

Abstract
5-hydroxymethylcytosine (5hmC), the first oxidized form of the well-known epigenetic modification 5-methylcytosine, is an independent regulator of gene expression and therefore a potential marker for disease. Here, we report on methods developed for a selective solid-state nanopore assay that enable direct analysis of global 5hmC content in human tissue. We first describe protocols for preparing genomic DNA derived from both healthy breast tissue and stage 1 breast tumor tissue and then use our approach to probe the net abundance of the modified base in each cohort. Then, we employ empirical data to adjust for the impact of nanopore diameter on the quantification. Correcting for variations in nanopore diameter among the devices used for analysis reveals no detectable difference in global 5hmC content between healthy and tumor tissue. These results suggest that 5hmC changes may not be associated with early-stage breast cancer and instead are a downstream consequence of the disease.
Keywords: Solid-state nanopore, hydroxymethylcytosine, cancer, epigenetics
Graphical abstract:
A selective solid-state nanopore assay is used to examine global 5-hydroxymethylcytosine (5hmC) content in human genomic DNA. 5hmC has been shown to be strongly downregulated in diverse cancers, but it is less clear whether the decrease is a cause or an effect of disease initiation. To investigate this, we compare DNA from healthy breast tissue with that derived from stage 1 breast tumor tissue. We show that no statistical difference is observed between the two even after implementing technical corrections to maximize quantitative accuracy. Our results suggest that 5hmC downregulation is not a driver of tumorigenesis but a result, indicating that the potential for the modification as a cancer biomarker may be diminished in early stage disease.
Background
5-Methylcytosine (5mC) is the most widely-studied DNA epigenetic modification with impacts on gene expression and protein binding that impact diverse disease states1,2. For example, 5mC can be broadly dysregulated in cancer, frequently resulting in hypermethylation of CpG dinucleotides found in gene regions as well as hypomethylation of repetitive regions like heterochromatic tandem repeats, retrotransposons, and other parasitic elements3. Consequently, both global and site-specific 5mC have been investigated as potential bioindicators4–7. A number of approaches have been employed for probing 5mC in DNA including most prevalently bisulfite sequencing8, which entails exposure to the chemical sodium bisulfite to convert unmethylated cytosines into uracils selectively while leaving 5mC unconverted. The differences can subsequently be determined by conventional DNA sequencing techniques. Other techniques that are currently being used or are being developed for 5mC analysis include methylation-sensitive restriction digestion9, immunoprecipitation10, and a range of emerging next-generation sequencing (NGS) technologies that can distinguish 5mC directly11–13. However, despite the challenges of bisulfite – especially the widespread damage it induces in DNA and the resulting large sample mass that it necessitates – it remains the gold-standard method.
With the elucidation of the demethylation pathway14,15, the additional modified base 5-hydroxymethylcytsoine (5hmC) has gained significant attention as a potential independent regulatory element that may have distinct value as a biomarker in its own right16. In cancer specifically, there have been reports suggesting that it is strongly and consistently depleted across tumor types17,18 and can serve as an indicator of progression19 and prognosis20–22. However, there is still a lack of clarity about whether 5hmC loss is a cause or an effect of tumorigenesis and the regulatory changes that accompany it23. This question could have consequences for the ultimate utility of 5hmC as a bioindicator, especially in early-stage disease. Unfortunately, distinguishing 5hmC is currently challenging: bisulfite treatment cannot discriminate it from 5mC24, methylation-sensitive restriction enzymes are not sensitive to its presence, and NGS technologies have demonstrated only initial indications of success beyond 5mC25,26. In response, sequencing techniques like oxidative bisulfite sequencing (oxBS-seq27) and TET-assisted bisulfite sequencing (TAB-seq28) have been developed to probe 5hmC. While highly effective, both of these options still utilize bisulfite and thus retain the inherent challenges associated with exposure to that chemical, leading to the development of other emerging techniques29. Several non-sequencing approaches to 5hmC assessment have also been reported, including both optical30,31 and mass spectrometric32 techniques.
As an alternative, we recently reported a novel method for global 5hmC quantification based on solid-state (SS-) nanopore technology33. SS-nanopores consist of a single, artificial aperture fabricated in a thin membrane that is employed as a barrier between two basins of electrolyte solution. Upon the application of a voltage across the membrane, a strong electric field is generated at the pore that both produces an ionic current signal and can be used to transport charged molecules through the opening electrophoretically. During a translocation, the ionic current is temporarily reduced by the transiting molecule, yielding a brief reduction in the measured ionic current (a resistive pulse, or ‘event’) that can be analyzed to quantify and characterize populations of molecules.
Despite its elegance, a major challenge with this general approach is that all molecules in a heterogeneous mixture produce events, necessitating downstream analytical differentiation that can be difficult to achieve. In response to this challenge, we developed34 a novel SS-nanopore assay (Figure 1a) in which event generation is dependent on the binding of target nucleic acid fragments to a protein (monovalent streptavidin, or MS35,36) via a high-affinity tag (biotin). By combining our assay with an innovative enzymatic 5hmC biotin-tagging protocol (Figure 1b) developed by Song, et al.37, we demonstrated its utility to quantify global 5hmC in genomic DNA derived from mouse brain tissue33. Because mouse brain DNA is known38 to contain relatively high amounts of 5hmC, this was a reasonable initial target to validate the measurement. However, genomic DNA with more typical physiological hmC content is critical for the translation of the technology.
Figure 1.
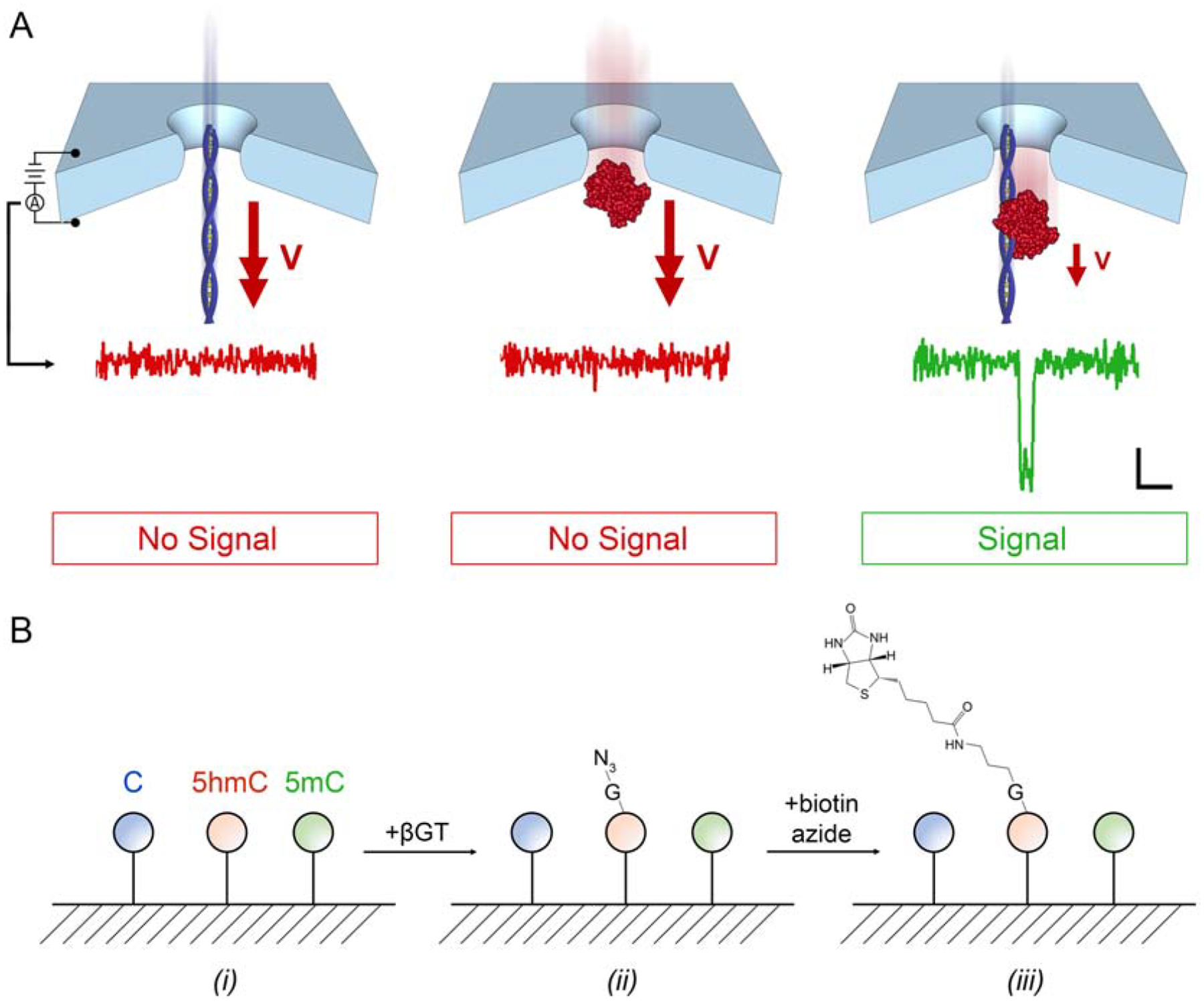
(A) Selective SS-nanopore assay. Voltage-driven translocations of either a short (biotinylated) DNA fragment (left) or MS (center) alone do not yield measurable electrical signals (below, showing conductance traces) because of their high threading velocity. However, once bound, the nucleoprotein complex (right) has a reduced velocity due to interactions with the pore walls during transit, consequently producing an electrical signal, or ‘event’. Scale bar applies to all traces (1 nS vertical, 500 μs horizontal). (B) Schematic of 5hmC biotinylation technique. DNA (i) is treated with β-glucosyltransferase (βGT) that attaches a glucose (G) featuring an azide (N3) moiety to 5hmC selectively (ii). The azide is subsequently reacted with biotin azide using copper-free click chemistry to attach a single affinity tag to the G (iii).
Here, we apply the approach instead to human genomic material. We first demonstrate sufficient resolution with the approach to measure 5hmC abundance in DNA derived from biopsies of human breast tissue, which has been shown to have low density of the modification39. We then implement improvements to our analytical strategy to account for device variability and compare 5hmC content between normal and stage 1 invasive breast carcinoma. Ultimately, we observe no statistical difference between cohorts, supporting the concept that reported 5hmC reductions may be the consequence of expression changes that accompany disease progression rather than an early cause.
Methods
Biomolecules
Synthetic monobiotinylated 60 nt DNA oligos with the sequence ATC AAC TGT TTC AGC CAC TGC TTC GCA GGC TGA CGT ATC TGA CGT GGT GCC AGC GAC GGA (where T indicates biotinylated thymine) were purchased commercially (Integrated DNA Technologies, Coralville, IA, USA) and used to produce duplex constructs via annealing in a 1:1 mixture with an unmodified homologous oligo. 75, 150, 200, and 250 bp monobiotinylated constructs were produced via PCR using lambda phage DNA (New England BioLabs, Ipswich, MA, USA) as a template. Primers were purchased (Integrated DNA Technologies) with a common reverse primer sequence of CAG TTG AGG ATC CCC ATA ATG CGG CTG TTT TCT G and forward primer sequences of GCA GCC GGA CGT GAA CGC GCA G (75 bp), AAC AAC TGT TTC AGC CAC TGC TTC (150 bp), AAA TCC CGT CTC TCA GGA GG (200 bp), and ATG AAC TTG TTA ACA GCA CCG ATG (250 bp). 150 bp mono-5hmC DNA was prepared the same way except using a reverse primer with the sequence CAG TTG AGG ATC CCC ATA ATG CGG CTG TTT TCT G (where C indicates 5hmC). All PCR and annealing products were confirmed by loading the products on a 2.5% agarose gel prepared in 1X TBE with GelRed nucleic acid stain (Phenix Research Products, Candler, NC, USA) and imaged using a Gel Doc™ system (BioRad, Hercules, CA, USA). Genomic DNA derived from the metastatic breast cancer cell line MFC7 was purchased commercially (BioChain Institute, Newark, CA, USA). Monovalent streptavidin35,36 (MS) featuring a single high-affinity biotin-binding domain was generously provided by the laboratory of Dr. Mark Howarth (Oxford University).
Patient-derived biospecimen preparation
Human breast biopsy tissues were obtained from females within the Wake Forest Baptist Health patient population found through histological assessment to have either (1) no cancer present (control) or (2) stage 1 invasive breast carcinoma. Specimens were collected by the Wake Forest Comprehensive Cancer Center tumor tissue bank as part of a study approved internally by the Wake Forest School of Medicine Institutional Review Board (IRB00031548) and all procedures followed were in accordance with institutional guidelines. Tissues were supplied frozen to the laboratory with sample masses of at least 150 mg. For each specimen, total genomic DNA was isolated using a commercial kit (Qiagen, Hilden, Germany) and the yield was quantified through photospectroscopy (NanoDrop, Thermo Fisher Scientific, Waltham, MA, USA). The isolate was suspended in a final volume of 50 μL in a microTUBE AFA fiber snap-cap (Covaris, Woburn, MA, USA) and then sheared mechanically to a mean length of <250 bp using a Covaris S220 focused-ultrasonicator operated at a peak incident power of 175 W and a duty factor of 10%, with 200 cycles per burst for a total of 280 s. We found that mechanical shearing was dependent on several extrinsic factors including net DNA concentration, so mean fragment lengths were confirmed by gel electrophoresis using a 4% agarose gel stained with GelRed nucleic acid stain (Biotium, Fremont, CA, USA) by comparing to a DNA length standard (GeneRuler 1 kb Plus DNA Ladder, Thermo Fisher Scientific). Quantitation was performed via image analysis with ImageJ software40.
5hmC labeling
5hmC bases were labeled with the protocol developed by Song et al.41 The reaction was performed in buffer containing 50 mM HEPES buffer at pH 7.9 (Thermo Fisher Scientific), 25 mM MgCl2 (Thermo Fisher Scientific), 1mM DDT (Thermo Fisher Scientific), 250 μM UDP-azide-gluocose (Active Motif, Carlsbad, CA, USA), 0.5 μM of T4-βGT (New England BioLabs), and at most 100 ng/μL DNA and incubated at 37° C for 2 hours. 350 μM sulfo-dibenzocyclooctyne-biotin (Sigma-Aldrich, St. Louis, MO, USA) was then added to the solution and incubated at 37° C overnight. Labeled DNA was purified using a QIAquick purification kit and eluted in pure deionized water. Labeling yield Y was determined by EMSA (see below) using 150 bp mono-5hmC that was labeled simultaneously with all patient biospecimens under identical conditions.
EMSA procedure
Biotin-labeled DNA was incubated in 1X PBS with a ~1.5X molar excess of MS for 10 min at room temperature. Samples were loaded on a 2.5% agarose gel with GelRed nucleic acid stain for visualization and run and imaged as described above. The buffer reservoirs of the electrophoresis unit were submerged in ice to reduce band diffusion and dissociation of the protein-DNA complex. Gel shift efficiency was determined by comparative band intensity analysis using ImageJ software40.
SS-nanopore fabrication and assay
SS-nanopore devices were fabricated using a Helium ion milling technique described elsewhere42. Briefly, a commercial (Norcada, Edmonton, AB, Canada) silicon chip (4.4 mm × 4.4 mm) supporting a small (8–20 μm), thin (30 nm) window of free-standing silicon nitride (SiN) was loaded into the sample chamber of a Helium ion microscope (HIM, Orion Plus, Carl Zeiss, Peabody, MA, USA). The HIM beam (5 pA, 20 um aperture) was focused and destigmated on an area of the chip near the window after which the beam was blanked, repositioned onto the window, and then exposed for a set amount of time to form a single nanopore. The prepared nanopore device was treated with a 10 W air plasma (Harrick Plasma, Ithaca, NY, USA) for 4 min before being loaded into a custom flow cell. A buffer solution consisting of 1 M NaCl and 1X PBS was introduced to the cis- and trans- chambers of the flow cell. A driving voltage was applied through Ag/AgCl electrodes inserted in the respective reservoirs. Ionic current was measured with a commercial patch clamp amplifier (Axopatch 200B, Molecular Devices, San Jose, CA, USA) using a collection rate of 200 kHz with a 100 kHz Bessel filter and recorded with custom data acquisition software (LabView, National Instruments, Austin, TX, USA). An additonal low-pass filter of 20 kHz was applied digitally. The open-pore current-voltage (I-V) response of a device was used to determine the diameter of the nanopore by employing the expression43
where μCAT and μAN are the mobilities of the cation and anion, respectively, derived from concentration-dependent, empirical measurements44, n is the number density of the ions, e is the elementary charge, d is nanopore diameter, and T is channel thickness (equal43 to 1/3 the full thickness of the membrane).
After introduction of biomolecules to the cis- chamber and application of a positive bias to the trans-chamber, translocation events manifested as brief interruptions in the measured current. Events were included in analyses only if their peak magnitudes were more than 4.5 standard deviations of the root-mean-square noise level from the baseline and their durations were between 20–2500 μs. Event rates were determined by considering at least 100 discrete 3.2 ms blocks of the uniterrupted current trace with error bars representing the standard deviation across all blocks.
Results
SS-nanopore determination of global 5hmC content in human genomic DNA
We first employed our SS-nanopore technique to probe the abundance of 5hmC in human DNA derived from healthy tissue. For this, 8 normal breast biopsies were obtained and processed (see Supplementary Information) to extract total genomic DNA. The net concentration co of each DNA isolate was determined by spectrophotometry. Because of the heterogenous nature of breast tissue in particular (featuring high adipocyte content), the yield varied significantly from sample to sample. However, all reported samples generated at least 1.5 μg of isolated DNA, which was suitable for our subsequent analyses. Since our SS-nanopore assay is viable only for short fragments (<250 bp typically33), we next mechanically sheared each DNA isolate to reduce their average length (Figure 2a(i)). Gel electrophoresis was performed (Figure 2b) along with a DNA standard to determine their individual mean fragment length l and thus account for run-to-run variations that can arise in the shearing process. Indeed, we observed that l ranged from 99 to 155 bp (see Table 1), and that the distribution of each was relatively narrow. While the differences were thus relatively minor, individual measurements of the samples enabled specific l values to be used, increasing measurement precision.
Figure 2.
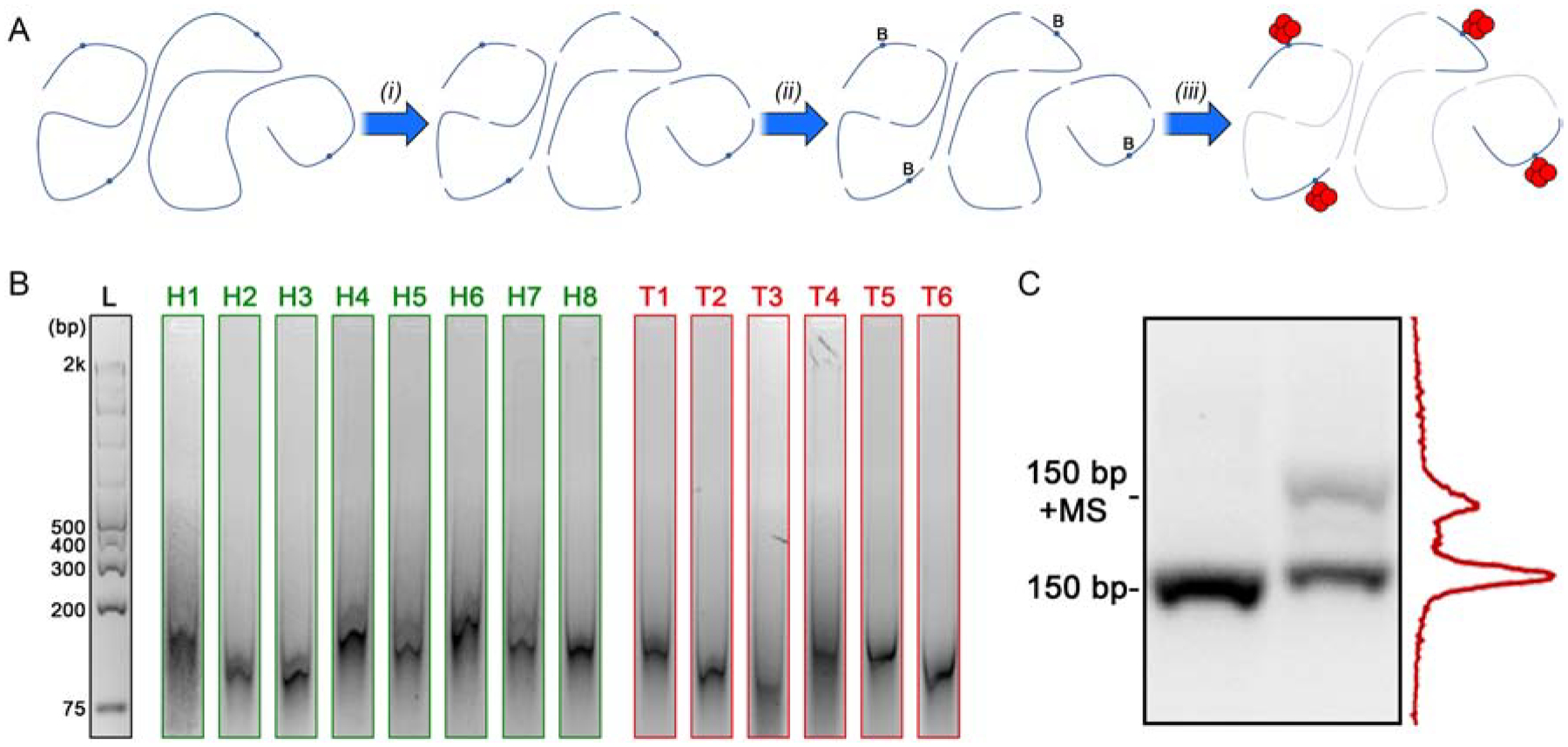
(A) Schematic representation of DNA treatment. Genomic DNA containing 5hmC (blue circles) is mechanically sheared (i) and then biotin-labeled (‘B’) at the 5hmC sites (ii). Labeled fragments are then bound (iii) by MS (red) for selective SS-nanopore analysis. Unlabeled fragments (faded) are not detected. (B) Gel images of all fragmented genomic DNA samples used in this study. Full gels shown in Supplementary Figure S1. (C) Gel analysis of a 150 bp mono-5hmC DNA construct following enzymatic labeling both alone (lane 1) and with bound MS (lane 2). Band intensity analysis of lane 2 (right) indicated a labeling yield of 35%.
Table 1.
Characteristics of DNA fragments derived from healthy breast tissue, stage 1 breast cancer tissue, and MCF7 cells as well as the resistance and diameter of the SS-nanopore(s) used to probe their 5hmC content.
| Sample # | Mean Length l (bp) | Nanopore Diameter d (nm) | |
|---|---|---|---|
| Healthy | H1 | 145 | 11.9 |
| H2 | 103 | 10.6 | |
| H3 | 99 | 10.7 | |
| H4 | 138 | 11.2 | |
| H5 | 123 | 12.6 | |
| H6 | 155 | 11.0 | |
| H7 | 134 | 10.6 | |
| H8 | 126 | 12.0 | |
| Stage 1 BC | T1 | 127 | 13.0 |
| T2 | 99 | 14.1 | |
| T3 | 93 | 13.3 | |
| T4 | 121 | 10.3 | |
| T5 | 117 | 12.2 | |
| T6 | 100 | 9.6 | |
| MFC7 Cell Line | - | 166 | 9.8 |
Each sample was then processed with a labeling approach (see Figure 1b) pioneered by Song, et al.41 and employed previously by us33 for this SS-nanopore assay. Briefly, the enzyme β-glycosyltransferase (β-GT) was used to attach a glucose moiety specifically to 5hmCs in the fragment sequences. The glucose was subsequently reacted with UDP-6 deoxy-6-azido-α-D-glucopyranoside (6-N3-Glu) to create a site for azide biotin incorporation (Figure 2a(ii)) through copper-free click chemistry, thereby providing a means for MS binding to labeled fragments (Figure 2a(iii)). Simultaneous with sample processing, we performed an identical treatment on a synthetic 150 bp DNA construct with a single 5hmC engineered into its sequence and probed it via an electromobility shift assay (EMSA, Figure 2c). By measuring the intensity of the shifted (i.e. biotin labeled) population relative to the total, we determined an analytical yield for the labeling protocol, Y, of 0.35. Since all labeling treatments were performed simultaneously using identical conditions, we used this value for Y across all samples.
With the fragmented, labeled product, we finally performed a series of SS-nanopore measurements, first on the as-prepared material alone and then on the same material incubated with MS to promote selective detection. For this study, we used 14 individual SS-nanopore devices ranging in diameter from 9.6–14.1 nm (see Table 1) and performed all analyses under 400 mV applied voltage. We observed that the as-prepared material yielded some events, attributable to the variation in fragment sizes resulting from mechanical shearing; in effect, some small fraction of the DNA in solution was large enough in size to produce events without MS conjugation. We considered these event rates as baselines to which the MS-bound measurements could be compared directly to determine the signal associated with the labeled material alone. When conjugated to MS, all samples produced significantly higher event rates (Figure 3), reflecting the selective detection of DNA fragments containing 5hmC. As a negative control, we also treated and prepared an aliquot of DNA (150 bp) with no 5hmC and observed no events for either as-prepared or MS-incubated material (data not shown).
Figure 3.
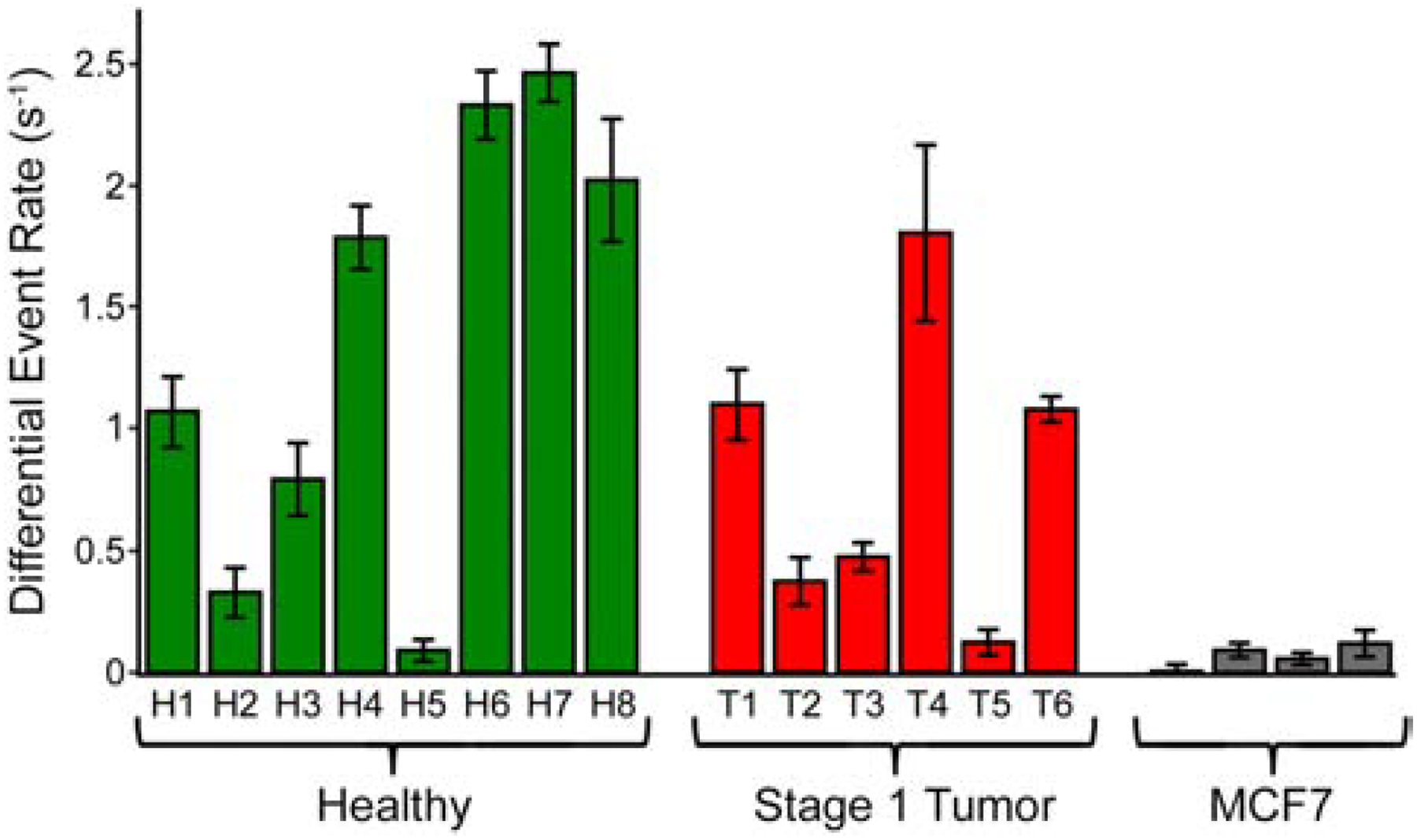
Differential SS-nanopore event rates (rates for fully labeled fragments bound with MS minus baseline rates) measured at an applied voltage of 400 mV for fragmented genomic DNA derived from healthy breast tissue (left, green), stage 1 breast cancer tissue (center, red), and MCF7 cell line DNA (right, grey).
SS-nanopore event rate, r, is known to scale with analyte concentration33,43, allowing data to be converted through the use of a simple calibration curve. Notably, r is also strongly impacted by DNA length33 in our assay, and so the observed variation in shearing efficiency for the patient-derived DNA investigated in this study (see Figure 2a and Table 1) necessitated consideration of construct size as a variable for accurate conversion. To address this, we performed a series of concentration-dependent measurements of translocation rate (400 mV) on four nanopore devices ranging in diameter from 8.6–11.9 nm and probed four different sizes of monobiotinylated DNA bound to MS: 75, 150, 200, and 250 bp (Figure 4a). Past measurements by ourselves33 and others43 have demonstrated that event rate varies as a function of analyte concentration according to a power law, but with an exponent that is close to unity. Consequently, these trends were described well by simple linear fits, the slopes of which changed regularly with construct length (Figure 4b). This provided a means by which to infer corresponding calibration curves for any intermediate size through interpolation. We were therefore able to use the mean fragment length of each individual sample to convert its associated event rate to an equivalent measured concentration, c’.
Figure 4.
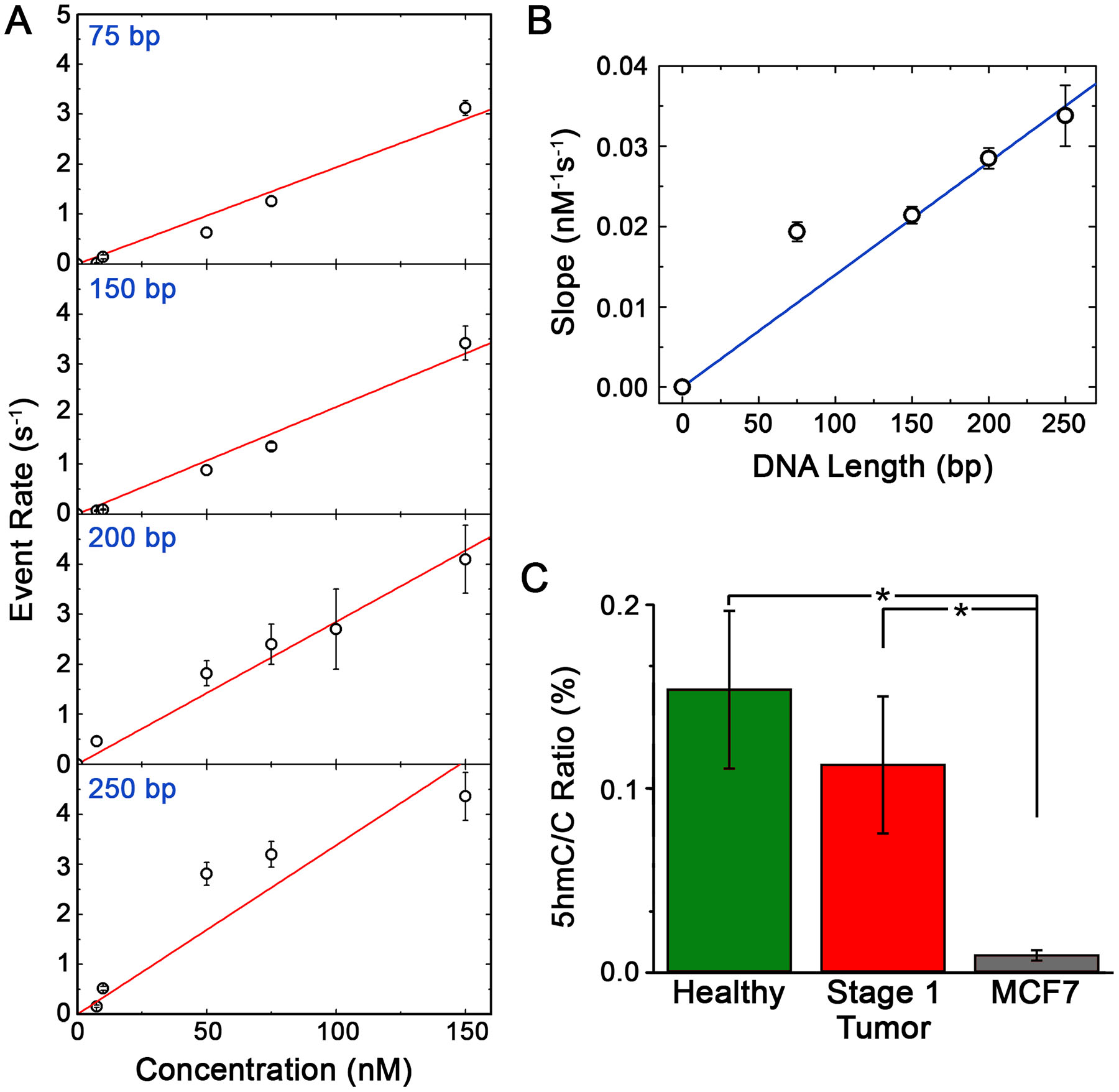
(A) SS-nanopore event rates (400 mV applied voltage) as a function of concentration for monobiotinylated dsDNA constructs of different length (75, 150, 200, and 250 bp) bound to MS. Red lines are linear fits to each data set. (B) Dependence of the slopes of the fits in (a) as a function of construct length. Blue line is a linear fit to the data. (C) 5hmC ratio R for genomic DNA isolated from healthy breast tissue, stage 1 breast cancer tissue, and MCF7 cells taking into account the length-dependent calibration from (B). * p < 0.05.
Ultimately, we sought to probe global 5hmC abundance. In our past work33, we used a simple expression to determine R, or the ratio of 5hmC bases to the total number of cytosines:
Here, α is the fractional cytosine content of the DNA, which we took to be 0.39 based on the average G+C content of the human genome45. The biotin labeling yield, Y, was derived from the EMSA analysis of synthetic oligonucleotides labeled simultaneously with our samples (see Figure 2b). Finally, the measured partial concentration of 5hmC fragments c’, total DNA concentration co, and mean fragment length l were determined independently for each sample as described above using SS-nanopore analysis, spectrophotometric analysis, and gel electrophoresis analysis, respectively. Note that this expression assumes that DNA fragments will contain either one modified base or none at all, as supported by the low abundance of 5hmC in the human genome in general. Combining all of the above factors, we calculated R for each healthy breast tissue DNA sample (Figure 4c, white), yielding a mean value of 0.15 ± 0.08% 5hmC/C.
In addition to the healthy breast tissue specimens, we also performed identical processing and assessments using 6 tumor tissue biopsies derived from stage 1 breast cancer patients. Total DNA yield from these tissues were comparable to healthy tissue and its mechanical shearing (see Figure 2a and Supplementary Figure S1) produced mean fragment lengths l that were consistent with those of the healthy tissues above (93–127 bp, see Table 1). Enzymatic labeling of tumor DNA fragments was performed simultaneously with all healthy DNA, enabling the same labeling yield Y to be applied. By again using spectrophotometry to determine co for each tumor DNA sample, we could then incorporate empirical results for c’ (using the interpolated calibrations from Figure 4b, as above) and l into our expression for 5hmC/C ratio to calculate R for each sample (Figure 4c, black). Ultimately, this yielded a mean R of 0.11 ± 0.07% 5hmC/C for the stage 1 breast cancer-derived genomic DNA. This value was somewhat less than that of the normal tissue DNA and could suggest a minor decrease in 5hmC abundance that accompanied the initial emergence of breast cancer, but an unpaired two-tailed t-test showed the difference to be statistically insignificant (p = 0.37).
Because 5hmC is known to be downregulated broadly in cancer DNA17,18, confirmation of its depletion in advanced breast cancer would serve to validate our measurement. Therefore, we finally sought to contrast our healthy and stage 1 results with global 5hmC content in genomic DNA isolated from MCF7 cells46: an aggressive human breast adenocarcinoma cell line that is a standard model for metastatic (i.e. late-stage) breast cancer47. To achieve this, we performed an identical treatment and analysis as was used for patient-derived DNA. Mechanical shearing of the material resulted in a mean fragment length of 166 bp (Supplementary Figure S2a) and 5hmC labeling – completed in an independent procedure (Supplementary Figure S2b) – produced a comparable yield Y (0.26) as that described above. SS-nanopore event rates collected from four separate measurements of labeled, MS-conjugated fragments were only slightly higher than unconjugated fragments (Figure 3, grey), consistent with the low 5hmC abundance known to be present in MCF748. Indeed, the mean R calculated from these measurements was 0.009 ± 0.003% (Figure 4c, grey), or about an order of magnitude less than either healthy or stage 1 breast cancer DNA. These data indicated a significant reduction compared to each patient cohort (p<0.05).
Implementing empirical data to account for diameter dependence of selective SS-nanopore quantification
In our past work, we employed SS-nanopores with a narrow range of diameters and assumed a consistent behavior from device to device. However, a diameter dependence should reasonably exist that could impact the comparison of results. For example, considering the extremes, a small diameter would promote detection of all passing constructs and thus yield a high event rate while a large diameter would be expected to produce few if any events due to limited pore wall interactions and reduced signal-to-noise ratio. Indeed, other groups have demonstrated that molecular translocations go largely undetected at a given measurement bandwidth when they are small relative to the SS-nanopore through which they are threaded49,50. Consequently, to investigate the influence of pore diameter quantitatively, we performed a series of SS-nanopore measurements in which only diameter was changed while all other conditions were kept constant, including applied voltage (400 mV) and analyte concentration (100 nM). Using multiple independent SS-nanopore devices, we first determined nanopore diameter of each empirically from the ionic current (see Supplementary Information) and then measured the event rate for a model monobiotinylated DNA construct (60 bp) bound to MS. Plotting the results (Figure 5a), we observed that the event rate r reduced dramatically as nanopore diameter d was increased from 8.0 to 14.9 nm. The data were fit well by an exponential trend (y = kxn, where k and n are fit variables). Control measurements of either DNA or MS alone did not yield events within the same size range. Note that below ~7.5 nm diameter, constructs clogged the pore rapidly due to steric hinderance since the pore diameter and the hydrodynamic diameter of the nucleoprotein complex were similar.
Figure 5.
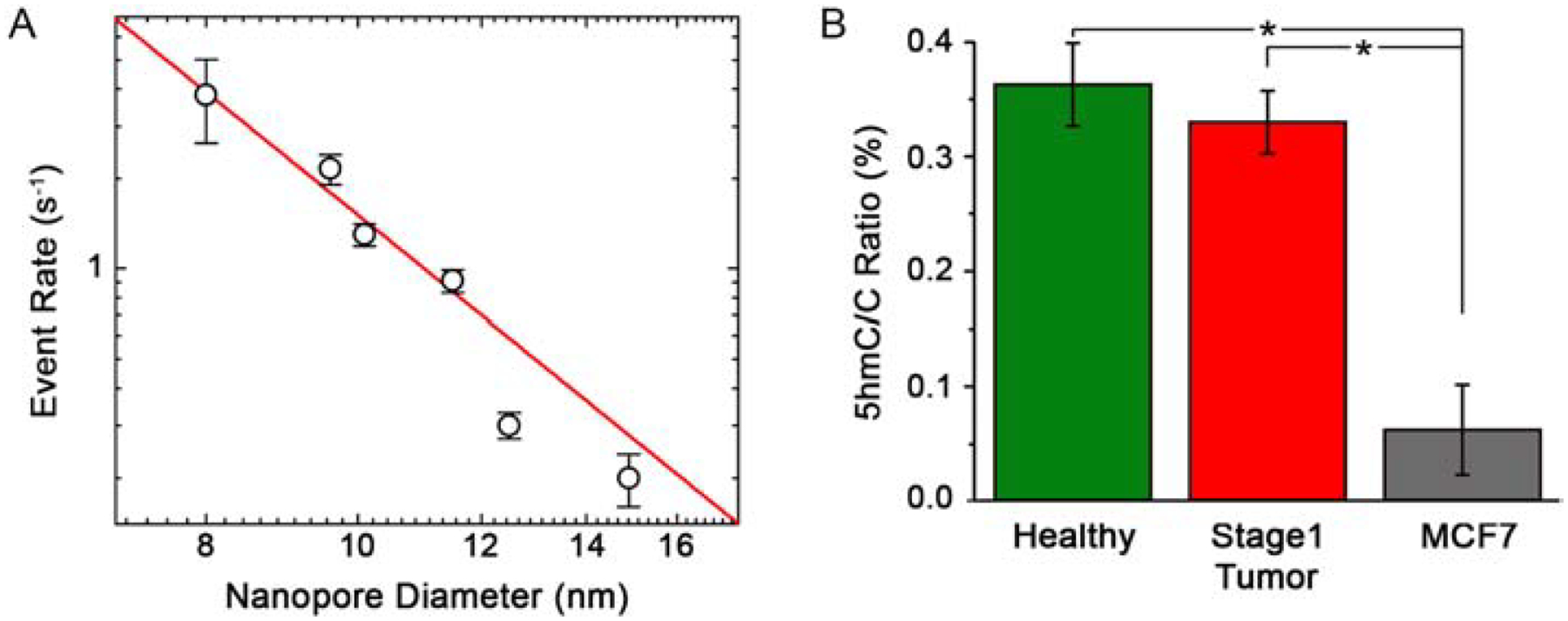
(A) Event rate for a 100 nM concentration of 60 bp dsDNA bound to MS measured at 400 mV through a range of SS-nanopore diameters. The red line is an exponential fit to the data (see text). (B) 5hmC ratios R corrected for device-to-device variation in pore diameter and normalized to 9.0 nm. * p < 0.001.
Elucidation of this dependence ultimately enabled rate results – and thus 5hmC/C ratios – to be adjusted to a single nanopore diameter by adjusting for the reduction in r observed with increasing d. Accordingly, we returned to our SS-nanopore measurements and converted the translocation rate of each healthy and stage I breast cancer DNA specimen to reflect results from a SS-nanopore of 9.0 nm, since this was the smallest diameter pore used in our measurements (see Table 1). Recalculating R for each patient sample with the corrected values, we found that the small decrease in 5hmC abundance between healthy and stage 1 breast cancer observed in our initial analysis (see Figure 4) was not altered meaningfully (Figure 5b), yielding corrected 5hmC/C values of 0.36 ± 0.03% for healthy and 0.33 ± 0.05% for tumor tissue. This again showed that no statistical difference (p = 0.37) could be identified between cohorts. Both of these values were of the same order as reported measurements of 5hmC abundance in normal breast tissue DNA39. Recalculation of R values for advanced stage MCF7 genomic DNA yielded a corrected mean value of 0.06 ± 0.04%, demonstrating a highly significant difference compared to both cohorts above (p<0.001, Figure 5b, grey).
Discussion
Our initial measurements of 5hmC abundance in DNA derived from healthy breast tissues and from stage 1 breast cancer tissues yielded mean R values of 0.15 ± 0.08% and 0.11 ± 0.07% 5hmC/C, respectively, which indicated no significant reduction in 5hmC abundance associated with the earliest stage of the disease. This contrasted with the significant reduction in 5hmC observed in DNA derived from a metastatic (i.e. late-stage) breast cancer cell line, which yielded a mean R value of 0.009 ± 0.003%. These analyses did not take into account signal differences stemming from the varying SS-nanopore diameters used for the measurements. Because our measurement mechanism is contingent on steric interactions between a bulky nucleoprotein complex and the nanopore walls, pore diameter should be a critical variable controlling outcomes. Indeed, conducting a series of measurements on SS-nanopores of different size using a model DNA-MS complex yielded an exponential dependence of event rate on diameter. We subsequently used this dependence to standardize our genomic DNA data by adjusting all acquired event rates to those that would arise from a single SS-nanopore diameter (9.0 nm, the smallest diameter pores used for analysis). However, even considering this adjustment, we found no difference in 5hmC levels between healthy and stage 1 breast cancer DNA with corrected values of 0.36 ± 0.03% and 0.33 ± 0.05%, respectively. Both of these adjusted values still differed significantly from that of the late-stage breast cancer cell line DNA, which yielded a much lower R of 0.06 ± 0.04%.
The intermediate nature of 5hmC makes its global reduction, which has been reported across many tumor types17, a common feature of cytosine demethylation pathway dysregulation. For example, in hypermethylated DNA, a lack of TET activity can prevent oxidation of 5mC, while in hypomethylated DNA, high TET activity (and/or low methyltransferase activity) can restrain cytosines to their canonical form. What has been less clear is whether the reduced occupancy at 5hmC is the result or the source of dysregulated transcription and signaling pathways associated with cancer development. Our results suggest that 5hmC changes known to be associated with breast cancer are a downstream effect of disease progression rather than a cause. Consequently, while global 5hmC variation could have value as a bioindicator of advanced progression, transformation, or therapeutic efficacy, it may not be a reliable marker of disease emergence in particular.
In this report, we applied a SS-nanopore assay capable of detecting protein-conjugated DNA to the assessment of the epigenetic modification 5hmC in human genomic DNA. By employing an enzymatic protocol for the specific labeling of 5hmC with a biotin moiety, we could facilitate selective binding of MS, providing a pathway to discrimination and ultimately to quantification. We performed analyses on DNA derived from both healthy breast tissue and from stage 1 breast cancer tissues. Our initial results suggested an insignificant reduction in global 5hmC in the disease state and after correcting for the effects of device-to-device diameter variance through empirical data, this observation was confirmed. In contrast, a highly significant reduction in 5hmC was found in late-stage breast cancer DNA compared to both healthy and stage 1 breast cancer using the MCF7 cell line as a model, in agreement with observations from the literature17,18. This suggests that 5hmC may be less reliable as a biomarker for the earliest stages of breast cancer than for advanced stages. More generalizable results can be achieved in the future through consideration of larger cohorts, more disease stages, and different cancer types.
Our results are also noteworthy for SS-nanopore analyses in two specific ways. First, they demonstrate quantitative epigenetic analysis of human genomic DNA, extending an approach that had previously been applied only to murine DNA33. Despite the analytical promise of SS-nanopores as a platform, they have only rarely been applied successfully to physiologically-derived samples43,51, and never to human genomic DNA of any kind to our knowledge. This is a critical hurdle to the ultimate translation of the technology. Second, the R values measured here were at least an order of magnitude lower than our previous results on murine brain DNA33; a model system that was chosen in part because of its high abundance of 5hmC, which provided a relatively sizeable initial measurement target. The results reported here are therefore indicative of the high 5hmC sensitivity achievable with our assay.
Supplementary Material
Acknowledgements
The authors wish to thank the Howarth Lab (Oxford University) for supplying monovalent streptavidin. This work was supported by NIH grants R21CA193067 and R33CA246448 (to A.R.H.). F.W. was supported by a predoctoral fellowship through the Redox Biology and Medicine Training Program (T32GM127261). Tissue collection was performed through support for the Wake Forest Comprehensive Cancer Center (P30CA012197).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
A.R.H declares the following competing interest: he is listed as inventor on a patent covering the SS-nanopore assay. The other authors declare no competing interests.
References
- 1.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Bergman Y & Cedar H DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol 20, 274–281 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Ehrlich M DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Srinivas PR, Kramer BS & Srivastava S Trends in biomarker research for cancer detection. Lancet Oncol 2, 698–704 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Esteller M, Corn PG, Baylin SB & Herman JG A gene hypermethylation profile of human cancer. Cancer Res 61, 3225–3229 (2001). [PubMed] [Google Scholar]
- 6.Belinsky SA Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer 4, 707–717 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Wilson AS, Power BE & Molloy PL DNA hypomethylation and human diseases. Biochim. Biophys. Acta-Rev. Cancer 1775, 138–162 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Frommer M et al. A Genomic Sequencing Protocol That Yields a Positive Display of 5-Methylcytosine Residues in Individual Dna Strands. Proc. Natl. Acad. Sci. U. S. A 89, 1827–1831 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu M, Yao J & Polyak K Methylation-specific digital karyotyping. Nat. Protoc 1, 1621–1636 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Jacinto FV, Ballestar E & Esteller M Methyl-DNA immunoprecipitation (MeDIP): Hunting down the DNA methylome. Biotechniques 44, 35–+ (2008). [DOI] [PubMed] [Google Scholar]
- 11.Flusberg BA et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–U72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laszlo AH et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. U. S. A 110, 18904–18909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson JT et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–+ (2017). [DOI] [PubMed] [Google Scholar]
- 14.Ito S et al. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 333, 1300–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohli RM & Zhang Y TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeifer GP, Kadam S & Jin SG 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics Chromatin 6, 10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin S-G et al. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res 71, 7360–7365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32, 663–669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z et al. Decreased 5-hydroxymethylcytosine levels correlate with cancer progression and poor survival: a systematic review and meta-analysis. Oncotarget 8, 1944–1952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Y et al. Low level of 5-Hydroxymethylcytosine predicts poor prognosis in non-small cell lung cancer. Oncol. Lett 11, 3753–3760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang F et al. 5-hydroxymethylcytosine loss is associated with poor prognosis for patients with WHO grade II diffuse astrocytomas. Sci. Rep 6, 20882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y et al. Decreased 5-Hydroxymethylcytosine (5-hmC) predicts poor prognosis in early-stage laryngeal squamous cell carcinoma. Am. J. Cancer Res 6, 1089–1098 (2016). [PMC free article] [PubMed] [Google Scholar]
- 23.Ficz G & Gribben JG Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics 104, 352–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PloS One 5, e8888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber J et al. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc. Natl. Acad. Sci. U. S. A 110, 18910–18915 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Z et al. Detecting DNA Modifications from SMRT Sequencing Data by Modeling Sequence Context Dependence of Polymerase Kinetic. PLoS Comput Biol 9, e1002935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Booth MJ et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc 8, 1841–1851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu M et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat. Protoc 7, 2159–2170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schutsky EK et al. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat. Biotechnol (2018) doi: 10.1038/nbt.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilat N et al. Single-molecule quantification of 5-hydroxymethylcytosine for diagnosis of blood and colon cancers. Clin. Epigenetics 9, 70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margalit S et al. 5-Hydroxymethylcytosine as a clinical biomarker: Fluorescence-based assay for high-throughput epigenetic quantification in human tissues. Int. J. Cancer 146, 115–122 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Shahal T, Koren O, Shefer G, Stern N & Ebenstein Y Hypersensitive quantification of global 5-hydroxymethylcytosine by chemoenzymatic tagging. Anal. Chim. Acta 1038, 87–96 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Zahid OK, Zhao BS, He C & Hall AR Quantifying mammalian genomic DNA hydroxymethylcytosine content using solid-state nanopores. Sci. Rep 6, 29565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlsen AT, Zahid OK, Ruzicka JA, Taylor EW & Hall AR Selective Detection and Quantification of Modified DNA with Solid-State Nanopores. Nano Lett 14, 5488–5492 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Howarth M et al. A monovalent streptavidin with a single femtomolar biotin binding site. Nat. Methods 3, 267–273 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fairhead M, Krndija D, Lowe ED & Howarth M Plug-and-Play Pairing via Defined Divalent Streptavidins. J. Mol. Biol 426, 199–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song CX et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 29, 68–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Globisch D et al. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS ONE 5, e15367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W & Liu M Distribution of 5-Hydroxymethylcytosine in Different Human Tissues. J. Nucleic Acids 2011, 1–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins TJ. ImageJ for microscopy. BioTechniques 43, 25–30 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Song CX et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 29, 68–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J et al. Rapid and precise scanning helium ion microscope milling of solid-state nanopores for biomolecule detection. Nanotechnology 22, 285310 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Wanunu M et al. Rapid electronic detection of probe-specific microRNAs using thin nanopore sensors. Nat. Nanotechnol 5, 807–814 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Haynes WM CRC Handbook of Chemistry and Physics, 97th Edition. (CRC Press, Boca Raton, FL, 2016). [Google Scholar]
- 45.Kong A et al. A high-resolution recombination map of the human genome. Nat. Genet 31, 241–247 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Levenson AS & Jordan VC MCF-7: the first hormone-responsive breast cancer cell line. Cancer Res 57, 3071–3078 (1997). [PubMed] [Google Scholar]
- 47.Dai X, Cheng H, Bai Z & Li J Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 8, 3131–3141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bachman M et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem 6, 1049–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plesa C et al. Fast Translocation of Proteins through Solid State Nanopores. Nano Lett 13, 658–663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larkin J, Henley RY, Muthukumar M, Rosenstein JK & Wanunu M High-Bandwidth Protein Analysis Using Solid-State Nanopores. Biophys. J 106, 696–704 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivas F et al. Label-free analysis of physiological hyaluronan size distribution with a solid-state nanopore sensor. Nat. Commun 9, 1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.