL1 as an important mediator of ALI.
Keywords: adherens junction, claudin-5, endothelial cells, lung vascular permeability, tight junction, UCHL1, VE-cadherin
INTRODUCTION
Acute lung injury (ALI) is a debilitating condition encountered in critically ill patients and represents a significant cause of ICU-related morbidity and mortality (1). Aberrant vascular permeability and inflammation that impair alveolar gaseous exchange are critical elements of ALI. Mechanical ventilation, frequently utilized in critically ill patients, can precipitate and aggravate ALI due to excessive mechanical stress, a condition known as ventilation-induced lung injury (VILI) (2). Despite improved recognition of this syndrome, effective therapies remain lacking, and ALI is estimated to account for ∼75,000 deaths and millions of hospital days per year in the United States (3). Moreover, ALI survivors have been found to have impaired lung function, neuropsychological deficits, and decreased quality of life in the long term (4, 5). Thus, novel therapeutic targets and strategies aimed at improving pulmonary endothelial barrier function may prove beneficial in mitigating the effects of increased vascular leak associated with ALI and may ultimately lead to improved clinical outcomes.
Utilizing global gene expression profiling, we previously identified growth arrest and DNA damage-inducible 45-α (GADD45α) as a significantly upregulated gene in preclinical models of VILI (6). We subsequently reported increased VILI susceptibility of GADD45α−/− mice (7). Further in silico analysis identified reduced levels of ubiquitin carboxyl-terminal hydrolase 1 (UCHL1), a deubiquitinating enzyme (DUB), in GADD45α−/− animals. Decreased UCHL1 levels were found to be due to the hypermethylation of the UCHL1 promoter, which, in turn, is associated with increased Akt K48 ubiquitination and subsequent proteasomal degradation. In addition, treatment of GADD45α−/− mice with 5-aza-deoxycytidine (5-AZA-DC), a DNA methyl transferase inhibitor, both significantly increased UCHL1 levels and attenuated VILI susceptibility.
Functionally, UCHL1 removes or edits poly- or monoubiquitin chains from ubiquitinated proteins that may regulate a range of cellular processes, including cell cycle, DNA repair, cell-cell communication, and apoptosis (8, 9). Although UCHL1 has been implicated in a variety of diseases including neurodegenerative disorders, lung cancer, and colon cancer (10, 11), its role in ALI had not previously been reported. We investigated the role of UCHL1 in regulation of endothelial cell (EC) permeability. We studied the effects of UCHL1 silencing and inhibition on human pulmonary artery EC permeability by measuring transendothelial resistance (TER). We also explored the role of UCHL1 in regulating adherens and tight junctional proteins, VE-cadherin and claudin-5, respectively. Finally, we investigated the potential effects of LDN-57444 (LDN), a pharmacological UCHL1 inhibitor, in murine VILI.
MATERIALS AND METHODS
Antibodies and Reagents
Antibodies and reagents were purchased commercially consistent with our prior reports (8, 12). Antibodies against UCHL1 (Cell Signaling, Danvers, MA), VE-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA), phosphor-Y658 VE-cadherin (Biosource, Grand Island, NY), β-catenin (Santa Cruz Biotechnology), FoxO1 (Cell Signaling, Danvers, MA), phospho-FoxO 1 (Cell Signaling), β-actin (Sigma, St. Louis, MO), claudin-5 (Cell Signaling), NF-κB (Cell Signaling), phospho-NF-κB p65 (Cell Signaling), ICAM-1 (Santa Cruz Biotechnology), VCAM-1 (Cell Signaling), horseradish peroxidase-conjugated anti-mouse, and anti-rabbit secondary antibodies (Cell Signaling) were purchased from the indicated vendors. A smart pool of silencing RNA (siRNA) specific for UCHL1, FoxO1, and nonspecific siRNA was purchased from GE Dharmacon (Lafayette, CO). Other reagents used include the proteasome inhibitor MG132 (Calbiochem, San Diego, CA), LDN-57444 (LDN, Calbiochem), hepatocyte growth factor (HGF) (Peprotech, Rocky Hill, NJ), and thrombin (Sigma).
Endothelial Cell Culture
Human pulmonary artery endothelial cells (ECs) were cultured in essential growth medium (EGM-2) containing 10% fetal bovine serum (Clonetics, Walkersville, MD). Cells were placed in an incubator at 37°C, 5% CO2, and 95% humidity to achieve contact-inhibited monolayers. Select experiments utilized human lung microvascular endothelial cells (HLMVECs, Lonza, Hayward, CA).
Endothelial Cell siRNA Transfection and Treatment with a UCHL1 Inhibitor
ECs were transfected with siRNA (100 nM, GE Dharmacon) specific for UCHL1, FoxO1, or nonspecific scrambled sequence using transfection reagent siPORT Amine (Ambion, Austin, TX) in serum-free conditions according to the manufacturer’s protocol. The medium was changed to EGM-2 containing 2% fetal bovine serum after 24 h of transfection, and protein silencing was checked after 72 h of transfection. Silenced cells were plated on the electric cell-substrate impedance system (ECIS) polycarbonate wells and Transwell cell culture insert after 48 h of transfection, and assays were performed after 24 h. UCHL1 inhibitor LDN was dissolved in 60% DMSO, and ECs were treated (5 µM) for 1.5 h. Also, 60% DMSO was used as a vehicle control.
Transendothelial Electrical Resistance Measurement
ECs were plated into polycarbonate wells containing evaporated gold microelectrodes to measure TER that evaluates real-time changes in cell morphology, attachment, and locomotion using an electric cell-substrate impedance system (ECIS) (Applied Biophysics, Troy, NY), as previously described (13). Cells were grown to confluence in EBM-2 containing 2% serum. TER values from each microelectrode were pooled at discrete timepoints and plotted versus time as the means ± SE. EC monolayers were treated with hepatocyte growth factor (HGF, 25 ng/mL) or thrombin (1 U/mL), agonists that enhance or disrupt barrier integrity, respectively.
Immunoblotting and Immunoprecipitation
As we have previously described (7), total proteins were extracted using NP-40 lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, and 5 mM ethylenediaminetetraacetic acid) supplemented with 40 mM sodium fluoride, 0.1 M sodium orthovanadate, 0.2 mM phenylmethylsulfonyl fluoride, 10 mM N′-ethyl maleimide, and protease and phosphatase inhibitor cocktail (Calbiochem, San Diego, CA). Lung homogenates and cell lysates were briefly sonicated and were subjected to cycles of thawing and freezing on dry ice. The protein concentrations were measured using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Western blotting was performed using standard protocols, and band densities were determined using ImageJ (National Institutes of Health, http://imagej.nih.gov/ij/). For immunoprecipitation, proteins were extracted in RIPA buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS with protease, and phosphatase inhibitor cocktails), and immunoprecipitation was performed, after preclearing with protein A/G Sepharose beads, by incubating protein extracts with primary antibodies (2 µg) overnight at 4°C. Immunocomplexes were collected by incubating with protein A/G Sepharose beads for 1 h at 4°C and were resolved by polyacrylamide gel electrophoresis followed by Western blotting.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (pH 7.0) for 20 min, permeabilized with 0.2% Triton X-100, and blocked with 5% bovine serum albumin for 1 h, followed by overnight incubation at 4°C with primary antibodies, as we have previously described (7, 13). Cells were incubated with goat anti-rabbit Alexa Fluor 635 and goat anti-mouse Alexa Fluor 488 secondary antibodies for 1 h. Nuclei were stained with Hoechst 33342, and coverslips were mounted on slides using mounting solution ProLong Gold. Cells were then imaged using a Carl Zeiss LSM 510 laser scanning confocal microscope (Göttingen, Germany).
Preclinical VILI Model
Male, 8–12-wk-old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were anesthetized with an intraperitoneal mix of ketamine (150 mg/kg) and xylazine (15 mg/kg) before intubation with a 20-gauge angiocatheter that was then attached to a small-animal mechanical ventilator (Harvard Apparatus, Holliston, MA) with VT = 30 mL/kg, 65 breaths/min, and positive end-expiratory pressure of 0 cm H2O for 4 h, as previously described (5, 14). Mice were treated with LDN (5 mg/kg ip) or vehicle 24 h and 1 h before VILI challenge. Notably, this dosing of LDN is consistent with dosing used by other investigators in vivo (15, 16). Bronchoalveolar lavage (BAL) fluid was collected and assessed for total protein as well as total and differential cell counts, as previously described (7, 13). All experiments and animal care procedures were approved by the University of Illinois at Chicago Animal Care and Use Committee and were performed in accordance with the Guide for the Care and Use of Laboratory Animals, Eighth Edition published by the Institute for Laboratory Animal Research.
Murine Lung Histology
As we have previously reported (13), lung tissue samples were fixed in formalin, embedded in paraffin, cut into 10-μm sections, and stained with hematoxylin and eosin (H&E). Histology slides were scanned and evaluated with ImageScope (Aperio, Vista, CA). H&E-stained lung sections (n = 6–10 per condition) were visualized, and representative images are provided.
Statistics
Unless otherwise stated, Student’s t test was used to compare the means of data from two experimental groups, whereas significant differences (P < 0.05) among multiple group comparisons were confirmed by one-way ANOVA followed by Tukey’s range test, consistent with our prior reports (7, 13). Results are expressed as means ± SE.
RESULTS
UCHL1 Knockdown and EC Barrier Function
To determine the role of UCHL1 in regulating EC permeability, human pulmonary artery ECs were transfected with siRNA specific for UCHL1 or a control siRNA and then grown to confluence overlying gold-plated microelectrodes. Cells were then treated with either HGF (25 ng/mL) or thrombin (1 U/mL) to effect barrier enhancement or disruption, respectively, and normalized TER was measured (Fig. 1, A and B). A significant increase in TER was observed in control ECs after HGF. However, peak HGF response (at 45 min) was significantly attenuated in UCHL1-silenced cells compared with controls. Similarly, TER nadir and time to recovery after treatment with thrombin (1 U/mL), a barrier-disrupting agent, were significantly increased in UCHL1-silenced cells compared with unsilenced cells. Moreover, UCHL1 silencing was associated with a significant reduction in basal TER measurements compared with control cells. Notably, these experiments were repeated using human lung microvascular endothelial cells with the same experimental conditions and confirmed a similar effect of UCHL1 knockdown, both qualitatively and quantitatively, on microvascular EC barrier regulation (Fig. 1C).
Figure 1.

Ubiquitin carboxyl terminal hydrolase 1 (UCHL1) knockdown negatively impacts endothelial cell (EC) barrier regulation. A: human pulmonary artery ECs were transfected with siRNA specific for UCHL1 (siUCHL1, 100 nM, 3 days) or nonspecific siRNA and were then plated on polycarbonate wells containing evaporated gold microelectrodes followed by treatment with hepatocyte growth factor (HGF; 25 ng/mL), a barrier-enhancing agonist, or vehicle with transendothelial resistance (TER) measured over time. At peak effect, HGF-induced barrier enhancement was significantly attenuated in UCHL1-silenced cells compared with controls (bar graph, *P < 0.05, n = 3). Western blotting for UCHL1 confirmed silencing (representative blot shown). B: in similar experiments, cells were treated with thrombin (1 U/mL), a barrier-disruptive agonist, or vehicle and TER was measured. At the nadir of TER changes, thrombin-induced barrier disruption was significantly increased (i.e., more disruption) in UCHL-1-silenced cells (bar graph, *P < 0.05, n = 3). C: experiments were repeated using human lung microvascular ECs (HLMVECs), which confirmed similar effects on barrier regulation (representative tracings shown).
UCHL1 Inhibition with LDN and EC Barrier Function
Next, experiments were repeated using LDN-57444 (LDN), a pharmacological inhibitor of UCHL1 activity (Fig. 2). TER measurements of ECs treated with LDN (5 µM, 1.5 h) before either HGF (25 ng/mL) or thrombin (1 U/mL) demonstrated a significant attenuation of HGF-induced barrier enhancement and augmentation of thrombin-induced disruption. Baseline TER measurements were unchanged in LDN-treated cells compared with controls. Together, these results highlight the negative regulatory effects of UCHL1 silencing and inhibition on EC barrier function.
Figure 2.

Ubiquitin carboxyl terminal hydrolase 1 (UCHL1) inhibition negatively impacts endothelial cell barrier regulation. A: endothelial cells were plated on polycarbonate wells containing evaporated gold microelectrodes followed by treatment with the UCHL1 inhibitor LDN 57444 (LDN, 5 µM) 1.5 h prior to hepatocyte growth factor (HGF; 25 ng/mL) and transendothelial resistance (TER) was measured over time. At peak effect, HGF-induced barrier enhancement was significantly attenuated in LDN-treated cells compared with untreated controls (bar graph, *P < 0.05, n = 3). B: in similar experiments, LDN-treated and controls cells were administered thrombin (1 U/mL) or vehicle and TER was measured. At the nadir of TER changes, thrombin-induced barrier disruption was significantly increased in UCHL1-silenced cells (bar graph, *P < 0.05, n = 3).
Redistribution of VE-Cadherin and β-Catenin in UCHL1-Silenced ECs
ECs maintain cell-cell contact and monolayer integrity via both adherens junction (AJ) and tight junctions (TJ). We next studied the role of UCHL1 in the spatial localization of VE-cadherin and β-catenin, two major AJ proteins, using confocal microscopy. Immunofluorescence images of control ECs revealed prominent membrane co-localization of VE-cadherin and β-catenin (Fig. 3A). However, UCHL1 silencing was notable for reduced levels of VE-cadherin and β-catenin at the membrane with a relative increase in the cytoplasmic distribution of both. In addition, Western blotting confirmed significantly reduced total VE-cadherin levels in UCHL1-silenced ECs, although no significant change was observed in β-catenin levels (Fig. 3B).
Figure 3.

Redistribution of VE-cadherin and β-catenin in ubiquitin carboxyl terminal hydrolase 1 (UCHL1)-silenced endothelial cells (ECs). A: immunofluorescence imaging of ECs transfected with control siRNA (nsRNA) or siUCHL1 (100 nM, 3 days) demonstrated co-localization of VE-cadherin and β-catenin at the cell periphery in control cells (arrows). In contrast, UCHL1 silencing demonstrated reduced levels of VE-cadherin and β-catenin at the membrane with redistribution to the cytoplasm (representative images shown). B: notably, Western blotting of whole cell lysates confirmed significantly reduced total VE-cadherin levels in UCHL1-silenced ECs (representative blot shown, *P < 0.05, n = 3).
Role of UCHL1 in VE-Cadherin Phosphorylation and Ubiquitination
Junctional weakness and VE-cadherin internalization are associated with tyrosine phosphorylation of VE-cadherin (17). In response to UCHL1 silencing, we next studied VE-cadherin phosphorylation at tyrosine 658 (Y658) and investigated the mechanism underlying the associated decreased VE-cadherin expression levels. We observed a significant increase in Y658 phosphorylation of VE-cadherin in UCHL1-silenced cells with a concomitant decrease in total VE-cadherin. Subsequently, we treated UCHL1-silenced ECs with chloroquine (20 μM, 2 h) or MG132 (10 μM, 2 h), lysosome and proteasome inhibitors, respectively, as VE-cadherin is known to be degraded by both the endosomal/lysosomal pathway (18) and the ubiquitin-proteasomal pathway (19). While no changes in VE-cadherin levels were observed with chloroquine treatment, the decreased levels of VE-cadherin in UCHL1-silenced cells were significantly rescued by MG132 (Fig. 4, A and B). Furthermore, increased ubiquitination of VE-cadherin was evident upon the immunoprecipitation of VE-cadherin in UCHL1-silenced ECs (Fig. 4C). Collectively, these results implicate UCHL1 as important for the maintenance and regulation of AJ via effects on VE-cadherin, including its cellular localization, phosphorylation, and its ubiquitination and proteasomal degradation. Although we did not investigate the mechanisms underlying increased VE-cadherin phosphorylation despite decreased total VE-cadherin in these experiments, we speculate that increased proteasomal degradation of VE-cadherin promotes its internalization from the membrane, an event known to be associated with increased phosphorylation (20).
Figure 4.

Proteasomal degradation of VE-cadherin is increased in response to ubiquitin carboxyl terminal hydrolase 1 (UCHL1) silencing. A: endothelial cells (ECs) transfected with siRNA (100 nM, 3 days) or nonspecific RNA (control siRNA) were treated with chloroquine (20 µM, 2 h), a lysosomal inhibitor, prior to Western blotting. In response to UCHL1 silencing, levels of VE-cadherin Y658 phosphorylation were significantly increased, whereas total VE-cadherin levels were significantly decreased (*P < 0.05 compared with respective controls transfected with nonspecific siRNA). However, chloroquine did not have a significant effect on either total VE-cadherin or phospho-VE-cadherin levels. B: in similar experiments, ECs transfected with siRNA (100 nM, 3 days) or nonspecific RNA (control siRNA) were treated with MG132 (10 µM, 2 h), a proteasomal inhibitor, prior to Western blotting. In response to UCHL1 silencing, levels of VE-cadherin Y658 phosphorylation were significantly increased (*P < 0.05 compared with respective controls transfected with nonspecific RNA). However, while silencing UCHL1 was associated with significantly decreased VE-cadherin phosphorylation at Y658, these effects were abrogated by pretreatment with MG132 (**P < 0.05) (n = 3/experimental condition). C: immunoprecipitation of VE-cadherin after UCHL1 silencing (100 nM, 3 days) followed by Western blotting for ubiquitin demonstrated an increased association of ubiquitin with VE-cadherin (representative blots shown).
Tight Junctional Protein Expression and Regulation in UCHL1-Silenced ECs
Claudins are the major transmembrane proteins of TJ that regulate paracellular permeability, cell polarity, and signal transduction (21). VE-cadherin spatial distribution is known to mediate claudin-5 expression regulated by the transcription factor FoxO1 (13, 20). Accordingly, we next assessed the role of UCHL1 in the regulation of claudin-5 expression. Western blotting revealed significantly reduced claudin-5 protein levels in UCHL1-silenced ECs compared with unsilenced controls (Fig. 5A). Furthermore, fluorescent images confirmed a significant reduction of claudin-5 expression at the cell periphery in UCHL1-silenced cells compared with unsilenced cells (Fig. 5B). Thus, VE-cadherin cytoplasmic distribution in UCHL1-silenced ECs correlates with reduced claudin-5 expression, consistent with our prior report (13).
Figure 5.
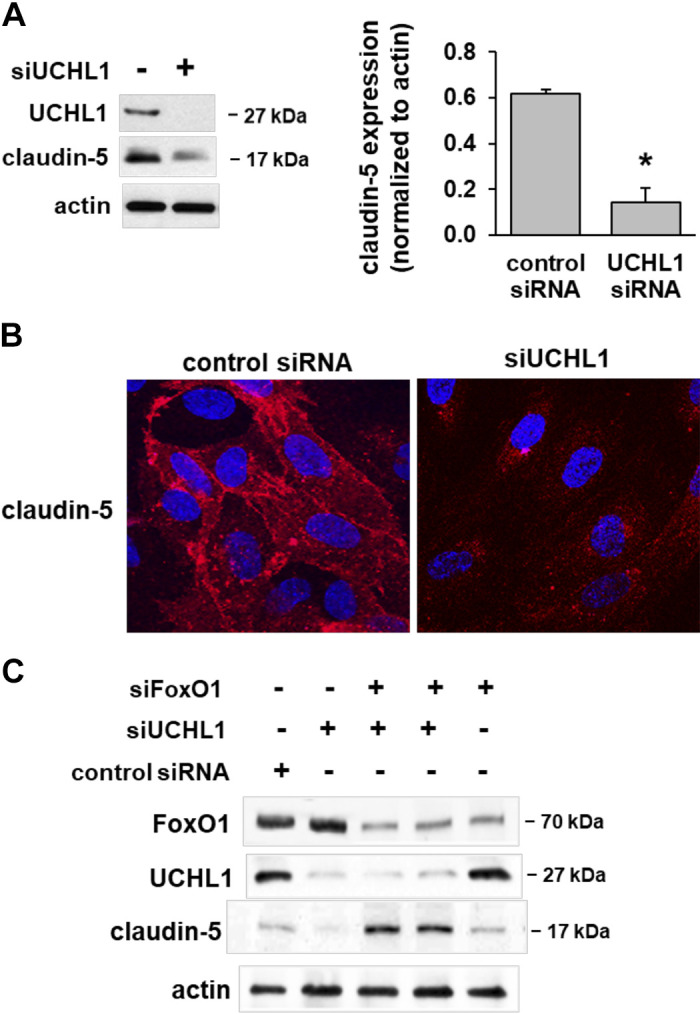
Claudin-5 expression is decreased in ubiquitin carboxyl terminal hydrolase 1 (UCHL1)-silenced ECs. A: Western blotting of UCHL1-silenced ECs (100 nM, 3 days) demonstrated decreased claudin-5 expression levels compared with controls transfected with control siRNA (*P > 0.05, n = 3). B: immunofluorescence of UCHL1-silenced ECs revealed decreased claudin-5 specifically localized at the cell membrane compared with controls. C: silencing of FoxO1 (siFoxO1, 100 nM, 3 days) alone had no effect on claudin-5 expression, whereas the combined effects of UCHL1 and FoxO1 silencing resulted in increased claudin-5 expression compared with controls (representative blots shown).
VE-cadherin internalization and degradation are known to promote accumulation of β-catenin and unphosphorylated FoxO1 in the nucleus, which transcriptionally inhibit claudin-5 expression (20). To study the regulation of claudin-5 by FoxO1 in UCHL1-silenced ECs, we silenced FoxO1 and UCHL1, separately and in combination, and assessed claudin-5 expression levels by Western blotting. FoxO1 silencing by itself, with only partial knockdown achieved, had little effect on claudin-5 expression, but the combination of UCHL1 and FoxO1 silencing resulted in significantly increased claudin-5 expression (Fig. 5C). These results may be consistent with a net effect of increased transcription due to decreases in FoxO1 (siRNA) and decreased nuclear translocation of β-catenin, which is known to be promoted directly by UCHL1 (22).
Effect of UCHL1 Silencing on NF-κB, ICAM-1, and VCAM-1
Prior studies have reported activation of the NF-κB pathway following UCHL1 inhibition in various cell types, including ECs (23, 24). We performed additional experiments to investigate the effects of UCHL1 silencing, alone and after thrombin stimulation, on NF-κB activation (NF-κB p65) as well as expression levels of ICAM-1 and VCAM-1, EC adhesion molecules known to mediate inflammation (Fig. 6). Although thrombin did affect increased NF-κB p65 phosphorylation, there was no effect on NF-κB associated with knockdown of UCHL1. There were also no effects on ICAM-1 or VCAM-1 expression levels after UCHL1 knockdown, although we did not investigate changes in expression levels after more than 15 min of thrombin that could be attributed to NF-κB activation.
Figure 6.

Ubiquitin carboxyl terminal hydrolase 1 (UCHL1) silencing does not affect NF-κB activation, ICAM-1, or VCAM-1 expression levels. ECs were transfected with siRNA specific for UCHL1 (siRNA, 100 nm, 3 days) or scramble siRNA (scr siRNA) prior to treatment with thrombin (1 U/mL, 15 min). Whole cell lysates were then used for Western blotting for NF-κB, phospho-NF-κB p65, ICAM-1, or VCAM-1 (representative blots shown).
Role of UCHL1 in the Elaboration of Murine VILI
To study the role of UCHL1 in the elaboration of vascular permeability and inflammation associated with VILI in vivo, wild-type C57Bl/6 mice were treated with LDN-57444 (5 mg/kg ip) or vehicle 24 h and again 1 h before VILI challenge (VT, 30 mL/kg, 4 h). A small but significant increase in BAL fluid protein levels was observed in control (spontaneously breathing) mice treated with LDN compared with untreated controls, whereas there was no difference in BAL total cell counts or neutrophils. Most notably, among VILI-challenged mice, LDN-treated mice had significantly increased BAL fluid total protein levels, total cell counts, and polymorphonuclear neutrophils (PMNs), compared with vehicle-treated controls (Fig. 7). In addition, evaluation of lung histology of VILI-challenged mice also confirmed significantly increased number of alveolar and interstitial inflammatory cells in LDN-treated mice compared with vehicle-treated controls (Fig. 8). These data support the idea that inhibition of UCHL1 in vivo increases VILI susceptibility, as characterized by augmented lung vascular permeability and inflammation, an observation consistent with our in vitro findings.
Figure 7.

Ubiquitin carboxyl terminal hydrolase 1 (UCHL1) inhibition is associated with increased VILI: BAL fluid analysis. C57Bl/6 mice were pretreated with the UCHL1 inhibitor LDN-57444 (LDN, 5 mg/kg ip) or vehicle 24 h and again 1 h before VILI challenge (VT, 30 mL/kg, 4 h). Bronchoalveolar lavage (BAL) was then collected for analysis of total protein (A) and total cell counts (B) as well as differential cell counts (C). Compared with VILI-challenged control animals, LDN-treated mice had significantly increased total protein and cell counts with significant increase specifically in PMNs (*P < 0.05, n = 3 animals/condition).
Figure 8.

Ubiquitin carboxyl terminal hydrolase 1 (UCHL1) inhibition is associated with increased VILI: lung histology. C57Bl/6 mice were pretreated with the UCHL1 inhibitor LDN-57444 (LDN, 5 mg/kg ip) or vehicle 24 h and again 1 h before VILI challenge (VT, 30 mL/kg, 4 h). Histology of lungs collected from select animals demonstrated interstitial edema and infiltration of inflammatory cells associated with VILI (bottom left) that was augmented by LDN treatment (bottom right; arrows indicate areas of edema and inflammatory cell infiltration; representative images shown).
DISCUSSION
Research aimed at characterizing the molecular mechanisms that regulate pulmonary endothelial barrier function offers the promise of identifying novel therapeutic targets to treat the dire clinical effects of increased vascular leak associated with ALI. We previously identified UCHL1 as significantly upregulated in response to excessive mechanical stress in a preclinical VILI model (7). Moreover, increased VILI susceptibility was associated with reduced UCHL1 expression, as we reported in GADD45α−/− mice. These animals express decreased UCHL1 due to promoter hypermethylation, whereas GADD45α−/− mice treated with a DNA methylase inhibitor were found to have increased UCHL1 levels and reduced VILI susceptibility (7). Our results now strongly support an important role for UCHL1 in EC barrier regulation and suggest that UCHL1 may represent a novel therapeutic target in ALI.
UCHL1 is a deubiquitinating enzyme that negatively regulates protein ubiquitination and thus has effects on ubiquitin-dependent signaling, protein stability, and ubiquitin homeostasis (8). Expression of UCHL1 is most prevalent in the brain, comprising as much as 5%–10% of neuronal proteins (25), and it has been implicated as a pathogenic molecule in neurodegenerative disorders including both Alzheimer’s and Parkinson’s diseases as well as Lewy body disease (26–28). While low levels of UCHL1 expression are found in other tissues, increased expression has been reported in a variety of cancers, including pancreatic, colorectal, and breast cancer (14, 22, 29). Conversely, decreased UCHL1 expression due to hypermethylation of the promoter has been linked to ovarian, nasopharyngeal, and gastric cancers, among others (25, 30, 31). Interestingly, increased UCHL1 expression has also been linked to improved outcomes in patients with neuroblastoma (32). Although there is evidence that vascular remodeling may be attenuated by UCHL1 through inhibition of NF-ĸB (24), the role of UCHL1 in vascular biology in general has been poorly characterized.
We found that UCHL1 is protective in models of increased lung vascular permeability, as both knockdown of UCHL1 (siRNA) and its pharmacologic inhibition (LDN) were associated with increased vascular leak both in vitro and in vivo. Our findings were also associated with increased ubiquitination and decreased expression levels of VE-cadherin, an adherens junction protein that regulates vascular permeability via Y658 phosphorylation (33). Furthermore, UCHL1 knockdown was associated with decreased expression of claudin-5, an effect reversed by silencing of the transcriptional repressor FoxO1 (Fig. 9). These data are consistent with the known regulation of claudin-5 expression by VE-cadherin via the inhibition of the nuclear accumulation of FoxO1 (20).
Figure 9.

Proposed schema of EC barrier regulation by ubiquitin carboxyl terminal hydrolase 1 (UCHL1). UCHL1 promotes VE-cadherin expression levels via inhibition of its deubiquitination and subsequent proteasomal degradation. In turn, VE-cadherin promotes claudin-5 expression by preventing the nuclear accumulation of β-catenin and FoxO1, transcriptional repressors of claudin-5. The combined effects of increased VE-cadherin and claudin-5 at the membrane augment EC barrier function. Conversely, decreased UCHL1 expression is associated with increased proteasomal degradation of VE-cadherin, which is preceded by its phosphorylation and internalization events associated with adherens junction disassembly and decreased EC barrier function.
Of note, although basal EC barrier function was reduced in UCHL1-silenced human pulmonary artery ECs over time, we observed no effect by LDN in this regard, nor was there an effect on basal barrier function in UCHL1-silenced HLMVECs. These observations, however, are consistent with our prior finding of precise effects of claudin-5 knockdown on EC barrier function in that basal permeability was not significantly affected after silencing of claudin-5, whereas a size-selective effect was observed with respect to monolayer permeability, as measured by Transwell flux of a small-molecular-weight marker (sodium fluorescein, 376 Da) but not a larger molecule (FITC-dextran, 2,000 kDa) (13).
One limitation of our study is the use of siRNA and pharmacologic inhibitors, both of which have potential nonspecific or off-target effects. However, the comparable results we observed using these separate strategies support the likelihood that these results are in fact due to decreased UCHL1 activity. Another potential limitation is the fact that it could be argued that our in vivo results do not confirm effects of LDN on lung vascular permeability directly. However, the observation of increased levels of both protein and inflammatory cells in the BAL from VILI-challenged animals that received LDN is undoubtedly strong evidence of increased lung vascular permeability due to LDN treatment since these effects simply could not occur otherwise.
Separately, although our data support the idea that UCHL1 regulates VE-cadherin and claudin-5, another limitation of the current study is that these effects were also not confirmed in our in vivo experiments. However, we previously found significant increases in expression levels of both VE-cadherin and claudin-5 in lung homogenates from GADD45α−/− mice associated with overexpression of UCHL1. Moreover, the importance of VE-cadherin, claudin-5, and FoxO1 as mediators of lung vascular permeability is supported by our prior work related to simvastatin, an HMG-CoA reductase inhibitor that continues to have tantalizing yet unproven potential as a treatment for ALI (13, 32). Notably, mice with a targeted deletion of FoxO1 in the endothelium were found to have decreased lung vascular leak and mortality in a preclinical sepsis model (Regmi et al., preprint article). In addition, numerous investigators have reported promising findings related to the therapeutic potential of a variety of agonists in lung injury models that also affect these molecules and their associated signaling. One recent example is nicorandil, a nitrate used to treat angina and ATP-sensitive K+ channel agonist, that was reported to both be protective in a preclinical ALI model and increase VE-cadherin expression in lung EC (12). Nonetheless, the realization of a safe and effective pharmacological treatment for ALI remains an unmet goal.
We have for the first time, to our knowledge, identified the effects on the elaboration of lung injury associated with the targeted inhibition of UCHL1, a DUB that has previously garnered much interest in the cancer literature but has otherwise not previously been studied in this context. Although these findings are novel, we recognize that the immediate translational applications may be limited. Nonetheless, it is clear that research focused on strategies aimed at effecting increased UCHL1, either its expression or activity, are now needed, and investigations in this area may ultimately yield effective therapeutic targets for patients with or at risk for ALI.
GRANTS
This research was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01 HL147942 (to J.R.J., Y.E., and W.C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M., J.G.N.G., and J.R.J. conceived and designed research; S.M., Y.E., S.S., H.Q., W.C., and M.B. performed experiments; S.M., Y.E., S.S., and M.B. analyzed data; S.M., Y.E., A.A.D., and J.G.N.G. interpreted results of experiments; S.M., Y.E., and W.C. prepared figures; S.M. drafted manuscript; Y.E. and J.R.J. edited and revised manuscript; S.M., A.A.D., J.G.G., and J.R.J. approved final version of manuscript.
REFERENCES
- 1.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS. Acute respiratory distress syndrome. Nat Rev Dis Primers 5: 18, 2019. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajic O, Dara SI, Mendez JL, Adesanya AO, Festic E, Caples SM, Rana R, St Sauver JL, Lymp JF, Afessa B, Hubmayr RD. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit Care Med 32: 1817–1824, 2004. doi: 10.1097/01.CCM.0000133019.52531.30. [DOI] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 4.Dowdy DW, Eid MP, Dennison CR, Mendez-Tellez PA, Herridge MS, Guallar E, Pronovost PJ, Needham DM. Quality of life after acute respiratory distress syndrome: a meta-analysis. Intensive Care Med 32: 1115–1124, 2006. doi: 10.1007/s00134-006-0217-3. [DOI] [PubMed] [Google Scholar]
- 5.Mikkelsen ME, Christie JD, Lanken PN, Biester RC, Thompson BT, Bellamy SL, Localio AR, Demissie E, Hopkins RO, Angus DC. The adult respiratory distress syndrome cognitive outcomes study: long-term neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med 185: 1307–1315, 2012. doi: 10.1164/rccm.201111-2025OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigoryev DN, Ma SF, Irizarry RA, Ye SQ, Quackenbush J, Garcia JG. Orthologous gene-expression profiling in multi-species models: search for candidate genes. Genome Biol 5: R34, 2004. doi: 10.1186/gb-2004-5-5-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitra S, Sammani S, Wang T, Boone DL, Meyer NJ, Dudek SM, Moreno-Vinasco L, Garcia JG, Jacobson JR. Role of growth arrest and DNA damage-inducible alpha in Akt phosphorylation and ubiquitination after mechanical stress-induced vascular injury. Am J Respir Crit Care Med 184: 1030–1040, 2011. doi: 10.1164/rccm.201103-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clague MJ, Coulson JM, Urbe S. Cellular functions of the DUBs. J Cell Sci 125: 277–286, 2012. doi: 10.1242/jcs.090985. [DOI] [PubMed] [Google Scholar]
- 9.Pfoh R, Lacdao IK, Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr Relat Cancer 22: T35–T54, 2015. doi: 10.1530/ERC-14-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jara JH, Frank DD, Ozdinler PH. Could dysregulation of UPS be a common underlying mechanism for cancer and neurodegeneration? Lessons from UCHL1. Cell Biochem Biophys 67: 45–53, 2013. doi: 10.1007/s12013-013-9631-7. [DOI] [PubMed] [Google Scholar]
- 11.Tokumaru Y, Yamashita K, Kim MS, Park HL, Osada M, Mori M, Sidransky D. The role of PGP9.5 as a tumor suppressor gene in human cancer. Int J Cancer 123: 753–759, 2008. doi: 10.1002/ijc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He M, Shi W, Yu M, Li X, Xu J, Zhu J, Jin L, Xie W, Kong H. Nicorandil attenuates LPS-induced acute lung injury by pulmonary endothelial cell protection via NF-κB and MAPK pathways. Oxid Med Cell Longev 2019: 4957646, 2019. doi: 10.1155/2019/4957646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Sharma R, Rizzo AN, Siegler JH, Garcia JG, Jacobson JR. Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am J Respir Cell Mol Biol 50: 328–336, 2014. doi: 10.1165/rcmb.2013-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyoshi Y, Nakayama S, Torikoshi Y, Tanaka S, Ishihara H, Taguchi T, Tamaki Y, Noguchi S. High expression of ubiquitin carboxy-terminal hydrolase-L1 and -L3 mRNA predicts early recurrence in patients with invasive breast cancer. Cancer Sci 97: 523–529, 2006. doi: 10.1111/j.1349-7006.2006.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cartier AE, Ubhi K, Spencer B, Vazquez-Roque RA, Kosberg KA, Fourgeaud L, Kanayson P, Patrick C, Rockenstein E, Patrick GN, Masliah E. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One 7: e34713, 2012. doi: 10.1371/journal.pone.0034713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynolds JP, Jimenez-Mateos EM, Cao L, Bian F, Alves M, Miller-Delaney SF, Zhou A, Henshall DC. Proteomic analysis after status epilepticus identifies UCHL1 as protective against hippocampal injury. Neurochem Res 42: 2033–2054, 2017. doi: 10.1007/s11064-017-2260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 179: 4053–4064, 2007. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 18.Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, Kowalczyk AP. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J Biol Chem 278: 19199–19208, 2003. doi: 10.1074/jbc.M211746200. [DOI] [PubMed] [Google Scholar]
- 19.Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M, Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3: 1208, 2012. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10: 923–934, 2008. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 21.Bazzoni G. Endothelial tight junctions: permeable barriers of the vessel wall. Thromb Haemost 95: 36–42, 2006. doi: 10.1160/TH05-07-0488. [DOI] [PubMed] [Google Scholar]
- 22.Zhong J, Zhao M, Ma Y, Luo Q, Liu J, Wang J, Yuan X, Sang J, Huang C. UCHL1 acts as a colorectal cancer oncogene via activation of the beta-catenin/TCF pathway through its deubiquitinating activity. Int J Mol Med 30: 430–436, 2012. doi: 10.3892/ijmm.2012.1012. [DOI] [PubMed] [Google Scholar]
- 23.Gu Y, Ding X, Huang J, Xue M, Zhang J, Wang Q, Yu H, Wang Y, Zhao F, Wang H, Jin M, Wu Y, Zhang Y. The deubiquitinating enzyme UCHL1 negatively regulates the immunosuppressive capacity and survival of multipotent mesenchymal stromal cells. Cell Death Dis 9: 459, 2018. doi: 10.1038/s41419-018-0532-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takami Y, Nakagami H, Morishita R, Katsuya T, Cui TX, Ichikawa T, Saito Y, Hayashi H, Kikuchi Y, Nishikawa T, Baba Y, Yasuda O, Rakugi H, Ogihara T, Kaneda Y. Ubiquitin carboxyl-terminal hydrolase L1, a novel deubiquitinating enzyme in the vasculature, attenuates NF-κB activation. Arterioscler Thromb Vasc Biol 27: 2184–2190, 2007. doi: 10.1161/ATVBAHA.107.142505. [DOI] [PubMed] [Google Scholar]
- 25.Day IN, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurobiol 90: 327–362, 2010. doi: 10.1016/j.pneurobio.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 279: 13256–13264, 2004. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 27.Ragland M, Hutter C, Zabetian C, Edwards K. Association between the ubiquitin carboxyl-terminal esterase L1 gene (UCHL1) S18Y variant and Parkinson’s disease: a HuGE review and meta-analysis. Am J Epidemiol 170: 1344–1357, 2009. doi: 10.1093/aje/kwp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Q, Liao L, Cheng D, Duong DM, Gearing M, Lah JJ, Levey AI, Peng J. Proteomic identification of novel proteins associated with Lewy bodies. Front Biosci 13: 3850–3856, 2008. doi: 10.2741/2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finnerty BM, Moore MD, Verma A, Aronova A, Huang S, Edwards DP, Chen Z, Seandel M, Scognamiglio T, Du YN, Elemento O, Zarnegar R, Min IM, Fahey TJ. UCHL1 loss alters the cell-cycle in metastatic pancreatic neuroendocrine tumors. Endocr Relat Cancer 26: 411–423, 2019. doi: 10.1530/ERC-18-0507. [DOI] [PubMed] [Google Scholar]
- 30.Jin C, Yu W, Lou X, Zhou F, Han X, Zhao N, Lin B. UCHL1 is a putative tumor suppressor in ovarian cancer cells and contributes to cisplatin resistance. J Cancer 4: 662–670, 2013. doi: 10.7150/jca.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita K, Park HL, Kim MS, Osada M, Tokumaru Y, Inoue H, Mori M, Sidransky D. PGP9.5 methylation in diffuse-type gastric cancer. Cancer Res 66: 3921–3927, 2006. doi: 10.1158/0008-5472.CAN-05-1511. [DOI] [PubMed] [Google Scholar]
- 32.Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M; Irish Critical Care Trials G, , et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 6: 691–698, 2018. doi: 10.1016/S2213-2600(18)30177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, Nottebaum AF, Vestweber D. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol 15: 223–230, 2014. doi: 10.1038/ni.2824. [DOI] [PubMed] [Google Scholar]