

Abstract
Objective:
The aim of the study was to investigate the effects of methacrylate-functionalized proanthocyanidins (MAPAs) on dentin collagen’s bio-stabilization against enzymatic degradation and crosslinking capability.
Methods:
Three MAPAs were synthesized via varying methacrylate (MA) to proanthocyanidins (PA) feeding ratios of 1:2, 1:1, and 2:1 to obtain MAPA-1, MAPA-2, and MAPA-3, respectively. The three MAPAs were structurally characterized by proton nuclear magnetic resonance (1H NMR) and Fourier transform infrared (FTIR) spectroscopic methods. 5-μm-thick dentin films were microtomed from dentin slabs of third molars. Following demineralization, films or slabs were treated with 1% MAPAs or PA in ethanol for 30 s. Collagen bio-stabilization against enzymatic degradation was analyzed by weight loss (WL) and hydroxyproline release (HYP) of films, as well as scanning electron microscopy (SEM) on dentin slabs. Crosslinking capacity and interactions of MAPAs with collagen were investigated by FTIR. Data were analyzed by ANOVA and Tukey’s test (α = 0.05%).
Results:
MA:PA feeding ratios affected MAPAs’ chemical structures which in turn led to different collagen stabilization efficacy against degradation and varied collagen crosslinking capabilities. Higher collagen stabilization efficacy was detected using MAPA-1 (WL 10.52%; HYP 13.53 μg/mg) and MAPA-2 (WL 5.99%; HYP 11.02 μg/mg), which was comparable to that using PA (WL 8.79%; HYP 13.17 μg/mg) (p > 0.05), while a lower collagen stability occurred in MAPA-3 (WL 38.48%; HYP 29.49 μg/mg), indicating over MA-functionalization would compromise its stabilization efficacy. In comparison, complete digestion was detected for untreated collagen (WL 100%; HYP 102.76 μg/mg). The above results were consistent with collagen crosslinking efficacy of the three MAPAs revealed by SEM and FTIR.
Significance:
A new class of novel polymerizable collagen cross-linkers MAPAs was synthesized and shown that, when appropriate MA:PA ratios were applied, the resulting MAPAs could render high collagen stability and the ability to copolymerize with resin monomers, overcoming the drawbacks of PA. These new polymerizable crosslinkers, when included in adhesives, could lead to long-lasting dentin bonding.
Keywords: Proanthocyanidins, methacrylate functionalization, collagen cross-linking, dentin collagen, hydroxyproline
1. INTRODUCTION
Resin-dentin bonding relies on the formation of hybrid layer in which there is micromechanical interlocking among demineralized collagen fibrils and infiltrated methacrylate adhesive resin. In the hybrid layer, minerals surrounding collagen fibrils are removed by acid etching and replaced/infiltrated by resin monomers. The bio-stability of both the underlying demineralized collagen and infiltrated resin is considered one of the most important factors for success of dental restorations [1, 2]. Despite recent advances, limited durability of resin-dentin interfaces is still an unsolved problem. Hydrolysis of sub-optimally polymerized adhesive resin and enzymatic degradation of unprotected demineralized collagen by host-derived enzymes (MMPs and cysteine cathepsins) lead to interface breakdown, lowering service life of restorations [2, 3]. Developing strategies that can reinforce both dentin collagen and resin from degradation is therefore a viable approach to enhancing the stability of the resin-dentin interface [1, 4].
Collagen bio-modification mediated by bioactive agents has emerged as a powerful tool for improving dentin’s mechanical, anti-enzymatic and bonding properties [5–9]. Among various bio-modification approaches, the exogenous collagen cross-linkers, especially natural polyphenolics such as proanthocyanidins (PA), have shown some promising applications due to their high bioactivity, absence of toxicity and wide availability [4, 8, 9]. However, in spite of encouraging collagen stabilization results with a treatment time even as short as tens of seconds, the use of PA in a clinically acceptable setting remains a challenge [10]. For example, the use of PA as a primer has been widely investigated [7–9, 11]. However, due to its antioxidant nature, PA needs to be rinsed off after priming, which is time-consuming and adds an extra step in bonding protocols. The inclusion of PA in etchants has also been tried [12, 13]; however, PA has poor solubility in etchant acids, requiring the addition of volatile solvents such as ethanol or acetone which results in short shelf life of etchants due to fast solvent evaporation. In addition, PA, like most polyphenolics in general, is susceptible to auto-oxidation reactions, constantly altering its structures and properties [14, 15]. Consequently, preparation of fresh solutions is required before every application. Furthermore, all those strategies, whether using PA as a primer or in an etchant, limit PA’s usage to the etch-and-rinse mode only, and cannot be applied to the self-etch mode.
In light of this, many attempts have been made to directly include PA into adhesives. Unfortunately, a negative effect of PA on the photo-polymerization of adhesive monomers [11, 16, 17] has been frequently observed. PA is a well-known free radical scavenger, hindering molecular chain initiation and propagation during polymerization [17, 18]. Poor polymerization of adhesive monomers results in reduced resin quality, breakdown of bonding interfaces as demonstrated in laboratorial studies [11, 16]. A recent two-year clinical evaluation showed that PA included in adhesives adversely affected polymerization of adhesive resin, causing higher loss rate of restorations and jeopardizing the longevity of composite restorations [19], further confirming the limitations of using PA directly in a clinical setting.
Grafting of additional functional unit onto methacrylate-based monomers has led to the development of multifunctional dental monomers. In addition to the polymerization function, these monomers may possess other functions such as self-etching ability, chemical interactions to dental substrates [20, 21], antibacterial and anti-MMPs activity [22, 23], etc. Specific functional unit is often incorporated into methacrylate (MA) through the ester linkage, thus maintaining polymerization ability of the vinyl moiety in MA groups [24, 25]. This strategy inspired us to develop a novel class of polymerizable collagen crosslinker methacrylate-functionalized proanthocyanidins (MAPAs). Following a one-step synthesis reaction, MAPAs have been synthesized aiming to combine the collagen cross-linking ability of PA and MA’s capability to co-polymerize with resin monomers into the same MAPA molecule. This strategy would reinforce both dentin collagen (via crosslinking) and resin (via co-polymerization) at the same time, therefore is a novel approach to enhancing stability of the resin-dentin interfaces.
During functionalization, the phenol hydroxyl (OH) groups in PA may be consumed and replaced by MA. Since the cross-linking capability of PA is attributed to its phenolic structures such as catechol with ortho-phenol OH groups, it is unclear if the MA grafting/replacement would affect/sacrifice MAPA’s collagen crosslinking ability. Effects of three MA:PA feeding ratios on the chemical structures of MAPAs and the stabilization and cross-linking of dentin collagen after MAPA treatment have therefore been evaluated in this study. The bio-stability of dentin collagen against collagenase degradation was assessed quantitatively by weight loss (WL) and hydroxyproline release (HYP), and qualitatively by scanning electron microscopy (SEM). The cross-linking efficacy and chemical interactions with collagen were studied by Fourier-transform infrared (FTIR) spectroscopy. The null hypotheses tested were that regardless of MA:PA feeding ratios, the synthesized MAPAs would not be able to (1) induce collagen bio-stabilization against degradation or (2) promote cross-linking and chemical interactions with collagen.
2. MATERIAL AND METHODS
All chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. The PA from grape seed extract (GSE) was generously donated by Mega Natural (Madera, CA, USA). Three polymerizable collagen crosslinkers derived from PA (MAPA-1, MAPA-2 and MAPA-3) were synthesized as further described below (Scheme 1). Catechin is the major monomeric unit for PA and the mole of PA samples was thus calculated as the mole of catechin units. All the treatment solutions (PA, MAPA-1, MAPA-2 and MAPA-3) were prepared at 1% concentration in pure ethanol (w/w). Collagenase (type I, from Clostridium histolyticum, ≥125 U/mg) solution was made at 0.1% (w/v) in TESCA buffer (50 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid, 0.36 mM CaCl2, pH = 7.4).
2.1. Synthesis of polymerizable collagen crosslinkers MAPAs
The MAPAs were synthesized from grape seed extract through a one-step reaction. Sample of PA (0.90 g, 3.1 mmol of catechin) was suspended in N,N-dimethylformamide (10 mL)/triethylamine (0.31 g, 3.1 mmol for MAPA-1; 0.63 g, 6.2 mmol for MAPA-2; 1.25 g, 12.4 mmol for MAPA-3) at 0 °C. To the above solution, methacryloyl chloride (0.16 g, 1.5 mmol for MAPA-1; 0.32 g, 3.1 mmol for MAPA-2; 0.65 g, 6.2 mmol for MAPA-3) was added dropwise with the MA:PA (based on catechin unit) molar ratio being 1:2, 1:1 and 2:1 (0.5 eq., 1 eq, and 2 eq) for MAPAs-1, −2, and −3, respectively. The resulting reaction mixture was stirred for another 1 h at 0 °C, and then stirred at room temperature overnight. Water was then added to the reaction mixture. After stirring under N2 for at least 2 h, the mixture was centrifuged, rinsed with distilled water twice, and the final product was collected and dried under vacuum for at least 48 h. Yield: 0.25 g, 24.3% for MAPA-1; 0.50 g, 44.8% for MAPA-2; 0.79 g, 59.7% for MAPA-3. The lower yields for MAPA-1 and MAPA-2 are due to their higher solubility in water, which led to product loss during the water rinsing step. The final products were characterized by 1H NMR (Fig. 1) and FTIR (Fig. 2).
Fig. 1.
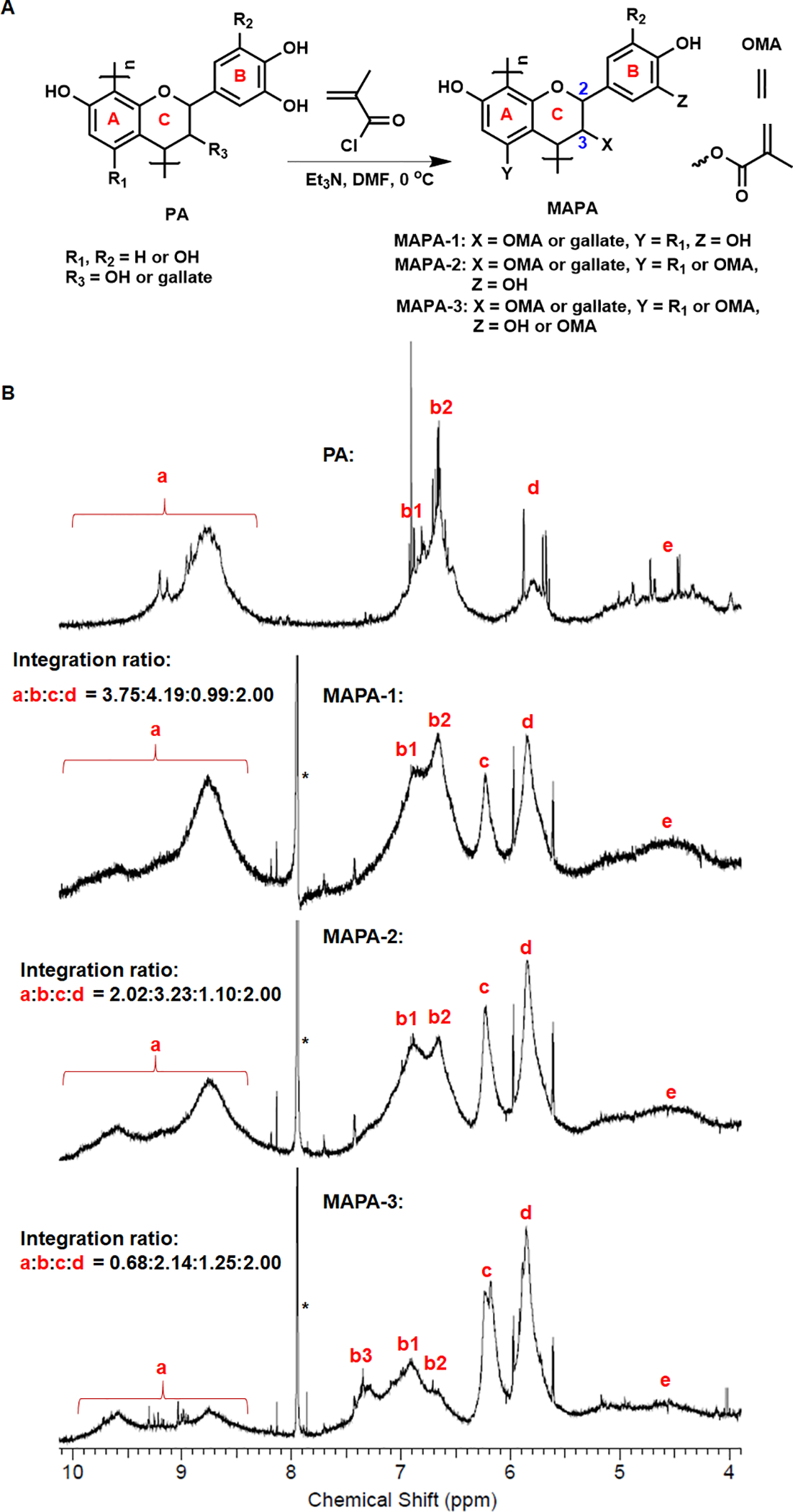
Schematic illustration of MAPA synthesis via chemical modification to PA (A). 1H NMR spectra of PA, MAPA-1, MAPA-2 and MAPA-3 (B).
Fig. 2.
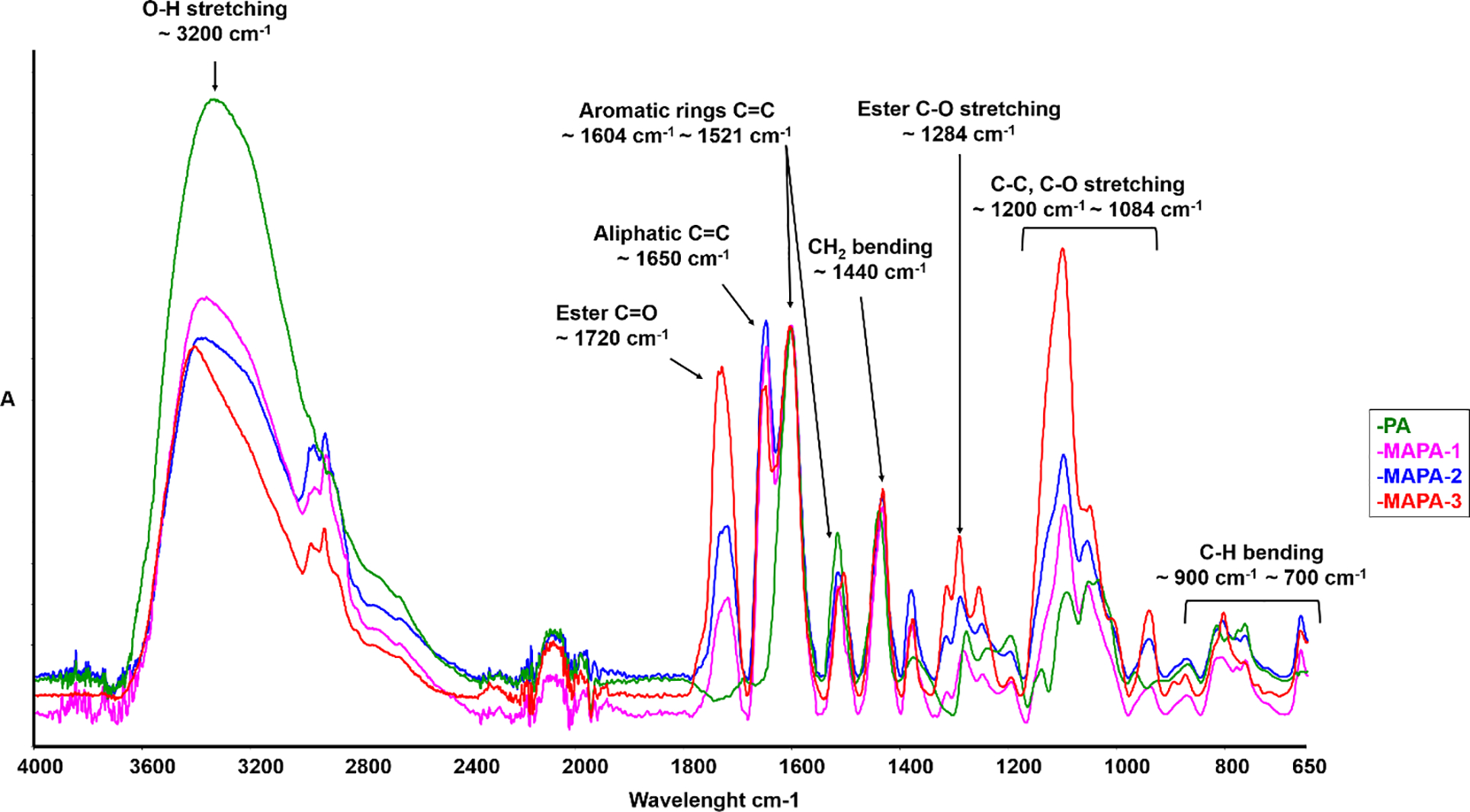
Representative FTIR spectra of PA and three synthesized MAPA-1, MAPA-2 and MAPA-3.
2.2. Dentin films preparation and MAPAs treatment
Sixteen non-carious human third molars were collected under the approved protocol by the University of Missouri-Kansas City Adult Heath Sciences Institutional Review Board (IRB). After storage in phosphate-buffered saline containing 0.002% sodium azide at 4 °C, the occlusal enamel and roots were removed from the teeth using a water-cooled diamond saw (Buehler, Lake Bluff, IL, USA). Six teeth were sectioned into dentin blocks (6 × 6 × 5 mm), which were further processed into 350 ultrathin dentin films (5 μm thick) with a tungsten carbide knife mounted on SM2500S microtome (Leica, Deerfield, IL, USA). These films were randomly assigned into 5 groups (n = 70/group) according to the cross-linkers investigated: PA, MAPA-1, MAPA-2, MAPA-3 and an untreated group as a negative control (solvent only). All the cross-linkers were prepared as treatment solutions at 1% in ethanol (w/w). Each of the dentin films was first demineralized with 10% phosphoric acid for 30 min, rinsed with deionized water for 30 min and spread on a plastic cover slip (Fisher Scientific, Pittsburgh, PA, USA) using a paintbrush. After blotting away excessive water, 30 μL of selected treatment solution was dropped onto each of the demineralized dentin films. After 30 s, the films were rinsed with absolute ethanol for 30 min (3 times × 10 min) in order to remove any residual treatment solution. The films were then dried for 48 h under vacuum. The treated and dried films were subsequently evaluated for collagen biostability – weight loss (WL) and hydroxyproline release assay (HYP) (n=60 per treatment group) and the remaining 10 films per group was further analyzed by FTIR.
2.3. Collagen biostability against collagenase digestion by weight loss and HYP release measurements of dentin films treated with MAPAs
The 60 treated dentin films from each treatment group were randomly assigned into 6 specimen sub-groups containing 10 films in each group. The films were immersed in 300 μL of 0.1% bacterial collagenase solution in TESCA at 37 °C for 1 h. The WL percentage was determined by the dry weight change before (W0) and after (W1) collagenase digestion for each specimen using an analytical balance (d = 0.01 mg, Mettler Toledo AG285, Zurich, Switzerland), and was calculated by the following equation: WL%=(W0-W1)/W0×100%.
For the HYP assay, the digestion solution was collected and hydrolyzed in 6M HCl at 110 °C for 24 h. The dry residue (free hydroxyproline and other amino acids) of each specimen was pre-treated by neutralization, oxidation and subjected to 5% Ehrlich’s reagent to develop the color. The absorbance was measured at 555 nm with a microplate reader (Biotek Instruments, Winooski, VT, USA). The trans-4-hydroxy-L-proline (analytical standard, Sigma-Aldrich) was used as the standard to obtain the working curve for quantifying the HYP release (n = 6) from each micro gram of demineralized dentin films during the digestion.
2.4. SEM morphology of demineralized dentin layer treated with MAPAs
Ten non-carious human third molars were processed into dentin slabs as follows. After crown removal, a uniform smear layer was created on the dentin surface utilizing wet 600-grit SiC sandpaper (Buehler) for 60 s. Further sections were made in the occlusal-apical direction at increments of 1 mm, followed by one cut parallel to and ~1.5 mm below the abraded surface to free the slabs. Slabs for SEM were notched at the middle position from the side opposite to the abraded surface for the purpose of subsequent fracturing and visualization. The abraded surfaces of notched slabs were etched with 32% phosphoric acid gel (Scotchbond Universal Etchant, 3M ESPE, St. Paul, MN, USA) for 15 s and rinsed with deionized water for 30 s. After being blot-dried, the demineralized dentin surfaces were treated for 30 s with each of the treatment solutions and rinsed three times (10 min each time) with ethanol. Eight dentin slabs were used for each treatment group, 4 of which were not subject to collagenase digestion, whereas the other 4 experienced 1 h of digestion at 37°C in 0.1 % collagenase solution. After rinsing, all the slabs were fixed in 2.5% glutaraldehyde buffered with 0.1 M sodium cacodylate for 1 h, dehydrated in graded solutions of ethanol (33%, 67%, 85%, 95% and 100%) for 2 h each and dried overnight in vacuum desiccator. The slabs were fractured, mounted on aluminum stubs, and coated with carbon. The fractured cross-sections were examined in a FEI/Philips XL30 Field-Emission Environmental SEM (Philips, Eindhoven, Netherlands) at 2500, 5000 and 10000 x magnification.
2.5. Collagen cross-linking effect and chemical interaction of MAPAs by FTIR
The remaining 10 treated and dried dentin films from each group were submitted to FTIR analysis (Spectrum One, Perkin-Elmer, Waltham, MA, USA) on a BaF2 disc, in order to detect collagen cross-linking effect and chemical interaction of MAPAs. Each FTIR spectrum was collected in the 650 – 4000 cm−1 wavenumber range at a resolution of 4 cm−1 and 64 scans. For spectral analysis, the spectrum of untreated dentin film was subtracted from the spectra of treated films to elucidate the interactions between the cross-linkers and collagen. The integration areas of the band at 1604 cm−1 (associated with PA components) of the resultant difference spectra were calculated to show the extent of chemical interactions with collagen, while the integration band area at 1235 cm−1 (amide III) from untreated collagen was used as an internal standard/reference. Thus, a band ratio (1604 cm−1 / 1235 cm−1) was calculated for each of the treatment groups.
2.6. Statistical analysis
After normality of distribution and homogeneity of variances (Komolgorov Smirnov and Levene tests, respectively), the averages were subjected to one-way analysis of variance and Tukey post hoc test (5%).
3. RESULTS
The synthesis of MAPAs was shown in Fig. 1A. Functionalization of PA with MA was achieved in one step by reacting PA with varied amounts of methacryloyl chloride. Depending on the mole ratios of MA applied, various OH groups in the PA structure could react to form methacrylate ester groups (OMA) as shown as X, Y, Z in Fig. 1A.
The 1H-NMR spectra of PA and MAPAs in DMSO-d6 were shown in Fig. 1B. Characteristically broad multiple-peak signals associated with the phenol OH protons (labeled as “a”), aryl protons (labeled as “b”), and the protons on the C2 and C3 of the C-ring (labeled as “d”) were identified for PA. While for MAPAs, the clear appearance of a new broad peak at δ = 6.0–6.4 ppm attributed to the MA vinyl protons (labeled as “c”) confirms the successful incorporation of MA into MAPAs. The integration ratios of signals a:b:c:d for MAPA-1, MAPA-2, and MAPA-3 were estimated to be 3.75:4.19:0.99:2.00, 2.02:3.23:1.10:2.00, and 0.68:2.14:1.25:2.00, respectively.
Representative FTIR spectra of PA and MAPAs are shown in Fig. 2. Compared to the spectrum of PA, the appearance of new bands at ~1720 cm−1 (C=O, ester), ~1650 cm−1 (C=C, aliphatic), and 1284 cm−1 (ester C-O stretching) of all three MAPAs is attributed to the MA functional group, confirming the grafting of MA onto PA. Meanwhile, the band intensities at 3200 cm−1 (O-H stretching) for the three MAPAs were all reduced, reflecting the partial consumption of hydroxyl groups after reacting with MA. Among the three MAPAs, the relative intensities of the bands associated with MA functional group (i.e., 1720 cm−1) increased from MAPA-1 to MAPA-2 and to MAPA-3, indicating increased amount of MA grating, which was consistent with the 1H NMR studies.
The stability results of dentin films, after treated with PA and MAPAs, against collagenase digestion were presented in Fig 3. The weight loss (WL) results (Fig. 3A) showed that all the cross-linkers led to significantly improved collagen biostability when compared to the untreated control (p < 0.001). While the collagen in the untreated group was completely digested (WL 100%), those treated with MAPAs showed significantly lower weight loss. Collagen treated with MAPA-1 (WL 10.52%) and MAPA-2 (WL 5.99%) showed the highest bio-stability, statistically comparable to that of PA (WL 8.79%) (p > 0.05). Collagen treated with MAPA-3, however, exhibited much lower biostability (WL 38.48%) (p < 0.001). Similar results were observed using HYP release measurements (Fig. 3B). HYP release is the highest in the untreated control (102.76 μg/mg), followed by MAPA-3 (29.49 μg/mg), while MAPA-2 (11.02 μg/mg) and MAPA-1 (13.53 μg/mg) showed the lowest (p < 0.001) and comparable HYP release that was statistically similar to that of PA (13.17 μg/mg) (p > 0.05).
Fig. 3.
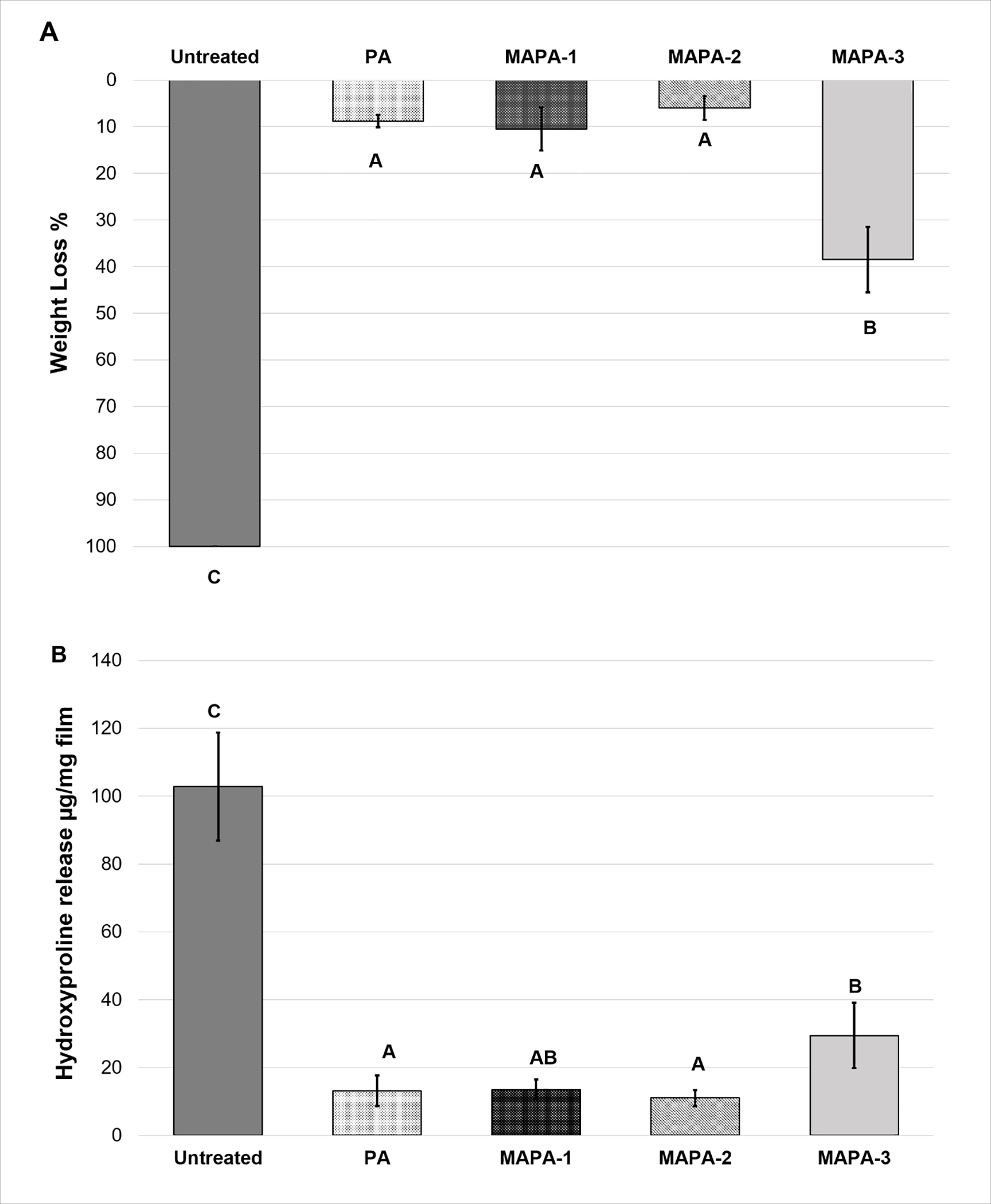
Means and standard-deviations of collagen resistance against collagenase digestion measured by (A) weight loss (%) and (B) hydroxyproline proline release (μg/mg film). Different capital letters demonstrate statistically significant difference among groups (p < 0.001).
Representative SEM micrographs of demineralized dentin layers treated with PA and MAPAs were presented in Fig 4. Before digestion, the cross-sections of etched dentin showed similar morphology regardless of crosslinker treatment, in which a dense layer of demineralized dentin (DD) collagen fibrils is seen under secondary electron imaging (SE-SEM) while the DD layer is more obviously shown in dark color under backscattered-electron mode (BSE-SEM). After 1 h of collagenase digestion, the DD layer of the untreated group completely disappeared, while the DD layer remained evident for specimens treated with PA or MAPAs for 30 s. The DD collagen layer appeared unchanged or intact before and after digestion for MAPA-1, MAPA-2 and PA. For MAPA-3, slight degradation after digestion was observed: there was a loss of approximately 0.9 μm-thick of DD layer in all MAPA-3 specimens.
Fig. 4.
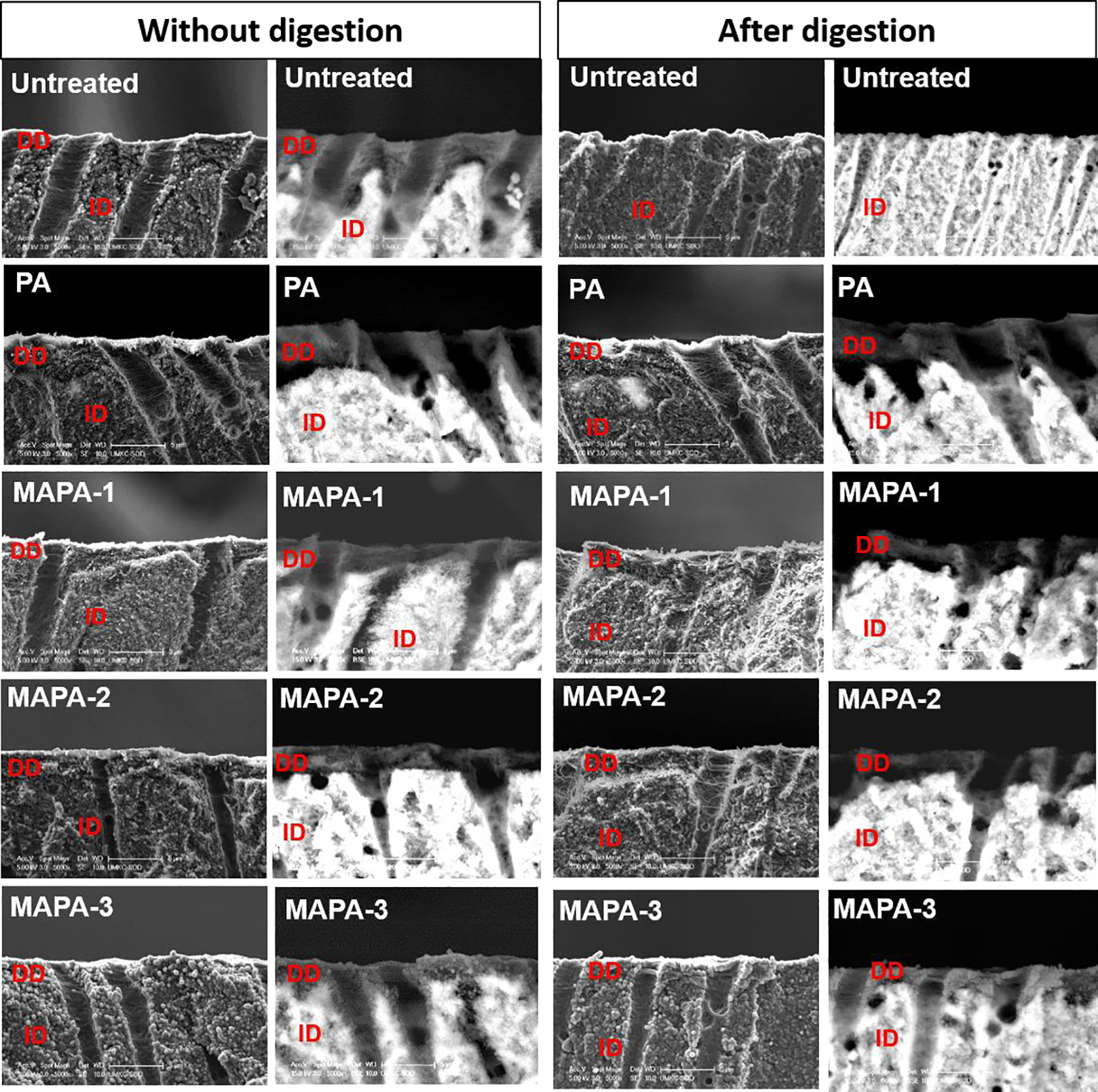
Representative photomicrographs (5000 x) of demineralized dentin slabs for all the treatment groups before and after digestion obtained from SEM in secondary-electron (SE) and backscattered-electron (BSE) modes. DD: demineralized dentin; ID: intact dentin.
Representative FTIR spectra of demineralized dentin films with and without crosslinking treatment were shown in Fig. 5A. The characteristic bands related to dentin collagen were assigned, such as C=O stretching for amide I (at ~1660 cm−1), out-of-phase combination of N-H bending and C-N stretching for amide II (at ~1544 cm−1), CH2 bending (at ~1450 cm−1) and in-phase combination of N-H bending and C-N stretching (at ~1235 cm−1) for amide III [26, 27]. Although some spectral bands from MAPAs and PA (shown in Fig. 5C) overlapped with those from collagen, there were still pronounced spectral changes when comparing the treated with untreated collagen. For example, the emerged ester band C=O at ~1720 cm−1 was clearly visible in MAPAs treated collagen, which was related to MA functional group. The broadening of amide I (~1660 cm−1) to lower wavenumbers, intensity decrease and shoulder formation of amide II (~1544 cm−1), increase of CH2 bending (~1450 cm−1) as well as decrease in intensity at ~1400 cm−1 were evidently identified after only 30 s treatment of PA or MAPAs. A closer peek at the spectra ranging from 1590–1360 cm−1 (the top right insert) showed that the extent of the above changes was dependent on the type of crosslinkers used. When comparing to the untreated control, more manifest changes were detected in PA, MAPA-1 and MAPA-2, while less pronounced changes were identified in MAPA-3.
Fig. 5.
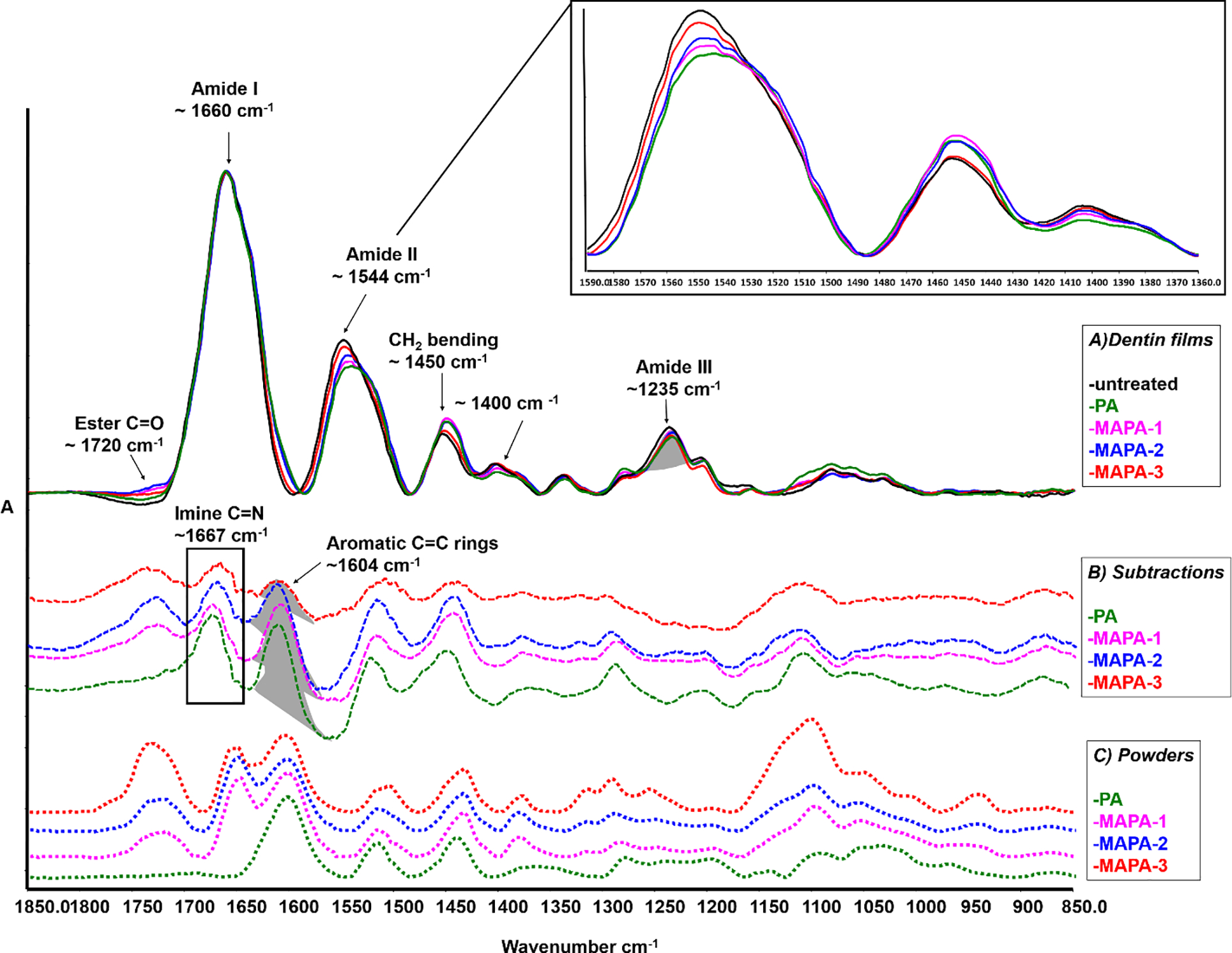
A) FITR spectra of untreated and treated dentin collagen films, in which main band assignments and associated spectral changes are identified after PA and MAPAs treatments (top spectra, solid lines), a closer peek at the spectra ranging from 1590–1360 cm−1 is in the top right corner. B) Difference FTIR spectra of treated dentin films with the spectrum of untreated collagen subtracted (middle spectra, dashed lines). C) FTIR spectra of PA and MAPAs powders (bottom spectra, dotted lines). The highlighted band area ratios at ~1604 cm−1 of C=C aromatic rings (resulting difference spectra) and at ~1235 cm−1 of amide III (untreated collagen) were calculated and shown in Table 1.
To further explore the potential collagen crosslinking and chemical interactions with different crosslinkers, the spectrum of untreated collagen was subtracted from the spectra of collagen specimens that were treated with PA or MAPAs. Using the amide III band as the internal standard for subtraction, the difference spectra are shown in Fig. 5B along with the spectra of PA and MAPA powders (Fig. 5C). Most of the bands in difference spectra could find matching counterparts in the spectra of powders, except for one band at ~1667 cm-1. The ratio of the band at 1604 cm−1 (associated with aromatic C=C rings in PA or MAPAs) and amide III band at 1235 cm−1 was used to calculate extent of interactions with collagen (Table 1). Those ratios obtained for collagen specimens treated with MAPA-1 and MAPA-2 were similar to each other and statistically comparable to that of PA (P > 0.05). Collagen treated with MAPA-3, however, showed a significantly lower 1604/1235 band ratio (p < 0.001).
Table 1.
Means and standard-deviations of the band area ratio (A1604cm −1/A1235cm −1) of collagen with different treatment group.*
| Treatment group | Band ratio (A1604cm −1/A1235cm −1) |
|---|---|
| PA | 1.11 ± 0.19 A |
| MAPA-1 | 0.88 ± 0.27 A |
| MAPA-2 | 0.86 ± 0.21 A |
| MAPA-3 | 0.29 ± 0.12 B |
Different capital letters demonstrate statistically significant difference among the groups (p < 0.001).
4. DISCUSSION
Over the past years, PA from GSE has been established as an effective collagen cross-linker, which could potentially enhance mechanical properties and bio-stability of dentin collagen matrix in the hybrid layer [4, 9, 28, 29]. However, like other collagen cross-linkers, PA is only able to bind to collagen, but cannot integrate/interact with adhesive resin. Moreover, PA’s negative effect on photo-polymerization of adhesive monomers remains unsolved, making the sole collagen cross-linking approach less clinically acceptable [11, 17],[19]. MAPAs synthesized in this study may provide a novel class of polymerizable collagen crosslinkers with the potential to reinforce dentin collagen via cross-linking and at the same time forming strong binding with adhesive resin through co-polymerization with adhesive monomers. These polymerizable crosslinkers could directly be included in the adhesive resin, overcoming the PA’s negative effect on polymerization and enhancing collagen-resin interactions.
PAs from grape seed extract are rich in oligomers of flavonoids (70~80% of n = 2~7 in Fig. 1A). Their flavonoid building blocks may have complicated chemical compositions but are dominated by catechins, epicatechins, and their gallate derivatives. All flavonoids contain the same basic three-ring skeleton (see Fig. 1A). Rings A and B can contain two or more hydroxyl groups while C-ring may contain a hydroxyl group at the C-3 position. When treated with methacryloyl chloride, ester bond formation would probably occur first with the hydroxyl group on the C-ring (C3 position) since alkyl alcohol is generally much more reactive then phenol hydroxyl groups toward ester bond formation. When a MA:PA ratio of 1:2 is used, as in MAPA-1, all MA groups are likely attached to the C-ring. In other words, aryl hydroxyl groups in rings A and B are not affected. When a MA:PA ratio of 1:1 is used, as in MAPA-2, all C3-hydroxyl groups in the C-ring would react and form ester bonds with MA units. Since only a portion of the C3 positions have OH groups (the rest are gallates), there would be excess methacryloyl chloride remaining after all C3-OH groups have reacted. The extra metahcryloyl chloride would then react with aryl hydroxyl groups on A-ring or B-ring. When more than 1 equivalent of MA is added, as in the case of MAPA-3 where the MA:PA ratio is 2:1, more phenolic hydroxyl groups would react once all C3-hydroxyl groups in the C-rings have been consumed.
As shown in Fig. 1B, the 1H NMR spectrum of PA in the chemical shift range of 4 ~ 10 ppm displayed four broad peaks, labeled as signals “a” (8~10 ppm), “b” (6.5~7.5 ppm), “d” (5.5~6.0 ppm ) and “e” (4.0~5.5 ppm), respectively. Those peaks can be attributed to aryl hydroxyl protons, aryl protons, C-ring protons (C2, C3), and C-4 protons, respectively. The 1H NMR spectra of all three MAPAs showed a distinct new peak at δ = 6.0–6.4 ppm, which is attributed to the vinyl protons in the MA unit, unambiguously confirming the attachment of MA units in all three MAPAs. A careful inspection reveals that signal “b” in PA contains two overlapping peaks “b1” and “b2”, which may be assigned to aryl protons in the B-ring and A-ring, respectively. When an MA unit is attached to the A-ring, the signal associated with aryl protons in A-ring will shift downfield, resulting in decreased intensity in signal “b2”. If MA is attached to the B-ring, aromatic protons associated with B-ring will shift further downfield, resulting in a new peak and decreased intensity in “b1” peak. Indeed, the 1H NMR spectra of MAPA-3 showed three “b” peaks. The appearance of the new downfield-shifted “b”-peak (“b3” in Fig. 1) indicates that some MA units are indeed attached to the B-ring in MAPA-3. For MAPA-1 and MAPA-2, no such new “b” peak is observed, indicating no MA is attached to the B-ring. It is noted that the “b2” peak intensity is slightly decreased from MAPA-1 to MAPA-2, indicating that there are some attachments of MA to the A-ring in MAPA-2. Based on the integration ratios of signal “c” versus signal “b”, one could estimate the MA:PA ratios in the synthesized MAPAs. The c/b integration ratios for MAPA-1, MAPA-2 and MAPA-3 are 0.99:4.19, 1.10:3.23, and 1.25:2.14, respectively, from which the estimated MA:PA ratios in MAPAs are 0.6, 0.9 and 1.5 for MAPA-1, MAPA-2, and MAPA-3, respectively. The lower MA:PA ratios in MAPA-2 and MAPA-3 than their reactant feeding ratios is likely due to the low reactivity of aryl hydroxyl groups. In summary, 1H NMR results confirmed the attachment of MA to PA in MAPAs. For MAPA-1, MA is attached to the C3-position. For MAPA-2, MA is attached to both the C3 position and some A-ring hydroxyl groups. While for MAPA-3, MA is attached to C3, some A-ring, and some B-ring hydroxyl groups.
The FTIR spectra of PA and MAPAs offer structural insights as well. A glance of the spectra of MAPAs reveals that there are distinct and strong new bands at 1720 and 1650 cm−1, attributable to carbonyl stretching and C=C stretching, respectively, which are associated with the MA unit. The band intensity at 1720 cm−1 increases from MAPA-1 to MAPA2 and to MAPA-3, consistent with the increasing content of MA in those MAPAs. It is worth noting that the band intensity at 1650 cm−1 does not following the same increasing pattern as that of band 1720 cm−1, especially for MAPA-3 whose band intensity at 1650 cm−1 is the lowest even though its MA content is supposed to be the highest. This is likely due to the presence of different amount of moisture in the samples since water exhibits an IR band near 1650 cm-1. Compared to MAPA-1 and MAPA-2, MAPA-3 contains more MA groups and fewer phenolic hydroxyl groups and is therefore much more hydrophobic. The broad band at ~ 3200 cm−1 is attributed to OH stretching. There are both aliphatic OH and phenolic OH in PA, and in general, phenolic OH stretching band is broader and extending further to the lower wavenumber region. As shown in Figure 2, the band maximum corresponding to O-H stretching shifted from 3200 cm−1 of PA to 3250 cm−1 of MAPAs and a significant O-H stretching band narrowing was observed for MAPA-3, indicating that there may be less hydrogen bonding in MAPA-3 due to MA functionalization.
Effects of MAPAs on collagen bio-stability were assessed using weight loss and HYP assay (Fig. 3). In this study, dentin collagen films were treated with MAPAs or PA for as short as 30 s and subsequently challenged by aggressive collagenase digestion. The WL and HYP results indicated that the attachment of MAs in MAPA-1 and MAPA-2 did not at all affect their ability to protect collagen from digestion. However, when higher MA;PA ratios were applied, as in MAPA-3, significant decrease in collagen stabilization against digestion was detected, although it was still better than untreated control. The above findings suggested that all MAPAs were able to provide some collagen stabilization; therefore, the first null hypothesis was rejected.
As mentioned earlier, when MA:PA ratios are less than 1:1, MAs are predominantly attached to the C3-OH groups, leaving phenolic hydroxyl groups intact. Since collagen crosslinking is mostly caused by phenolic units (i.e., catechol units), attaching MAs to C3-OH only as in MAPA-1, or to C-3 and some A-ring as in MAPA-2, would not affect its crosslinking capability. When further higher MA:PA ratios are used, however, some phenolic hydroxyl groups in B-ring would start to react with the excess MA units. In other words, in MAPA-3 some catechol OH groups would have reacted with MA as revealed in 1H NMR studies, therefore reducing its crosslinking capability, which is exactly what have been observed in the collagen stability studies.
PA and MAPAs could improve collagen biostability against collagenase degradation through three possible pathways. First, it may be related to the cross-linking effect exerted by PA component on collagen through various chemical reactions, initiated at specific telopeptides in collagen molecules which increases the formation of inter and intra-fibrillary crosslinks and promotes conformational changes of collagen, consequently leading to more resistance against unwinding[8]. Second, triple helix strengthened after cross-linking could possess fewer cleavage sites recognized by collagenase [30]. Third, even without crosslinking, physical presence of the PA component may exert additional effect directly on collagenase/enzymes via hydrogen bonding and hydrophobic interactions [30], reducing collagenase’s molecular mobility and causing changes in the catalytic region [10]. Considering that all these potential effects are dependent on interactions between the PA component and collagen or collagenase/enzymes, it is reasonable to expect that a dramatic reduction of collagen reactive phenol hydroxyl (OH) groups as in MAPA-3 could reduce its the stabilization ability.
To simulate a more clinically relevant condition, dentin surface was etched by phosphoric acid gel for 15 s to create a demineralized layer, subsequently treated with one of the cross-linkers for 30 s as a primer, and then submitted to collagenase degradation. It is worth pointing out that after 30 s crosslinking treatment, an extended rinse with ethanol was applied to completely remove any un-bonded MAPAs in the layer. In fact, this extended rinse (3 times, 10 min each) with good solvent (ethanol) was applied to both dentin films and demineralized layers after crosslinking treatment to avoid the presence of MAPAs that are either left-over residues or weakly/reversibly bound with collagen. This thorough-rinse procedure would help to evaluate whether the collagen stabilization by MAPAs against digestion was due to strong, irreversible crosslinking interactions, or via the above-mentioned third pathway of direct collagenase/enzymes inhibition by residual MAPAs. SEM results (Fig. 4) showed that all MAPAs were able to penetrate into demineralized dentin layer and protect collagen against digestion within a quick, clinically relevant time (30 s). After digestion, the DD collagen layer in MAPA-1, MAPA-2 appeared intact, similar to PA, while slight degradation was seen in MAPA-3, which was consistent with the above quantitative results on dentin films. It is worth to point out that MAPAs show less discoloration effect on demineralized dentin layer than PA; however, this effect needs to be evaluated over time for potential clinical applications.
FTIR spectra of untreated and treated collagen were compared to understand the specific interactions between collagen and MAPAs. As shown in Fig. 3A, spectral changes were generally more pronounced in MAPAs-1 and −2, and were comparable to those of PA; while they were less evident in MAPA-3 when compared to untreated control. The spectral changes might be related to the following possible interactions. First, broadening of amide I (~1660 cm−1) and a decrease in intensity and shoulder formation of amide II (~1544 cm−1) are likely attributed to hydrogen bonding via amino and amide groups of collagen and phenolic hydroxyl (OH) groups of PA component [31]. Some studies have suggested that hydrogen bonding is the main mechanism of interactions between collagen and PAs [5, 31]. However, it was also proposed that the principal driving force towards interactions could be the “hydrophobic effects”, which were enhanced by “hydrogen bonding” of the phenolic groups to prolines on collagen [32]. The second possible interaction is related to dehydration of collagen (result of crosslinking), which is evidenced by an intensity decrease of ~1400 cm−1 [28, 33] and according to Miles et al., dehydration provides strong evidence of cross-linking effect and major reason for enhanced collagen stability [28, 33]. When the untreated collagen spectrum was subtracted from those of treated collagen, the resultant difference spectra (Fig. 5B) further verified the incorporation of MAPAs in collagen. Most of the bands in difference spectra could find matching parts in the powders’ spectra (Fig. 5C). There is a new band at ~1667 cm−1 that could not be detected in powders. This band likely indicates the third possible interaction: covalent bonding [32, 34], which has been attributed to the imine (C=N) stretching of the Schiff base formed between PA component and collagen [28, 29]. Such a covalent bond formation involves auto-oxidation of catechol moieties to ortho-quinone groups which subsequently react with free amine groups on proteins [35]. All these evidences indicate strong interactions of MAPAs with dentin collagen, and the second null hypothesis has to be rejected.
The band ratio (1604 cm−1/1235 cm−1) was used to quantify the extent of MAPAs’ interaction/incorporation with collagen (Fig. 5, Table 1). The logic for choosing the band at ~1604 cm−1 was that the C=C of aromatic rings was a functional group present only in PA component, while the ~1235 cm−1 amide III of collagen was not affected after cross-linking treatment. Consequently, the band ratio (1604 cm−1/1235 cm−1) can reflect the amount of MAPAs incorporated in dentin collagen. The higher the ratio, the higher the interactions between collagen and PA component. MAPA-1 and MAPA-2 showed higher ratios, statistically similar to PA, while MAPA-3 showed a reduced ratio. These FTIR results are consistent with collagen stabilization outcomes measured both qualitatively and quantitatively.
5. CONCLUSION
MAPAs with MA units covalently linked through ester bonds were successfully synthesized. MA units were linked to PA predominantly through C3-hydroxyl groups when the MA:PA ratios were less than 1:1, leaving phenolic hydroxyl groups intact. Consequently, the resulting MAPAs, as in MAPA-1 and MAPA-2, maintained comparable collagen crosslinking capability to that of PA. Collagen treated with MAPA-1 or MAPA-2 for a period as short as 30 s showed remarkable biostability against collagenase digestion that was also comparable to PA. However, when more than one equivalent MA was used, as in MAPA-3, some phenolic hydroxyl groups also reacted to form ester bonds with MA. Thus, MAPA-3 may show better polymerization capability with adhesive monomers, but its collagen crosslinking property was shown to be compromised and reduced, although still much better than the control. With the superior collagen crosslinking properties for MAPA-1 and MAPA-2 demonstrated, the next aim to validate the dual functionality of MAPAs is to evaluate their copolymerization capability with adhesive monomers. The reported MAPAs may represent a novel class of polymerizable collagen crosslinkers with the potential to reinforce dentin collagen and co-polymerize with adhesives simultaneously. The above promising results warrant further studies on the effect of MAPAs’ inclusion into adhesives on polymerization and dentin bonding.
ACKNOWLEDGMENTS
This study was supported by Research Grant R01-DE027049 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Breschi L, Maravic T, Cunha SR, Comba A, Cadenaro M, Tjaderhane L, et al. Dentin bonding systems: From dentin collagen structure to bond preservation and clinical applications. Dent Mater 2018;34:78–96. 10.1016/j.dental.2017.11.005. [DOI] [PubMed] [Google Scholar]
- [2].Breschi L, Mazzoni A, Ruggeri A, Cadenaro M, Di Lenarda R, De Stefano Dorigo E. Dental adhesion review: aging and stability of the bonded interface. Dent Mater 2008;24:90–101. 10.1016/j.dental.2007.02.009. [DOI] [PubMed] [Google Scholar]
- [3].Liu Y, Tjaderhane L, Breschi L, Mazzoni A, Li N, Mao J, et al. Limitations in bonding to dentin and experimental strategies to prevent bond degradation. J Dent Res 2011;90:953–68. 10.1177/0022034510391799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bedran-Russo AK, Pauli GF, Chen SN, McAlpine J, Castellan CS, Phansalkar RS, et al. Dentin biomodification: strategies, renewable resources and clinical applications. Dent Mater 2014;30:62–76. 10.1016/j.dental.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bedran-Russo AK, Pashley DH, Agee K, Drummond JL, Miescke KJ. Changes in stiffness of demineralized dentin following application of collagen crosslinkers. J Biomed Mater Res B Appl Biomater 2008;86:330–4. 10.1002/jbm.b.31022. [DOI] [PubMed] [Google Scholar]
- [6].Castellan CS, Bedran-Russo AK, Karol S, Pereira PN. Long-term stability of dentin matrix following treatment with various natural collagen cross-linkers. J Mech Behav Biomed Mater 2011;4:1343–50. 10.1016/j.jmbbm.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cova A, Breschi L, Nato F, Ruggeri A Jr., Carrilho M, Tjaderhane L, et al. Effect of UVA-activated riboflavin on dentin bonding. J Dent Res 2011;90:1439–45. 10.1177/0022034511423397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hass V, Luque-Martinez IV, Gutierrez MF, Moreira CG, Gotti VB, Feitosa VP, et al. Collagen cross-linkers on dentin bonding: Stability of the adhesive interfaces, degree of conversion of the adhesive, cytotoxicity and in situ MMP inhibition. Dent Mater 2016;32:732–41. 10.1016/j.dental.2016.03.008. [DOI] [PubMed] [Google Scholar]
- [9].Liu Y, Dusevich V, Wang Y. Proanthocyanidins rapidly stabilize the demineralized dentin layer. J Dent Res 2013;92:746–52. 10.1177/0022034513492769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tjäderhane L, Nascimento FD, Breschi L, Mazzoni A, Tersariol IL, Geraldeli S, et al. Strategies to prevent hydrolytic degradation of the hybrid layer-A review. Dent Mater 2013;29:999–1011. 10.1016/j.dental.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Green B, Yao X, Ganguly A, Xu C, Dusevich V, Walker MP, et al. Grape seed proanthocyanidins increase collagen biodegradation resistance in the dentin/adhesive interface when included in an adhesive. J Dent 2010;38:908–15. 10.1016/j.jdent.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hass V, Luque-Martinez I, Munoz MA, Reyes MF, Abuna G, Sinhoreti MA, et al. The effect of proanthocyanidin-containing 10% phosphoric acid on bonding properties and MMP inhibition. Dent Mater 2016;32:468–75. 10.1016/j.dental.2015.12.007. [DOI] [PubMed] [Google Scholar]
- [13].Liu Y, Dusevich V, Wang Y. Addition of Grape Seed Extract Renders Phosphoric Acid a Collagen-stabilizing Etchant. J Dent Res 2014;93:821–7. 10.1177/0022034514538972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Le Bourvellec C, Renard CM. Interactions between polyphenols and macromolecules: quantification methods and mechanisms. Crit Rev Food Sci Nutr 2012;52:213–48. 10.1080/10408398.2010.499808. [DOI] [PubMed] [Google Scholar]
- [15].Millet M, Poupard P, Guilois-Dubois S, Zanchi D, Guyot S. Self-aggregation of oxidized procyanidins contributes to the formation of heat-reversible haze in apple-based liqueur wine. Food Chem 2019;276:797–805. 10.1016/j.foodchem.2018.09.171. [DOI] [PubMed] [Google Scholar]
- [16].Hechler B, Yao X, Wang Y. Proanthocyanidins alter adhesive/dentin bonding strengths when included in a bonding system. Am J Dent 2012;25:276–80. [PMC free article] [PubMed] [Google Scholar]
- [17].Liu Y, Wang Y. Effect of proanthocyanidins and photo-initiators on photo-polymerization of a dental adhesive. J Dent 2013;41:71–9. 10.1016/j.jdent.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Quideau S, Deffieux D, Douat-Casassus C, Pouysegu L. Plant polyphenols: chemical properties, biological activities, and synthesis. Angew Chem Int Ed Engl 2011;50:586–621. 10.1002/anie.201000044. [DOI] [PubMed] [Google Scholar]
- [19].de Souza LC, Rodrigues NS, Cunha DA, Feitosa VP, Santiago SL, Reis A, et al. Two-year clinical evaluation of proanthocyanidins added to a two-step etch-and-rinse adhesive. J Dent 2019;81:7–16. 10.1016/j.jdent.2018.12.012. [DOI] [PubMed] [Google Scholar]
- [20].Feitosa VP, Ogliari FA, Van Meerbeek B, Watson TF, Yoshihara K, Ogliari AO, et al. Can the hydrophilicity of functional monomers affect chemical interaction? J Dent Res 2014;93:2016. 10.1177/0022034513514587. [DOI] [PubMed] [Google Scholar]
- [21].Yoshihara K, Hayakawa S, Nagaoka N, Okihara T, Yoshida Y, Van Meerbeek B. Etching Efficacy of Self-Etching Functional Monomers. J Dent Res 2018;97:1010–6. 10.1177/0022034518763606. [DOI] [PubMed] [Google Scholar]
- [22].Cheng L, Weir MD, Zhang K, Arola DD, Zhou X, Xu HH. Dental primer and adhesive containing a new antibacterial quaternary ammonium monomer dimethylaminododecyl methacrylate. J Dent 2013;41:345–55. 10.1016/j.jdent.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tezvergil-Mutluay A, Agee KA, Uchiyama T, Imazato S, Mutluay MM, Cadenaro M, et al. The inhibitory effects of quaternary ammonium methacrylates on soluble and matrix-bound MMPs. J Dent Res 2011;90:535–40. 10.1177/0022034510389472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Van Landuyt KL, Snauwaert J, De Munck J, Peumans M, Yoshida Y, Poitevin A, et al. Systematic review of the chemical composition of contemporary dental adhesives. Biomaterials 2007;28:3757–85. 10.1016/j.biomaterials.2007.04.044. [DOI] [PubMed] [Google Scholar]
- [25].Puertas-Bartolome M, Vazquez-Lasa B, San Roman J. Bioactive and Bioadhesive Catechol Conjugated Polymers for Tissue Regeneration. Polymers (Basel) 2018;10. 10.3390/polym10070768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu Y, Acharya G, Lee CH. Effects of dialdehyde starch on calcification of collagen matrix. J Biomed Mater Res A 2011;99:485–92. 10.1002/jbm.a.33209. [DOI] [PubMed] [Google Scholar]
- [27].Barth A, Zscherp C. What vibrations tell us about proteins. Q Rev Biophys 2002;35:369430. 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- [28].Liu Y, Chen M, Yao X, Xu C, Zhang Y, Wang Y. Enhancement in dentin collagen’s biological stability after proanthocyanidins treatment in clinically relevant time periods. Dent Mater 2013;29:485–92. 10.1016/j.dental.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vidal CM, Zhu W, Manohar S, Aydin B, Keiderling TA, Messersmith PB, et al. Collagen-collagen interactions mediated by plant-derived proanthocyanidins: A spectroscopic and atomic force microscopy study. Acta Biomater 2016;41:110–8. 10.1016/j.actbio.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Madhan B, Krishnamoorthy G, Rao JR, Nair BU. Role of green tea polyphenols in the inhibition of collagenolytic activity by collagenase. Int J Biol Macromol 2007;41:16–22. 10.1016/j.ijbiomac.2006.11.013. [DOI] [PubMed] [Google Scholar]
- [31].He L, Mu C, Shi J, Zhang Q, Shi B, Lin W. Modification of collagen with a natural crosslinker, procyanidin. Int J Biol Macromol 2011;48:354–9. 10.1016/j.ijbiomac.2010.12.012. [DOI] [PubMed] [Google Scholar]
- [32].Haslam E, Lilley TH. Interactions of natural phenols with macromolecules. Prog Clin Biol Res 1986;213:53–65. 10.1042/bj1390285. [DOI] [PubMed] [Google Scholar]
- [33].Miles CA, Avery NC, Rodin VV, Bailey AJ. The increase in denaturation temperature following cross-linking of collagen is caused by dehydration of the fibres. J Mol Biol 2005;346:551–6. 10.1016/j.jmb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- [34].Beart JE, Lilley TH, Polyphenol interactions EH. Part 2.1 covalent binding of procyanidins to proteins during acid-catalysed decomposition; observations on some polymeric proanthocyanidins. J Chem Soc Perkin1 1985;2:1439–43. 10.1039/P29850001439. [DOI] [Google Scholar]
- [35].Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007;318:426–30. 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]