

Abstract
Atopic dermatitis (AD) is a common immune-medicated skin disease. Previous studies have explored the relationship between Human Leukocyte Antigen (HLA) allelic variation and AD with conflicting results. The aim was to examine HLA Class I genetic variation, specifically peptide binding groove variation, and associations with AD. A case-control study was designed to evaluate HLA class I allelic variation and binding pocket polymorphisms, using next generation sequencing on 464 subjects with AD and 388 without AD. Logistic regression was used to evaluate associations with AD by estimating odds ratios (95% confidence intervals). Significant associations were noted with susceptibility to AD (B*53:01) and protection from AD (A*01:01, A*02:01, B*07:02 and C*07:02). Evaluation of polymorphic residues in Class I binding pockets revealed six amino acid residues conferring protection against AD: A9F (HLA-A, position 9, phenylalanine) [pocket B/C], A97I [pocket C/E], A152V [pocket E], A156R [pocket D/E], B163E [pocket A] and C116S [pocket F]. These findings demonstrate that specific HLA class I components are associated with susceptibility or protection from AD. Individual amino acid residues are relevant to protection from AD and set the foundation for evaluating potential HLA Class I molecules in complex with peptides/antigens that may initiate or interfere with T-cell responses.
Keywords: Atopic Dermatitis, Case-Control Study, Human Leukocyte Antigen, Allergy
1. Introduction
Atopic dermatitis (AD) is a common and chronic, itchy inflammatory skin disorder, which has been associated with genetic risk factors.[1–4] Several small studies using older immunogenotyping technology have explored the relationship between Human Leukocyte Antigen (HLA) allelic variation and AD with conflicting results.[5–9] Independently, several large genome wide association studies (GWAS) that relied on imputation protocols, of Europeans and Asians with AD, have reported associations between HLA genes and AD.[6–9] These reports identify HLA regions associated with AD or atopic illnesses such as asthma and food allergy which are commonly seen with AD. Reporting was based on single nucleotide changes such as rs4713555 (located between DRB1 and DQA1)[6, 7], rs28383201 (between DRB1 and DQA1)[10], rs9368677 (between HLA-B and HLA-C)[11], rs9469099 (TSBP1)[11], rs2251396 (between HLA-B and MICA)[9], and rs6474 (complement region)[9]. A careful assessment of the RSID (reference SNP cluster ID) variants listed and noted above reveal that by themselves these single nucleotide polymorphisms (SNPs) do not uniquely describe HLA allelic variants, as they are not located within HLA genes, but they denote the relevance of a region as being implicated in the disease process.[6–9, 12, 13] Considering the critical role of HLA genes for the immune response, these findings indicate the need for identifying specific HLA polymorphisms relevant to AD and the potential role that they may play in disease susceptibility/protection.
AD, classically, has been associated with T-cell dysregulation[14–16], which is most often assumed to be with T-cells that skew to TH2 pathways, involving CD4 T-cells associated with allergic disease.[15, 16] However, TH1 pathways most often involve HLA Class I interacting with CD8 T-cells, which is frequently associated with viral and autoimmune illnesses.[17–19] Considering that AD has been associated with viral syndromes, including severe viral infections like eczema herpeticum, and with autoimmune diseases, CD8 cells which have been shown to be decreased in those with AD, may also play a role in the disease process.[20–27] Of note is a recent study demonstrating that while individuals with AD do generally have active TH2 pathways, TH1 pathways are also active in adults with AD.[28] Independently, several studies including a recent study by Mack et al. have associated AD with NK cell function.[21, 29–32] NK cells function by directly interacting with HLA Class I. It is, therefore, meaningful to investigate the possible role of HLA Class I polymorphisms in the AD disease process.
Using next generation sequencing (NGS)-based technologies that allow for the comprehensive and detailed characterization of HLA genes (including exonic and intronic sequences) we determined allelic variations within the AD and control groups. Thereafter, and based on the allelic variation, we assessed the differential frequency of specific amino acid residues withing the binding pocket, critical for peptide binding.[33–35] Our primary objective was to examine as to whether HLA Class I genetic variation, specifically within the peptide binding groove, is associated with AD. To our knowledge, no previous studies have explored the role of HLA class I binding site variation and its association with AD disease risk.
2. Materials and Methods:
2.1. Population
The primary source of subjects for this study was the Genetics of Atopic Dermatitis (GAD) cohort. All subjects were examined by dermatologists with expertise in the diagnosis of AD from the following Dermatology practice locations: University of Pennsylvania Perelman School of Medicine, Children’s Hospital of Philadelphia, Pennsylvania State University/Hershey Medical Center, and Washington University School of Medicine in St Louis. All subjects had a history and an exam consistent with AD (cases) or no history of AD by history and examination (controls). There was no age restriction for enrollment. All subject related information was obtained using a standard case report form that was completed by the subject, after subject interview, and/or medical record review.
All subjects or legal guardians provided written informed consent or, if appropriate, assent approved by their appropriate Institutional Review Board.
2.2. DNA Analysis
DNA was collected using Oragene DNA collection kits (DNA Genotek, Ottawa Canada) as previously reported.[36] The three classical HLA Class I genes (−A, −B, −C) for individuals in the GAD cohort were sequenced using targeted amplicon-based NGS with Omixon Holotype HLA™ V2 kits (Budapest, Hungary). HLA genes were amplified (Qiagen LR PCR kits, Valencia, CA) on a Veriti thermal cycler (ThermoFisher, Waltham, MA), then amplicons from each gene were pooled per sample with library preparation occurring according to the manufacturer’s protocol. The final library was sequenced on an Illumina MiSeq (San Diego, CA) using paired-end 2×150 V2 chemistry. Omixon Twin™ (7,000 pairs/locus, v, 2.5.1) and GenDx NGSengine® (300,000 pairs/sample) analyzed each set of Fastq files. Genotyping was conducted in the Immunogenetics Laboratory of Children’s Hospital of Philadelphia, a CLIA and ASHI accredited clinical laboratory, using clinical protocols with appropriate quality controls and standards.
2.3. Analysis
Analyses were conducted based on HLA allelic variation induced changes to protein residues in exonic coding regions as denoted in receptor binding regions. Protein (reside) variation were determined based on IMGT/HLA data. We focused on specific pocket residues as being critical for peptide binding and therefore possibly involved in disease processes, as previously reported.[33] This decision was based on Simpson Reciprocal Index (SRI).[33] As a result, we focused only on those residues with the highest SRI (≥2.4, i.e. the residues that exert the highest influence in peptide binding accounting for the number of amino acid substitutions at each of these positions and the relevant frequency of alleles with the particular amino acid in the particular positional residue).[33] We, therefore, evaluated positions 9, 97, 114, 152 and 156 of HLA-A; 67, 97, 116, and 163 of HLA-B; and positions 9, 116 and 156 of HLA-C (Table 1).
Table 1:
Simpson reciprocal index (SRI)[33] is presented for the full GAD cohort for residues known to occur within HLA Class I binding pockets. SRI that are greater than or equal to the threshold of 2.4 are bolded.
| Simpson reciprocal index | |||
|---|---|---|---|
| Position | HLA-A | HLA-B | HLA-C |
| 9 | 2.86 | 1.81 | 2.62 |
| 63 | 1.54 | 1.99 | 1 |
| 66 | 1.86 | 1.16 | 1.36 |
| 67 | 1.29 | 3.77 | 1 |
| 70 | 1.75 | 1.76 | 1 |
| 74 | 1.60 | 1.82 | 1 |
| 77 | 2.07 | 2.00 | 1.94 |
| 80 | 1.38 | 2.22 | 1.94 |
| 81 | 1.38 | 1.83 | 1 |
| 97 | 2.98 | 2.84 | 1.52 |
| 99 | 1.26 | 1.02 | 1.89 |
| 114 | 3.16 | 2.14 | 1.68 |
| 116 | 2.31 | 3.66 | 2.51 |
| 152 | 2.52 | 1.89 | 1.83 |
| 156 | 2.43 | 2.27 | 3.11 |
| 163 | 1.53 | 2.59 | 1.57 |
| 167 | 1.56 | 1.31 | 1 |
Residue frequencies (RF) were based on the number of chromosomes with alleles that coded for the residue variant and were estimated with 95% confidence interval (CI). RF was estimated separately for those with and without AD. The odds ratio (OR) of having AD was estimated using an additive model for the residue variation and logistic regression comparing those with and without AD. OR were not adjusted in that conceptually HLA variation is a somatic genetic trait. Secondary analyses included estimates adjusted by ancestry and gender as well as estimates by ancestry. The effect estimates were not adjusted for other atopic illnesses like asthma, seasonal allergies or food allergies because these illness are likely on the same causal pathway as atopic dermatitis as noted in studies of the atopic march.[37] Our final analysis is at the amino acid level within the binding pocket of the HLA molecules (e.g., a phenotype).
Correction of p-values for multiple comparisons is not straightforward.[38, 39] The goal behind correction is to control the type I error rate (α) and thereby improve the reproducibility of investigations.[38, 40, 41] Our investigation was conducted because previous studies indicated interest in Class I HLA genes with respect to AD. As a result, we report Bonferroni correction (pcorr) by number of variants or residues analyzed per HLA gene. We also report the uncorrected p-value. All statistical analyses were conducted using Stata Version 16.1 (College Station, TX).
3. Results
3.1. Characteristics of study subjects
HLA genotyping was performed on 464 individuals with AD (cases) and 388 without (controls) members of GAD. Within the full GAD cohort, 503 (59.0%) were of European-Ancestry, 318 (37.3%) were of African-Ancestry, 479 (56.2%) were female, 291 (39.3%) had asthma, 342 (40.1%) had seasonal allergies, and 205 (24.1%) had both. Of the 464 subjects in the GAD cohort with AD, 232 (50.0%) were of European-Ancestry, 203 (43.8%) were of African-Ancestry, 303 (65.3%) were female, 254 (54.7%) had asthma, 290 (62.5%) had seasonal allergies, and 190 (40.9%) had both. For this group, the median age of AD onset was 0.75 years (interquartile range: 0.25 – 11). Of the 388 subjects in the GAD cohort without AD (controls), 271 (69.8%) were of European-Ancestry, 115 (30.0%) were of African-Ancestry, 176 (45.4%) were female, 49 (12.7%) had asthma, 63 (16.3%) had seasonal allergies, and 15 (3.8%) had both. As expected, the presence of asthma and seasonal allergies were significantly different between the case and control groups (p <0.001)
3.2. HLA class I allelic sequencing
For the 852 members of the GAD cohort, the results for alleles with allelic frequency of ≥ 0.05, are presented in Table 2, and include four HLA-A, six HLA-B and six HLA-C alleles. As expected, AF varied by ancestry (Table 2). To be complete, all genotyping results are available in the supplement Table 1.
Table 2:
HLA allelic frequencies for the full GAD cohort, GAD by race and GAD by case/control status for alleles with an allelic frequency of ≥ 0.05. See supplemental Table 1 for all alleles.
| Full GAD n=852 | European Ancestry n=503 | African Ancestry n=318 | All Controls n=388 | All Cases n=464 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Allele | n | AF (95% CI) | n | AF (95% CI) | n | AF (95% CI) | n | AF (95% CI) | n | AF (95% CI) |
| A*01:01 | 182 | 0.11 (0.09,0.12) | 158 | 0.16 (0.14,0.18) | 20 | 0.03(0.02,0.05) | 115 | 0.15 (0.12,0.18) | 67 | 0.07 (0.06,0.09) |
| A*02:01 | 305 | 0.18 (0.16,0.20) | 241 | 0.24 (0.21,0.27) | 59 | 0.09(0.07,0.12) | 162 | 0.21 (0.18,0.24) | 143 | 0.15 (0.13,0.18) |
| A*03:01 | 215 | 0.13 (0.11,0.14) | 150 | 0.15 (0.13,0.17) | 63 | 0.10(0.08,0.12) | 102 | 0.13 (0.11,0.16) | 113 | 0.12 (0.10,0.14) |
| A*24:02 | 106 | 0.06 (0.05,0.07) | 81 | 0.08 (0.06,0.10) | 15 | 0.02(0.01,0.04) | 55 | 0.07 (0.05,0.09) | 50 | 0.05 (0.04,0.07) |
| B*07:02 | 162 | 0.09 (0.08,0.11) | 103 | 0.10 (0.08,0.12) | 52 | 0.08(0.06,0.11) | 94 | 0.12 (0.10,0.15) | 68 | 0.07 (0.06,0.09) |
| B*08:01 | 120 | 0.07 (0.06,0.08) | 101 | 0.10 (0.08,0.12) | 17 | 0.03(0.02,0.04) | 60 | 0.08 (0.06,0.10) | 60 | 0.06 (0.05,0.08) |
| B*35:01 | 95 | 0.06 (0.05,0.07) | 53 | 0.05 (0.04,0.07) | 41 | 0.06(0.05,0.09) | 46 | 0.06 (0.04,0.08) | 49 | 0.05 (0.04,0.07) |
| B*44:02 | 102 | 0.06 (0.05,0.07) | 87 | 0.09 (0.07,0.11) | 14 | 0.02(0.01,0.04) | 51 | 0.07 (0.05,0.09) | 51 | 0.05 (0.04,0.07) |
| B*44:03 | 80 | 0.05 (0.04,0.06) | 45 | 0.04 (0.03,0.06) | 36 | 0.06(0.04,0.08) | 28 | 0.04 (0.02,0.05) | 52 | 0.06 (0.04,0.07) |
| B*53:01 | 77 | 0.05 (0.04,0.06) | 10 | 0.01 (0.00,0.02) | 66 | 0.10(0.08,0.13) | 22 | 0.03 (0.02,0.04) | 55 | 0.06 (0.04,0.08) |
| C*03:04 | 104 | 0.06 (0.05,0.07) | 55 | 0.05 (0.04,0.07) | 41 | 0.06(0.05,0.09) | 41 | 0.05 (0.04,0.07) | 63 | 0.07 (0.05,0.09) |
| C*04:01 | 233 | 0.14 (0.12,0.15) | 124 | 0.12 (0.10,0.15) | 107 | 0.17(0.14,0.20) | 98 | 0.13 (0.10,0.15) | 135 | 0.15 (0.12,0.17) |
| C*05:01 | 106 | 0.06 (0.05,0.07) | 86 | 0.09 (0.07,0.10) | 19 | 0.03(0.02,0.05) | 55 | 0.07 (0.05,0.09) | 51 | 0.05 (0.04,0.07) |
| C*06:02 | 115 | 0.07 (0.06,0.08) | 83 | 0.08 (0.07,0.10) | 29 | 0.05(0.03,0.06) | 65 | 0.08 (0.07,0.11) | 49 | 0.05 (0.04,0.07) |
| C*07:01 | 203 | 0.12 (0.10,0.14) | 154 | 0.15 (0.13,0.18) | 47 | 0.07(0.05,0.10) | 98 | 0.13 (0.10,0.15) | 105 | 0.11 (0.09,0.14) |
| C*07:02 | 175 | 0.10 (0.09,0.12) | 118 | 0.12 (0.10,0.14) | 46 | 0.07(0.05,0.10) | 97 | 0.13 (0.10,0.15) | 78 | 0.08 (0.07,0.10) |
With respect to associations with AD, five alleles maintained significance after adjusting for multiple comparisons: A*01:01, A*02:01, B*07:02, B*53:01, and C*07:02. Only B*53:01 was associated with an increased susceptibility for AD (Table 3). While individual allelic frequencies could vary by ancestry, the effect estimates changed minimally after adjustment for gender and race and changed minimally when evaluated within European or African ancestry (Tables 2 & 3). B*07:02 and C*07:02 were in linkage disequilibrium (LD) (r2=0.69) and this was noted in those of European (r2=0.85) and African ancestry (r2=0.45). B*07:02~C*07:02 represents a known and common haplotype. The other alleles reported above were not in LD.
Table 3:
Association of AD and HLA Class I allelic variation presented as odds ratios with 95% confidence intervals (OR > 1 represents increased risk). ORs presented for the full cohort. The full cohort adjusted by ancestry and for just those of European or African ancestry. The p-value correction (in manuscript) is based on analyses per gene; HLA-A 4, HLA-B 6 and HLA-C 6.
| Allele | OR | p-value | Adjusted OR | European Ancestry | African Ancestry |
|---|---|---|---|---|---|
| A*01:01 | 0.46 (0.33, 0.63) | 1.35e-06 | 0.55(0.40, 0.77) | 0.54 (0.37, 0.77) | 0.54(0.22, 1.35) |
| A*02:01 | 0.71 (0.56, 0.90) | .0049846 | 0.82(0.63, 1.06) | 0.94 (0.70, 1.24) | 0.64(0.38, 1.11) |
| A*03:01 | 0.92 (0.70, 1.21) | .5642952 | 1.05(0.79, 1.41) | 0.94 (0.67, 1.31) | 1.31(0.76, 2.26) |
| A*24:02 | 0.75 (0.51, 1.20) | .1549402 | 0.89(0.59, 1.34) | 1.09 (0.70, 1.71) | 0.13(0.04, 0.47) |
| B*07:02 | 0.60 (0.43, 0.82) | .0015001 | 0.56(0.40, 0.78) | 0.48 (0.31, 0.75) | 0.77(0.44, 1.34) |
| B*08:01 | 0.84 (0.59, 1.20) | .3335906 | 1.03(0.71, 1.50) | 1.02 (0.68, 1.52) | 1.03(0.43, 2.46) |
| B*35:01 | 0.88 (0.59, 1.34) | .5634918 | 0.88(0.57, 1.35) | 0.62 (0.34, 1.12) | 1.36(0.70, 2.66) |
| B*44:02 | 0.83 (0.57, 1.23) | .3660473 | 1.06(0.71, 1.60) | 1.10 (0.71, 1.69) | 0.77(0.28, 2.13) |
| B*44:03 | 1.60 (1.00, 2.58) | .0516518 | 1.62(0.99, 2.66) | 1.52 (0.82, 2.81) | 2.06(0.92, 4.63) |
| B*53:01 | 1.98 (1.22, 3.22) | .005361 | 1.43(0.87, 2.36) | 1.77 (0.49, 6.36) | 1.51(0.88, 2.60) |
| C*03:04 | 1.31 (0.87, 1.98) | .1919211 | 1.23(0.80, 1.90) | 1.35 (0.77, 2.36) | 1.10(0.57, 2.11) |
| C*04:01 | 1.17 (0.89, 1.54) | .2586665 | 1.12(0.84, 1.50) | 0.80 (0.55, 1.16) | 2.10(1.28, 3.42) |
| C*05:01 | 0.77 (0.52, 1.13) | .1841949 | 0.97(0.64, 1.45) | 0.92 (0.59, 1.43) | 0.97(0.37, 2.54) |
| C*06:02 | 0.60 (0.41, 0.89) | .0109947 | 0.66(0.44, 0.98) | 0.66 (0.41, 1.06) | 0.80(0.38, 1.69) |
| C*07:01 | 0.89 (0.67, 1.18) | .4195639 | 1.06(0.78, 1.42) | 1.06 (0.76, 1.47) | 1.00(0.53, 1.88) |
| C*07:02 | 0.66 (0.49, 0.90) | .0084513 | 0.66(0.48, 0.91) | 0.58 (0.39, 0.86) | 0.88(0.48, 1.60) |
3.3. HLA Class 1 residue analysis
SRI analysis revealed four HLA-A residue positions with 19 variants, four HLA-B residue positions with 19 variants and three HLA-C residues with 11 variants that had scores of greater than or equal to 2.4 (Table 1). The ORs for association with AD are presented in Table 4. The amino acid residues associated with prevention of AD after adjustment for multiple testing included A9F (pcorr =2.2×10−4), A97I (pcorr =0.034), A152V (pcorr =0.038), A156R (pcorr =1.9×10−4), B163E (pcorr =0.017), and C116S (pcorr =0.030). None of these residues are in LD (i.e., all r2 < 0.20). The effect estimates for association for the residues A9F, A152V, A156R, B163E, and C116S were similar in those of African and European ancestry. The effect estimates for association for A97I and A152V were likely not the same for those of African and European ancestry (Table 4). None were associated with an increased susceptibility. HLA-A*01:01, as well as several other alleles, is one of the alleles that generates the A9F, A97I, and A156R residue phenotypes. HLA-A*02:01 as well as several other alleles, is one of the alleles that generates the A152V residue phenotype. HLA-B*07:02 as well as several other alleles, is one of the alleles that produces the B163E residue phenotype. HLA-C*07:02 as well as several other alleles, is one of the alleles that generates the C116S residue phenotype. All known alleles associated with these residues are presented in Supplemental Table 2.
Table 4:
Association of AD and HLA Class I residue variation presented as odds ratios with 95% confidence intervals (OR > 1 represents increased risk). ORs presented for the full cohort as well as by European or African ancestry. The corrected p-value (in manuscript) is based on analyses per residue; HLA-A 19, HLA-B 19 and HLA-C 11.
| Allele | Full GAD | p-value | European Ancestry | African Ancestry |
|---|---|---|---|---|
| A9T | 1.55 (1.11,2.16) | .0104174 | 1.40 (0.84,2.34) | 1.23 (0.77,1.96) |
| A9Y | 0.98 (0.77,1.25) | .8759459 | 1.01 (0.73,1.41) | 0.81 (0.55,1.18) |
| A9S | 1.12 (0.88,1.43) | .3475483 | 1.13 (0.81,1.58) | 0.83 (0.57,1.23) |
| A9F | 0.64 (0.53,0.77) | 1.91e-06 | 0.72 (0.56,0.91) | 0.89 (0.63,1.25) |
| A97M | 1.16 (0.93,1.44) | .196362 | 1.32 (0.98,1.77) | 0.79 (0.56,1.14) |
| A97I | 0.73 (0.60,0.89) | .0018121 | 0.60 (0.46,0.79) | 1.15 (0.82,1.60) |
| A114R | 0.61 (0.43,0.87) | .0067458 | 0.39 (0.22,0.72) | 0.81 (0.50,1.30) |
| A114H | 0.67 (0.48,0.93) | .0150107 | 1.00 (0.58,1.71) | 0.54 (0.35,0.83) |
| A114Q | 1.24 (0.94,1.64) | .125937 | 1.37 (0.95,1.97) | 1.04 (0.65,1.64) |
| A114E | 1.38 (0.96,2.00) | .0861521 | 1.18 (0.59,2.37) | 1.09 (0.68,1.75) |
| A152A | 0.63 (0.38,1.03) | .0674447 | 0.38 (0.18,0.80) | 0.82 (0.38,1.75) |
| A152E | 0.92 (0.73,1.16) | .4897042 | 1.01 (0.76,1.34) | 1.09 (0.72,1.65) |
| A152R | 1.81 (1.05,3.13) | .0333442 | 2.08 (0.60,7.18) | 1.27 (0.67,2.38) |
| A152W | 1.12 (0.68,1.84) | .6674959 | 0.97 (0.41,2.29) | 0.94 (0.49,1.80) |
| A152V | 0.63 (0.46,0.85) | .0028012 | 1.01 (0.60,1.70) | 0.51 (0.34,0.76) |
| A156L | 1.03 (0.86,1.24) | .7397109 | 1.09 (0.86,1.39) | 1.05 (0.77,1.45) |
| A156W | 1.15 (0.87,1.52) | .3152551 | 1.34 (0.89,2.00) | 0.81 (0.54,1.22) |
| A156Q | 0.77 (0.57,1.03) | .074487 | 0.84 (0.59,1.20) | 0.56 (0.26,1.19) |
| A156R | 0.51 (0.37,0.69) | .0000103 | 0.55 (0.38,0.79) | 0.70 (0.36,1.38) |
| B67C | 0.69 (0.50,0.95) | .0246945 | 0.81 (0.56,1.19) | 0.81 (0.41,1.60) |
| B67F | 0.94 (0.75,1.17) | .5577738 | 0.86 (0.64,1.14) | 1.31 (0.89,1.92) |
| B67M | 1.02 (0.72,1.45) | .9191346 | 0.61 (0.36,1.04) | 1.38 (0.79,2.41) |
| B67S | 1.02 (0.84,1.23) | .8488475 | 1.35 (1.05,1.73) | 0.82 (0.59,1.15) |
| B67Y | 0.75 (0.57,0.99) | .0412008 | 0.63 (0.43,0.93) | 0.79 (0.51,1.23) |
| B97R | 1.14 (0.95,1.37) | .171127 | 1.27 (1.00,1.63) | 1.10 (0.81,1.49) |
| B97S | 0.71 (0.57,0.90) | .0037412 | 0.74 (0.55,0.99) | 0.74 (0.49,1.12) |
| B97T | 0.74 (0.52,1.05) | .093219 | 0.83 (0.55,1.26) | 0.63 (0.29,1.37) |
| B97V | 0.86 (0.55,1.35) | .5138767 | 0.50 (0.26,0.95) | 2.43 (0.98,6.03) |
| B97N | 0.44 (0.22,0.87) | .0176398 | 0.55 (0.26,1.14) | 0.56 (0.08,4.05) |
| B97W | 0.92 (0.56,1.53) | .7530051 | 1.06 (0.57,1.95) | 0.92 (0.37,2.28) |
| B116D | 0.91 (0.69,1.20) | .5071285 | 0.99 (0.71,1.38) | 1.41 (0.80,2.49) |
| B116F | 0.76 (0.54,1.08) | .1252513 | 0.99 (0.67,1.47) | 0.72 (0.33,1.54) |
| B116L | 1.07 (0.74,1.55) | .7064316 | 1.28 (0.80,2.06) | 0.83 (0.45,1.52) |
| B116Y | 0.78 (0.64,0.95) | .013964 | 0.76 (0.59,0.99) | 0.88 (0.62,1.26) |
| B116S | 1.05 (0.85,1.29) | .6725844 | 0.95 (0.72,1.27) | 1.05 (0.76,1.46) |
| B163E | 0.67 (0.53,0.85) | .0008871 | 0.66 (0.49,0.89) | 0.63 (0.40,0.97) |
| B163L | 1.10 (0.92,1.31) | .3057666 | 0.95 (0.75,1.20) | 1.26 (0.92,1.73) |
| B163T | 0.91 (0.73,1.12) | .3654661 | 1.24 (0.95,1.62) | 0.74 (0.49,1.10) |
| C9D | 0.79 (0.65,0.96) | .0182299 | 0.78 (0.61,1.00) | 1.07 (0.74,1.54) |
| C9F | 1.83 (0.84,3.98) | .1298465 | 1.78 (0.78,4.07) | NA |
| C9S | 1.21 (0.94,1.58) | .1435786 | 0.85 (0.60,1.21) | 2.07 (1.30,3.28) |
| C9Y | 1.01 (0.84,1.21) | .9144933 | 1.10 (0.87,1.40) | 0.92 (0.68,1.26) |
| C116F | 1.23 (1.00,1.50) | .0479706 | 0.98 (0.75,1.29) | 1.79 (1.27,2.52) |
| C116L | 0.59 (0.28,1.27) | .1801457 | 0.70 (0.31,1.59) | 0.56 (0.03,9.11) |
| C116S | 0.75 (0.62,0.91) | .0027865 | 0.81 (0.63,1.03) | 0.81 (0.58,1.12) |
| C116Y | 1.25 (0.91,1.72) | .1671472 | 1.29 (0.87,1.92) | 1.25 (0.68,2.29) |
| C156E | 1.04 (0.74,1.46) | .8112022 | 1.25 (0.77,2.04) | 0.78 (0.48,1.26) |
| C156L | 1.27 (0.92,1.74) | .1466801 | 1.18 (0.78,1.79) | 1.16 (0.66,2.04) |
| C156T | 0.84 (0.69,1.02) | .0810395 | 0.69 (0.51,0.91) | 1.50 (1.11,2.03) |
4. Discussion
We conducted high-resolution NGS sequencing of genes in the HLA Class I region of individuals with AD. The study focused on European Americans and African Americans with AD using a case-control study design. HLA allelic variation and protein residue changes related to these variants were assessed. From the HLA Class I region, A*01:01, A*02:01, B*07:02, and C*07:02 prevented and B*53:01 increased the susceptibility of AD. Amino acid residues, A9F, A97I, A152V, A156R, B163E, and C116S, which are from binding sites in the Class I molecule, were associated with prevention of AD.
The assessment of individual positional residues and amino acids aims at focusing on specific sub-molecular entities within HLA molecules and their association to AD. A residue can be common to multiple different HLA alleles as such each individual allele might not show significant associations, but the residue across alleles (i.e., it is a composite of alleles) can denote a phenotype that is associated with disease.[34] In our study, individual residues associated with HLA-A confer protection from AD. These residues were at positions 9 (pockets C/B), 97 (pockets C/E), 152 (pocket E) and 156 (pocket E/D). For HLA-B, residue at position 163 (pocket A) and for HLA-C residue located at position 116 (pocket F) were protective of AD. Alleles B*07:02 and C*07:02 are in LD. Admittedly, none of the aforementioned pocket residues within each individual Class I alleles is expected to operate independently from each other, however none of these pocket residues are in LD (data not shown) within different class 1 isotypes. Since the residue variants influence peptide binding, their identification is critically important as we search for peptides/antigens that bind to HLA receptors with relevance to AD.[20] By analyzing the amino acid polymorphisms involved in the HLA binding pockets, we evaluated biologically relevant functionality and our analysis was consistent with the widely accepted notion that HLA disease associations can be defined much more sensitively at the amino acid level than at the classical HLA allelic level.[33, 34]
Several immunologic functions can be influenced by HLA Class I activation. HLA has been shown to be associated with multiple disease states, with some alleles being protective and others conferring susceptibility. The HLA-B allele has been associated with autoimmune and inflammatory diseases such as ankylosing spondylitis, thyroid disease, Type 1 diabetes, and psoriasis.[19] In fact, recent research suggests that ~90% of causal autoimmune disease variants reside within non-coding regions of the human genome and therefore it is very likely that multiple elements (like regulatory miRNAs) within the HLA genes could be important.[42]
As with all epidemiologic studies, this study has limitations. Although this study is the largest study of its kind, we were under-powered to evaluate less common HLA alleles and for this reason we a priori limited our analyses to alleles with a frequency of ≥0.05. Restricting our analyses to more common alleles may limit a fuller understanding of the underlying mechanisms. However, alleles that might have been too uncommon to be individually evaluated would have been included in our analyses of protein residue variation, since this included groups of alleles that share individual amino acids at specific positional residues. Our ability to trace the phenotype of the likely protein residue alteration of the allelic variants’ effect to the receptor antigen interface is physiologically important and confirms the significance of our initial allelic findings. The study cohort was derived from academic dermatology offices and as such patients were likely more severe, so it is possible that our results may not generalize to other sites.
In summary, we conducted a case-control study of individuals with AD and controls using NGS of the three Classic HLA Class I alleles. HLA Class I allelic variation was demonstrated to be associated with AD, B*53:01, and protective for AD with respect to A*01:01, A*02:01, B*07:02, and C*07:02 as well as the specific protein residues, A9F, A97I, A152V, A156R, B163E and C116S, associated with these alleles. The identification of these sub-molecular HLA elements (residues) associated with a particular phenotype (disease progression, protection or susceptibility), are therefore good probes for dissecting the physiology of reported associations. Further studies of HLA Class I including the identification of peptides from known antigens implicated in AD that interact with these alleles as well as known antigens will lead to a better understanding of the immunologic pathology of AD and has the potential to lead to new therapeutic models.
Supplementary Material
Fig. 1.
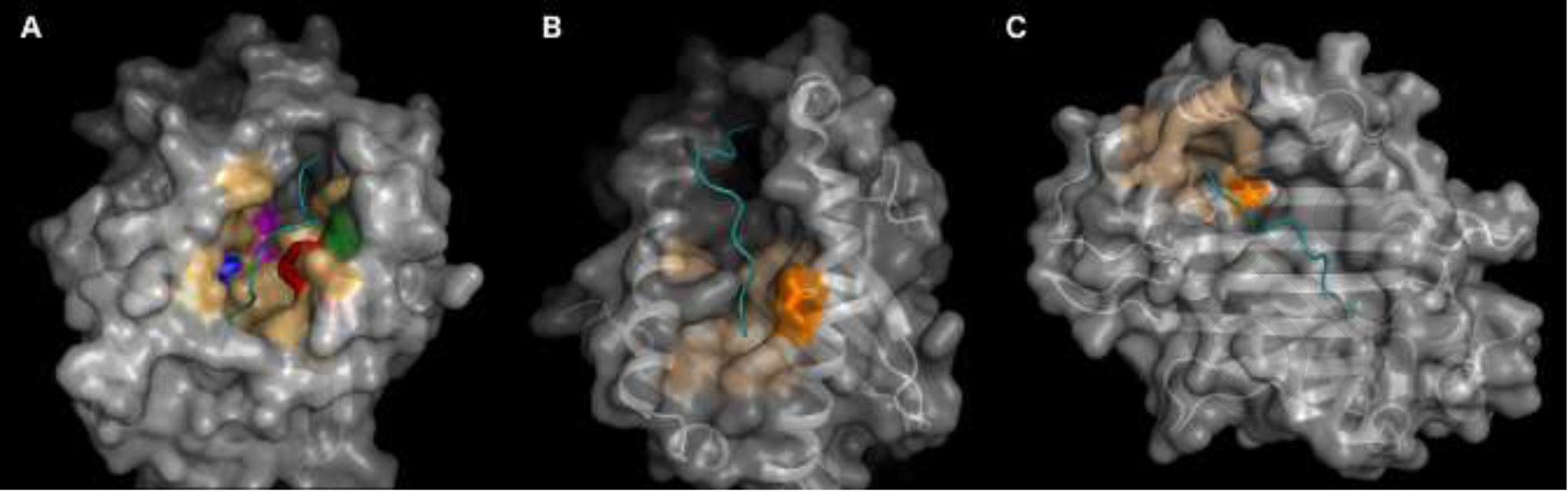
Locations of the significant residues. The 3-dimensional structure of each HLA Class I molecule is shown, highlighting the significant residues and pockets in which they participate. In all images the HLA protein is shown with a carbon backbone overlayed with the surface. The peptide presented by the HLA molecule is shown in cyan, and the non-significant HLA pocket residues are shown in light orange. The significant residue(s) for each HLA molecule is shown in a bold color and with the side chains as sticks. Beta-2 microglobulin is present as part of each crystal structure and has been hidden. A) HLA-A, based on an HLA-A*01:01 crystal structure (Protein Data Bank (PDB): 6AT9), has been mutated to show a Valine residue at position 152. Each significant residue is shown separately: A9F is blue, A97I is magenta, A152V is green and A156R is red. B) HLA-B, based on a HLA-B*27:05 crystal structure (PDB ID: 2BSS) shows B163E as orange. C) HLA-C, based on a HLAC*06:02 crystal structure (PDB ID: 5 W67), shows C116S also as orange. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Acknowledgments and role of funding source:
This work was supported in part by grants from the National Institutes for Health (NIAMS) R01-AR060962 (PI: Margolis) and R01-AR070873 (MPI: Margolis/Monos). The sponsors had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; and the decision to submit the manuscript for publication.
Potential Conflicts of Interest:
David Margolis is or recently has been a consultant for Pfizer, Leo, and Sanofi with respect to studies of atopic dermatitis and serves on an advisory board for the National Eczema Association. Dr. Chiesa Fuxench has served as a consultant for the National Eczema Association, Asthma and Allergy Foundation of America, Pfizer, Abbvie, and Sanofi receiving honoraria, and receives or has received research grants (to the Trustees of the University of Pennsylvania) from Regeneron, Sanofi, Leo, Tioga, Vanda pharmaceuticals, and Menlo Therapeutics for clinical trials related to atopic dermatitis; and has received payment for continuing medical education work related to atopic dermatitis that was supported indirectly by Sanofi and Regeneron. Dimitri S. Monos is Chair of the Scientific Advisory Board of Omixon and owns options in Omixon. Dimitri Monos, Deborah Ferriola, and Jamie Duke receive royalties from Omixon. Brian S. Kim has served as a consultant to AbbVie, Almirall, Amagma, Cara Therapeutics, Daewoong, Incyte, OM Pharma, and Pfizer; has served on advisory boards for AstraZeneca, Boehringer Ingelheim, Cara Therapeutics, Celgene Corporation, Regeneron Pharmaceuticals, Sanofi Genzyme, and Trevi Therapeutics; is a shareholder in Locus Biosciences; has a pending patent for JAK inhibitors in chronic itch.
Abbreviations:
- AF
Allelic frequency
- AD
Atopic dermatitis
- CI
Confidence interval
- DNA
Deoxyribonucleic acid
- GAD
Genetics of Atopic Dermatitis
- GWAS
Genome wide association study
- HLA
Human Leukocyte Antigen
- LD
Linkage disequilibrium
- NMDP
National Marrow Donor Program
- NGS
Next generation sequencing
- KIR
Killer cell Ig-like receptor family
- OR
Odds Ratio
- p
p-value
- SNP
Single nucleotide polymorphisms
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability: Genotyping frequency results are available in the supplement and on request.
References:
- [1].Abuabara K, Magyari A, McCulloch CE, Linos E, Margolis DJ, Langan SM: Prevalence of Atopic Eczema Among Patients Seen in Primary Care: Data From The Health Improvement Network. Ann Intern Med 2019;170:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abuabara K, Ye M, McCulloch CE, Sullivan A, Margolis DJ, Strachan DP et al. : Clinical onset of atopic eczema: Results from 2 nationally representative British birth cohorts followed through midlife. J Allergy Clin Immunol 2019;144:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chiesa Fuxench ZC, Block JK, Boguniewicz M, Boyle J, Fonacier L, Gelfand JM et al. : Atopic Dermatitis in America Study: A Cross-Sectional Study Examining the Prevalence and Disease Burden of Atopic Dermatitis in the US Adult Population. J Invest Dermatol 2019;139:583. [DOI] [PubMed] [Google Scholar]
- [4].Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH et al. : Atopic Dermatitis in US Adults: From Population to Health Care Utilization. J Allergy Clin Immunol Pract 2019;7:1524. [DOI] [PubMed] [Google Scholar]
- [5].Margolis DJ, Mitra N, Kim B, Gupta J, Hoffstad OJ, Papadopoulos M et al. : Association of HLA-DRB1 genetic variants with the persistence of atopic dermatitis. Human Immunology 2015;76:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paternoster L, Standl M, Chen CM, Ramasamy A, Bonnelykke K, Duijts L et al. : Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nature Genetics 2012;44:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP et al. : Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015;47:1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun LD, Xiao FL, Li Y, Zhou WM, Tang HY, Tang XF et al. : Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nature Genetics 2011;43:690. [DOI] [PubMed] [Google Scholar]
- [9].Weidinger S, Willis-Owen SA, Kamatani Y, Baurecht H, Morar N, Liang L et al. : A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Human Molecular Genetics 2013;22:4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baurecht H, Hotze M, Brand S, Buning C, Cormican P, Corvin A et al. : Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am J Hum Genet 2015;96:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M et al. : Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nature Genetics 2012;44:1222. [DOI] [PubMed] [Google Scholar]
- [12].Ellinghaus D, Baurecht H, Esparza-Gordillo J, Rodriguez E, Matanovic A, Marenholz I et al. : High-density genotyping study indentifies four new susceptibility loci for atopic dermatitis. Nature Genetics 2013;45:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Esparza-Gordillo J, Marenholz I, Lee YA: Genome-wide approaches to the etiology of eczema. [Review]. Current Opinion in Allergy & Clinical Immunology 2010;10:418. [DOI] [PubMed] [Google Scholar]
- [14].Leung DY, Bieber T: Atopic dermatitis. [Review] [100 refs]. Lancet 2003;361:151. [DOI] [PubMed] [Google Scholar]
- [15].Agrawal R, Wisniewski JA, Woodfolk JA: The role of regulatory T cells in atopic dermatitis. Curr Probl Dermatol 2011;41:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Harris VR, Cooper AJ: Atopic dermatitis: the new frontier. Med J Aust 2017;207:351. [DOI] [PubMed] [Google Scholar]
- [17].Busch R, Kollnberger S, Mellins ED: HLA associations in inflammatory arthritis: emerging mechanisms and clinical implications. Nat Rev Rheumatol 2019;15:364. [DOI] [PubMed] [Google Scholar]
- [18].Debebe BJ, Boelen L, Lee JC, Sanders EJ, Anzala O, Kamali A et al. : Identifying the immune interactions underlying HLA class I disease associations. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gough SC, Simmonds MJ: The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr Genomics 2007;8:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gong JJ, Margolis DJ, Monos DS: Predictive in silico binding algorithms reveal HLA specificities and autoallergen peptides associated with atopic dermatitis. Arch Dermatol Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Katsuta M, Takigawa Y, Kimishima M, Inaoka M, Takahashi R, Shiohara T: NK Cells and γδ+ T Cells Are Phenotypically and Functionally Defective due to Preferential Apoptosis in Patients with Atopic Dermatitis. The Journal of Immunology 2006;176:7736. [DOI] [PubMed] [Google Scholar]
- [22].Tang TS, Bieber T, Williams HC: Does “autoreactivity” play a role in atopic dermatitis? J Allergy Clin Immunol 2012;129:1209. [DOI] [PubMed] [Google Scholar]
- [23].Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH et al. : Association of atopic dermatitis with allergic, autoimmune, and cardiovascular comorbidities in US adults. Ann Allergy Asthma Immunol 2018;121:604. [DOI] [PubMed] [Google Scholar]
- [24].Langan SM, Abuabara K, Henrickson SE, Hoffstad O, Margolis DJ: Increased Risk of Cutaneous and Systemic Infections in Atopic Dermatitis-A Cohort Study. J Invest Dermatol 2017;137:1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sanyal RD, Pavel AB, Glickman J, Chan TC, Zheng X, Zhang N et al. : Atopic dermatitis in African American patients is T(H)2/T(H)22-skewed with T(H)1/T(H)17 attenuation. Ann Allergy Asthma Immunol 2019;122:99. [DOI] [PubMed] [Google Scholar]
- [26].Czarnowicki T, He H, Krueger JG, Guttman-Yassky E: Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol 2019;143:1. [DOI] [PubMed] [Google Scholar]
- [27].Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R et al. : Phenotype of atopic dermatitis subjects with a history of eczema herpecticum. Journal of Allergy & Clinical Immunology 2009;124:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Renert-Yuval Y, Del Duca E, Pavel AB, Fang M, Lefferdink R, Wu J et al. : The molecular features of normal and atopic dermatitis skin in infants, children, adolescents and adults. J Allergy Clin Immunol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mack MR, Brestoff JR, Berrien-Elliott MM, Trier AM, Yang TB, McCullen M et al. : Blood natural killer cell deficiency reveals an immunotherapy strategy for atopic dermatitis. Sci Transl Med 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hall TJ, Rycroft R, Brostoff J: Decreased natural killer cell activity in atopic eczema. Immunology 1985;56:337. [PMC free article] [PubMed] [Google Scholar]
- [31].Kabashima K, Weidinger S: NK cells as a possible new player in atopic dermatitis. J Allergy Clin Immunol 2020;146:276. [DOI] [PubMed] [Google Scholar]
- [32].Niepieklo-Miniewska W, Majorczyk E, Matusiak L, Gendzekhadze K, Nowak I, Narbutt J et al. : Protective effect of the KIR2DS1 gene in atopic dermatitis. Gene 2013;527:594. [DOI] [PubMed] [Google Scholar]
- [33].van Deutekom HW, Kesmir C: Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most? Immunogenetics 2015;67:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zerva L, Cizman B, Mehra NK, Alahari SK, Murah R, Zmijewski CM et al. : Arginine at positions 13 or 70–71 in pocket 4 of HLA-DRB1 is associated with susceptibility to tuberculoid leprosy. J Exp Med 1996;183:829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X et al. : Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012;44:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Margolis DJ, Apter AJ, Gupta J, Hoffstad O, Papadopoulos M, Campbell LE et al. : The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol 2012;130:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kapoor R, Menon C, Hoffstad O, Warren B, Leclerc P, Margolis DJ: The prevalence of atopic triad in children wiht physician-confirmed atopic dermatitis. J Am Acad Dermatol 2008;58:68. [DOI] [PubMed] [Google Scholar]
- [38].Armstrong RA: When to use the Bonferroni correction. Ophthalmic Physiol Opt 2014;34:502. [DOI] [PubMed] [Google Scholar]
- [39].Newsom RB: Frequentist q-values for mulitple-test procedures. The Stata Journal 2010;10:568. [Google Scholar]
- [40].Martínez-Camblor P, Pérez-Fernández S, Díaz-Coto S: The role of the p-value in the multitesting problem. Journal of Applied Statistics 2019:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Perneger TV: What’s wrong with Bonferroni adjustments. Bmj 1998;316:1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S et al. : Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.