

Abstract
Chemical warfare nerve agent exposure leads to status epilepticus (SE) that may progress to epileptogenesis and severe brain pathology when benzodiazepine treatment is delayed. We evaluated the dose–response effects of delayed midazolam on toxicity induced by soman (GD) in the plasma carboxylesterase knockout (ES1−/−) mouse, which, similar to humans, lacks plasma carboxylesterase. Initially, we compared the median lethal dose (LD50) of GD exposure in female ES1−/− mice across estrous with male mice and observed a greater LD50 during estrus compared with proestrus or to males. Subsequently, male and female GD-exposed ES1−/− mice treated with a dose range of midazolam 40 min after seizure onset were evaluated for survivability, seizure activity, and epileptogenesis. GD-induced neuronal loss and microglial activation were evaluated 2 weeks after exposure. Similar to our previous observations in rats, delayed treatment with midazolam dose-dependently increased survival and reduced seizure severity in GD-exposed mice, but was unable to prevent epileptogenesis, neuronal loss, or gliosis. These results suggest that midazolam is beneficial against GD exposure, even when treatment is delayed, but that adjunct therapies to enhance protection need to be identified. The ES1−/− mouse GD exposure model may be useful to screen for improved medical countermeasures against nerve agent exposure.
Keywords: chemical warfare nerve agent, midazolam, refractory status epilepticus, carboxylesterase knockout mouse, epileptogenesis
Introduction
Exposure to chemical warfare nerve agents (CWNA) can lead to an acute hyperactivation of the cholinergic system with physiological repercussions that include miosis, hypersecretions, muscle fasciculation, severe seizures progressing to status epilepticus (SE), and fatal respiratory complications (reviewed in Ref. 1). Standard treatment available in the form of autoinjectors includes a muscarinic acetylcholine (ACh) receptor antagonist (e.g., atropine sulfate), an oxime to reactivate nonfunctional synaptic acetylcholinesterase (AChE; e.g., 2-PAM or HI-6), and a benzodiazepine (e.g., diazepam or midazolam) as an anticonvulsant. Though effective at terminating seizure when administered promptly, the anticonvulsant efficacy of benzodiazepines decreases when such treatment is delayed. Prolonged SE brings about changes in the composition of neuronal excitatory and inhibitory receptors that underlie the mechanism of their progression to pharmacoresistant seizures. The internalization of synaptic γ-aminobutyric acid (GABA) receptors is believed to underlie the time-dependent reduction of the anticonvulsant potency of benzodiazepines (reviewed in Refs. 2–4). In line with the latter mechanism, in both rats and guinea pigs, the benzodiazepines midazolam and diazepam are less effective in terminating GD- or sarin-induced seizure when treatment is delayed to 30–60 min after seizure activity when compared with treatment 1–5 min after exposure; in addition, higher doses are needed to alleviate seizure activity.5–8 The delayed administration of midazolam or diazepam to GD- or sarin-exposed rats does not prevent the development of spontaneous recurrent seizure (SRS), or prevent the brain damage, neuroinflammatory responses, and behavioral deficits caused by CWNA-induced seizure.3, 4, 8–15 However, although delayed midazolam treatment fails to quickly terminate seizure and is not fully neuroprotective, it may reduce seizure severity in the first hour after treatment and dose-dependently reduce mortality compared with rats that were not given midazolam.3, 15, 16
Although the rat and other rodent models of GD exposure have traditionally been used for the identification of novel therapies that counteract the aforementioned effects of CWNA-induced SE, the presence of plasma carboxylesterase activity in such animals may confound results. Because GD binds irreversibly to carboxylesterase it reduces the systemic concentration of the organophosphorus agent that is freely available to inhibit AChE,17 thereby providing some protection against lethal doses of certain organophosphorus compounds, including GD.18 This species difference in the expression of plasma carboxylesterase has been eliminated by the development of a conditional carboxylesterase gene knockout (ES1−/−) mouse that specifically lacks the activity of the enzyme in plasma, while maintaining its activity in other tissues.19 Delayed midazolam administered 15 min after seizure onset in male ES1−/− mice exposed to high-dose GD is unable to rapidly terminate acute seizure activity or prevent SRS, brain pathology, or microglial activation.20
Much of the research for the identification of novel medical countermeasures makes use of adult male animal models that may not necessarily represent the range of responses to CWNA exposure of a heterogeneous population. In general in preclinical models, females are more sensitive to the toxic effects of exposure to toxic chemicals.21 However, the effects of sex are only sporadically reported in the literature and are, at times, contradictory, as age, route of exposure, species or stage of estrous may impact the outcome. A few studies report female rats to be more sensitive to the lethal effects of GD or sarin compared with male rats.22–26 The aforementioned studies, however, were conducted without taking into consideration the estrous cycle stage at the moment of CWNA exposure, which may impact response to toxic exposure. In rats, the lethal effects of subcutaneous (SC) exposure to sarin vary depending on the stage of the estrous cycle, with female rats having lower susceptibility during proestrus.27 The etiology for the observed sexual dimorphism in CWNA toxicity is not fully understood, but the inclusion of female subjects in preclinical research of medical countermeasures against CWNA exposure should be of paramount importance as they are in general more susceptible to the toxic effects (reviewed in Ref. 21) and as sex may influence neuropathological outcomes and the effectiveness of therapeutics.
In the present work, we expand our previous studies and report on the dose-response effects of delayed midazolam treatment in male and female ES1−/− mice when midazolam is administered at 40 min after the onset of SE. The extended treatment time point is to model a more realistic therapeutic window for first responders in the event of civilian casualties following CWNA exposure. Results from this study provide the groundwork for a human-relevant mouse model to use to identify adjunct therapies that enhance or complement the effectiveness of benzodiazepines against CWNA exposure.
Materials and methods
Animals
Male (24–−28 g) and female (17–22 g) ES1−/− mice were obtained at 8–9 weeks of age from a breeding colony established at the United States Army Medical Research Institute of Chemical Defense (USAMRICD). Animals were single-housed, with food and water available ad libitum, on a 12 h:12 h light-dark cycle with lights on at 0600. Determination of estrous cycle stage was performed by vaginal cytology approximately 1 h before exposure, following previously described methods.28 Prior to exposure, cage bedding was removed and replaced an iso-PAD™. Mice were weighed daily. The experimental protocol was approved by the Institutional Animal Care and Use Committee at USAMRICD, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89–544), as amended.
Experiment 1: Median lethal (LD50) dose of GD across estrous and comparison with male ES1−/− mice
Prior to exposure, mice were divided into one of three groups: estrus, diestrus, or proestrus. To determine the LD50 of GD, female ES1−/− mice were exposed SC to GD (5 mL/kg) in a stage-wise adaptive dose design using previously described methods.29 The LD50 was estimated using a probit transformation in a nonlinear regression analysis with maximum likelihood procedures to the combined data from all dosing stages. For each estrous stage, the stage-wise approach of exposures continued until the ratio of the half-width of the 95% confidence interval (CI) (CI: defined as (Upper bound – Lower bound)/(2 × LD50)) for the LD50 was less than 0.40. The 95% CI was calculated using the delta method. The LD50 in female ES1−/− mice was compared with the previously reported LD50 in male ES1−/− mice.20
Experiment 2: Dose–response evaluation of midazolam against GD exposure in male and female ES1−/− mice
Surgeries and EEG recording.
Mice were implanted under 2–5% isoflurane with ETA-F10 telemetry transmitters from Data Sciences International (DSI™; St. Paul, MN) to record an electroencephalogram (EEG) and body temperature as previously described.30 Briefly, after SC implantation of transmitters, wires were wrapped around two cortical stainless steel screw electrodes placed at 1.5 mm right of the midline, and 1.5 mm anterior, and 3.0 mm posterior to bregma. Buprenex® SR (ZooPharm) was administered (0.5 mg/kg, SC) to minimize pain. All mice were given 1–2 weeks of recovery from surgery before exposure to saline or GD. EEG data were continuously recorded at least 24 h before GD exposure up to study endpoint (14 days after exposure) using Dataquest ART Acquisition software (DSI™) and digitized at 250 Hz to evaluate seizure onset, acute seizure, and the development of SRS for 2 weeks after exposure.
GD exposures and treatments.
Male (n = 37) and female (n = 28) ES1−/− mice were exposed SC to either saline (No GD; control group) or 82 μg/kg GD (pinacolyl methylphosphonofluoridate; the U.S. Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD), as previously described.20 GD-exposed mice were treated intraperitoneally (IP) with an admix of atropine sulfate (4 mg/kg) and HI-6 (50 mg/kg) at 1 min after exposure, whereas saline-exposed mice received saline injections in lieu of an atropine/HI-6 combination since atropine dries secretions and may lead to complications in animals not exposed to agent. Following exposures, EEG activity was monitored in real-time, and seizure onset was defined as the appearance of rhythmic high-amplitude spikes (>2 × baseline) that lasted at least 10 s (based on Ref. 31). Midazolam (1, 3, or 9 mg/kg; IP) was administered at 40 min after seizure onset.
EEG scoring and power spectra analysis.
EEG activity was processed as previously described.20 Briefly, seizure activity was determined in 2-s epochs following detection thresholds that were based on the slope of a linear robust fit applied to a fast Fourier transform (0.1 Hz resolution with Nyquist frequency of 125 Hz) and normalized power spectra (0.1–10 Hz) during 24 h of baseline. The EEG signal was filtered using a Butterworth filter (passband of 0.1–125 Hz; notch filter of 60 Hz), as previously described.32 During a seizure event, the slope of the linear robust fit increases because of an increase in the power spectra magnitude and a shift of dominant frequencies. When both power spectra and slope reach threshold, the 2-s epochs are marked as potential seizures. All events identified during processing were visually inspected by a blinded and unbiased scorer to either confirm or reject their classification as seizures. A 60-s epoch was used to integrate the power spectra calculated through fast Fourier transform and obtain power spectrum density (μV2/Hz; 0.1–125 Hz). Finally, full EEG power spectrum data were further reduced by extracting the median power (20 min bins) in 60-min intervals to obtain EEG power spectral density values at baseline (24 h prior to GD or saline exposure), SE (20 min before treatment), and 1 and 6 h after treatment.
Brain tissue collection and immunohistochemistry.
Two weeks after exposure, mice were injected with sodium pentobarbital (75 mg/kg, IP, Fatal Plus®; Patterson Veterinary) and perfused with heparinized phosphate buffer followed by a 4% paraformaldehyde solution as previously described.20 Brains were removed and kept in 4% paraformaldehyde for 6 h, after which time the solution was changed to 20% sucrose for at least 72 h for cryoprotection of the tissue. Sectioning and staining of tissue was performed by FD Neurotechnologies (Columbia, MD) using methods described in Ref. 33. Brain sections were coronally cut at a thickness of 30 μm, and immunohistochemistry was performed using antibodies against the neuronal nuclear protein (NeuN mouse anti-NeuN IgG 1:600; Millipore, Billerica, MA) and the ionized calcium–binding adaptor molecule 1 (Iba1; rabbit anti-Iba1 IgG 1:6,000; Wako Chemicals, Richmond, USA) to visualize neurons and microglia, respectively. For Iba1, cresyl violet was used as a counterstain for visualization of anatomic landmarks.
Cell counts.
Immunostained slides were scanned using an Olympus BX61IVS microscope with a Pike F-505 camera (Allied Vision, Exton, PA). Image-Pro® Plus (Media Cybernetics, Inc., Rockville, MD) was used to trace regions of interest (the lateral thalamus, medial thalamus, basolateral amygdala, layer III of the piriform cortex, and the hilus region of the hippocampus) in coverslip-mounted brain slices and obtain counts of NeuN-positive cells using the particle analysis function. For analysis of highly dense NeuN-positive neurons in the CA1 region of the hippocampus, stereology using Stereo Investigator® software (MBF Bioscience) was performed in five sections per mouse using the optical fractionator method.34 Analysis of the density and changes in morphology (i.e., cell body-to-cell size ratio) of Iba1-positive cells, an indication of microglial cell activation,35, 36 was performed in brain regions using the ImageJ software (National Institutes of Health, Bethesda, MD) and methods modified from Ref. 35 and described in Ref. 20. Brain regions evaluated included the lateral thalamus, medial thalamus, CA1 region of the hippocampus, basolateral amygdala, and layer III of the piriform cortex. For each brain tissue slice, areas of interest were traced using anatomic landmarks in the region between −1.06 to −1.94 mm from bregma.
Data analysis
To identify significant differences in LD50 values among GD-exposed female mice in different stages of the estrous cycle (proestrus, estrus, and diestrus) and male mice, the ratio of the LD50 from each group was calculated along with a 95% CI for the ratio. A significant difference between LD50s was identified when the 95% CI of the ratio did not include the value of 1. To determine the main effect of sex, midazolam dose, and time on the severity of GD-induced body weight, body temperature, and EEG power spectral density, a general linear model analysis with a repeated measures paradigm was used. Cox regression analysis was performed on survival data to determine the effects of sex and midazolam dose on survival over time, followed by Kaplan–Meier analysis and log-rank test for pairwise group comparisons. The effects of sex and midazolam dose on the percent of survival at study endpoint were evaluated by logistic regression followed by a Chi-square or Fisher’s exact test for dose group comparisons. Cox regression analysis was performed on the time to the onset of the first SRS of animals to determine the effect of sex and midazolam dose on epileptogenesis incidence curves. A two-way ANOVA was used to determine the effects of sex, midazolam dose, and their interaction on the duration of seizure activity, number of SRS, NeuN, and Iba1 cell density, as well as changes in cell body-size-to-cell-size ratio of Iba1-immunoreactive cells. Tukey’s test was used for post-hoc analysis. Differences were considered statistically significant when P < 0.05.
Results
Median lethal (LD50) dose of GD in female and male ES1−/− mice
We have previously determined the dose-lethality response distribution of SC exposure to GD in male ES1−/− mice,20 which is shown for comparison purposes (Fig. 1 and Table 1). Data from the present dose–lethality response study show that at 23.6 μg/kg, the estimated LD50 in female ES1−/− mice at estrus is significantly higher than that at proestrus (19.8 μg/kg) and also higher compared with males (19.2 μg/kg). See Table 1 for the GD LD10, LD50, and LD90 as well as the 95% CI and the slope of the dose–response curve in male and female mice across estrous.
Figure 1.

Dose–response curve for GD-induced lethality in adult male and female ES1−/− mice. Each point in the graph represents the average estimated percent of lethality for adult female ES1−/− mice exposed SC to a particular dose of GD at different estrous cycle stages. At 23.6 μg/kg (95% CI: 20.8–26.7 μg/kg), the LD50 of GD for female ES1−/− mice in estrus (n = 17) was 1.14 and 1.19 times higher than that of female ES1−/− mice in diestrus (n = 18) and proestrus (n = 14), respectively. The LD10 and LD90 for mice in estrus, diestrus, and proestrus are also shown in the graph. The dose–lethality curve for GD toxicity in male ES1−/− mice (gray diamond) from Ref. 20 was used for comparison.
Table 1.
The 24-h LD50 and 95% CI for male and female mice SC exposed to soman (GD)
| Group | LD10 (μg/kg) (95% CI) |
LD50 (μg/kg) (95% CI) |
LD90 (μg/kg) (95% CI) |
Slope |
|---|---|---|---|---|
| Female | ||||
| Proestrus | 16.6 (13.5–20.3) | 19.8 (17.7–22.1)* | 23.6 (19.4–28.6) | 16.7 |
| Estrus | 19.4 (16.0–23.4) | 23.6 (20.8–26.7) | 28.7 (22.9–36.0) | 15.0 |
| Diestrus | 17.8 (15.3–20.7) | 20.8 (18.9–22.8) | 24.2 (20.1–29.2) | 19.1 |
| Male | 17.2 (15.2–19.5) | 19.2 (18.0–20.5)* | 21.4 (19.1–24.0)* | 27.1 |
P < 0.05 compared with estrus
Survival, body temperature and weight loss following GD exposure in mice treated with delayed midazolam
Assessment of survival data by Cox regression analysis revealed a significant effect of midazolam dose on mean survival in 14 days following GD exposure, with no effect of sex and no interaction between sex and dose. Since no effect of sex, survival data of male and females were subsequently combined for each treatment group to perform multiple group comparisons. Kaplan–Meier analysis with a log-rank test detected a dose-dependent increase in mean survival times when midazolam was given as a delayed anticonvulsant treatment after GD exposure (Fig. 2). Mice exposed to GD and treated with 1 mg/kg of midazolam showed a mean survival time of 3.3 days (95% CI: 0.2–6.3 days), which was significantly different from the mean survival times of No GD (14 days (all animals survived up to endpoint, so no 95% CI is reported), P < 0.001) and mice that were exposed to GD and received 9 mg/kg of midazolam (11.5 days (95% CI, 9–14 days); P < 0.01). Mice exposed to GD and treated with 3 mg/kg of midazolam also showed a mean survival time 7.5 days (95% CI: 5.1–9.9 days) that was statistically different (P < 0.01) from the No GD group and from mice that were exposed to GD and received 9 mg/kg of midazolam (P < 0.05). The mean survival time of the group that was exposed to GD and treated with 9 mg/kg midazolam was not significantly different from the mean survival time of the No GD control group. At the time of study endpoint, GD-exposed mice that received 1, 3, and 9 mg/kg midazolam showed 18.2%, 46.2%, and 78.6% survival, respectively. All mice in the No GD control group survived up to study endpoint (100% survival). Previous observations of factor effects were corroborated by logistic regression analysis of survival at study endpoint; no significant effects of sex or sex by dose interaction were found. A Chi-square or Fischer’s exact test for dose group comparisons found a significant difference in endpoint survival percentage between No GD and GD-exposed 1 (P < 0.001) and 3 mg/kg (P < 0.01) midazolam treatment groups. Survival for GD-exposed with 9 mg/kg midazolam treatment was not significantly different from the No GD control group. For body temperature, there was a significant interaction between time (average of 60 min bins) and group, independent of sex. Comparisons of the group at each time point revealed that mice exposed to GD and treated with midazolam had reduced body temperature with the highest dose of midazolam (9 mg/kg) resulting in the greatest and more prolonged decrease in body temperature compared with No GD control and to the lower doses of midazolam (Fig. 3). GD-exposed mice also had a significant loss of body weight on days 1–3 after exposure compared with baseline (P < 0.05), with body weight returning to baseline within 1 week of exposure.
Figure 2.
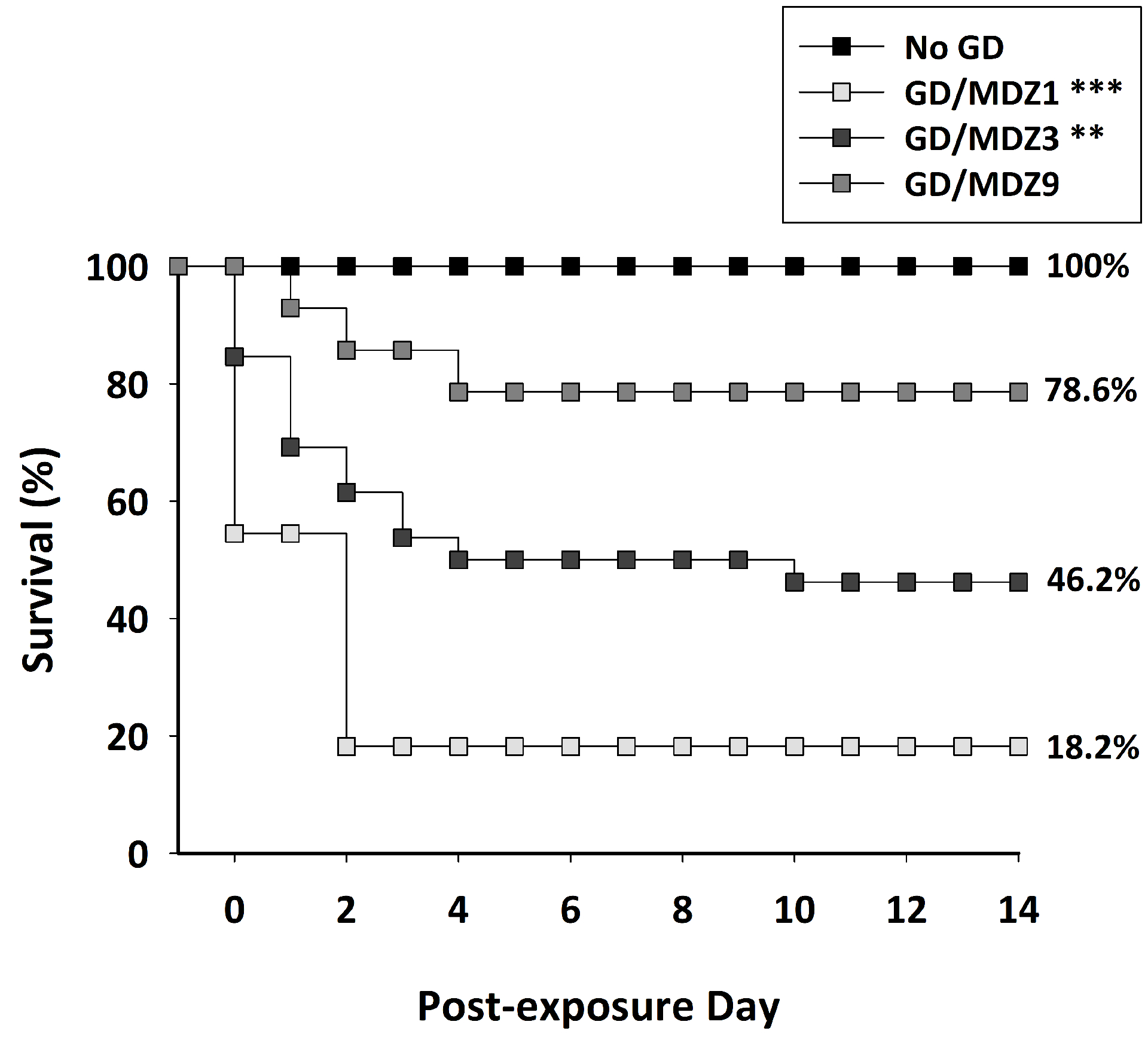
Dose-dependent effect of midazolam (MDZ) on survival following GD-induced status epilepticus in ES1−/− mice. Mice exposed SC to 82 μg/kg of GD and treated with 1 mg/kg MDZ (GD/MDZ1; P < 0.001; n = 11) or 3 mg/kg MDZ (GD/MDZ3; P = 0.001; n = 26) had a significantly lower mean survival time compared with control (No GD; n = 14), while those treated with 9 mg/kg (GD/MDZ9; n = 14) were not significantly different from No GD control. The GD/MDZ1 and GD/MDZ3 groups also had a significantly lower mean survival time compared with the GD/MDZ9 group of mice. The percent of survival at study endpoint is also shown in graph. GD/MDZ1 (***P < 0.001) and GD/MDZ3 (**P < 0.01) had significantly lower survival compared with No GD, while the GD/MDZ9 group was not significantly different from No GD control. The dose-dependent effects of midazolam were independent of sex.
Figure 3.
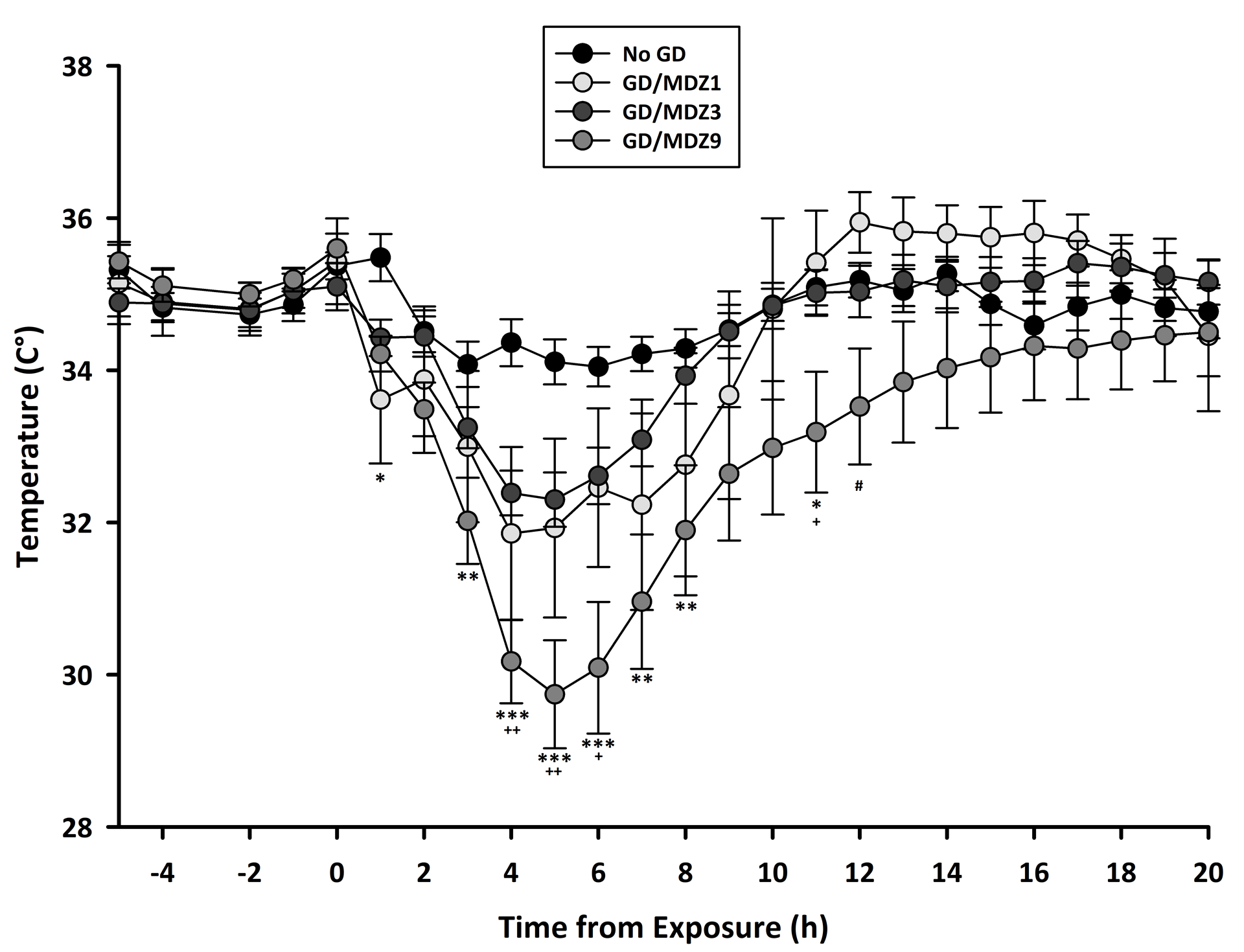
Dose-dependent effect of midazolam (MDZ) on body temperature following GD exposure. Mice exposed SC to 82 μg/kg of GD and treated with MDZ had reduced body temperature in the hours after exposure, with a greater and more prolonged decrease in those that received the highest dose (9 mg/kg) of MDZ (GD/MDZ9; n = 13) compared with those that received 1 mg/kg MDZ (GD/MDZ1; #P < 0.05; n = 6) or 3 mg/kg MDZ (GD/MDZ3; +P < 0.05; ++P < 0.01; n = 16) or with control (No GD; ***P < 0.001; **P < 0.01; *P < 0.05; n = 13) that received 3 mg/kg MDZ, with effects independent of sex. Data shown are combined temperature data in male and female mice. Values shown are hourly averages.
Behavioral and EEG seizure following GD exposure in mice treated with delayed midazolam
Subcutaneous exposure to a seizure-inducing dose of GD resulted in severe cholinergic toxic signs consisting of ataxia, head bobs, and body tremors within 10 min of exposure; these toxic signs reached a maximum median severity of 3 in male and 3.5 in female ES1−/− mice during the first 40 min following exposure. Behavioral seizure progressed into SE in both male and female mice with an average onset of 4.4 ± 1.4 and 3.9 ± 2.2 min, respectively. Administration of all doses of midazolam at 40 min after the onset of GD-induced SE failed to terminate seizure activity. Male ES1−/− mice administered 1, 3, or 9 mg/kg of midazolam spent an average of 873.2 ± 317.9, 622.5 ± 411.9, and 581.0 ± 437.9 min (±SD) in seizure, respectively, during the first 24 h after GD exposure. Seizure activity in female ES1−/− mice during the first 24 h after GD exposure lasted an average of 556.7 ± 320.1 min (± SD) in the group that received 3 mg/kg midazolam and an average of 632.0 ± 545.2 min (± SD) in the group that received 9 mg/kg midazolam. Since female ES1−/− mice that received 1 mg/kg midazolam after GD exposure did not survive past the end of the day of exposure, their average time spent in seizure activity during the first 24 h after exposure is not included. There was no significant effect of sex or midazolam dose on the duration of acute seizure activity following GD exposure.
A full-spectral power analysis of EEG activity was performed following previously described procedures.32 For the EEG power spectral density there was a significant interaction between midazolam dose and the time period following GD exposure. Therefore each time point (SE, 1 and 6 h after SE) was evaluated separately for each dose, and each treatment dose was evaluated separately over time. Comparing groups at each time revealed that GD-exposed mice had increased EEG power spectral density during SE (***P < 0.001; Fig. 4). GD-exposed mice had increased power spectral density compared with No GD during the 1-h time period after midazolam (1, 3, and 9 mg/kg) treatment but during the 6-h posttreatment time period, only the two lower doses of midazolam (1 and 3 mg/kg) were significantly different from No GD, demonstrating that 9 mg/kg of midazolam reduced seizure severity. Evaluating each treatment dose over time revealed that while the 3 and 9 mg/kg midazolam-treated mice had reduced EEG power spectral density at the 1 and 6 h posttreatment times compared with the SE period (+++P < 0.001), the 1 mg/kg midazolam dose was less effective at reducing seizure severity and at the 1-h postexposure time point was not significantly different from the SE period. In sum, there was a dose-dependent decrease in severity in midazolam-treated mice, even when treatment was delayed 40 min after seizure onset as indicated by reduced EEG power spectral density.
Figure 4.
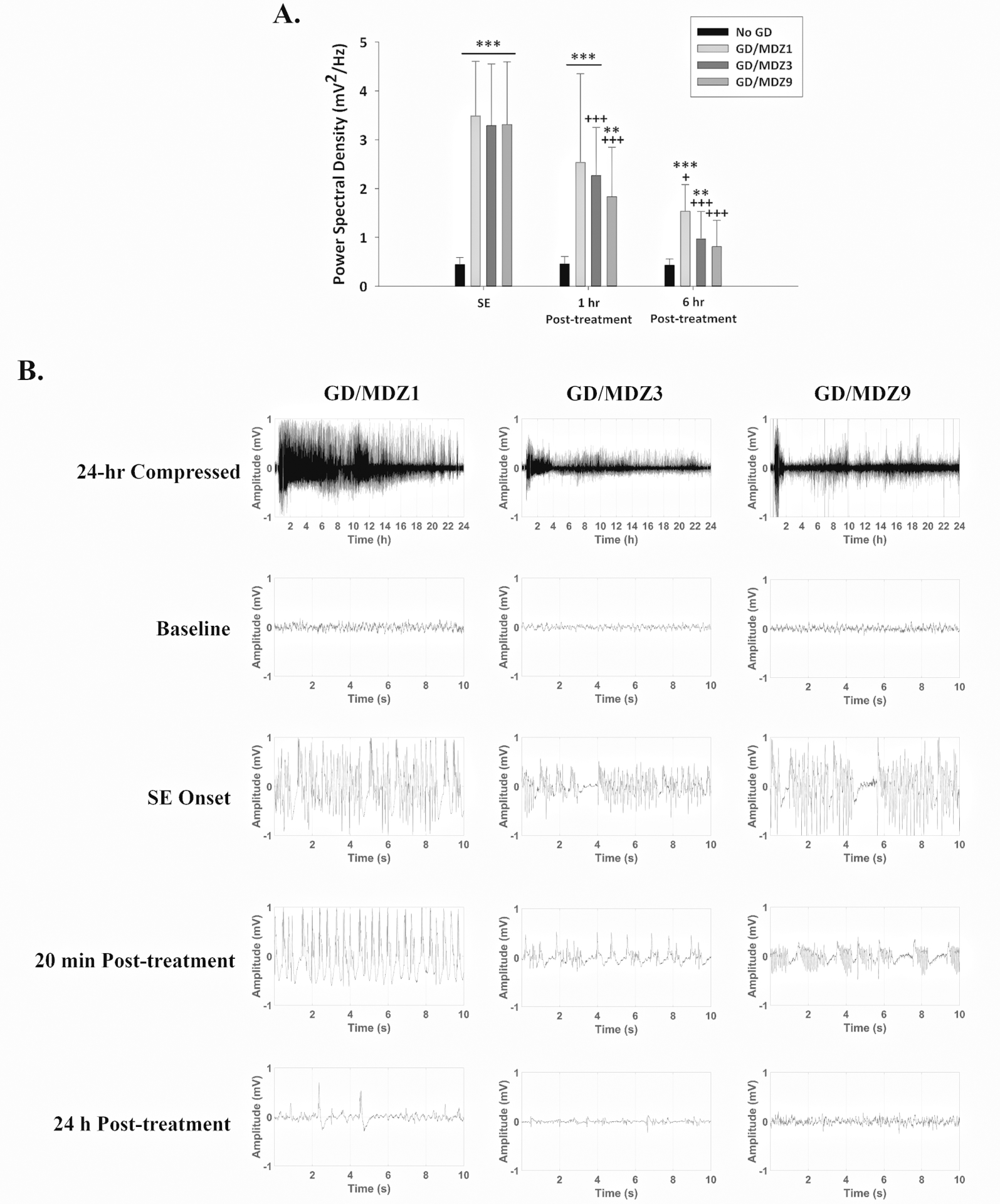
(A) GD (82 μg/kg; SC) exposure increased EEG power spectral density, a marker of seizure severity, during SE, which continued to be increased 1 h after exposure compared with control (No GD; n = 13). Midazolam (3 (GD/MDZ3; n = 19) and 9 (GD/MDZ9; n = 13) mg/kg) reduced EEG power spectral density at 1 and 6 h compared with SE, while 1 mg/kg (GD/MDZ1; n = 5) was only effective at 6 hours. Treatment with 9 mg/kg MDZ reduced EEG power spectral density 6 h after GD exposure and was not significantly different from the No GD control at this time. ***P < 0.001; **P < 0.01 significantly different from No GD. +P < 0.05, +++P < 0.001 significantly different from SE time point after MDZ treatment. (B) Representative EEG tracings of baseline (prior to GD exposure), the onset of SE, 20 min and 24 h after MDZ treatment.
Midazolam did not prevent the development of SRS in the days following GD-induced SE (Fig. 5). In surviving GD-exposed male ES1−/− mice that showed GD-induced SE, 87.5% (n = 7 out of 8) and 75% (n = 3 out of 4) of those receiving 3 and 9 mg/kg of midazolam, respectively, developed SRS (Fig. 5A). Epileptogenesis was also observed in surviving GD-exposed female ES1−/− mice; an incidence of 100% (n = 4 out of 4) was observed in the group that received 3 mg/kg of midazolam, while 83.3% (n = 5 out of 6) of female mice treated with 9 mg/kg of midazolam developed SRS. No effect of sex or midazolam dose on the SRS incidence was observed. In animals that developed SRS, there was no effect of sex or midazolam dose in the number of SRS detected in 2 weeks following GD-induced SE (Fig. 5B); we observed an average (± SD) of 11.7 ± 11.7 SRS in males in the 3 mg/kg midazolam group, 11.3 ± 9.3 SRS in males in the 9 mg/kg midazolam group, 10.5 ± 2.4 SRS in females in the 3 mg/kg midazolam group, and 6.8 ± 5.8 events in females in the 9 mg/kg midazolam group.
Figure 5.
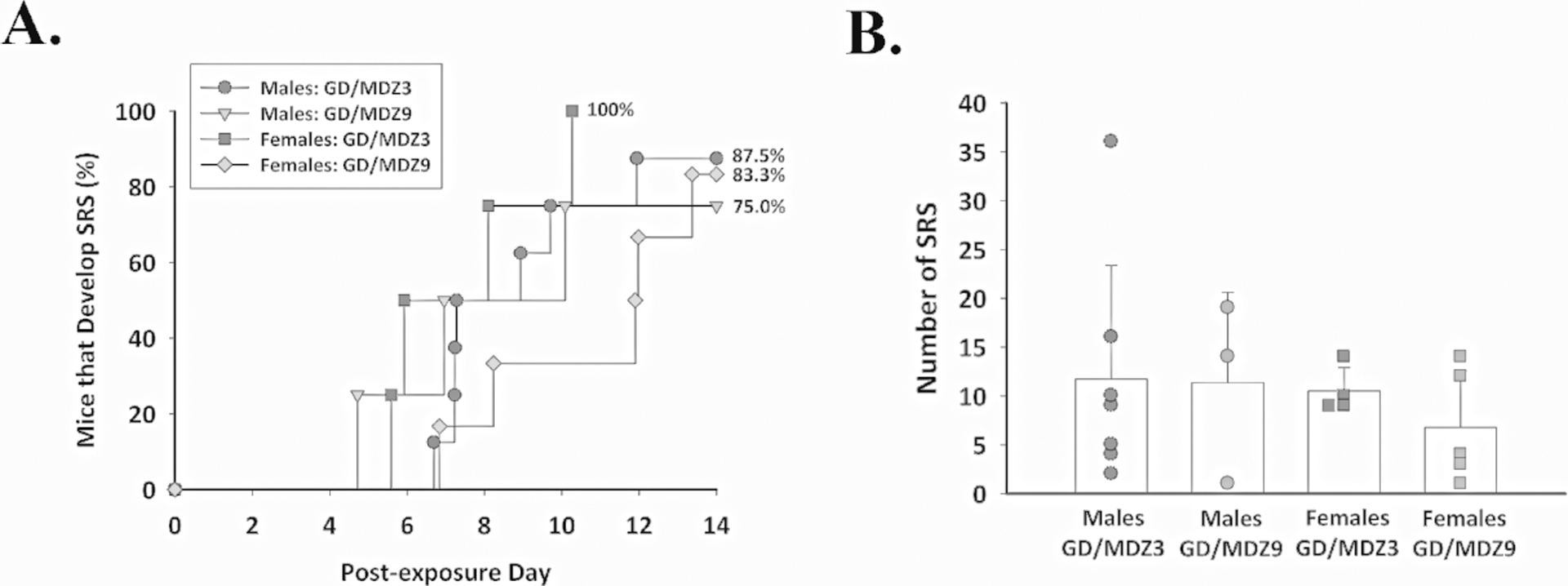
Delayed midazolam (MDZ) failed to prevent the development of spontaneous recurrent seizures (SRS) following GD-induced status epilepticus, independent of sex. Male and female ES1−/− mice were exposed to 82 μg/kg of GD and treated with 3 mg/kg (GD/MDZ3; n = 11) or 9 mg/kg (GD/MDZ9; n = 10) of MDZ at 40 min after seizure onset, and EEG activity was monitored for 14 days after exposure. (A) The onset of the first detected SRS for each surviving animal is graphed to indicate the percentage of mice in each group that developed SRS in the days following GD-induced SE and delayed MDZ treatment. (B) The average (± SD) number of SRS events is graphed for each group.
Neuronal cell survival and microglia activation 2 weeks after GD exposure in mice treated with delayed midazolam
GD-exposed ES1−/− mice treated with delayed midazolam (3 or 9 mg/kg) had a loss of neurons 2 weeks after GD exposure, compared with no agent control mice (Fig. 6). There was no effect of sex on neuronal cell density in any of the brain regions analyzed. Significant loss of neurons occurred in the dorsomedial thalamus (3 and 9 mg/kg, P < 0.001), dorsolateral thalamus (3 and 9 mg/kg, P < 0.001), basolateral amygdala (3 and 9 mg/kg, P < 0.001), layer 3 of the piriform cortex (P < 0.001, 3 mg/kg; P = 0.01, 9 mg/kg), and the hilus of the hippocampus (3 and 9 mg/kg, P < 0.001) of midazolam-treated GD-exposed mice, demonstrated by fewer NeuN-positive cells, compared with the no agent controls. Using stereology, loss of neurons was also confirmed in the CA1 of the hippocampus in GD-exposed mice treated with 3 (P = 0.001) and 9 mg/kg (P < 0.01) of midazolam.
Figure 6.

Midazolam (MDZ) treatment delayed to 40 min after GD-induced seizure failed to prevent neuronal loss in ES1−/− mice, independent of sex. (A) Coronal section in control (No GD) mouse showing regions of interest. (B) The average (± SD) density of NeuN-positive cells is shown for the dorsomedial thalamus (Med Thalamus), dorsolateral thalamus (Lat Thalamus), basolateral amygdala (Amygdala), layer 3 of the piriform cortex, and the CA1 of the hippocampus in coronal sections in the bregma range from −1.28 to −1.64 millimeters. Data shown is combined male and female mice exposed SC to GD (82 μg/kg) and treated with 3 mg/kg MDZ (GD/MDZ3; n = 10) or 9 mg/kg MDZ (GD/MDZ9; n = 11) at 40 min after seizure onset compared with No GD (n = 14). (C) Representative images of NeuN-immunostained brain samples are shown. **P < 0.01, and ***P < 0.001, compared with control.
GD-exposed ES1−/− mice treated with delayed midazolam (3 or 9 mg/kg) had increased microgliosis and microglial cell activation 2 weeks after GD exposure, compared with no agent control mice (Fig. 7A). GD-exposed mice showed Iba1-positive cells with significantly higher cell body-to-cell size ratio in the dorsomedial thalamus (3 and 9 mg/kg, P < 0.001), dorsolateral thalamus (3 and 9 mg/kg, P < 0.001), basolateral amygdala (3 and 9 mg/kg, P < 0.001), the piriform cortex (3 and 9 mg/kg, P < 0.001), and the CA1 region of the hippocampus (3 and 9 mg/kg, P < 0.001) compared with no agent control mice (Fig. 7B). In the CA1 region, we detected a lower average cell body-to-cell size ratio in the 9 mg/kg midazolam group compared with the 3 mg/kg midazolam group (P < 0.05). An increase in microgliosis was also observed in GD-exposed mice, demonstrated by an increase in the density of Iba1-positive cells in the dorsomedial thalamus (3 mg/kg, P < 0.01; 9 mg/kg, P < 0.001), dorsolateral thalamus (3 and 9 mg/kg, P < 0.001), basolateral amygdala (3 and 9 mg/kg, P < 0.001), piriform cortex (3 mg/kg, P < 0.01; 9 mg/kg, P < 0.05), and the CA1 region of the hippocampus (3 mg/kg, P < 0.001; 9 mg/kg, P < 0.01) compared with no agent control mice (Fig. 7C). There were no sex differences in microglial morphological and density responses.
Figure 7.
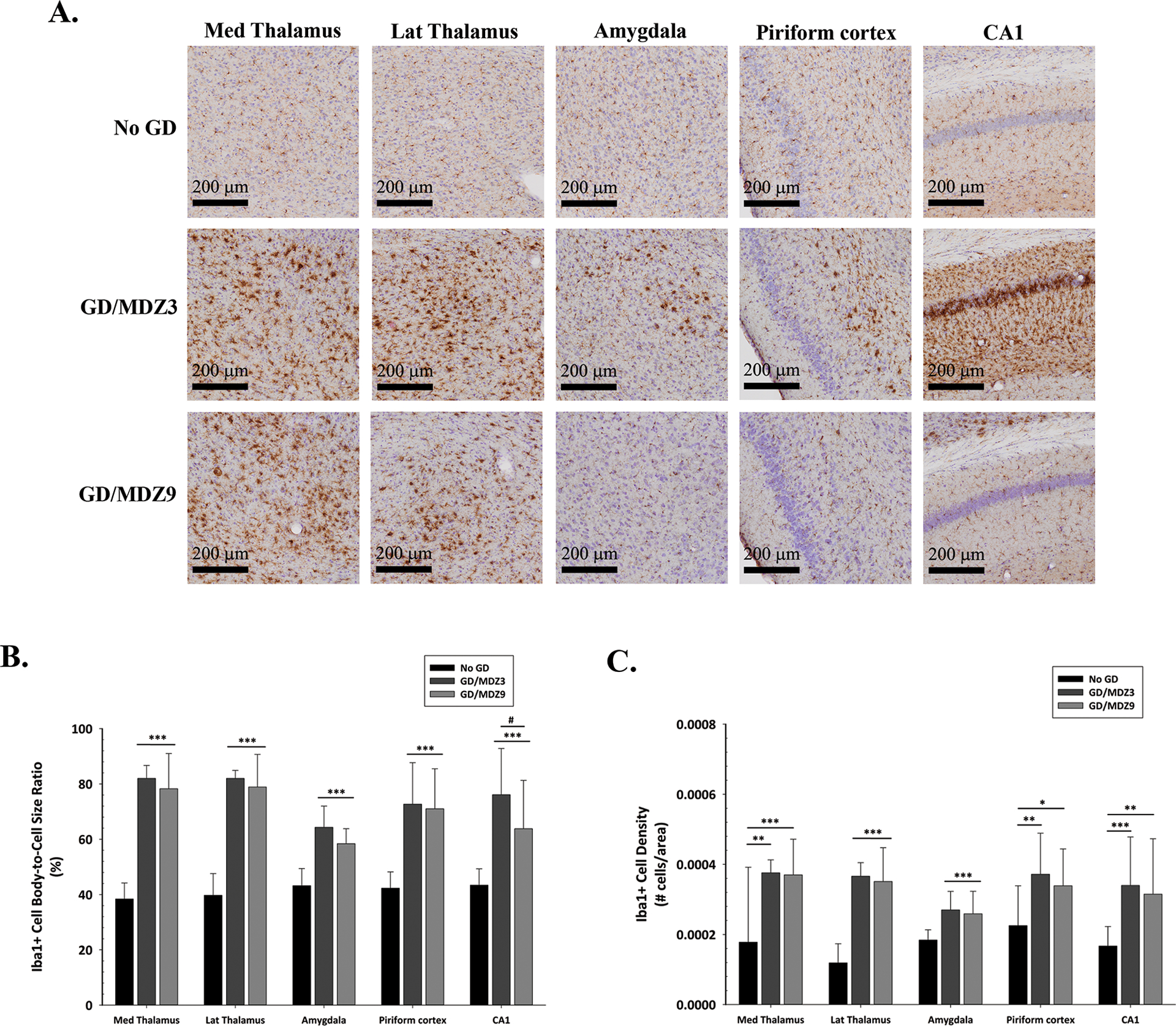
Midazolam (MDZ) treatment delayed to 40 min after GD-induced seizure failed to prevent microglial activation in ES1−/− mice, independent of sex. (A) Representative images of Iba1-immunostained brain samples from mice exposed SC to GD (82 μg/kg) that received 3 mg/kg MDZ (GD/MDZ3; n = 10) or 9 mg/kg (GD/MDZ9; n = 11) of MDZ 40 min after seizure onset, compared with control (No GD; n = 14) mice. (B) The average (± SD) cell-body-to-cell-size ratios and (C) density of ionized calcium–binding adaptor molecule 1 (Iba1), a marker for microglia, are shown for the dorsomedial thalamus (Med Thalamus), dorsolateral thalamus (Lat Thalamus), basolateral amygdala (Amygdala), layer 3 of the piriform cortex, and the CA1 of the hippocampus. Cresyl violet (purple) counterstain was used for visualization of anatomic landmarks. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the No GD group. #P < 0.05, GD/MDZ3 compared with the GD/MDZ9 group.
Discussion
We report on the dose–response efficacy of midazolam in reducing acute and subchronic effects of GD-induced SE in male and female ES1−/− mice when treatment is administered at a delayed time point. Benzodiazepines are commonly used as the first line of pharmacological intervention against seizure activity as they greatly improve survival. Our current study demonstrates that midazolam treatment delayed to 40 min after seizure onset dose-dependently increases survival following GD exposure in male and female ES1−/− mice. Similar to humans ES1−/− mice lack plasma carboxylesterase and may be a useful animal model to predict the efficacy of medical countermeasures in humans. The percent of survival of ES1−/− mice following GD-induced SE was dependent on the dose of midazolam that was administered, with 9 mg/kg providing the highest survival. These findings are in agreement with our previous findings in male rats in which midazolam (1–9 mg/kg, IP) at 40 min after GD-induced SE dose-dependently increased survival.15 Survival data from the present mouse study show that when midazolam treatment is delayed to 40 min after SE onset, a dose of at least 9 mg/kg is needed to obtain a survival percentage (78.6%) that is comparable to the results of survival (66.7%) from a prior study in which 5 mg/kg (IP) of midazolam was administered 15 min after SE in male mice.20 The current study to include a delay of 40 min for the administration of midazolam after the onset of CWNA-induced seizure is highly relevant to a time point when treatments may be provided by first responders in a mass casualty CWNA attack.
These results are also in line with previous studies in GD-exposed guinea pigs that required higher doses of midazolam in response to an increase in treatment delay to increase the efficacy of seizure control and survival.6, 16 Although seizure duration was not reduced by midazolam in the current study, high-dose midazolam treatment reduced EEG power spectral density, a marker of seizure severity, while 1 mg/kg midazolam was ineffective at reducing seizure severity in the first hour after treatment. This finding is similar to previous observations in GD-exposed rats in which midazolam (3 mg/kg) reduced power spectral density during the first hour following treatment.15 In the present study, survival data throughout 14 days following GD-induced SE represent a key contrast from former studies that focused on the lethality effect during the first 24 h after the toxic insult. Although several animal models of chemically-induced SE have shown that the anticonvulsive effectiveness of benzodiazepines significantly decreases with increasing delay in administration,37 our current findings demonstrate the benefit to midazolam administration even when treatment is delayed in a human-relevant mouse model.
Consistent with our former observations in rats,7, 11, 15 the present study demonstrated that in GD-exposed ES1−/− mice, midazolam treatment delayed to 40 min after seizure onset was unable to significantly reduce seizure activity, or prevent epileptogenesis or neuronal loss. The current findings are also in agreement with our previous study in male ES1−/− mice in which midazolam (5 mg/kg) at 15 min after GD-induced seizure onset was unable to prevent SRS or brain pathology.20 The current study is novel in that, in addition to including a dose range analysis of midazolam efficacy against GD, it includes both male and female mice. The inclusion of female subjects in preclinical research of medical countermeasures against CWNA exposure should be considered as they are in general more susceptible to the toxic effects (reviewed in Ref. 21).
Limited data on sexually dimorphic responses to CWNA toxicity exist. The only observed sex difference in the current study was on the lethal effects of GD. Female rats are more sensitive than males to lethality resulting from subcutaneous exposure to GD;26 increased susceptibility to GD also occurs in pubertal female rats.38 Female adult rats may be more susceptible to the lethal effects of intramuscular (IM) sarin exposure39 or whole-body exposure to sarin or cyclosarin22, 23, 25; however, our laboratory observed no sex difference in subcutaneous (SC) sarin exposure in rats.23 Interestingly, while female rats are reported to be more sensitive to the lethal effects of GD and sarin,22–26 in multiple mouse strains, female mice are less sensitive to the lethal effects of sarin.40 In the present study, female mice in estrus were less sensitive to the lethal effects of GD toxicity compared with male mice, as well as compared with female mice in proestrus. By contrast, female rats in proestrus are less sensitive to the lethal effects of sarin compared with female rats in estrus, and females do not differ from males.27 One proposed mechanism for this protection during estrus is that brain levels of progesterone and the neurosteroid allopregnanolone are elevated.41 Neurosteroids such as allopregnanolone are modulators of the major inhibitory GABAA receptor (reviewed in Ref. 42), as well as anticonvulsive properties,43, 44 and these effects may contribute to the increased protection against GD lethality that we observed in the female ES1−/− mice in estrus. Further research on species differences in the impact of sex on the toxic outcome of CWNA exposure may be warranted.
Our finding that GD exposure transiently reduced body temperature in ES1−/− mice in the hours after exposure is similar to the effects of GD on body temperature in other mouse strains and rats.11, 45 Atropine, as a standard medical countermeasure in the current study partially antagonizes reduction in body temperature caused by acute GD poisoning.46 In the current study, ES1−/− mice treated with the highest dose of midazolam had a greater and more prolonged reduction in body temperature compared with those that received 1 and 3 mg/kg midazolam and control mice that were not exposed to GD but received only midazolam. In clinical studies, midazolam dose-dependently interferes with thermoregulation.47, 48 Clinically, mild hypothermia reduces mortality and in animal models of epileptic, ischemic and traumatic brain injury is neuroprotective (reviewed in Ref. 49). In pilocarpine-exposed rats, deep hypothermia reduces SE, and moderate hypothermia reduces mortality, prevents the calcium plateau and reduces hippocampal injury.50 Hypothermia alters numerous mechanisms that may contribute to neuroprotection, including a reduction in cerebral metabolic rate and oxygen consumption, alteration of ion pumps and channels, and slowing the release of excitatory neurotransmitters, all of which may inhibit seizure activity.51 Treatment with the benzodiazepine diazepam reduces body temperature,52 and mild hypothermia in combination with the diazepam reduces performant pathway–induced SE. Of potential interest, in rats, mild hypothermia decreases the clearance of midazolam.53
In summary, these findings of midazolam increase in survival and reduction in seizure severity but failure to prevent epileptogenesis or brain pathology support the advantages of midazolam as a medical countermeasure against GD exposure, but also indicate the critical need to identify effective adjunct therapies to mitigate the toxic and neuropathological effects of GD exposure. The present study further supports the ES1−/− mouse GD exposure model as a useful tool to screen for improved medical countermeasures against the effects of acute CWNA exposure. The advancement of improved treatments against CWNA-induced SE, as well as treatments that can be given as adjunct to benzodiazepines, can greatly benefit from well-characterized preclinical animal models of CWNA exposure.
Acknowledgements
This research was supported by NINDs R21 NS103820-02 to Dr. Lucille A. Lumley-Lange and the Geneva Foundation. E.K. and K.W. were supported in part by an appointment to the Research Participation Program for the U.S. Army Medical Research and Development Command administered by the Oak Ridge Institute for Science and Education through an agreement between the U.S. Department of Energy and U.S. Army Medical Research and Development Command. The authors acknowledge Ms. Zora-Maya Keith for her assistance in data compilation and Dr. Linn Cadieux, Ms. Sandra DeBus, Mr. Erik Matson, and Mr. Timothy Barry, II for management of the USAMRICD ES1−/− mouse colony.
Footnotes
Competing interests
The authors declare no competing interests. M.A.F. conducted EEG analysis under a contract with BioSEad but was blinded to the treatment groups.
Disclosure
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
References
- 1.Jett DA & Spriggs SM. 2018. Translational research on chemical nerve agents. Neurobiol Dis: 104335. [DOI] [PubMed] [Google Scholar]
- 2.Niquet J, Baldwin R, Suchomelova L, et al. 2016. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann N Y Acad Sci. 1378(1): 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niquet J, Lumley L, Baldwin R, et al. 2019. Early polytherapy for benzodiazepine-refractory status epilepticus. Epilepsy Behav: 106367. [DOI] [PubMed] [Google Scholar]
- 4.Niquet J, Lumley L, Baldwin R, et al. 2019. Rational polytherapy in the treatment of cholinergic seizures. Neurobiol Dis: 104537. [DOI] [PubMed] [Google Scholar]
- 5.McDonough JH Jr., McMonagle J, Copeland T, et al. 1999. Comparative evaluation of benzodiazepines for control of soman-induced seizures. Arch Toxicol. 73(8–9): 473–8. [DOI] [PubMed] [Google Scholar]
- 6.McDonough JH, McMonagle JD, and Shih TM. 2010. Time-dependent reduction in the anticonvulsant effectiveness of diazepam against soman-induced seizures in guinea pigs. Drug Chem Toxicol. 33(3): 279–83. [DOI] [PubMed] [Google Scholar]
- 7.Schultz MK, Wright LK, Stone MF, et al. 2012. The anticholinergic and antiglutamatergic drug caramiphen reduces seizure duration in soman-exposed rats: synergism with the benzodiazepine diazepam. Toxicol Appl Pharmacol. 259(3): 376–86. [DOI] [PubMed] [Google Scholar]
- 8.Chapman S, Yaakov G, Egoz I, et al. 2015. Sarin-induced brain damage in rats is attenuated by delayed administration of midazolam. Neurotoxicology. 49: 132–8. [DOI] [PubMed] [Google Scholar]
- 9.Chapman S, Kadar T, and Gilat E. 2006. Seizure duration following sarin exposure affects neuro-inflammatory markers in the rat brain. Neurotoxicology. 27(2): 277–83. [DOI] [PubMed] [Google Scholar]
- 10.de Araujo Furtado M, Lumley LA, Robison C, et al. 2010. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia. 51(8): 1503–10. [DOI] [PubMed] [Google Scholar]
- 11.Schultz MK, Wright LK, de Araujo Furtado M, et al. 2014. Caramiphen edisylate as adjunct to standard therapy attenuates soman-induced seizures and cognitive deficits in rats. Neurotoxicol Teratol. 44: 89–104. [DOI] [PubMed] [Google Scholar]
- 12.Moffett MC, Schultz MK, Schwartz JE, et al. 2011. Impaired auditory and contextual fear conditioning in soman-exposed rats. Pharmacol Biochem Behav. 98(1): 120–9. [DOI] [PubMed] [Google Scholar]
- 13.Langston JL, Wright LK, Connis N, et al. 2012. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: increased startle reactivity and perseverative behavior. Pharmacol Biochem Behav. 100(3): 382–91. [DOI] [PubMed] [Google Scholar]
- 14.Marrero-Rosado B, Rossetti F, Rice MW, et al. 2018. Age-Related Susceptibility to Epileptogenesis and Neuronal Loss in Male Fischer Rats Exposed to Soman and Treated With Medical Countermeasures. Toxicol Sci. 164(1): 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumley LA, Rossetti F, de Araujo Furtado M, et al. 2019. Dataset of EEG power integral, spontaneous recurrent seizure and behavioral responses following combination drug therapy in soman-exposed rats. Data Brief. 27: 104629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih TM, Duniho SM, and McDonough JH. 2003. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol Appl Pharmacol. 188(2): 69–80. [DOI] [PubMed] [Google Scholar]
- 17.Maxwell DM, Brecht KM, and O’Neill BL. 1987. The effect of carboxylesterase inhibition on interspecies differences in soman toxicity. Toxicol Lett. 39(1): 35–42. [DOI] [PubMed] [Google Scholar]
- 18.Maxwell DM 1992. The specificity of carboxylesterase protection against the toxicity of organophosphorus compounds. Toxicol Appl Pharmacol. 114(2): 306–12. [DOI] [PubMed] [Google Scholar]
- 19.Duysen EG, Koentgen F, Williams GR, et al. 2011. Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chem Res Toxicol. 24(11): 1891–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marrero-Rosado B, de Araujo Furtado M, Schultz CR, et al. 2018. Soman-induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia. 59(12): 2206–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipnick RL, Cotruvo JA, Hill RN, et al. 1995. Comparison of the up-and-down, conventional LD50, and fixed-dose acute toxicity procedures. Food Chem Toxicol. 33(3): 223–31. [DOI] [PubMed] [Google Scholar]
- 22.Mioduszewski R, Manthei J, Way R, et al. 2002. Interaction of exposure concentration and duration in determining acute toxic effects of sarin vapor in rats. Toxicol Sci. 66(2): 176–84. [DOI] [PubMed] [Google Scholar]
- 23.Wright LK, Lumley LA, Lee RB, et al. 2017. Younger rats are more susceptible to the lethal effects of sarin than adult rats: 24 h LC50 for whole-body (10 and 60 min) exposures. Drug Chem Toxicol. 40(2): 134–139. [DOI] [PubMed] [Google Scholar]
- 24.Wright LKM, Lee RB, Clarkson ED, et al. 2016. Female rats are less susceptible during puberty to the lethal effects of percutaneous exposure to VX. Toxicol Rep. 3: 895–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anthony JS, Haley M, Manthei J, et al. 2004. Inhalation toxicity of Cyclosarin (GF) vapor in rats as a function of exposure concentration and duration: potency comparison to sarin (GB). Inhal Toxicol. 16(2): 103–11. [DOI] [PubMed] [Google Scholar]
- 26.Sket D 1993. Efficacy of antidotes against soman poisoning in female physostigmine-protected rats. Pharmacol Toxicol. 72(1): 25–30. [DOI] [PubMed] [Google Scholar]
- 27.Smith CD, Wright LK, Garcia GE, et al. 2015. Hormone-dependence of sarin lethality in rats: Sex differences and stage of the estrous cycle. Toxicol Appl Pharmacol. 287(3): 253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLean AC, Valenzuela N, Fai S, et al. 2012. Performing vaginal lavage, crystal violet staining, and vaginal cytological evaluation for mouse estrous cycle staging identification. J Vis Exp(67): e4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feder PI, Hobson DW, Olson CT, et al. 1991. Stagewise, adaptive dose allocation for quantal response dose-response studies. Neurosci Biobehav Rev. 15(1): 109–14. [DOI] [PubMed] [Google Scholar]
- 30.Lundt A, Wormuth C, Siwek ME, et al. 2016. EEG Radiotelemetry in Small Laboratory Rodents: A Powerful State-of-the Art Approach in Neuropsychiatric, Neurodegenerative, and Epilepsy Research. Neural Plast. 2016: 8213878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nissinen J, Lukasiuk K, and Pitkanen A. 2001. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus. 11(3): 299–310. [DOI] [PubMed] [Google Scholar]
- 32.de Araujo Furtado M, Zheng A, Sedigh-Sarvestani M, et al. 2009. Analyzing large data sets acquired through telemetry from rats exposed to organophosphorous compounds: an EEG study. J Neurosci Methods. 184(1): 176–83. [DOI] [PubMed] [Google Scholar]
- 33.Hsu SM, Raine L, and Fanger H. 1981. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 29(4): 577–80. [DOI] [PubMed] [Google Scholar]
- 34.Bonthius DJ, McKim R, Koele L, et al. 2004. Use of frozen sections to determine neuronal number in the murine hippocampus and neocortex using the optical disector and optical fractionator. Brain Res Brain Res Protoc. 14(1): 45–57. [DOI] [PubMed] [Google Scholar]
- 35.Hovens IB, Nyakas C, and Schoemaker RG. 2014. A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol Neuroinflammation. 1(2): 82–8. [Google Scholar]
- 36.Tynan RJ, Naicker S, Hinwood M, et al. 2010. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 24(7): 1058–68. [DOI] [PubMed] [Google Scholar]
- 37.Kuruba R, Wu X, and Reddy DS. 2018. Benzodiazepine-refractory status epilepticus, neuroinflammation, and interneuron neurodegeneration after acute organophosphate intoxication. Biochim Biophys Acta Mol Basis Dis. 1864(9 Pt B): 2845–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wright LK, Lee RB, Vincelli NM, et al. 2016. Comparison of the lethal effects of chemical warfare nerve agents across multiple ages. Toxicol Lett. 241: 167–74. [DOI] [PubMed] [Google Scholar]
- 39.Pittel Z, Grauer E, Gez R, et al. 2018. Sex modulated effects of sarin exposure in rats: Toxicity, hypothermia and inflammatory markers. Neurotoxicology. 66: 121–127. [DOI] [PubMed] [Google Scholar]
- 40.Matson LM, Lee-Stubbs RB, Cadieux CL, et al. 2018. Assessment of mouse strain differences in baseline esterase activities and toxic response to sarin. Toxicology. 410: 10–15. [DOI] [PubMed] [Google Scholar]
- 41.Corpechot C, Collins BE, Carey MP, et al. 1997. Brain neurosteroids during the mouse oestrous cycle. Brain Res. 766(1–2): 276–80. [DOI] [PubMed] [Google Scholar]
- 42.Paul SM & Purdy RH. 1992. Neuroactive steroids. FASEB J. 6(6): 2311–22. [PubMed] [Google Scholar]
- 43.Kokate TG, Svensson BE, and Rogawski MA. 1994. Anticonvulsant activity of neurosteroids: correlation with gamma-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 270(3): 1223–9. [PubMed] [Google Scholar]
- 44.Lumley L, Miller D, Muse WT, et al. 2019. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long-term effects of whole-body sarin exposure in rats. Epilepsia Open. 4(3): 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clement JG 1991. Effect of a single dose of an acetylcholinesterase inhibitor on oxotremorine- and nicotine-induced hypothermia in mice. Pharmacol Biochem Behav. 39(4): 929–34. [DOI] [PubMed] [Google Scholar]
- 46.Clement JG 1993. Pharmacological nature of soman-induced hypothermia in mice. Pharmacol Biochem Behav. 44(3): 689–702. [DOI] [PubMed] [Google Scholar]
- 47.Kurz A, Sessler DI, Annadata R, et al. 1995. Midazolam minimally impairs thermoregulatory control. Anesth Analg. 81(2): 393–8. [DOI] [PubMed] [Google Scholar]
- 48.Matsukawa T, Hanagata K, Ozaki M, et al. 1997. I.m. midazolam as premedication produces a concentration-dependent decrease in core temperature in male volunteers. Br J Anaesth. 78(4): 396–9. [DOI] [PubMed] [Google Scholar]
- 49.Motamedi GK, Lesser RP, and Vicini S. 2013. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia. 54(6): 959–70. [DOI] [PubMed] [Google Scholar]
- 50.Phillips KF, Deshpande LS, and DeLorenzo RJ. 2018. Hypothermia Reduces Mortality, Prevents the Calcium Plateau, and Is Neuroprotective Following Status Epilepticus in Rats. Front Neurol. 9: 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niquet J, Baldwin R, Gezalian M, et al. 2015. Deep hypothermia for the treatment of refractory status epilepticus. Epilepsy Behav. 49: 313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mailliet F, Galloux P, and Poisson D. 2001. Comparative effects of melatonin, zolpidem and diazepam on sleep, body temperature, blood pressure and heart rate measured by radiotelemetry in Wistar rats. Psychopharmacology (Berl). 156(4): 417–26. [DOI] [PubMed] [Google Scholar]
- 53.Empey PE, Miller TM, Philbrick AH, et al. 2012. Mild hypothermia decreases fentanyl and midazolam steady-state clearance in a rat model of cardiac arrest. Crit Care Med. 40(4): 1221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]