

Abstract
Great strides have been made in cancer immunotherapy including the breakthrough successes of anti-PD-(L)1 checkpoint inhibitors. In Merkel cell carcinoma (MCC), a rare and aggressive skin cancer, PD-(L)1 blockade is highly effective. Yet, ~50% of patients either do not respond to therapy or develop PD-(L)1-refractory disease and, thus, do not experience long-term benefit. For these patients, additional or combination therapies are needed to augment immune responses that target and eliminate cancer cells.
Therapeutic vaccines targeting tumor-associated antigens (TAA), mutated self-antigens, or immunogenic viral oncoproteins are currently being developed to augment T cell responses. Approximately 80% of MCC cases in the United States are driven by the ongoing expression of viral T-antigen (T-Ag) oncoproteins from genomically integrated Merkel cell polyomavirus (MCPyV). Since T-Ag elicits specific B- and T-cell immune responses in most persons with virus-positive MCC (VP-MCC), and ongoing T-Ag expression is required to drive VP-MCC cell proliferation, therapeutic vaccination with T-Ag is a rational potential component of immunotherapy. Failure of the endogenous T-cell response to clear VP-MCC (allowing clinically evident tumors to arise) implies that therapeutic vaccination will need to be potent and synergize with other mechanisms to enhance T-cell activity against tumor cells. Here, we review the relevant underlying biology of VP-MCC, potentially applicable therapeutic vaccine platforms, and antigen delivery formats. We also describe early successes in the field of therapeutic cancer vaccines and address several clinical scenarios in which VP-MCC patients could potentially benefit from a therapeutic vaccine.
Keywords: Merkel cell carcinoma, MCPyV, cancer therapeutic vaccine, immunotherapy
Introduction
Merkel cell carcinoma (MCC) is a rare neuroendocrine malignancy that typically occurs in the skin (recently reviewed by Harms et al.1). The MCC recurrence rate ranges from 25–75% depending on the stage2, leading to a 33–46% disease-specific mortality3, 4. The current incidence of MCC in the United States is ~2500 cases/year. However, a recent report documented a 95% increase in MCC incidence between the years 2000–2013, with ~3200 cases/year expected by 20255. MCC has higher incidence rates among immunosuppressed populations and older persons with particularly elevated risk among Caucasian men1. The majority of MCC cases (~80%) are associated with clonal integration of the Merkel cell polyomavirus (MCPyV) into the host genome6. The remaining ~20% of cases are caused by UV mutations alone and are associated with prolonged UV-exposure1, 7, 8.
Primary MCPyV infection occurs during childhood. Infection is thought to be chronic, with viral DNA detected on the skin in >50% of healthy individuals9–11,12. The cell types infected13 and possible health effects of commensal MCPyV infection require further study.
The most successful cancer vaccines to date are preventive vaccines for HPV and HBV. Because such a small minority of MCPyV-infected persons develop VP-MCC, a preventive vaccine for MCPyV is likely not cost-effective. In contrast, a therapeutic vaccine targeting the relevant MCPyV proteins may prevent disease recurrence and be helpful in specific populations with both limited and relapsed VP-MCC as detailed below.
The biology of Polyomavirus-driven Merkel cell carcinoma
Seroprevalence and viral detection studies indicate that MCPyV is widespread in healthy individuals (Figure 1). At baseline, healthy people have antibodies to certain MCPyV proteins, as indicated by the high prevalence of antibodies to viral capsid proteins14, 15. In contrast, healthy individuals rarely have detectable antibody responses to MCPyV T-Ag (~1% of the population have borderline positive titers). This low immunogenicity of T-Ag may in part be due to limited expression of the T-Ag in the course of routine infection, and also the nuclear localization signal (NLS) within the C terminal domain of the large T-Ag (LT) oncoprotein16. Nuclear targeting reduces antigen processing and presentation by HLA-class I, potentially accounting for low CD8 T cell responses to T-Ag in healthy persons. In contrast to healthy individuals, patients with VP-MCC tumors often have T cells specific for MCPyV oncoproteins17 and also robust anti-T-Ag humoral immune responses15, 18. Importantly, the magnitude of antibodies to MCPyV T antigen oncoproteins (but not capsid proteins) correlates directly with tumor burden18. This association is the basis for a clinical test for tumor recurrence15, 18. Such antibody responses suggest T-Ag oncoproteins are likely coordinated between helper CD4+ T cells and B cells, either in draining lymph nodes or potentially in organized lymphoid structures within or near tumors19–21.
Figure 1: Etiology of virus-positive MCC and potential clinical scenarios for a therapeutic vaccine.
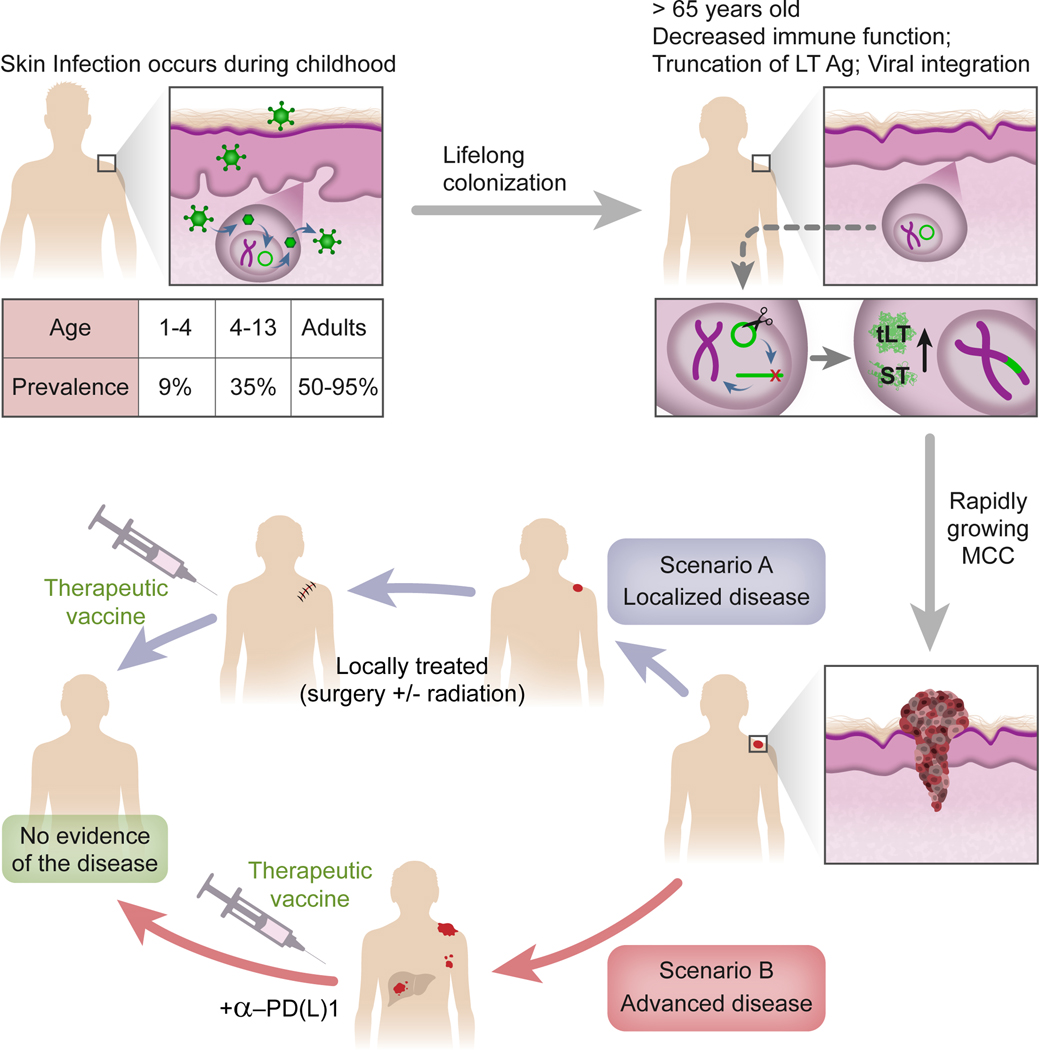
Seroprevalence of polyomavirus-specific capsid antibodies indicates that MCPyV is common in the general population starting in childhood9, 125. MCPyV then colonizes skin throughout life without causing known disease, except in rare cases when it integrates into the human genome and leads to MCC. This occurs mainly in persons > 65 and is likely driven by immune senescence and rare mutational events. Following viral integration, two oncoproteins, truncated Large T (tLT) and Small T (ST) antigens, are expressed and promote carcinogenesis. A therapeutic vaccine could potentially be tested in localized or advanced disease.
Some studies demonstrate the presence of B cells in MCC tumors22, 23 and tertiary lymphoid structures (TLS), typically enriched with B cells, were identified in the periphery of MCC tumors23, 24. The TLS in these MCC tumors contained CD4, CD8 and CD20-expressing cells (T and B cells), and correlated with recurrence-free survival23. The presence of TLS in VP-MCC may underlie the observed direct correlation of T-Ag antibodies with tumor burden, if in fact T-Ag-specific B cells are activated within or reside in or near tumors. However, the roles of B cells and TLS remain understudied and further investigation is required in order to understand their function in MCC.
In order for MCPyV to cause VP-MCC, two rare events are required: 1) truncation of the Large T (LT) antigen proximal to the C-terminal domains involved in viral DNA replication and 2) linearization and clonal integration of MCPyV into the host genome. After genome integration, VP-MCC tumors express a truncated form of LT protein, as well as full-length small T (ST) protein. Both T-Ag isoforms are involved in MCC tumor persistence and proliferation25, 26, and drive tumorgenicity by distinct mechanisms. Truncation of the LT antigen deletes the helicase domain required for viral replication, preventing destruction of infected host cells through inappropriate DNA replication initiated within the viral sequence itself. Additionally, loss of the NLS27 results in redistribution of LT to the cytoplasm28, where it is accessible to the antigen processing machinery. Although MCPyV LT truncation mutations vary from patient to patient, the highly conserved N-terminal LXCXE motif that binds and inhibits the retinoblastoma tumor suppressor (Rb) is invariably preserved29, 30. Of interest, this motif may also bind the Stimulator of Interferon genes (STING) protein, and is preserved in other pathogenic viruses such as cancer-related human papillomavirus strains 31. The ST antigen promotes cell survival and proliferation by multiple, incompletely understood mechanisms. For example, the LT-Stabilization Domain (LSD) contributes by stabilization of truncated LT and induction of oncoproteins such as c-Myc and Cyclin E32. A different mechanism of action to stabilization of the truncated LT by ST antigen was demonstrated in a recent study33. Additionally, ST can inhibit p53 activity in MCC cells via the canonical regulator of p53, MDM434.
It is clear that VP-MCC survival is dependent on the continued expression of MCPyV T antigens. Indeed, many MCC patients have T cell responses against ST and LT35–37, but tumors also downregulate class-I HLA38, suggesting not only dependence on oncoprotein T-antigens for ongoing replication and survival but also active escape from immune pressure. This reliance on T-Ag expression and presence of compensatory mechanisms suggests that MCPyV T-antigens provide an ideal target for a therapeutic vaccine if tumor immune evasion mechanisms can be overcome.
MCPyV oncoproteins as promising targets for immunotherapy
One of the major limitations in tumor immunology is identification of appropriate tumor-specific immunological targets. Tumor neoepitopes are typically discovered through whole-exome sequencing (WES), mRNA quantitation and in silico prediction using patient HLA types. This complex, customized, and expensive process has a challenging candidate-to-hit ratio.
Virus-induced cancers express “non-self” viral antigens that are foreign to the host, potentially increasing their inherent immunogenicity compared to overexpressed non-mutated tumor associated antigens, such as NYESO-1. With a combined T-antigen oncoprotein size of approximately 400 amino acids and very little variation between MCPyV strains, several groups have used standard immunologic approaches to detect T cell responses to MCPyV T-Ag35–37, 39. Indeed, CD8 T cells appear to play a significant role in controlling MCC as patients who have brisk tumoral CD8 T cell infiltration and a cytotoxic T cell profile experience markedly improved outcomes40, 41. Moreover, patients with greater intratumoral T cell receptor diversity among their MCPyV-specific T cells also have significantly improved MCC-specific survival37.
The abundance of circulating MCPyV-specific T cells as measured by peptide-HLA tetramers generally tracks with tumor burden, often being elevated at diagnosis when a larger tumor burden is present and decreasing following successful reduction of the tumor by surgery or other modalities36. This fluctuation of MCPyV-specific T cells may very well be a reflection of the amount of tumor-viral antigen available and is consistent with poor transition to long-lived memory cells. As noted above, the expansion of anti-MCPyV T cell responses in MCC patients is supported by detection of MCPyV-specific CD8 T cells in patients but not in healthy individuals17, 36.
As observed in many cancers and infections, MCPyV-specific T cells are generally enriched at the site of disease, albeit blood is readily obtainable and thus the focus of many studies. Using an HLA-A*24:02-restricted epitope (LT92–101) a tetramer was developed which enabled recognition of MCPyV-specific T cells in the PBMC of 7 of 11 (64%) HLA-A*24:02-positive patients35, 36. Another study of 27 individuals used a tetramer-enrichment strategy and identified 9 potential T-Ag T cell epitopes restricted by several population-prevalent HLA alleles (HLA-A*01, HLA-A*02, HLA-A*03, HLA-A*11 or HLA-B*07), exclusively in the PBMC of MCC patients and not in healthy individuals17.
While studying circulating T cells is important for understanding the immunogenicity and T cell specificity, blood-based lymphocytes do not provide an accurate picture of their roles within the “battlefield” of the tumor microenvironment. Accordingly, studies have focused on detecting specific tumor-infiltrating lymphocyte (TIL) responses by both measuring cytokine production from CD8 T cells upon recognition of tumor-associated antigens and via HLA-peptide tetramers. The apparent proportion of VP-MCC TIL that include T-Ag-specific CD8 T cell responses appears to vary with the technology used for detection (Figure 2). When a single HLA-appropriate tetramer was used in one report, 5 of 24 patients’ TIL had detectable antigen-specific responses37. In another study, 6 of 21 TIL from VP-MCC subjects were positive when assayed with a limited panel of patient-matching artificial antigen presenting cells (aAPC)39. Recently, we used the aAPC approach to probe patients’ TIL for multiple relevant HLA-A and B allelic variants42. This work detected T-Ag-specific CD8 TIL in the majority of biopsies (9 of 12) from persons with VP-MCC. While the true proportion of tumors infiltrated with T-Ag-specific CD8 T cells may vary between populations and assay methods, the failure of these cells to clear tumors by definition indicates that further augmentation of the endogenous response may be required.
Figure 2: MCPyV Large and Small T oncoproteins and immune “hot-zones.”.
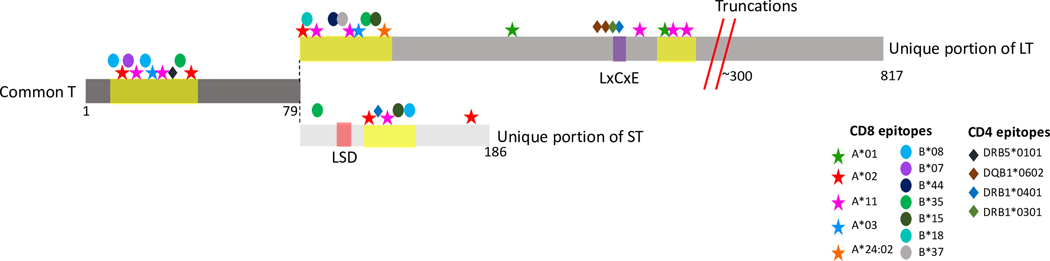
The common T sequence (middle-left) is shared between Large T and Small T. Top: Unique portion of Large T (LT). Bottom: unique portion of small T (ST). Common T encodes the region recognized by human antibodies to T-Ag.
Yellow highlighted areas indicate hot zones of enriched T-cell specific epitopes as detailed in the text. Documented HLA-restricting alleles are marked with either asterisks (HLA-A), ovals (HLA-B) or diamonds (HLA-DR/DQ).
Large T truncations (red diagonal lines) occur at approximately amino acid 300; the exact location varies from patient to patient. The LxCxE motif (purple) is the Rb binding domain. The small T LSD domain (orange) is an LT-stabilization domain that also mediates other oncoprotein functions.
CD4 T-cell responses, which are both essential for mediating humoral responses and for dendritic cell ‘licensing’ (promoting the capacity of dendritic cells to stimulate cytotoxic T cells) and subsequent CD8 T-cell activation, have also been identified within TIL from VP-MCC. Indeed, a total of 6 distinct CD4 T cell epitopes were detected within a limited region of LT. One epitope (LT209–228) was highly prevalent among MCC patients, as tetramer-positive cells were detected in 14 of 18 patients43 (Figure 2).
These studies demonstrate that TIL from MCC tumors are often specific for MCPyV-oncoproteins and that MCPyV specific T-cells can also circulate. However, patient T cell responses against epitope-HLA combinations predicted to be immunogenic are often not detectable. While knowledge of and assays for HLA-appropriate T cell responses are rapidly advancing, it is likely that in some patients, certain HLA-peptide T-cell responses are not only below current detection thresholds but are actually absent. Indeed, the presence of clinically evident tumors implies that T cell responses are frequently ineffective. A therapeutic vaccine could provide support to these cancer-specific T cells and promote anti-tumor immunity. Unlike preventive vaccines, a therapeutic vaccine is thought to improve immunity by supporting insufficiently primed T cells in addition to possibly stimulating naïve CD8 T cells. Supporting ineffectively primed T cells is particularly important since naïve T cells are produced in low number in older patients44 and existing CD8 T cells have likely encountered cognate tumor antigen due to the large antigen burden of cancer. Improperly primed CD8 T cells have not seen their cognate antigen on dendritic cells with costimulatory signals. They exhibit decreased effector function and a state similar to exhaustion45, 46. This is supported by recent studies showing therapeutic vaccination prior to immunotherapy preserves T cell function and mediates tumor regression via induction of early exhausted (stem-like) CD8 T cells that respond to immune therapy47. Each of these concepts provide support for development of a therapeutic vaccine that may enable tumor-specific T cell responses to reach a functionally effective threshold, especially if combined with measures to enhance trafficking to tumors, overcome tumor endogenous immune escape factors and promote persistence of T cells.
Vaccine Design
Target antigens
One of the first tasks when designing a therapeutic cancer vaccine is identifying an optimal antigen. In general, tumor antigens can be divided into cancer-associated antigens, and cancer-specific antigens48. Cancer-associated antigens such as cancer-testis antigens (i.e. MAGE-A3) and over-expressed proteins (i.e. HER-2/neu) are germline-coded antigens that may be expressed at low levels in healthy tissues or in gonadal tissues. Although these antigens are attractive vaccine targets because they are shared across many patients and cancers, they have numerous drawbacks including their dispensable nature to most cancers and frequent toxicity of immune responses to healthy tissues49, 50. In contrast, tumor-specific antigens such as oncoviral- and neoantigens are attractive since they are exclusively expressed in tumor tissue. Some oncoviral-antigens in particular are ideal vaccine targets since they drive carcinogenesis and thus cannot be downregulated to avoid immune destruction. Oncoviral-antigens are also shared between patients so a vaccine would not need to be personalized for each patient. VP-MCC has two such oncoviral-antigens, the MCPyV small and large T antigens (ST, LT). As noted above, there are well-documented T cell responses to these antigens, and autologous T cell therapies targeting these antigens have induced clinical responses38, 51. However, the endogenous T cell response is often suboptimal with many patients having undetectable T cell responses against known ST or LT antigens (see section above). When present, the T cell responses exhibit characteristics of dysfunction36. These observations suggest that MCPyV ST and LT antigens are appealing candidates for a therapeutic MCC vaccine.
Methods of Antigen Delivery: Protein and Peptides
One of the simplest ways to deliver tumor antigens is polypeptides or whole protein. This approach is used for some preventive and therapeutic vaccines for infectious disease52 and cancer53, 54. A key benefit of the simple approach is that patients would not develop immunity against other vaccine components such as vector proteins. This allows multiple rounds of vaccination without limiting boosts due to the development of anti-vector adaptive immune responses. This is in contrast to viral-vectored vaccines where pre-existing immunity to viral-vector is associated with a weaker immune response, relating to adaptive immunity preventing or limiting vector infection and transduction55, 56. However, since peptides alone are prone to induce tolerance, a danger signal must also be administered, typically an adjuvant compound (discussed below). Overall, the simplicity and safety of this approach make it an appealing option for a therapeutic vaccine (Table 1).
Table 1.
Comparison of antigen delivery platforms
| Modality | Type | Rationale | Benefits | Drawbacks | Relevant citations or trials |
|---|---|---|---|---|---|
| Peptide | Neoantigen | Cancers often have mutations in proteins that can be recognized as non-self by T cells | Can target proteins necessary for cancer survival | Patient-specific, difficult to identify neoantigens that would drive strong immune responses | 19, 117 |
| Viral tumor antigen | Viral proteins are highly immunogenic, Sometimes required for tumor growth | Shared across patients, truly foreign, highly immunogenic, often required for carcinogenesis | Not applicable for most cancers | 98 | |
| Tumor associated antigen | Many cancers express antigens which would only otherwise be expressed in embryonic or gonadal tissues or lowly expressed in healthy tissues | Shared across patients | Since these are often not required for cancer survival, tumors can down regulate these proteins to evade an immune response | 118 | |
| Nucleic Acid | DNA | Putative tumor antigens are encoded into DNA constructs which can then be delivered to patients where the antigens are transcribed and translated | Stable and inexpensive mechanism of antigen delivery, intrinsic adjuvanticity as can encode protein adjuvants | Difficulties in transfection, degradation by DNases | 99 |
| mRNA | Putative tumor antigens are encoded into mRNA constructs which can then be delivered to patients where the antigens are directly translated | RNA is highly immunogenic, can encode protein adjuvants | Also, difficulties in transfection, degradation by RNases | 60 | |
| Self-amplifying RNA | Viral RNA encodes an RNA polymerase in addition to tumor antigen(s), increasing both transcription and adjuvanticity | Higher levels of transcription and immunogenicity | Less tested | 54, 55 | |
| Cellular | Tumor cell | Tumor cells are expanded ex vivo and killed cells are injected with adjuvants. This does not require identifying individual neo-antigens | Does not require identification of tumor antigens, tumor cells can be engineered to express adjuvants or even co-stimulatory signals | Potential autoimmune side effects, low proportion of neoantigens | 119–121 |
| Dendritic cell | Dendritic cells are professional antigen presenting cells primarily responsible for strong T cell responses; can be expanded and treated with tumor antigens and infused into patients. | Allows direct presentation to T cells with co-stimulatory signals. | Difficult and expensive manufacturing. HLA Diversity precludes an “off-the-shelf” product | 122 | |
| Pathogen | Viral | Viral antigens can be encoded into viral vectors allowing efficient delivery to patients | More efficient delivery than nucleic acids | Immunity to vector complicates repeat vaccination | 68 |
| Bacteria phages | Phages are well known to display antigens on their capsids which could be sources of antigen in a vaccine | Simple manufacturing (bacterial culture) contain some PAMPS | No replication in eukaryotic cells | NCT04034225 | |
| Bacterial | Bacteria expressing tumor antigens can be killed and used as a vaccine | Bacteria encode numerous immune agonists, relatively simple manufacturing | Difficulty in precise control over antigen and adjuvant levels | 70, 71, 123 | |
| in situ | Adjuvant only vaccine | Intratumoral injection of adjuvant stimulates intratumoral DCs and T cells | Does not require identification of tumor antigens, tumor cells can be engineered to express adjuvants or even co-stimulatory signals | Less likely to develop adaptive immune responses, Intratumoral environment is suboptimal site to stimulate T cell responses due to suppressive characteristics | 83, 84 |
| Oncolytic Viruses | Viruses attenuated to replicate in cancer cells but not healthy cells could drive a systemic adaptive immune response against tumor antigens in addition to local immune effects | Agnostic to tumor type, augments both innate and adaptive immune responses, replicative capacity of virus increases immunogenicity | Typically requires intralesional delivery which is not possible in some cases | 124 | |
| Radiation | In rare circumstances, radiation can induce systemic responses through an abscopal effect | Radiation has well documented in-field tumorcidal effects independent of immunogenicity | Abscopal effect is rare and poorly understood | 125, 126 |
Whole proteins, or protein antigens conjugated to immune enhancing molecules, are preferred to peptides as they contain all potential epitopes within individual patients and the HLA-diverse population. Whole proteins also require endogenous antigen processing which promotes presentation of biologically relevant cleavage peptides in the proper HLA context. However, in the specific case of MCPyV T-Ag, there is the possible risk of promoting oncogenesis by delivering unmodified oncoproteins, a concern shared by some other vaccine platforms (discussed below). To avoid this, multiple synthetic long peptides (SLP) covering the oncoprotein sequence can be used. This avoids the risk of carcinogenesis, has been shown to induce both CD4 and CD8 T cell responses and to have anti-tumor activity for HPV57, 58, and remains an attractive strategy for VP-MCPyV. To date, SLP-based vaccines appear to be most effective in early stage cancers or carcinoma in situ57, 58. An additional concern is the processing and presentation of HLA-binding minimal epitopes from SLP could differ from those derived from whole protein59.
Peptide or protein antigens can also be modified to increase immunogenicity or selectively promote processing and presentation to T cells. One such approach is to covalently link the antigen of interest to an adjuvant. This ensures that the same APC that take up antigen receive additional danger signals and thus are competent to provide costimulatory signals to T cells. This approach has been successful in animal models that conjugate long peptides to a TLR agonists60–62. Peptides can also be expressed as fusions with proteins that direct them towards antigen processing machinery and HLA presentation63. Overall, polypeptides of various lengths are attractive for their general lack of toxicity, clinical experience, and ease of manufacturing. Several studies53, 54 have shown their ability to stimulate T cell responses against tumor antigens.
Methods of Antigen Delivery: Nucleic Acids
Tumor antigens can also be encoded in nucleic acid products to express tumor antigens in vivo. This approach shares some of the design constraints and attractive options of subunit protein vaccines, with additional challenges and potential. For example, antigens, adjuvants, and targeting moieties can be included in a single construct. Nucleic acids also have auto-adjuvant activity, which can be further enhanced by the insertion of specific, stimulatory sequences. Cytoplasmic dsDNA stimulates immune responses via the cGAMP/STING pathway and TLR9, while exogenous RNAs can signal through the RIG-I pathway and TLRs 3, 7, 864. These and other nucleic acid sensor pathways can promote robust immune responses against target antigens. A dose-sparing effect can be obtained using self-amplifying RNA molecules such as alphavirus replications, which encode an RNA-dependent RNA polymerase to increase the copy number of immune stimulatory and antigen-encoding RNA in transduced cells65, 66.
A DNA vaccine has been tested in an animal model of MCC. In these studies from the Hung lab, murine melanoma cells were engineered to express MCPyV tumor antigens67–69. Mice were then treated with a DNA vaccine expressing the ST or LT antigens, inducing tumor regression. Interestingly, initial studies using vaccination of LT antigen alone stimulated a strong CD4 response69. However, a follow up study in which the LT antigen was conjugated to calreticulin a much more robust CD8 T cell response was induced, possibly due to increased antigen uptake by cross-presenting dendritic cells68. These findings further support conjugating tumor antigens to other proteins that can modulate T cell responses and may be particularly relevant for MCC.
It is worth noting that nucleic acid-based vaccines can also incorporate features of peptide vaccines such as fusion to proteins that influence antigen trafficking, processing, and presentation or co-expression with a protein adjuvant. One example of how this could be pursued in MCC is a DNA vaccine that conjugates MCPyV ST or LT to lysosome-associated membrane glycoprotein (LAMP). This directs these antigens towards the lysosome and HLA-II presentation, in order to promote a CD4 response. This approach has been shown to be safe in phase I trials to induce allergen tolerance70. The use of LAMP to elicit functional anti-tumor responses to MCC is under active investigation in animal models63.
Despite their advantages, nucleic acid-based vaccines have not been reliably effective in humans. Several vaccines have had disappointing clinical trials with only a small fraction of patients developing T cell responses against target antigens71–73. These trials tested numerous tumor associated antigens in several cancer types including renal cell carcinoma, non-small cell lung cancer and melanoma. However, the vaccines induced weaker responses in humans than in animal models, possibly due to low antigen expression or differences in immune nucleic acid sensing pathways. As a result, no nucleic acid-based vaccine is currently licensed in humans. Promising adjuncts such as lipid nanoparticle or electroporation-based delivery systems and dose escalation can increase immunogenicity74. There is still substantial interest in nucleic acid-based vaccines75 and these platforms will likely continue to improve.
Methods of Antigen Delivery: Microbe Vectored-vaccines
Microbes can also be used to deliver tumor antigens and stimulate adaptive immune responses. This approach is attractive since microbes can naturally stimulate a strong immune response. In general, there is a correlation between the replication-competence of the vector and the strength of the immune response to the tumor antigens, as vector amplification increases stimulation of PAMPs and antigen dose. The tradeoff, however, is that replication competence may bring with it virulence, especially in immune compromised cancer patients, such that vector attenuation and safety need to be extremely carefully addressed in animal models and early-stage clinical trials. In addition, many pathogens have evolved immune evasion mechanisms, which need to be understood and engineered out of vaccine vectors to improve responses31, 76, 77. This issue, combined with microbe genome stability during manufacturing, and the complexity of manufacturing add considerable time and expense to microbe-vectored vaccine workflows.
Several therapeutic cancer vaccine trials using pox or adenoviral vectors have been conducted in humans with mixed results. A trial using vaccinia and fowlpox vectors to express the NY-ESO-1 tumor-antigen in melanoma patients induced CD8 T cell responses in a majority of patients (88%) by the conclusion of the trial. However, only 14% of patients had a clinical response to vaccination78.
Therapeutic vaccination vectors other than human viruses are also beginning to move into trials. The most common of these use Listeria monocytogenes engineered to express tumor antigens. An initial trial had sepsis-related adverse events but did induce T cell responses to the vaccine target antigens79. Subsequent trials with additional vector attenuation have proven safer80 and to improve survival in patients with refractory pancreatic cancer81. Another such approach uses bacteriophages to deliver cancer antigens of interest82, 83. Phages have been known to be capable of promoting immunity in vivo since 198584. An advantage of this approach is the ease and low cost of phage manufacture in bacterial culture. While at least one phage vaccine has reached clinical stage for cancer (NCT03120832), overall this platform remains unproven and requires more immunogenicity and safety work. If multiple doses are used, the development of binding antibodies, known to occur after administration of some phages85 could divert antigen either away or potentially even towards the antigen processing pathways required for T cell responses, adding further complexity. In summary, microbe-based vaccines have several advantages including self-adjuvanticity and the potential for intracellular delivery of antigens. Although less developed than other vaccine formats, these platforms are likely to be of further interest in the near future (Table 1).
Methods of Antigen Delivery: Cell-based vaccination
While adoptive cell transfer has recently been clinically approved for the delivery of redirected T cells, cellular-based vaccine therapies can alternatively be used to stimulate endogenous T cell responses. Tumor cells expanded and inactivated ex vivo can be reinfused back into patients where they can act as a source of antigen or even antigen presenting cells86. Alternatively, autologous dendritic cells loaded with tumor antigens have the potential to present tumor peptides in the correct HLA context, together with costimulatory signals.
For inactivated tumor cells vaccines, a tumor biopsy is obtained and tumor cells are then expanded ex vivo87, 88. The tumor cells can be modified to express adjuvants or co-stimulatory signals and then, they are killed or inactivated prior to administration back into patients. Large trials using this approach have shown modest efficacy in early stage cancers in the early 2000s, however the substantial cost in lower risk cancers has hindered further development87, 88.
Vaccination with dendritic cells (DC) includes loading antigen into DC ex vivo, which provides control over DC phenotype and maturation. Most such vaccines that now reach the clinical stage use cross-presenting dendritic cells, with the potential to stimulate CD8 T cells in vivo upon re-administration to the patient. A noteworthy example of DC vaccination is Sipuleucel-T, an FDA-approved vaccine for the treatment of castration-resistant metastatic prostate cancer. Although the clinical benefit of Sipuleucel-T is relatively modest (4 months improved survival89), these trials provide a strong rationale for future trials of dendritic cell vaccines. These agents are limited by costly, complex, and time-consuming manufacturing and individualized nature, and are thus most relevant for patients with particularly high-risk disease (Table 1).
Adjuvants and In Situ Vaccination
Adjuvant selection is thought to be as important as choosing an antigen delivery system. Adjuvants can be broken down into two major categories: Innate immune agonists that largely target innate immune cells and cytokines that typically support T cells or APC. Innate immune adjuvants are usually small molecules which mimic molecular patterns found in bacteria or viruses such as nucleic acids or TLR agonists which are thought to act by activating relevant antigen presenting cells64. Adaptive immune adjuvants are usually proteins which would normally be expressed by immune cells to support APC or T cell growth or differentiation90.
The importance of adjuvants can be illustrated by “adjuvant only” or in situ vaccines. In contrast to the above vaccination approaches which rely on delivering tumor antigens as part of a vaccine, in situ vaccines rely on endogenous antigens in the tumor and aim to improve responses to these antigens. Indeed, it is now recognized that some irradiation and cytotoxic chemotherapy regimens may work at least in part by promoting immunogenic cell death91, 92. These “adjuvant-only” concepts have had moderate success in small MCC trials. GLA-100 is a stable emulsion of a synthetic TLR4 agonist and was tested in 7 patients with metastatic MCC. This compound is thought to modulate innate immune cells such as macrophages in the tumor microenvironment so that they shift from a suppressive and towards an inflammatory phenotype. In this small trial, 2 patients had sustained partial responses in the target leision93. A separate trial tested an IL-12-expressing plasmid in MCC using DNA electroporation directly into tumors. IL-12 supports T cell differentiation and polarizes CD4 T cells towards a Th1 phenotype. In this trial, 3 of 10 patients with multiple metastatic lesions had partial responses, including in non-target tumors94.
Encouraged by these results, two newer trials delivering immune agonists into tumors are ongoing. One approach targets the stimulator of interferon genes (STING; NCT03172936). The STING pathway senses intracellular DNA and activates interferon signaling, known to be critical for immune responses following radiation95, 96. STING agonists have been shown to reverse anti-PD-1 therapy resistance in animal models97. A separate trial investigating AST-008, a nanoparticle- TLR9 agonist, is being studied for PD-(L)1-refractory MCC (NCT03684785). The nanoparticle formulation is designed to increase phagocytic APC uptake and delivery to endosomal TLR9.
In situ vaccine trials in MCC support the notion that adjuvants on their own can stimulate innate and adaptive immune cells. Combining these therapies with optimally delivered tumor antigen will likely further strengthen immune responses in MCC.
Early successes of therapeutic vaccines
Personalized neoantigens vaccines:
As mentioned above, tumor-associated antigens are often divided into two groups: 1) non-mutated self-antigens that may be tissue-specific and overexpressed in various cancers and 2) neoantigenic peptides derived from somatic mutations in cancer cell genomes98. Therapeutic cancer vaccines that target tumor-associated self-antigens often have low clinical efficacy and generate poor T-cell responses. Presumably, because it correlates with low avidity T cells, as high avidity T cells targeting self-antigens are eliminated during thymic selection98, 99. In contrast, neoantigens are ideal targets for therapeutic vaccines as the T-cell repertoire, including avidity, is not reduced by thymic selection100.
Because most cancer mutations are patient-specific, personalized approaches are appropriate for high tumor mutational burden (TMB) malignancies. Indeed, recent developments of in silico prediction approaches enable characterization and selection of neoantigens for personalized therapeutic vaccines53, 54, 101. Two groups provided clinical data demonstrating the promise of personalized neoantigen vaccines. Sahin et al.101 identified tumor-associated mutations in 13 patients with advanced melanoma. Using HLA data and epitope prediction tools, 10 peptides were selected per patient and administered as RNA vaccines. Eight patients (61%) that started the treatment without any detectable radiological lesions had a robust immune response and remained recurrence-free up to 23 months. The other patients relapsed after initiation of vaccine. Among these, one had a complete response after completion of the vaccine series and the second after receiving subsequent anti-PD-1 treatment.
Analyses of PBMC suggested that the majority of T cell responses to these vaccines were de novo rather than boosts of pre-existing responses. CD4 T cell responses dominated over CD8 recognition of vaccine epitopes, despite vaccine design targeting CD8 T cells as discussed above. In the 2nd study, Ott et al.53 tested a peptide-based vaccine in 6 patients with previously untreated advanced melanoma. Patient-specific somatic mutations were identified, and twenty neoantigens were selected with HLA prediction tools. Thirty amino acid long peptides were injected with poly IC:LC adjuvant. Four patients (66%), who entered after surgical resection, remained free from disease recurrence for 25 months. The other 2 patients who entered the study with lung metastases had complete responses after subsequent anti-PD-1 treatment. The vaccine stimulated both CD8 and CD4 PBMC interferon (IFN)-γ responses, with a preponderance of CD4 responses. T cell receptor (TCR) analyses suggested that vaccine-elicited cells included de novo T-cell clonotypes.
Another vaccine (based on 20 personalized long peptides per patient) was recently tested in glioblastoma54. Glioblastoma is an immunologically “cold” tumor with a relatively low mutational burden102. While this trial did not result in clinical benefit, a significant benchmark was achieved by the demonstration of specific T cell responses to a classically non-immunogenic tumor. In addition to generating CD4 T cell responses, patients had an increase in tumor-infiltrating T cells.
HPV therapeutic vaccines:
Human papillomaviruses (HPV) are circular DNA viruses. Some genotypes are associated with cervical, anal, vulval, and oropharyngeal cancers103. Similar to VP-MCC, a limited number of viral oncoproteins are persistently expressed in HPV-associated cancers and mechanistically drive cell proliferation. Prophylactic vaccination prevents HPV-associated neoplastic diseases when used before primary infection104. This great success has led to interest in therapeutic vaccination that is administered after disease presentation.
HPV16 and HPV18-driven malignant transformation are strongly dependent on the E6 and E7 oncoproteins105, 106. Many prototypes of therapeutic vaccines that induce T cell responses to E6 and E7 have been developed107–109. Vaccination with mixed SLP (ISA101 vaccine) from HPV E6 and E7 oncoproteins with adjuvant in women with HPV-16-positive vulvar intraepithelial neoplasia resulted in regression in 94% of patients, of whom 47% experienced a complete response108. This was associated with both CD8 and CD4 T cell responses. An E6 and E7 DNA vaccine (VGX-3100) was tested in a phase 2, double-blind trial in women with HPV16/18-positive precancerous conditions. Histopathological regression was observed in 49% of treated patients compared to 36% in the placebo group109. CD8 T cell and antibody responses correlated with the histopathological regression. The vaccine is currently in phase III trials [NCT03185013 and NCT03721978].
In contrast to clinical responses in precancerous conditions108, 109, E6 and E7 therapeutic cancer vaccines have yielded clinically disappointing results in patients with advanced or recurrent HPV 16-related cancers110, 111, despite enhancement of blood T cell responses. This suggests that vaccine-activated T cells are being inhibited by immunosuppression58. Therefore, synergism with approaches to overcome local and systemic immune suppression are rational to enhance the clinical activity of cancer vaccines in persons with established tumors. A phase 2 clinical trial that combined ISA101and PD-1 blockade, in patients with recurrent and invasive HPV 16 associated malignancies, resulted in a 33% overall response rate (ORR) (8/24 subjects), higher than the 16% to 22% ORR with PD-1 blockade alone58. Similar trials are underway [NCT03260023].
HPV murine models support such combination therapy. A therapeutic vaccine combined with anti-PD-1 resulted in a better tumor regression when compared to anti-PD-1 treatment alone112–114. Moreover, it was shown that vaccine alone increased tumor-infiltrating CD8 T cells but also raised PD-1 expression levels. Combination of the vaccine with anti-PD-1 preserved tumor-infiltrating CD8 cells and reduced the numbers of T cells expressing PD-1112, 114, and demonstrated synergistic anti-tumoral responses.
Biological considerations prior to clinical implementation
Selection of the appropriate treatment for MCC depends on many factors. Practice guidelines emphasize disease stage115, with early disease typically being managed with surgery and radiation while more advanced disease is managed with systemic immunotherapies such as anti-PD-(L)1 blockade. We envision possible roles for therapeutic vaccination at both ends of the VP-MCC clinical spectrum.
Adjuvant therapy for early MCC
Patients diagnosed with local MCC will often undergo surgical excision. Radiation of the excised area and the adjacent lymph nodes is also often included116. This combination is extremely effective in rendering patients free of detectable disease, but recurrences frequently arise near the primary tumor site, in the nodal region, and/or distantly1, 116. Vaccination after local treatment could generate or enhance a systemic immune response that recognizes early emerging microscopic tumors and eliminates them by efficient effector responses. Therefore, a safe therapeutic vaccine to augment MCPyV immunity would be useful to help prevent MCC recurrence. With regard to clinical trials, an appealing alternative to adjuvant studies is the neoadjuvant setting, in which vaccine is initiated in the short window between diagnostic biopsy and excision. This provide an opportunity for histopathlogic study of the removed tumor after potential immune boosting. The conserved nature of MCPyV T-Ag allows consideration of a neoantigen trial as the vaccine candidate product would be off-the-shelf and ready to administer.
Systemic/advanced disease
Patients with advanced disease currently receive PD-(L)1 blockade as the preferred first-line therapy. The response rates, 50% - 65%24, 117, are higher than those for most other cancers. Unfortunately, approximately half of all treated patients do not experience prolonged benefit1, 118. The fact that some MCC patients have modest or non-detectable T cell immune responses to MCPyV antigens36, 37 suggests that a paucity of antigen-specific T cells capable of being functionally augmented by anti-PD-(L)1 may contribute to suboptimal clinical responses. Therefore, a combination of a therapeutic vaccine and PD-(L)1 inhibitor could potentially be beneficial for VP-MCC patients. Indeed, several studies in mice and humans suggest it is possible to both stimulate T cell function and prevent the induction of T cell exhaustion. Such studies have demonstrated that vaccination increases the number of T cells infiltrating tumors54, 101, 112, 113. The increase in T cell number occurs by generating either de novo T cell responses or by augmenting existing T cell responses. However, in advanced disease, an increase of T cell numbers by a therapeutic vaccine is usually not sufficient to clear tumors completely. Furthermore, alongside the increase in T cell responses and infiltration into tumors, an increase in exhaustion markers, such as PD-1 on TIL and PD-L1 on tumor cells, is also often observed112, 113. Importantly, mice treated simultaneously with a therapeutic vaccine and either anti PD-1112, or anti-PD-L1113 demonstrated prolonged T cell stimulation and stronger suppression of tumor growth. Additionally, a clinical trial in men and women with recurrent HPV has tested anti-PD-1 treatment combined with a synthetic long peptide vaccine, leading to promising anti-tumor responses compared to PD-1 blockade alone58. These studies suggest that combining a cancer vaccine with PD-(L)1 blockade has significant clinical potential for VP-MCC.
The sequencing of vaccination and PD-(L)1 therapy was explored in a recent mouse study that showed how different treatment protocols result in distinct responses. Verma et al.47 showed that simultaneous PD-1 inhibitor and vaccination was beneficial, but pre-treatment with PD-1 blockade prior to combination therapy abrogated responses. The inhibitory effect of pretreatment with anti-PD-1 was mediated by PD-1+CD38hi CD8 T cells, the depletion of which resulted in a more robust anti-tumor response and improved mice survival.
In the difficult setting of patients whose tumors are already refractory to anti-PD-(L)1, it may be beneficial to add an additional therapy, such as a T cell modulatory cytokine. A rational candidate is the immunomodulator IL-12. IL-12 has been shown to be a potent producer of anti-tumor immunity119, which stimulates different effector cells (NK and T cells)120. IL-12 has been broadly tested in the clinic; several studies have demonstrated that IL-12 treatment increases tumor-infiltrating lymphocytes and the circulating T cell response in refractory settings121, 122. Potentially, such a cytokine could restore some of the immune functions and support further benefit from a combined therapeutic vaccine with PD-(L)1 blockade.
Potential clinical trial designs
It is mechanistically attractive to consider up-front combination of therapeutic vaccination with anti-PD-(L)1 therapy, but it is probably not feasible to carry out such a trial in a rare disease with a relatively high response rate to anti-PD-(L)1 monotherapy. The establishment of incremental benefit for a therapeutic vaccine would require a large and expensive trial. For example, to detect an increase in response rate from 60% to 80%, ~162 patients would be required for a randomized trial, with 80% power to detect a difference of that magnitude123. A 20-percentage point improvement in the response rate is a very optimistic estimate, representing a 50% decrease in the number of non-responders. The number of patients required for a trial is very sensitive to the assumed effect size. If the effect size is cut to 10% (60% response rate increases to 70% with vaccine) ~712 patients would be required for the trial123. Since the largest trial to date in advanced MCC (88 patients) required more than a dozen international sites124, it is likely impractical to explore first-line combination of a therapeutic vaccine with PD-(L)1 blockade. The greatest unmet need in MCC is to develop effective therapy for patients with PD-(L)1 refractory VP-MCC. Because anti-PD-(L)1 refractory metastatic MCC has no proven effective therapy, even occasional responses in this setting could be clinically meaningful and indicative of benefit of vaccine therapy.
For the adjuvant setting, clinical trial design is again challenging based on a relatively large number of patients required to determine efficacy. Statistically adequate sample size depends on the number of anticipated events (recurrences) which varies significantly based on stage at presentation. Higher-risk groups (e.g. stage IIIB disease) would require fewer patients to meet statistical significance because ~70% of these patients are expected to recur after initial treatment. In this scenario, only 62 patients would need to be randomized to vaccine versus placebo in order to assess clinical benefit, assuming the vaccine reduced the recurrence rate by one half123. In contrast, for stage I (~20% risk of recurrence), a clinical trial would require nearly 400 patients, assuming the vaccine also reduced recurrence rate by one half (to 10%) in this setting123. Since there are several other adjuvant trials being conducted (NCT03712605, NCT03271372), and they focus on higher risk patients, an adjuvant vaccine trial would likely need to preferentially enroll lower risk patients, meaning target trial size may need to be ~200, based on appropriate risk stratification for stage. Prior to embarking on a significant trial to assess efficacy in the adjuvant setting, a smaller feasibility and immunogenicity clinical trial would likely make sense, after a candidate vaccine has been evaluated in a pre-clinical model. In this initial human study, the extent and persistence of anti-MCPyV immune responses to a vaccine could be assessed as outlined in the MCPyV oncoproteins section above. This analysis will be facilitated by the fact that after tumor removal, these immune responses typically fall very rapidly.
Conclusions
The clinical efficacy of cancer vaccines has remained modest despite decades of effort. Different cancers pose distinct challenges that must be addressed in a customized manner. VP-MCC is an appealing candidate for a therapeutic vaccine because: 1) MCC is an inherently immune-sensitive cancer, strongly associated with baseline immune status, and generally responsive to immunotherapies. 2) The small and conserved antigenic space of the relevant MCPyV-encoded oncoproteins are amenable to vaccine construction and well-defined immune monitoring tools such as tetramers and TCR sequencing36, 37.3) MCPyV viral antigens are inherently immunogenic, leading to robust B and T lymphocyte responses that can be readily detected throughout the clinical course.
Several studies that have characterized MCPyV-specific T cells in MCC tumors clearly demonstrate that T cell infiltration, TCR diversity, and T cell frequency are associated with better patient outcomes36, 37, 40. However, in most cases these responses are not sufficient and T cells fail to clear tumors, indicating that augmentation of the endogenous response may be needed. Treatment with a therapeutic vaccine could potentially both enhance existing immune responses and induce de novo T cell responses. Depending on the clinical scenario evaluated, as discussed above, vaccination could prevent late recurrences by eliminating micrometastases or could rescue patients with advanced disease who do not have initial or sustained responses to checkpoint inhibition.
Several factors suggest that VP-MCC is an interesting model of therapeutic vaccination. The viral oncoprotein is T-cell immunogenic providing a trackable biomarker to measure responses. Since both T and B cell responses drop after tumor removal 18, 36, persistent responses from vaccine would contrast with naturally occurring transitory responses and help to differentiate the two. The clonotypic complexity of T cell responses to LT can readily be followed in blood, tumor biopsies, and TIL37, 42 providing another biomarker for vaccine responses.
Because of the unique advantages of this disease, it should be a high priority for the field to explore therapeutic vaccination in this setting. It is likely that clinical trials of a therapeutic vaccine for VP-MCC will yield valuable insights relevant to other immunogenic cancers that may be more common.
Acknowledgments
Funding: NIH-P01-CA225517, NIH-T32-CA080416, Prostate Cancer Foundation Kelsey Dickson Team Science Courage Research Team Award and UW MCC Patient Gift Fund.
Footnotes
Data statement: Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Conflict of Interest: DMK and PN are co-inventors on institutionally-owned US patent applications relevant to this article. DMK has received funding from Immunomics Therapeutics Incorporated relevant to this article. PN has received grant support from EMD Serono and Bristol Myers Squibb as well as honoraria from Merck and EMD-Serono.
References:
- 1.Harms PW, Harms KL, Moore PS, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nature reviews Clinical oncology. 2018;15(12):763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms KL, Healy MA, Nghiem P, et al. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Annals of surgical oncology. 2016;23(11):3564–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agelli M, Clegg LX, Becker JC, et al. The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer. 2010;34(1):14–37. [DOI] [PubMed] [Google Scholar]
- 4.Harms PW. Update on Merkel Cell Carcinoma. Clin Lab Med. 2017;37(3):485–501. [DOI] [PubMed] [Google Scholar]
- 5.Paulson KG, Park SY, Vandeven NA, et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. Journal of the American Academy of Dermatology. 2018;78(3):457–463 e452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goh G, Walradt T, Markarov V, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2016;7(3):3403–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong SQ, Waldeck K, Vergara IA, et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer research. 2015;75(24):5228–5234. [DOI] [PubMed] [Google Scholar]
- 9.Chen T, Hedman L, Mattila PS, et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol. 2011;50(2):125–129. [DOI] [PubMed] [Google Scholar]
- 10.Sourvinos G, Mammas IN, Spandidos DA. Merkel cell polyomavirus infection in childhood: current advances and perspectives. Arch Virol. 2015;160(4):887–892. [DOI] [PubMed] [Google Scholar]
- 11.Coursaget P, Samimi M, Nicol JT, et al. Human Merkel cell polyomavirus: virological background and clinical implications. Apmis. 2013;121(8):755–769. [DOI] [PubMed] [Google Scholar]
- 12.Schowalter RM, Pastrana DV, Pumphrey KA, et al. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7(6):509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, Yang R, Payne AS, et al. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe. 2016;19(6):775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carter JJ, Paulson KG, Wipf GC, et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J Natl Cancer Inst. 2009;101(21):1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulson KG, Lewis CW, Redman MW, et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer. 2017;123(8):1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Hein J, Richardson SC, et al. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J Biol Chem. 2011;286(19):17079–17090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyngaa R, Pedersen NW, Schrama D, et al. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(7):1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulson KG, Carter JJ, Johnson LG, et al. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer research. 2010;70(21):8388–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petitprez F, de Reynies A, Keung EZ, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. [DOI] [PubMed] [Google Scholar]
- 21.Seow DYB, Yeong JPS, Lim JX, et al. Tertiary lymphoid structures and associated plasma cells play an important role in the biology of triple-negative breast cancers. Breast Cancer Res Treat. 2020. [DOI] [PubMed] [Google Scholar]
- 22.Giraldo NA, Nguyen P, Engle EL, et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J Immunother Cancer. 2018;6(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behr DS, Peitsch WK, Hametner C, et al. Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in Merkel cell carcinomas. International journal of clinical and experimental pathology. 2014;7(11):7610–7621. [PMC free article] [PubMed] [Google Scholar]
- 24.Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. The New England journal of medicine. 2016;374(26):2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shuda M, Kwun HJ, Feng H, et al. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. The Journal of clinical investigation. 2011;121(9):3623–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houben R, Shuda M, Weinkam R, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84(14):7064–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houben R, Angermeyer S, Haferkamp S, et al. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int J Cancer. 2015;136(5):E290–300. [DOI] [PubMed] [Google Scholar]
- 28.Borchert S, Czech-Sioli M, Neumann F, et al. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J Virol. 2014;88(6):3144–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora R, Chang Y, Moore PS. MCV and Merkel cell carcinoma: a molecular success story. Curr Opin Virol. 2012;2(4):489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shuda M, Feng H, Kwun HJ, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105(42):16272–16277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau L, Gray EE, Brunette RL, et al. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350(6260):568–571. [DOI] [PubMed] [Google Scholar]
- 32.Kwun HJ, Shuda M, Feng H, et al. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe. 2013;14(2):125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dye KN, Welcker M, Clurman BE, et al. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and beta-TrCP. PLoS Pathog. 2019;15(1):e1007543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park DE, Cheng J, Berrios C, et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc Natl Acad Sci U S A. 2019;116(3):1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iyer JG, Afanasiev OK, McClurkan C, et al. Merkel cell polyomavirus-specific CD8(+) and CD4(+) T-cell responses identified in Merkel cell carcinomas and blood. Clin Cancer Res. 2011;17(21):6671–6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Afanasiev OK, Yelistratova L, Miller N, et al. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(19):5351–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller NJ, Church CD, Dong L, et al. Tumor-Infiltrating Merkel Cell Polyomavirus-Specific T Cells Are Diverse and Associated with Improved Patient Survival. Cancer Immunol Res. 2017;5(2):137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulson KG, Voillet V, McAfee MS, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. 2018;9(1):3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samimi M, Benlalam H, Aumond P, et al. Viral and tumor antigen-specific CD8 T-cell responses in Merkel cell carcinoma. Cellular immunology. 2019;344:103961. [DOI] [PubMed] [Google Scholar]
- 40.Paulson KG, Iyer JG, Tegeder AR, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 2011;29(12):1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson KG, Iyer JG, Simonson WT, et al. CD8+ lymphocyte intratumoral infiltration as a stage-independent predictor of Merkel cell carcinoma survival: a population-based study. American journal of clinical pathology. 2014;142(4):452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jing L, Ott M, Church C, et al. High prevalence and diverse HLA restriction of intratumoral oncoprotein-specific CD8 T cells in polyoma virus-driven Merkel cell carcinoma. Cancer Immunology Research. 2020;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longino NV, Yang J, Iyer JG, et al. Human CD4(+) T Cells Specific for Merkel Cell Polyomavirus Localize to Merkel Cell Carcinomas and Target a Required Oncogenic Domain. Cancer Immunol Res. 2019;7(10):1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.den Braber I, Mugwagwa T, Vrisekoop N, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36(2):288–297. [DOI] [PubMed] [Google Scholar]
- 45.Ahrends T, Spanjaard A, Pilzecker B, et al. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity. 2017;47(5):848–861.e845. [DOI] [PubMed] [Google Scholar]
- 46.Smith CM, Wilson NS, Waithman J, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5(11):1143–1148. [DOI] [PubMed] [Google Scholar]
- 47.Verma V, Shrimali RK, Ahmad S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol. 2019;20(9):1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Science translational medicine. 2013;5(197):197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chapuis AG, Afanasiev OK, Iyer JG, et al. Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol Res. 2014;2(1):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunningham AL, Lal H, Kovac M, et al. Efficacy of the Herpes Zoster Subunit Vaccine in Adults 70 Years of Age or Older. The New England journal of medicine. 2016;375(11):1019–1032. [DOI] [PubMed] [Google Scholar]
- 53.Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17(3):295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Vos van Steenwijk PJ, Ramwadhdoebe TH, Lowik MJ, et al. A placebo-controlled randomized HPV16 synthetic long-peptide vaccination study in women with high-grade cervical squamous intraepithelial lesions. Cancer Immunol Immunother. 2012;61(9):1485–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Massarelli E, William W, Johnson F, et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients With Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA oncology. 2019;5(1):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8(8):607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zom GG, Khan S, Britten CM, et al. Efficient induction of antitumor immunity by synthetic toll-like receptor ligand-peptide conjugates. Cancer Immunol Res. 2014;2(8):756–764. [DOI] [PubMed] [Google Scholar]
- 61.Kastenmuller K, Wille-Reece U, Lindsay RW, et al. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest. 2011;121(5):1782–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lynn GM, Sedlik C, Baharom F, et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heiland T, Rosean CB, Sinha P, et al. Abstract: Lysosomal-associated membrane protein-1-targeting of the large T antigen of Merkel cell polomavirus elicits potent CD4+ T cell responses. First International Symposium on Merkel Cell Carcinoma. 2019;Moffitt Cancer Center. [Google Scholar]
- 64.O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460. [DOI] [PubMed] [Google Scholar]
- 65.Vogel AB, Lambert L, Kinnear E, et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Molecular therapy : the journal of the American Society of Gene Therapy. 2018;26(2):446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brito LA, Kommareddy S, Maione D, et al. Self-amplifying mRNA vaccines. Advances in genetics. 2015;89:179–233. [DOI] [PubMed] [Google Scholar]
- 67.Gomez B, He L, Tsai YC, et al. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. 2013;3(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez BP, Wang C, Viscidi RP, et al. Strategy for eliciting antigen-specific CD8+ T cell-mediated immune response against a cryptic CTL epitope of merkel cell polyomavirus large T antigen. Cell & bioscience. 2012;2(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng Q, Gomez BP, Viscidi RP, et al. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine. 2012;30(7):1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su Y, Romeu-Bonilla E, Anagnostou A, et al. Safety and long-term immunological effects of CryJ2-LAMP plasmid vaccine in Japanese red cedar atopic subjects: A phase I study. Hum Vaccin Immunother. 2017;13(12):2804–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sebastian M, Schroder A, Scheel B, et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer immunology, immunotherapy : CII. 2019;68(5):799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rittig SM, Haentschel M, Weimer KJ, et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther. 2011;19(5):990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weide B, Carralot JP, Reese A, et al. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother. 2008;31(2):180–188. [DOI] [PubMed] [Google Scholar]
- 74.Meyer M, Huang E, Yuzhakov O, et al. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J Infect Dis. 2018;217(3):451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Servick K. This mysterious $2 billion biotech is revealing the secrets behind its new drugs and vaccines. Science: AAAS 2017. [Google Scholar]
- 76.Quakkelaar ED, Redeker A, Haddad EK, et al. Improved innate and adaptive immunostimulation by genetically modified HIV-1 protein expressing NYVAC vectors. PLoS One. 2011;6(2):e16819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene therapy. 2003;10(4):292–303. [DOI] [PubMed] [Google Scholar]
- 78.Odunsi K, Matsuzaki J, Karbach J, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci U S A. 2012;109(15):5797–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dattananda CS, Rajkumari K, Gowrishankar J. Multiple mechanisms contribute to osmotic inducibility of proU operon expression in Escherichia coli: demonstration of two osmoresponsive promoters and of a negative regulatory element within the first structural gene. J Bacteriol. 1991;173(23):7481–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le DT, Brockstedt DG, Nir-Paz R, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18(3):858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33(12):1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bao Q, Li X, Han G, et al. Phage-based vaccines. Adv Drug Deliv Rev. 2019;145:40–56. [DOI] [PubMed] [Google Scholar]
- 83.Karbach J, Neumann A, Brand K, et al. Phase I clinical trial of mixed bacterial vaccine (Coley’s toxins) in patients with NY-ESO-1 expressing cancers: immunological effects and clinical activity. Clin Cancer Res. 2012;18(19):5449–5459. [DOI] [PubMed] [Google Scholar]
- 84.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. [DOI] [PubMed] [Google Scholar]
- 85.Smith LL, Buckley R, Lugar P. Diagnostic Immunization with Bacteriophage PhiX 174 in Patients with Common Variable Immunodeficiency/Hypogammaglobulinemia. Front Immunol. 2014;5:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Enomoto Y, Bharti A, Khaleque AA, et al. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J Immunol. 2006;177(9):5946–5955. [DOI] [PubMed] [Google Scholar]
- 87.Vermorken JB, Claessen AM, van Tinteren H, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet. 1999;353(9150):345–350. [DOI] [PubMed] [Google Scholar]
- 88.Jocham D, Richter A, Hoffmann L, et al. Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: phase III, randomised controlled trial. Lancet. 2004;363(9409):594–599. [DOI] [PubMed] [Google Scholar]
- 89.Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17(11):3520–3526. [DOI] [PubMed] [Google Scholar]
- 90.Zuniga-Pflucker JC. T-cell development made simple. Nat Rev Immunol. 2004;4(1):67–72. [DOI] [PubMed] [Google Scholar]
- 91.Zhou J, Wang G, Chen Y, et al. Immunogenic cell death in cancer therapy: Present and emerging inducers. J Cell Mol Med. 2019;23(8):4854–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. [DOI] [PubMed] [Google Scholar]
- 93.Bhatia S, Miller NJ, Lu H, et al. Intratumoral G100, a TLR4 Agonist, Induces Antitumor Immune Responses and Tumor Regression in Patients with Merkel Cell Carcinoma. Clin Cancer Res. 2019;25(4):1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bhatia S, Longino NV, Miller NJ, et al. Intratumoral Delivery of Plasmid IL12 Via Electroporation Leads to Regression of Injected and Noninjected Tumors in Merkel Cell Carcinoma. Clin Cancer Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deng L, Liang H, Xu M, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5):843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee CH, Yelensky R, Jooss K, et al. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018;39(7):536–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pedersen SR, Sorensen MR, Buus S, et al. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: implications for cancer immunotherapy. J Immunol. 2013;191(7):3955–3967. [DOI] [PubMed] [Google Scholar]
- 100.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. [DOI] [PubMed] [Google Scholar]
- 102.Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19(8):1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Otter S, Whitaker S, Chatterjee J, et al. The Human Papillomavirus as a Common Pathogen in Oropharyngeal, Anal and Cervical Cancers. Clin Oncol (R Coll Radiol). 2019;31(2):81–90. [DOI] [PubMed] [Google Scholar]
- 104.Arbyn M, Xu L, Simoens C, et al. Prophylactic vaccination against human papillomaviruses to prevent cervical cancer and its precursors. Cochrane Database Syst Rev. 2018;5:Cd009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci. 2008;13:1003–1017. [DOI] [PubMed] [Google Scholar]
- 106.Vande Pol SB, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology. 2013;445(1–2):115–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grunwitz C, Salomon N, Vascotto F, et al. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology. 2019;8(9):e1629259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. The New England journal of medicine. 2009;361(19):1838–1847. [DOI] [PubMed] [Google Scholar]
- 109.Trimble CL, Morrow MP, Kraynyak KA, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386(10008):2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Driel WJ, Ressing ME, Kenter GG, et al. Vaccination with HPV16 peptides of patients with advanced cervical carcinoma: clinical evaluation of a phase I-II trial. Eur J Cancer. 1999;35(6):946–952. [DOI] [PubMed] [Google Scholar]
- 111.van Poelgeest MI, Welters MJ, van Esch EM, et al. HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J Transl Med. 2013;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rice AE, Latchman YE, Balint JP, et al. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015;22(9):454–462. [DOI] [PubMed] [Google Scholar]
- 113.Lin PL, Cheng YM, Wu DW, et al. A combination of anti-PD-L1 mAb plus Lm-LLO-E6 vaccine efficiently suppresses tumor growth and metastasis in HPV-infected cancers. Cancer Med. 2017;6(9):2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Z, Zhou H, Wang W, et al. A novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology. 2016;5(6):e1147641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.NCCN. Merkel Cell Carcinoma (Version 2.2019) Available: https://merkelcell.org/wp-content/uploads/2017/10/MCC_v.2.2019-2.pdf. Accessed January 31, 2020.
- 116.Becker JC, Stang A, DeCaprio JA, et al. Merkel cell carcinoma. Nat Rev Dis Primers. 2017;3:17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Topalian SL, Bhatia S, Hollebecque A, et al. Abstract CT074: Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in Merkel cell carcinoma (MCC). Cancer Research. 2017;77(13 Supplement):CT074. [Google Scholar]
- 118.Nghiem P, Bhatia S, Lipson EJ, et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J Clin Oncol. 2019;37(9):693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):155–168. [DOI] [PubMed] [Google Scholar]
- 120.Zundler S, Neurath MF. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015;26(5):559–568. [DOI] [PubMed] [Google Scholar]
- 121.Chiocca EA, Yu JS, Lukas RV, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med. 2019;11(505). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bekaii-Saab TS, Roda JM, Guenterberg KD, et al. A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol Cancer Ther. 2009;8(11):2983–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.SP K. Sample Size Calculator Available: https://clincalc.com/stats/samplesize.aspx.
- 124.Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology. 2016;17(10):1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pastrana DV, Tolstov YL, Becker JC, et al. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009;5(9):e1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Asa SL, Dardick I, Van Nostrand AW, et al. Primary thyroid thymoma: a distinct clinicopathologic entity. Hum Pathol. 1988;19(12):1463–1467. [DOI] [PubMed] [Google Scholar]
- 127.Vantomme V, Dantinne C, Amrani N, et al. Immunologic analysis of a phase I/II study of vaccination with MAGE-3 protein combined with the AS02B adjuvant in patients with MAGE-3-positive tumors. J Immunother. 2004;27(2):124–135. [DOI] [PubMed] [Google Scholar]
- 128.Harris JE, Ryan L, Hoover HC Jr., et al. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group Study E5283. J Clin Oncol. 2000;18(1):148–157. [DOI] [PubMed] [Google Scholar]
- 129.Berger M, Kreutz FT, Horst JL, et al. Phase I study with an autologous tumor cell vaccine for locally advanced or metastatic prostate cancer. J Pharm Pharm Sci. 2007;10(2):144–152. [PubMed] [Google Scholar]
- 130.Fishman M, Hunter TB, Soliman H, et al. Phase II trial of B7–1 (CD-86) transduced, cultured autologous tumor cell vaccine plus subcutaneous interleukin-2 for treatment of stage IV renal cell carcinoma. J Immunother. 2008;31(1):72–80. [DOI] [PubMed] [Google Scholar]
- 131.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24(19):3089–3094. [DOI] [PubMed] [Google Scholar]
- 132.Maciag PC, Radulovic S, Rothman J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: a Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine. 2009;27(30):3975–3983. [DOI] [PubMed] [Google Scholar]
- 133.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–2788. [DOI] [PubMed] [Google Scholar]
- 134.Schaub SK, Stewart RD, Sandison GA, et al. Does Neutron Radiation Therapy Potentiate an Immune Response to Merkel Cell Carcinoma? International Journal of Particle Therapy. 2018;5(1):183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cotter SE, Dunn GP, Collins KM, et al. Abscopal effect in a patient with metastatic Merkel cell carcinoma following radiation therapy: potential role of induced antitumor immunity. Arch Dermatol. 2011;147(7):870–872. [DOI] [PubMed] [Google Scholar]