

Abstract
Rationale & Objective:
Immune activation is fundamental to the pathogenesis of many kidney diseases. Innate immune molecules such as soluble urokinase-type plasminogen activator receptor (suPAR) have been linked to incidence and progression of chronic kidney disease (CKD). Whether other biomarkers of immune activation are associated with incident kidney failure with replacement therapy (KFRT) in African Americans with non-diabetic kidney disease is unclear.
Study Design:
Prospective cohort
Setting & Participants:
African American Study of Kidney Disease and Hypertension participants with available baseline serum samples for biomarker measurement.
Predictors:
Baseline serum soluble tumor necrosis factor receptor 1 and 2 (sTNFR1; sTNFR2), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ) levels.
Outcomes:
Incident KFRT, all-cause mortality.
Analytic Approach:
Cox proportional hazards models.
Results:
Among 500 participants with available samples, mean glomerular filtration rate was 44.7 ml/min/1.73 m2 and median urine protein-to-creatinine ratio was 0.09 g/g at baseline. Over a median follow-up 9.6 years, there were 161 (32%) KFRT and 113 (23%) death events. In models adjusted for demographic and clinical factors and baseline kidney function, each two-fold higher baseline level of sTNFR1, sTNFR2, and TNF-α was associated with 3.66 (95% CI: 2.31,5.80), 2.29 (95% CI: 1.60,3.29), and 1.35-fold (95% CI: 1.07,1.71) greater risks of KFRT, respectively; in comparison, the association between suPAR and KFRT was 1.39 (95% CI: 1.04,1.86). These three biomarkers were also significantly associated with death (up to 2.2-fold higher risks per 2-fold higher baseline level; p≤0.01). IFN-γ was not associated with either outcome. None of the biomarkers modified the association of APOL1 high-risk status (genetic risk factors for kidney disease among individuals of African ancestry) with KFRT (p-interaction>0.05).
Limitations:
Limited generalizability to other ethnic groups or CKD etiologies.
Conclusions:
Among African Americans with CKD attributed to hypertension, baseline levels of sTNFR1, sTNFR2, and TNF-α but not IFN-γ were associated with KFRT and mortality.
Keywords: sTNFR1, sTNFR2, TNF-α, IFN-γ, ESKD, biomarkers, APOL1, immune activation
INTRODUCTION
Chronic kidney disease (CKD) is an urgent public health problem that affects an estimated 697.5 million adults worldwide.1 Progression of CKD to kidney failure with replacement therapy (KFRT) is associated with increased morbidity and mortality.2,3 Understanding factors that promote kidney function decline is therefore paramount to improving the outcomes of patients with CKD.
Central to the pathogenesis of many types of kidney disease is activation of the innate and/or adaptive immune systems.4–6 In the African American Study of Kidney Disease and Hypertension (AASK), higher baseline levels of soluble urokinase-type plasminogen activator receptor (suPAR), a biomarker of immune activation, were associated with increased risks of CKD progression and incident KFRT.7 Biomarkers of the tumor necrosis factor signaling pathway also appear promising in identifying individuals at risk of progressive CKD.8–11 Among patients with type 1 diabetes, higher baseline plasma levels of soluble tumor necrosis factor receptors 1 and 2 (sTNFR1, sTNFR2) were associated with 2.5 to 3.0-fold higher risks of incident CKD Stage G3+ (comparing fourth vs. first through third quartiles)8 whereas in patients with type 2 diabetes, each quartile increase in baseline plasma sTNFR1 and sTNFR2 was associated with 9.8 and 6.0-fold higher risks of developing KFRT, respectively.9 In two large trials of patients with type 2 diabetes, baseline plasma levels of sTNFR1 and sTNFR2 significantly improved prediction of kidney function decline beyond traditional clinical factors.10 In the Chronic Renal Insufficiency Cohort (CRIC; 48% with diabetes), the highest quartile of baseline plasma tumor necrosis factor alpha (TNF-α) was associated with a 42% higher risk of CKD progression, defined as a ≥50% decline in estimated glomerular filtration rate (eGFR) or KFRT, compared to the first quartile.11 Although substantial, the associations of sTNFR1, sTNFR2, and TNF-α with CKD have primarily been reported in individuals with diabetes and of European descent.8–11 Whether these associations also exist in non-diabetic kidney disease and/or African Americans is unclear and warrants further investigation.
Immune activation may also have an important role in APOL1-associated kidney disease. Risk variants in the APOL1 gene (G1 and G2), are found almost exclusively in individuals of African ancestry and have emerged as risk factors for various kidney diseases (e.g., HIV-associated nephropathy, collapsing lupus glomerulopathy) and CKD progression.12–18 However, not all individuals with the APOL1 high-risk genotypes develop kidney disease, thus suggesting that a “second hit” is necessary.15,18,19 To date, in vitro and animal model studies have implicated that this “second hit” involves the activation of inflammatory pathways.4,20,21 In vivo, interferon gamma (IFN-γ) and TNF-α increase APOL1 expression in endothelial cells and podocytes.20–22 In mouse models, increased APOL1 expression, particularly the G1 and G2 risk variants, is associated with azotemia and albuminuria.21 To our knowledge, there have been no studies in humans examining whether IFN-γ and TNF-α modify the association of APOL1 with progressive kidney disease.
Utilizing data and stored serum samples from AASK, a cohort of African Americans with hypertension-attributed CKD, we measured baseline levels of sTNFR1, sTNFR2, TNF-α, and IFN-γ. We hypothesized that higher concentrations of each biomarker would be associated with greater risks of KFRT, CKD progression, and all-cause mortality. We further hypothesized that these biomarkers would augment APOL1-associated risks for KFRT and CKD progression.
METHODS
Study Population
We included 500 AASK trial participants with available baseline serum samples. Details regarding AASK have previously been described.23–25 Briefly, AASK was a 3×2 factorial, double-blinded, randomized, controlled trial wherein 1,094 African Americans with hypertension-attributed CKD were randomized to one of three blood pressure (BP) medications (ramipril, metoprolol, or amlodipine) and one of two BP goals (mean arterial pressure of 102–107 mmHg or ≤92 mmHg). Inclusion criteria included ages 18–70 years, diastolic BP >95 mmHg, and 125I-iothalamate GFR 20–65 ml/min/1.73 m2. Exclusion criteria included diabetes, urine protein-to-creatinine ratio (PCR) >2.5 g/g, or CKD etiology other than hypertension.23–25 Trial participants were enrolled from February 1995 to September 1998 and followed until September 2001, the pre-specified end date of the trial.23,24 Participants without KFRT were then invited to the cohort phase of AASK, which spanned from April 2002 to June 2007. During this second phase, all participants received ramipril with a BP goal of <140/90 mmHg, and after 2004, <130/80 mmHg due to changes in national guidelines.24 Informed consent was obtained and protocols were approved by institutional review boards at each participating site.23,24
Biomarker Measurements
We measured biomarkers of immune activation from stored serum samples collected at the AASK trial baseline visit. Measurements were performed from December 2019 to January 2020 using Meso Scale Discovery assays (Meso Scale Diagnostics; Rockville, Maryland), which combines electrochemiluminescence with multiarray technology, by personnel blinded to participant data. sTNFR1 and sTNFR2 were measured in a 2-plex plate; TNF-α and IFN-γ were measured together on a separate plate. Inter-assay coefficients of variation, determined from 6% duplicate samples, were: sTNFR1=3.33%, sTNFR2=2.96%, TNF-α=7.52%, and IFN-γ=6.17%. Baseline serum suPAR levels were measured in 2017 using the suPARnostic® ELISA kit (Virogates; Copenhagen, Denmark).7
Genotyping of APOL1 Risk Variants and Ancestry Informative Markers
A subset of participants were genotyped for the APOL1 risk variants and 140 ancestry informative markers in an ancillary study.15 Genotyping for G1 (rs73885319 and rs60910145) and G2 (rs71785313) were performed using ABI Taqman (Applied Biosystems; Foster City, California). APOL1 high-risk status was defined as 2 copies of the risk variants (i.e., G1/G1, G2/G2, or G1/G2) and low-risk status as 1 or no copies (i.e., G1/G0, G2/G0, or G0/G0).12–15,19 Percentage of European ancestry was estimated via ANCESTRYMAP.15
Outcomes
The primary outcome was incident KFRT, defined as initiation of chronic dialysis or kidney transplantation. Secondary outcomes included all-cause mortality and CKD progression, defined as a doubling of serum creatinine or KFRT. Serum creatinine was measured at a central laboratory at 6-month intervals during both phases of AASK.23,24
Covariates
At the screening visit (prior to randomization), each participant underwent 3 baseline BP measurements in a seated position after >5 minutes of rest using a Hawksley random zero sphygmomanometer. The latter two measurements were then averaged.23 GFR was ascertained via direct measurement of renal 125I-iothalamate clearance.23 Urine protein and urine creatinine were measured at a central laboratory using the modified Jaffe reaction and pyrogallol red technique, respectively, from 24-hour urine specimens and the ratio taken to determine urine PCR.24 Serum high-sensitivity c-reactive protein (hsCRP) was measured at a central laboratory by nephelometry (Dade Behring BN1).26
Statistical Analysis
Baseline characteristics by biomarker tertiles and APOL1 risk status were compared using ANOVA, Wilcoxon rank-sum, or Kruskal-Wallis tests for continuous variables and Pearson’s chi-squared test for categorical variables. Distributions of continuous variables were assessed and if skewed, log2-transformed to achieve more normal distributions (e.g., sTNFR1, sTNFR2, TNF-α, IFN-γ, suPAR, urine PCR, hs-CRP). To evaluate the association of each biomarker with KFRT, Cox models were constructed. Model 0 was unadjusted. Model 1 adjusted for demographic factors (age, sex). Model 2 further adjusted for clinical factors (baseline systolic BP, body mass index, current smoking). Model 3 further adjusted for baseline GFR. Model 4, our primary model, further adjusted for baseline log2-transformed urine PCR. Among individuals with APOL1 genotyping, Model 5 further adjusted for APOL1 risk status and European ancestry. Each analysis was repeated for the outcomes of all-cause mortality and CKD progression. In sensitivity analyses, we accounted for randomization groups and hsCRP, a general marker of inflammation. We also performed competing risks analyses, based on the method of Fine and Gray, treating death as a competing risk.27 For comparison purposes, we evaluated associations between suPAR and KFRT in our study population, using log2-transformed suPAR as the exposure. Participants were censored at death or on June 30, 2007.
We included interaction terms between each biomarker (as a log2-transformed variable) and categories of APOL1 risk status, age, sex, systolic BP, GFR, and urine PCR to evaluate for effect modification. To assess the predictive value of adding log2-transformed biomarkers to a clinical model, we calculated the Harrell’s C-statistic for the following models: 1) Clinical (Model 4); 2) Clinical+sTNFR1; 3) Clinical+sTNFR1+TNF-α; 4) Clinical+sTNFR1+suPAR; and 5) Clinical+sTNFR1+TNF-α+suPAR. Differences in the Harrell’s C statistic for each model with the biomarker(s) versus the clinical model were then determined. Analyses were conducted with Stata 15.1 software (StataCorp LLC; College Station, Texas).
RESULTS
Baseline Characteristics
Among 1,094 AASK trial participants, 500 had baseline serum samples available for biomarker measurement and comprised our study population (Figure 1). Participants without available samples had slightly higher mean GFR (46.4 vs. 44.7 ml/min/1.73 m2) and suPAR levels (4,487 vs. 4,417 pg/mL) compared to those included in our study. Otherwise, the two groups were alike (Supplementary Table 1).
Figure 1:
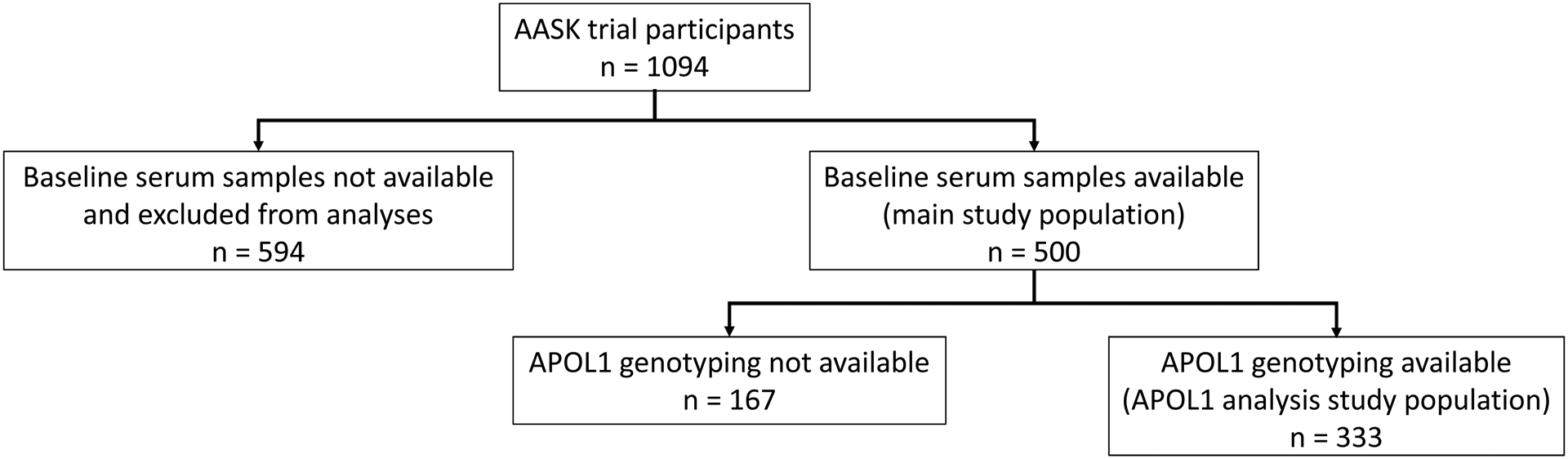
Flowchart of study population included in current study.
At baseline, the mean age was 54.1 ± 10.6 years, 37% were female, mean GFR was 44.7 ± 12.7 ml/min/1.73 m2, and median urine PCR was 0.09 (Interquartile range [IQR] 0.03 to 0.39) g/g. Median (IQR) levels of each biomarker were as follows: sTNFR1: 2,875 (2,170 to 3,905) pg/mL, sTNFR2: 13,021 (9,345 to 17,155) pg/mL, TNF-α: 2.92 (2.20 to 3.95) pg/mL, and IFN-γ: 5.51 (3.81 to 8.69) pg/mL. Participants in the highest sTNFR1 tertile were significantly younger and had worse kidney function measures (i.e., GFR, urine PCR) compared to participants in lower tertiles (Table 1). Higher tertiles of sTNFR1 also had higher median levels of other biomarkers of immune activation, including sTNFR2, TNF-α, and suPAR. Participant characteristics by tertiles of the other biomarkers are presented in Supplementary Tables 2–4. sTNFR1 and sTNFR2 were highly correlated with each other (correlation of 0.88) and moderately correlated with TNF-α and suPAR (correlations of 0.42 to 0.55; Table 2).
Table 1:
Baseline characteristics by sTNFR1 tertiles.
| Characteristic | Tertile 1 n=167 |
Tertile 2 n=167 |
Tertile 3 n=166 |
p-value |
|---|---|---|---|---|
| Range, pg/mL | 1,267 to 2,372 | 2,374 to 3,428 | 3,445 to 9,168 | |
| Age, years | 58.1 ± 9.0 | 53.1 ± 10.6 | 51.0 ± 10.9 | <0.001 |
| Female | 67 (40%) | 64 (38%) | 56 (34%) | 0.46 |
| APOL1 risk status | ||||
| Low | 92 (82%) | 83 (71%) | 71 (68%) | 0.05 |
| High | 20 (18%) | 34 (29%) | 33 (32%) | |
| % European ancestry | 17.9 ± 15.2 | 14.6 ± 10.6 | 17.8 ± 11.8 | 0.09 |
| Years of hypertension | 14.4 ± 9.8 | 14.5 ± 9.5 | 13.2 ± 10.5 | 0.40 |
| History of heart disease | 87 (52%) | 90 (54%) | 75 (45%) | 0.24 |
| Systolic BP, mm Hg | 149 ± 25 | 152 ± 25 | 153 ± 23 | 0.39 |
| BMI, kg/m2 | 30.1 ± 5.7 | 31.5 ± 6.4 | 31.0 ± 7.3 | 0.11 |
| Current smoking | 40 (24%) | 44 (26%) | 48 (29%) | 0.59 |
| GFR, ml/min/1.73 m2 | 54.7 ± 7.7 | 47.7 ± 9.1 | 31.7 ± 8.0 | <0.001 |
| Urine PCR, g/g | 0.03 (0.02 to 0.06) |
0.10 (0.03 to 0.32) |
0.37 (0.12 to 0.99) |
<0.001 |
| hsCRP, mg/dL | 0.32 (0.16 to 0.71) |
0.50 (0.24 to 0.97) |
0.46 (0.18 to 0.91) |
0.01 |
| sTNFR2, pg/mL | 8,651 (7,487 to 9,884) |
13,042 (10,962 to 14,735) |
18,972 (15,936 to 22,917) |
<0.001 |
| TNF-α, pg/mL | 2.40 (1.78 to 2.92) |
3.03 (2.27 to 3.93) |
3.74 (2.67 to 4.95) |
<0.001 |
| IFN-γ, pg/mL | 5.23 (3.72 to 7.55) |
5.55 (3.73 to 9.01) |
5.73 (3.95 to 11.68) |
0.12 |
| suPAR, pg/mL | 3,468 (2,821 to 4,334) |
4,463 (3,352 to 5,583) |
5,561 (4,618 to 7,116) |
<0.001 |
| Log2(sTNFR2) | 13.07 ± 0.32 | 13.65 ± 0.32 | 14.24 ± 0.39 | <0.001 |
| Log2(TNF-α) | 1.23 ± 0.61 | 1.56 ± 0.60 | 1.90 ± 0.67 | <0.001 |
| Log2(IFN-γ) | 2.47 ± 0.86 | 2.67 ± 1.15 | 2.86 ± 1.43 | 0.01 |
| Log2(suPAR) | 11.69 ± 0.61 | 12.11 ± 0.57 | 12.47 ± 0.55 | <0.001 |
| BP goal | ||||
| Intensive | 87 (52%) | 79 (47%) | 84 (51%) | 0.67 |
| Standard | 80 (48%) | 88 (53%) | 82 (49%) | |
| Drug group | ||||
| Ramipril | 69 (41%) | 59 (35%) | 67 (40%) | 0.70 |
| Metoprolol | 69 (41%) | 70 (42%) | 67 (40%) | |
| Amlodipine | 29 (17%) | 38 (23%) | 32 (19%) |
Data presented as mean ± standard deviation; number (percent); median (interquartile range).
Abbreviations: BP=blood pressure; BMI=body mass index; GFR=glomerular filtration rate; PCR=protein-to-creatinine ratio; hsCRP=high-sensitivity c-reactive protein; sTNFR1=soluble tumor necrosis factor receptor 1; sTNFR2=soluble tumor necrosis factor receptor 2; TNF-α=tumor necrosis factor alpha; IFN-γ=interferon gamma; suPAR=soluble urokinase-type plasminogen activator receptor.
Data missing for the following variables: APOL1 (n=167); European ancestry (n=167); Years of hypertension (n=3); suPAR (n=14); log2(suPAR) (n=14)
Table 2:
Pearson correlations of log2-transformed biomarkers of immune activation.
| sTNFR1 | sTNFR2 | TNF-α | IFN-γ | suPAR | |
|---|---|---|---|---|---|
| sTNFR1 | 1.00 | ||||
| sTNFR2 | 0.88 | 1.00 | |||
| TNF-α | 0.42 | 0.48 | 1.00 | ||
| IFN-γ | 0.14 | 0.28 | 0.28 | 1.00 | |
| suPAR | 0.53 | 0.55 | 0.31 | 0.22 | 1.00 |
Abbreviations: sTNFR1=soluble tumor necrosis factor receptor 1; sTNFR2=soluble tumor necrosis factor receptor 2; TNF-α=tumor necrosis factor alpha; IFN-γ=interferon gamma; suPAR=soluble urokinase-type plasminogen activator receptor.
Among participants with genotyping, 87 (26%) had APOL1 high-risk status and 246 (74%) had low-risk status. Participants with APOL1 high-risk status were younger, had lower mean systolic BP and GFR, and had higher median urine PCR, serum sTNFR1, sTNFR2, and suPAR compared to participants with low-risk status. Otherwise, the two APOL1 risk groups were similar at baseline, including with respect to TNF-α and IFN-γ levels (Supplementary Table 5).
Associations of Biomarkers with KFRT and CKD Progression
Over a median follow-up of 8.5 years, 161 participants developed KFRT. In unadjusted analyses, each 2-fold higher baseline level of sTNFR1 was associated with an 8.10-fold greater risk of incident KFRT (95% CI: 6.15, 10.66). This association was robust to adjustment for demographic and clinical factors, and attenuated but remained statistically significant upon further adjustment for baseline GFR and proteinuria (HR=3.66; 95% CI: 2.31, 5.80). Similarly, each 2-fold higher baseline level of sTNFR2 was associated with 5.09 (95% CI: 4.03, 6.43) and 2.29-fold (95% CI: 1.60, 3.29) greater risks of incident KFRT in unadjusted model and Model 4, respectively. The association of TNF-α with incident KFRT was smaller though still significant (HR=1.88 [95% CI: 1.54, 2.29] for unadjusted model; HR=1.35 [95% CI: 1.07, 1.71] for Model 4) whereas IFN-γ was not associated with incident KFRT in any of the models (all p>0.05; Table 3). In comparison, the association between suPAR and KFRT in this population was HR=1.39 (95% CI: 1.04, 1.86) per two-fold higher baseline level. Conclusions did not change upon further adjustment for randomized treatment groups or hs-CRP (Supplementary Table 6). In Kaplan-Meier curves, higher tertiles of baseline sTNFR1, sTNFR2, and TNF-α were associated with higher risk of incident KFRT (Figure 2). In additional analyses, the association of sTNFR2 with KFRT appeared stronger among older participants (p-interaction=0.008) and those without baseline proteinuria (p-interaction=0.045). Otherwise, associations did not differ significantly by subgroups (Supplementary Table 7). Finally, accounting for the competing risk of death yielded similar results with the exception of TNF-α, for which the subdistribution HR lost statistical significance but direction of association remained the same (Supplementary Table 8).
Table 3:
Associations of log2-transformed biomarkers of immune activation with kidney failure with replacement therapy in AASK.
| Model | n | events | sTNFR1 | sTNFR2 | TNF-α | IFN-γ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | |||
| Model 0: Unadjusted | 500 | 161 | 8.10 (6.15, 10.66) | <0.001 | 5.09 (4.03, 6.43) | <0.001 | 1.88 (1.54, 2.29) | <0.001 | 1.11 (0.98, 1.25) | 0.10 |
| Model 1: Adjusted for age and sex | 500 | 161 | 7.44 (5.59, 9.89) | <0.001 | 4.63 (3.63, 5.91) | <0.001 | 1.69 (1.39, 2.07) | <0.001 | 1.08 (0.96, 1.22) | 0.19 |
| Model 2: Model 1 + systolic BP, BMI, and current smoking | 500 | 161 | 8.30 (6.06, 11.35) | <0.001 | 4.98 (3.82, 6.50) | <0.001 | 1.69 (1.39, 2.07) | <0.001 | 1.08 (0.96, 1.22) | 0.19 |
| Model 3: Model 2 + GFR | 500 | 161 | 4.90 (3.16, 7.62) | <0.001 | 2.76 (1.97, 3.86) | <0.001 | 1.31 (1.05, 1.64) | 0.02 | 1.09 (0.96, 1.23) | 0.18 |
| Model 4: Model 3 + log2(urine PCR) | 500 | 161 | 3.66 (2.31, 5.80) | <0.001 | 2.29 (1.60, 3.29) | <0.001 | 1.35 (1.07, 1.71) | 0.01 | 1.03 (0.91, 1.16) | 0.69 |
| Model 5: Model 4 + APOL1 risk status and European ancestry | 333 | 112 | 3.83 (2.21, 6.61) | <0.001 | 2.53 (1.63, 3.95) | <0.001 | 1.28 (0.97, 1.70) | 0.09 | 1.02 (0.88, 1.18) | 0.81 |
Hazard ratios are per 2-fold higher baseline level of each biomarker.
Abbreviations: BP=blood pressure; GFR=glomerular filtration rate; PCR=protein-to-creatinine ratio; sTNFR1=soluble tumor necrosis factor receptor 1; sTNFR2=soluble tumor necrosis factor receptor 2; TNF-α=tumor necrosis factor alpha; IFN-γ=interferon gamma.
Figure 2:
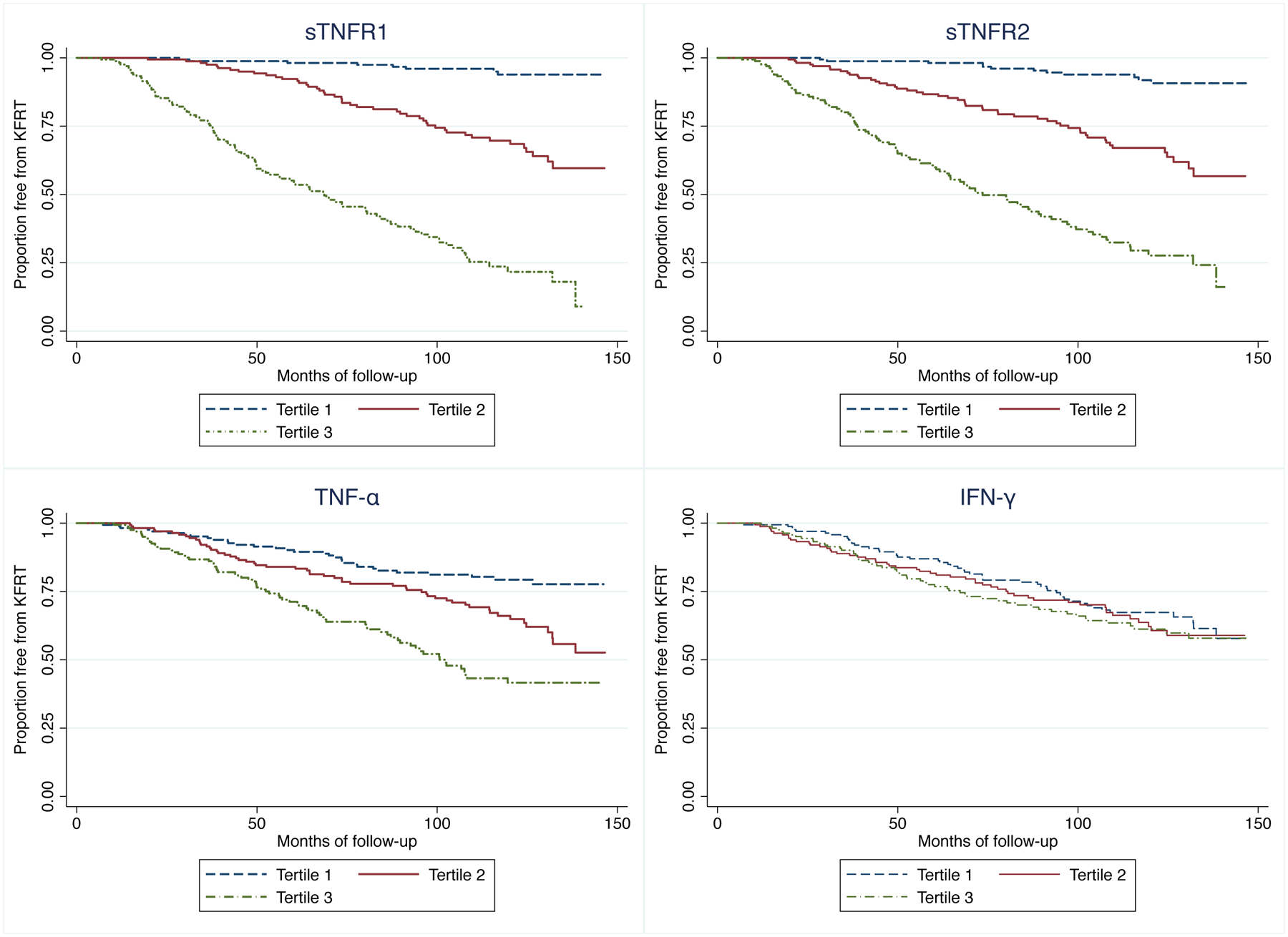
Kaplan-Meier survival estimates of kidney failure with replacement therapy, by biomarker tertiles.
When considering the secondary outcome of CKD progression, there were 196 events over a median follow-up 7.3 years. In unadjusted models, each 2-fold higher baseline level of sTNFR1, sTNFR2, and TNF-α was associated with 6.10 (95% CI: 4.76, 7.82), 4.00 (95% CI: 3.22, 4.95), and 1.76-fold (95% CI: 1.47, 2.11) greater risks for CKD progression, respectively. After adjusting for demographic/clinical factors and baseline kidney function, these associations were attenuated but remained significant (sTNFR1: HR=2.96 [95% CI 1.93, 4.55]; sTNFR2: HR=1.85 [95% CI: 1.32, 2.58]; TNF-α: HR=1.31 [95% CI: 1.06, 1.62] per 2-fold higher baseline level). As in our primary analyses, there was no association between IFN-γ and CKD progression (Supplementary Table 6 and 9).
Associations of Biomarkers with All-cause Mortality
There were 113 deaths over a median follow-up of 9.6 years. In unadjusted models, each 2-fold higher baseline level of sTNFR1, sTNFR2, and TNF-α was associated with 1.7 to 1.8-fold greater risks of all-cause mortality (all p≤0.002). After accounting for demographic/clinical factors and baseline kidney function, these associations strengthened, with each 2-fold higher baseline level of sTNFR1, sTNFR2, TNF-α being associated with 2.0 to 2.2-fold higher risks of death (all p≤0.01). Baseline IFN-γ was not associated with all-cause mortality (Table 4; Supplementary Table 6).
Table 4:
Associations of log2-transformed biomarkers of immune activation with all-cause mortality in AASK.
| Model | n | events | sTNFR1 | sTNFR2 | TNF-α | IFN-γ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | Hazard Ratio (95% CI) | p-value | |||
| Model 0: Unadjusted | 500 | 113 | 1.65 (1.20, 2.27) | 0.002 | 1.66 (1.23, 2.25) | 0.001 | 1.83 (1.42, 2.34) | <0.001 | 1.02 (0.87, 1.19) | 0.82 |
| Model 1: Adjusted for age and sex | 500 | 113 | 2.02 (1.46, 2.80) | <0.001 | 2.10 (1.54, 2.87) | <0.001 | 2.16 (1.64, 2.83) | <0.001 | 1.08 (0.92, 1.27) | 0.33 |
| Model 2: Model 1 + systolic BP, BMI, and current smoking | 500 | 113 | 1.90 (1.38, 2.63) | <0.001 | 1.95 (1.44, 2.65) | <0.001 | 2.07 (1.57, 2.74) | <0.001 | 1.10 (0.94, 1.30) | 0.23 |
| Model 3: Model 2 + GFR | 500 | 113 | 2.00 (1.18, 3.39) | 0.01 | 1.98 (1.29, 3.03) | 0.002 | 1.95 (1.44, 2.62) | <0.001 | 1.09 (0.93, 1.29) | 0.29 |
| Model 4: Model 3 + log2(urine PCR) | 500 | 113 | 2.21 (1.26, 3.85) | 0.01 | 2.07 (1.34, 3.20) | 0.001 | 1.95 (1.45, 2.62) | <0.001 | 1.09 (0.93, 1.29) | 0.29 |
| Model 5: Model 4 + APOL1 risk status and European ancestry | 333 | 55 | 2.88 (1.31, 6.35) | 0.01 | 2.44 (1.31, 4.54) | 0.01 | 2.14 (1.42, 3.23) | <0.001 | 1.23 (0.99, 1.52) | 0.06 |
Hazard ratios are per 2-fold higher baseline level of each biomarker.
Abbreviations: BP=blood pressure; GFR=glomerular filtration rate; PCR= protein-to-creatinine ratio; sTNFR1=soluble tumor necrosis factor receptor 1; sTNFR2=soluble tumor necrosis factor receptor 2; TNF-α=tumor necrosis factor alpha; IFN-γ=interferon gamma.
APOL1 Risk Genotypes and Immune Activation
Among participants with available genotyping (n=333), each 2-fold higher baseline level of sTNFR1 was associated with a significantly increased risk of KFRT (HR=3.83; 95% CI: 2.21, 6.61), CKD progression (HR=2.76; 95% CI: 1.68, 4.54), and mortality (HR=2.88; 95% CI: 1.31, 6.35), after adjusting for APOL1 risk status and European ancestry. Similar trends were observed for sTNFR2 and TNF-α but not IFN-γ (Tables 3–4; Supplementary Table 9). There was no evidence of interaction between APOL1 high-risk status and sTNFR1, sTNFR2, TNF-α, or IFN-γ for the outcomes of KFRT (p-interaction=0.51, 0.53, 0.98, and 0.43, respectively) and CKD progression (p-interaction=0.86, 0.92, 0.75, and 0.38, respectively).
Improvement in Prediction Model Discrimination by Biomarkers
The C-statistic for the fully adjusted clinical model in predicting KFRT was excellent at 0.849 (95% CI: 0.820, 0.878). Adding sTNFR1, the biomarker with the strongest association, to the clinical model led to a small but statistically significant improvement in the C-statistic at 0.860 (95% CI: 0.833, 0.887; difference of 0.011; 95% CI: 0.001, 0.021). Addition of TNF-α, suPAR, or both to the model did not further improve discrimination measures (C-statistics of 0.860, 0.860, and 0.860, respectively; Table 5).
Table 5:
Harrell’s C statistic for clinical models ± biomarkers in predicting KFRT.
| Model | Harrell’s C Statistic (95% CI) |
Difference in C Statistic (95% CI) |
|---|---|---|
| Clinical Model: adjusted for age, sex, systolic BP, BMI, current smoking, GFR, and log2(urine PCR) | 0.849 (0.820, 0.878) |
Ref |
| Clinical Model + log2(sTNFR1) | 0.860 (0.833, 0.887) |
0.011 (0.001, 0.021) |
| Clinical Model + log2(sTNFR1) + log2(TNF-α) | 0.860 (0.834, 0.887) |
0.011 (0.002, 0.021) |
| Clinical Model + log2(sTNFR1) + log2(suPAR) | 0.860 (0.833, 0.887) |
0.011 (0.001, 0.021) |
| Clinical Model + log2(sTNFR1) + log2(TNF-α) + log2(suPAR) | 0.860 (0.833, 0.887) |
0.011 (0.001, 0.021) |
Abbreviations: BP=blood pressure; GFR=glomerular filtration rate; PCR=protein-to-creatinine ratio; sTNFR1=soluble tumor necrosis factor receptor 1; TNF-α=tumor necrosis factor alpha; IFN-γ=interferon gamma; suPAR=soluble urokinase-type plasminogen activator receptor.
DISCUSSION
In this study of African Americans with hypertension-attributed CKD, higher baseline levels of sTNFR1, sTNFR2, TNF-α, but not IFN-γ were independently associated with increased risks of KFRT, CKD progression, and all-cause mortality. None of the biomarkers that we examined, however, modified the association of APOL1 high-risk status with KFRT or CKD progression. We also report that the addition of sTNFR1, the biomarker with the strongest associations in our study population, to a clinical model improved KFRT risk prediction, albeit by a small magnitude. Further inclusion of additional biomarkers did not. Taken together, our findings support the hypothesis that the tumor necrosis factor signaling pathway plays an important role in CKD progression in African Americans with non-diabetic kidney disease.
To date, few studies have investigated the clinical significance of sTNFR1 and sTNFR2 in non-diabetic kidney disease, with little representation of African Americans. In the Cholesterol and Recurrent Events trial (14% with diabetes; 2% African American), higher sTNFR1 were associated with faster eGFR decline (−0.49 ml/min/1.73 m2 per year comparing highest vs. lowest tertiles).28 Similarly, in the Multi-Ethnic Study of Atherosclerosis (27% with impaired fasting glucose or diabetes; 24% African American), each standard deviation higher sTNFR1 was associated with a 43% higher risk of 40% decline in eGFR.29 In the Beaver Dam CKD study (9% with diabetes; 98% Caucasian), the highest tertile of sTNFR2 was associated with a 2.1-fold higher risk of incident CKD compared to the lowest tertile.30 In contrast, Schei et al. reported in a general population cohort of Norwegians without CKD (0% diabetes) that higher baseline levels of sTNFR2 were associated with slower declines in GFR (+0.09 ml/min/1.73 m2/year per standard deviation increase).31 More recently, in the CKD in Children cohort (median age 11 years; 20% African American), the highest quartiles of sTNFR1 and sTNFR2 were associated with 4.1-fold and 2.6-fold greater risks of CKD progression, defined as a 50% decline in eGFR or KFRT, compared to the lowest quartiles.32 The present study adds to this literature by demonstrating strong associations of sTNFR1, sTNFR2, and TNF-α with risks of KFRT, CKD progression, and mortality among African Americans with non-diabetic kidney disease.
With a 2 to 4-fold higher lifetime risk of CKD G4+ and KFRT compared to Caucasians, African Americans carry an excess burden of progressive kidney disease.33 The APOL1 high-risk genotypes, present in ~13% of African Americans, account for some of the racial disparities in advanced CKD.12,13,34 Parsa et al. reported that AASK participants with the APOL1 high-risk status were 2.16 and 1.88-fold more likely to develop KFRT and CKD progression, respectively, compared to their counterparts with the low-risk status.15 Still, 42% of individuals with the APOL1 high-risk status did not experience CKD progression over a median follow-up of 9 years.15 We previously described potential interactive effects of suPAR with APOL1 risk status, where APOL1 high-risk status was associated with faster eGFR decline when suPAR was >3,000 pg/mL but not <3,000 pg/mL in AASK and the Emory Cardiovascular Biobank.4 However, suPAR did not modify the association of APOL1 high-risk status with KFRT or CKD progression in AASK alone.7 We expand upon these findings by reporting that higher baseline levels of other biomarkers of immune activation (i.e., sTNFR1, sTNFR2, TNF-α, IFN-γ) also did not modify the association of APOL1 high-risk status with KFRT or CKD progression, and that sTNFR1 and sTNFR2 in particular were moderately correlated with suPAR.
To our knowledge, only one other study has examined the association of sTNFR1 and sTNFR2 with kidney outcomes in the context of APOL1. Utilizing data from BioMe (16% with diabetes), an electronic-medical record-based retrospective cohort, Nadkarni et al. reported that each 2-fold higher baseline level of sTNFR1 and sTNFR2 was independently associated with a 2.0 and 1.5-fold higher risk of a composite renal outcome comprising of a sustained decline in eGFR by ≥40% or KFRT.35 Their study was limited to African Americans with the APOL1 high-risk status, and thus did not provide insight on whether sTNFR1 or sTNFR2 modify the association of APOL1 high-risk status with CKD progression.35 The results of our study, which included both individuals with the APOL1 high- and low-risk genotypes, suggest that the associations of sTNFR1 and sTNFR2 with progressive kidney disease may not differ by APOL1 risk status.
We hypothesized that higher baseline levels of IFN-γ would augment the APOL1-associated risk for worsening CKD because prior in vitro studies showed that this inflammatory cytokine increases APOL1 expression in endothelial cells and podocytes, both cell types found in the human kidney.20,21 Moreover, expression of the APOL1 G1 and G2 variants increases cytotoxicity in a dose-dependent manner.21 In patients with HIV-associated nephropathy and lupus collapsing glomerulopathy, two other entities known to be associated with APOL1 high-risk status, tubuloreticular inclusions are often seen on kidney biopsies.36–39 These electron-dense deposits are considered to be interferon footprints, likely reflecting high interferon states.36,37 Nichols et al. also described a series of 7 patients who developed focal segmental glomerulosclerosis after receiving interferon therapy and were all found to have 2 APOL1 risk alleles.20 Despite this prior evidence that IFN-γ may be a “second hit,” we found no association between baseline IFN-γ levels and KFRT, CKD progression, or all-cause mortality and no interactive effects of IFN-γ with APOL1 high-risk status for any of these outcomes. Perhaps, biomarkers measured at baseline may not be the biologically relevant time period to study with regards to APOL1-mediated kidney disease.
Our findings have potential implications. In the research arena, sTNFR1, sTNFR2, and/or TNF-α could be used to enrich recruitment of patients with CKD to clinical trials. Identifying individuals who are more likely to experience the outcome of interest (e.g., KFRT, CKD progression, or mortality) could reduce the number of participants needed or shorten the duration of the trial. Clinically, patients with higher levels of these biomarkers may benefit from intensification of conventional CKD treatments.
Strengths of our study include the prospective design of the AASK trial and cohort, long duration of follow-up (up to 12.2 years), direct measurement of GFR, consideration of multiple biomarkers, and evaluation of the interactive effects of immune activation with APOL1 risk status. Our study also has limitations. First, given that AASK comprised African Americans with hypertension-attributed CKD, our results may not be generalizable to other ethnic groups or CKD etiologies. Although AASK excluded individuals with baseline diabetes or urine PCR >2.5 g/g, prior studies have shown strong associations in these other populations.8–11,29 Second, our sample size was relatively small, especially for our analyses involving APOL1. We may have been underpowered to detect an interaction between our biomarkers of immune activation and APOL1 risk status. Third, a “second hit” occurring early in the disease process would not be captured in our study population because moderate to severe CKD was already present at the time of enrollment. Fourth, although the results suggest a strong association between biomarkers and adverse outcomes, they do not imply causality. An intervention which lowers these biomarkers would not necessarily be expected to improve CKD prognosis.
In conclusion, among African Americans with CKD attributed to hypertension, baseline serum levels of sTNFR1, sTFNR2, and TNF-α were associated with adverse kidney outcomes and mortality, with sTNFR1 appearing to have the strongest associations. Future studies are needed to determine the clinical utility of measuring and/or targeting these biomarkers in both patient care and clinical trials.
Supplementary Material
Supplementary Table 1: Baseline characteristics comparing AASK participants included vs. excluded from current study based on the availability of samples.
Supplementary Table 2: Baseline characteristics by sTNFR2 tertiles.
Supplementary Table 3: Baseline characteristics by TNF-α tertiles.
Supplementary Table 4: Baseline characteristics by IFN-γ tertiles.
Supplementary Table 5: Baseline characteristics by APOL1 risk status.
Supplementary Table 9: Associations of log2-transformed biomarkers of immune activation with chronic kidney disease progression (doubling of serum creatinine/KFRT) in AASK.
Supplementary Table 6: Associations of log2-transformed biomarkers of immune activation with KFRT, chronic kidney disease progression, and all-cause mortality in AASK in Model 4, further accounting for randomized treatment groups and high-sensitivity c-reactive protein.
Supplementary Table 7: Associations of log2-transformed biomarkers of immune activation with KFRT in Model 4, by subgroups of age, sex, systolic blood pressure, GFR, and urine PCR.
Supplementary Table 8: Associations of log2-transformed biomarkers of immune activation with KFRT in Model 4, accounting for the competing risk of death.
ACKNOWLEDGEMENTS:
We thank the participants of AASK. The African American Study of Kidney Disease and Hypertension (AASK) was conducted by the AASK Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The samples from the AASK trial reported here were supplied by the NIDDK Central Repositories via X01DK118497. The AASK trial and cohort were supported by institutional grants from the NIH and NIDDK (M01 RR-00080, M01 RR-00071, M0100032, P20-RR11145, M01 RR00827, M01 RR00052, 2P20 RR11104, RR029887, DK 2818-02, DK057867, and DK048689), and the following pharmaceutical companies (King Pharmaceuticals, Pfizer, AstraZeneca, GlaxoSmithKline, Forest Laboratories, Pharmacia, and Upjohn).
SUPPORT:
TKC is supported by a George M. O’Brien Center for Kidney Research Pilot and Feasibility Grant from Yale University (under Award Number NIH/NIDDK P30DK079310) and NIH/NIDDK K08DK117068. Biomarkers were measured by the Translational Research Core of the George M. O’Brien Kidney Center at Yale University. MEG and SL are supported by NIH/NIDDK R01DK108803. CRP is supported by NIH/NIDDK U01DK106962.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURE: CRP is a member of the advisory board of Renalytix AI and owns equity in the same. CRP serves on the Data Safety and Monitoring Board of Genfit. JR is a co-founder and shareholder of Trisaq, a bio-pharmaceutical company that develops novel therapies for renal diseases. The other authors declare that they have no relevant financial interests.
DISCLAIMER: This manuscript was not prepared in collaboration with Investigators of the AASK study and does not necessarily reflect the opinions or views of the AASK study, the NIDDK Central Repositories, the NIH, or the NIDDK.
REFERENCES
- 1.GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395:709–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coresh J, Turin TC, Matsushita K, et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA 2014;311:2518–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsushita K, Coresh J, Sang Y, et al. Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: a collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol 2015;3:514–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayek SS, Koh KH, Grams ME, et al. A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease. Nat Med 2017;23:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yatim KM, Lakkis FG. A brief journey through the immune system. Clin J Am Soc Nephrol 2015;10:1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gluba A, Banach M, Hannam S, Mikhailidis DP, Sakowicz A, Rysz J. The role of Toll-like receptors in renal diseases. Nat Rev Nephrol 2010;6:224–35. [DOI] [PubMed] [Google Scholar]
- 7.Luo S, Coresh J, Tin A, et al. Soluble Urokinase-Type Plasminogen Activator Receptor in Black Americans with CKD. Clin J Am Soc Nephrol 2018;13:1013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012;23:516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niewczas MA, Gohda T, Skupien J, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 2012;23:507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coca SG, Nadkarni GN, Huang Y, et al. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J Am Soc Nephrol 2017;28:2786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amdur RL, Feldman HI, Gupta J, et al. Inflammation and Progression of CKD: The CRIC Study. Clin J Am Soc Nephrol 2016;11:1546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010;329:841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010;128:345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipkowitz MS, Freedman BI, Langefeld CD, et al. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int 2013;83:114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsa A, Kao WH, Xie D, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013;369:2183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol 2013;24:722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 2011;22:2129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grams ME, Rebholz CM, Chen Y, et al. Race, APOL1 Risk, and eGFR Decline in the General Population. J Am Soc Nephrol 2016;27:2842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen TK, Choi MJ, Kao WH, et al. Examination of Potential Modifiers of the Association of APOL1 Alleles with CKD Progression. Clin J Am Soc Nephrol 2015;10:2128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 2015;87:332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017;23:429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy 2008;4:1079–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright JT Jr., Bakris G, Greene T, et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 2002;288:2421–31. [DOI] [PubMed] [Google Scholar]
- 24.Appel LJ, Wright JT Jr., Greene T, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 2010;363:918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright JT Jr., Kusek JW, Toto RD, et al. Design and baseline characteristics of participants in the African American Study of Kidney Disease and Hypertension (AASK) Pilot Study. Control Clin Trials 1996;17:3S–16S. [DOI] [PubMed] [Google Scholar]
- 26.Hung AM, Crawford DC, Griffin MR, et al. CRP polymorphisms and progression of chronic kidney disease in African Americans. Clin J Am Soc Nephrol 2010;5:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fine JP GR. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999;94:496–509. [Google Scholar]
- 28.Tonelli M, Sacks F, Pfeffer M, et al. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int 2005;68:237–45. [DOI] [PubMed] [Google Scholar]
- 29.Bhatraju PK, Zelnick LR, Shlipak M, Katz R, Kestenbaum B. Association of Soluble TNFR-1 Concentrations with Long-Term Decline in Kidney Function: The Multi-Ethnic Study of Atherosclerosis. J Am Soc Nephrol 2018;29:2713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankar A, Sun L, Klein BE, et al. Markers of inflammation predict the long-term risk of developing chronic kidney disease: a population-based cohort study. Kidney Int 2011;80:1231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schei J, Stefansson VT, Eriksen BO, et al. Association of TNF Receptor 2 and CRP with GFR Decline in the General Nondiabetic Population. Clin J Am Soc Nephrol 2017;12:624–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg JH, Abraham AG, Xu Y, et al. Plasma Biomarkers of Tubular Injury and Inflammation Are Associated with CKD Progression in Children. J Am Soc Nephrol 2020;31:1067–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3–5 in the United States. Am J Kidney Dis 2013;62:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 2013;24:1484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadkarni GN, Chauhan K, Verghese DA, et al. Plasma biomarkers are associated with renal outcomes in individuals with APOL1 risk variants. Kidney Int 2018;93:1409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chander P, Soni A, Suri A, Bhagwat R, Yoo J, Treser G. Renal ultrastructural markers in AIDS-associated nephropathy. Am J Pathol 1987;126:513–26. [PMC free article] [PubMed] [Google Scholar]
- 37.Willicombe M, Moss J, Moran L, et al. Tubuloreticular Inclusions in Renal Allografts Associate with Viral Infections and Donor-Specific Antibodies. J Am Soc Nephrol 2016;27:2188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudose S, Santoriello D, Bomback AS, Stokes MB, D’Agati VD, Markowitz GS. Sensitivity and Specificity of Pathologic Findings to Diagnose Lupus Nephritis. Clin J Am Soc Nephrol 2019;14:1605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jennette JC, Iskandar SS, Dalldorf FG. Pathologic differentiation between lupus and nonlupus membranous glomerulopathy. Kidney Int 1983;24:377–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Baseline characteristics comparing AASK participants included vs. excluded from current study based on the availability of samples.
Supplementary Table 2: Baseline characteristics by sTNFR2 tertiles.
Supplementary Table 3: Baseline characteristics by TNF-α tertiles.
Supplementary Table 4: Baseline characteristics by IFN-γ tertiles.
Supplementary Table 5: Baseline characteristics by APOL1 risk status.
Supplementary Table 9: Associations of log2-transformed biomarkers of immune activation with chronic kidney disease progression (doubling of serum creatinine/KFRT) in AASK.
Supplementary Table 6: Associations of log2-transformed biomarkers of immune activation with KFRT, chronic kidney disease progression, and all-cause mortality in AASK in Model 4, further accounting for randomized treatment groups and high-sensitivity c-reactive protein.
Supplementary Table 7: Associations of log2-transformed biomarkers of immune activation with KFRT in Model 4, by subgroups of age, sex, systolic blood pressure, GFR, and urine PCR.
Supplementary Table 8: Associations of log2-transformed biomarkers of immune activation with KFRT in Model 4, accounting for the competing risk of death.