

Abstract
Background and aims:
Understanding the mechanisms by which tumors adapt to therapy is critical for developing effective combination therapeutic approaches to improve clinical outcomes for patients with cancer.
Methods:
To identify promising and clinically actionable targets for managing colorectal cancer (CRC), we conducted a patient-centered functional genomics platform that includes approximately 200 genes and paired this with a high-throughput drug screen that includes 262 compounds in four patient-derived xenografts (PDXs) from patients with CRC.
Results:
Both screening methods identified exportin 1 (XPO1) inhibitors as drivers of DNA damage-induced lethality in CRC. Molecular characterization of the cellular response to XPO1 inhibition uncovered an adaptive mechanism that limited the duration of response in TP53mutated, but not in TP53-wild type CRC models. Comprehensive proteomic and transcriptomic characterization revealed that the ATM/ATR-CHK1/2 axes were selectively engaged in TP53mutant CRC cells upon XPO1 inhibitor treatment, and that this response was required for adapting to therapy and escaping cell death. Administration of KPT-8602, an XPO1 inhibitor, followed by AZD-6738, an ATR inhibitor, resulted in dramatic antitumor effects and prolonged survival in TP53-mutant models of CRC.
Conclusion:
Our findings anticipate tremendous therapeutic benefit and support the further evaluation of XPO1 inhibitors, especially in combination with DNA damage checkpoint inhibitors, to elicit an enduring clinical response in patients with CRC harboring TP53 mutations.
Keywords: CRC, PDX, combination therapy, genomic biomarker
One Sentence Summary:
Developing an approach that combines functional genomics, drug screening and patient-derived xenograft models, we uncovered novel rational drug combinations in colorectal cancer based on TP53 mutational status.
Graphical Abstract
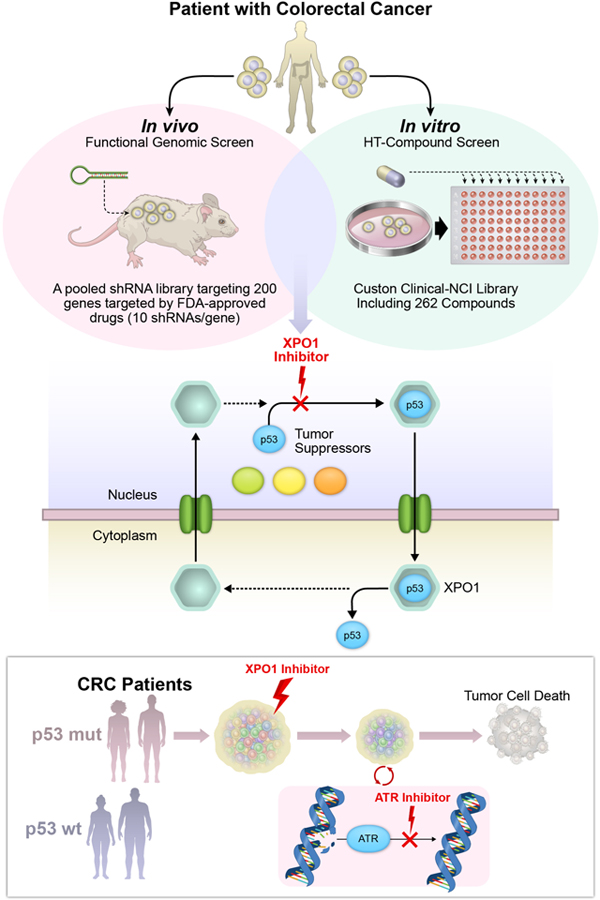
Introduction
Colorectal cancer (CRC) remains a leading cause of cancer-related morbidity and mortality.1 Tumor molecular profiling initiatives, including The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGA) efforts, through advanced sequencing technologies and data analyses, have identified clinically actionable alterations in genes and pathways in a broad range of cancer types, including CRC.2–7 Yet, the functional implications of these genetic lesions in CRC are incompletely understood, complicating the clinical positioning of available targeted agents. For instance, a recent trial failed to demonstrate clinical benefit of a BRAF inhibitor in CRC patients with BRAFV600E-mutant tumors.8
Similarly, comprehensive functional genomics screens conducted both in vitro and in vivo have identified numerous potential drug targets;9–12 however, the translation of screening outputs to robustly validated targets and the development of clinical therapeutics remains slow and resource intensive. To accelerate the identification of clinically actionable therapeutic opportunities in CRC, we combined in vivo functional genomics with in vitro drug screening in matched patient-derived tumor cell cultures and xenografts. We identified XPO1, a eukaryotic nuclear-cytoplasmic exporter and a validated drug target in multiple cancer indications13–15, as a novel therapeutic target in CRC. Recent preclinical and clinical studies with XPO1 inhibitors, KPT-330 and KPT-8602, have confirmed their potent activity in different cancer types.16–19 KPT-330 is now approved for treatment of multiple myeloma and, more recently, diffuse large B-cell lymphoma.20
Tumor plasticity and adaptation to targeted therapies contribute substantially to disease relapse/progression.21–25 Our characterization of tumor response to XPO1 inhibitor-induced DNA damage uncovered that TP53 wild type tumor cells recovered from XPO1 inhibitor treatment, likely due to arrest in the G1/S phase of the cell division cycle that facilitated DNA repair. In contrast, TP53-mutant tumors, which have a defective G1 checkpoint, experienced severe DNA damage accumulation and relied on activation of the G2/M DNA damage checkpoint via the ATM/ATR-Chk1/2 axes. Consistently, sequential administration of KPT-8602 followed by the ATR inhibitor, AZD-6738, yielded a synergistic survival benefit in CRC PDX models harboring TP53 mutations. Moreover, sequential treatment of XPO1 inhibitor-treated tumors with the CDK4/6 inhibitor, palbociclib, resulted in robust anti-tumor effects independent of TP53 mutational status.
Together, our data support the study of XPO1 inhibitor therapy alone and in combination with other DDR- and cell cycle-targeting drugs in subpopulations of patients with CRC.
Materials and Methods
In Vivo shRNA Screens
A custom library targeting 196 gene targets of FDA-approved targeted therapies was constructed using chip-based oligonucleotide synthesis and cloned into the pRSI16 lentiviral vector as a pool. PDX1 cells were infected using a multiplicity of infection (MOI) = 0.3 TU/cell. For the human PDX experiments, injections consisted of 3×106 cells to ensure coverage of 1,500 cells/barcode. Genomic DNA extraction, barcode amplification and preparation of sequencing libraries were performed according to our previously published protocol.11
shRNA Screen Hit Analysis
Illumina-generated sequences were processed using CASAVA (v.1.8.2), and resulting reads were processed using our in-house pipeline as previously described.11 For each sample log2 foldchange (FC) was calculated comparing tumors to the reference pellet. A summary measure per condition was derived using median of quantile transformed log2 FC across replicates. Thereafter, a modified version of RSA algorithm was used to derive a gene-level summary measure per condition.26
High Throughput (HT) Compound Screens
Screening of 262 drugs was accomplished at Texas A&M Health Science Center: Institute of Bioscience and Technology (IBT), GCC Screening Core. This library consisted of 150 Custom Clinical drugs and 112 NCI_AOD5 (National Cancer Institute Approved Oncology) drugs. The process is explained in details in supplementary methods and on their website in detail.27 All 4 CRC PDX lines were screened after optimization.
HT dose-response
Dose-response curves were based on a fraction affected (fa) calculation: fa = 1-Ti/C; where Ti is the cell count (or biochemical viability measurement) in the drug test well, and C is the cell count (or biochemical viability measurement) for vehicle-treated controls, with both measurements taken at the end of the assay.
After the drug concentration values have been transformed to their base-10 logarithm values, a four-parameter logistic (Hill) equation is fit to the data, and the following parameters are estimated:
E0 – the lowest value of fa, indicating the minimum effect of the drug.
Emax – the highest value of fa, indicating the maximum effect of the drug.
Log(EC50) – the base-10 logarithm of the concentration at which an effect of (EmaxE0)/2 is seen.
Slope – The slope of the transition region of the dose/response curve. Higher values indicate drugs which transition from having little effect to having a significant effect over a shorter range of doses, i.e., a steeper slope.
The area under the fitted curve was calculated using numerical integration then normalized to a value between 0 and 1 (AUCn). The mean-squared error (MSE) of all points relative to the fitted curve was also calculated. Classification of drug responses was defined as:
Class 1: AUCn >= 0.7, MSE < 0.01
Class 2: AUCn >= 0.4, MSE < 0.025
Class 3: AUCn > 0.1, MSE < 0.05
Class 4: all other drugs which fail to pass the criteria for Class 1 through
Human Colon Cell Lines and Culture Conditions
All cell lines were kept at 37°C in a humidified atmosphere with 5% CO2. SNU-C5 cells were cultured in RPMI-1640 with 10% FBS. SNU-61 cells were cultured in RPMI-1640 with HEPES supplemented with 10% of heat-inactivated FBS. Human colon cell lines and all other colon cancer cell lines were cultured in DMEM/F12 supplemented with 10% FBS. All cell lines were tested for mycoplasma contamination and fingerprinted. Mutation profiles for CRC-relevant cancer genes (http://www.cancer-genes.org/; FDR < 0.01) were obtained from Cancer Cell Line Encyclopedia (CCLE) (Supplementary Table 7). Mutations for WiDr and COLO320DM are not reported as they are derivatives of HT29 and COLO320, respectively.
In Vitro Treatments
Dose-response studies
Cell viability was assessed by measuring ATP content using the CellTiter-Glo luminescent assay in cells plated in black 96-well plates for 96h with or without drug treatment. The 50% effective concentration (EC50) values were calculated with Prism by means of a four-parameter, nonlinear regression analysis. Cytotoxicity was determined at the different time points and in the different cell lines to match cytotoxicity measurements with a given assay
Wash-out/Drug combination studies
Cells were exposed to DMSO or KPT-330 100nM for 48h cells. Cells were collected and replated on either 10-cm or 24-well plates with fresh medium and kept in culture for 300h. For drug combination studies, cells previously treated with XPO1 inhibitor were replated on 96-well plates and treated with palbociclib or AZD-6738 at different doses for 96h.
Tumor engraftment
All PDX models were generated and obtained from the laboratory of Dr. Scott Kopetz at UT MD Anderson Cancer Center.28 Rodent care and housing were in accordance with institutional guidelines and regulations as well as according to IACUC approved animal protocols.
For PDX-derived cell implantation, cells in log phase were trypsinized and resuspended as 2.0×107 cells/mL in 1:1 PBS:matrigel. For PDX pre-clinical studies, small tumor fragments (~0.1cm3) were collected from 1st or 2nd generation PDXs and transplanted into the right flank of recipient NSG mice. Animals were randomized to treatment based on tumor volume and tumor samples were collected when they reached 1500mm3, or when the tumor reached protocol limits with respect to ulceration.
In Vivo Drug Treatments
Mice were dosed orally once/day with DMSO (vehicle), KPT-8602, AZD-6738 or palbociclib for 5 days on/2 days off cycles. Drug dose was scaled to body weights of individual animals for final dosing volumes of: KPT-8602 5, 10 and 15ml/kg; AZD-6738 50ml/kg; and palbociclib 100ml/kg. 5-Fluorouracil (5-FU) was dosed intraperitoneally twice/week scaled to volume of 25ml/kg. For serial combination studies, mice were treated with each compound for 2 weeks. Mice were provided water and LabDiet 5053 chow ad libitum.
Statistical Analysis.
Data are presented as the mean ± S.D. of biological replicates. Statistical analyses were performed using a two-tailed Student’s t-test. Survival experiments were analyzed with a log-rank (Mantel-Cox) test and expressed as Kaplan–Meier survival curves.
Results
Integrated genetic and drug screening platforms identify targeted therapies for CRC
To identify therapeutic targets in CRC, we integrated functional genomics and drug screens in matched patient-derived tumor cells and xenografts (PDXs) (Fig. 1A). In vivo screens in PDX tumors were conducted using an shRNA library targeting 196 genes known to encode targets of drugs either approved by the U.S. Food and Drug Administration, or under clinical investigation (FDAome; 10 independent shRNAs/gene) (Fig. 1A and Supplementary Table 1). Concurrently and using the same models, we screened a custom clinical drug library comprised of 262 clinically available compounds in vitro in 2D-array format (CellTiter-Glo) (Fig. 1A and Supplementary Table 2). These orthogonal approaches identified genetic drivers essential for in vivo tumor maintenance with high translational potential in clinically relevant models.
Figure 1.

Integrated genomic and pharmacologic screening using CRC PDX models to identify therapeutic opportunities (A) Schematic of orthogonal screening platform: in vivo shRNA screens in CRC PDXs using a pooled genetic library targeting products of 196 FDA-approved or under clinical investigation genes was combined with in vitro high-throughput compound screens using a Custom Clinical NCI-library including 262 compounds. (B) Genetic landscape of 4 CRC PDXs in the in vitro and in vivo screening pipeline. Three out of four models displayed KRAS mutations (C0999, B1011 and C1047); one of the models harbored BRAF/PIK3CA mutation (B1003). (C) Density distribution of barcodes (shRNA) for transduced PDX cells (References) and three in vivo tumor replicates (Tx 1, 2 and 3) from 4 CRC PDXs infected with the FDAome shRNA lentiviral library. (D) Fraction of scoring genes in the library. Gene-rank analysis highlighting behavior of EGFR, AKT1, mTOR and PIK3CA hits in the FDAome in vivo screens executed in 4 independent CRC PDX models: C0999, B1003, B1011 and C1047 (RSA = redundant shRNA activity, logP). (E) Schematic of the high-throughput drug screen workflow and heatmap of the 30 most potent compounds and their AUCs for the 4 PDX models’ responses to drug exposure in vitro. Results were classified into 4 groups calculated by the extension of the area under the curve (AUCn; Class 1: AUCn≥0.7; Class2: AUCn≥0.4; Class3: AUCn>0.1; Class4: AUCn<0.1). (F) Topscoring genes and corresponding compounds were prioritized for investigation by integrating the orthogonal screening results with currently available clinical trial information in CRC.
Screens were conducted in 4 CRC PDX models and PDX-derived cell cultures (three KRAS mutant: C0999, B1011 and C1047; one BRAF mutant: B1003) (Fig. 1B and Supplementary Fig. 1A) were expanded for a maximum of 3 passages from excision. Each sample was molecularly and histologically characterized to confirm similarity with the tumors of origin.
In vivo screens employed our previously described two-step method for in vivo loss-of-function pooled shRNA screening. Briefly, to determine the engraftment efficiency of each CRC PDX to ensure adequate coverage of the molecular complexity of the library9–11, early passage cultures were transduced with a non-targeting 2.7K molecular barcode library (Empty BC) to “tag” individual cells. Barcode representation was then analyzed by deep sequencing and compared between transduced reference cells (PDX1 cells) and the barcoded cell population of tumors established in NSG mice (PDX2 tumors). Three models for which we confirmed statistically comparable barcode representation between reference PDX1 cells and PDX2 tumors, as well as among PDX2 tumor replicates (Supplementary Fig. 1B), were selected for screening. These findings support that complex libraries can be maintained in vivo in our CRC PDX models.
Again following our previously described protocol, the selected PDX models were screened using the FDAome library. Deep sequencing analysis identified barcodes depleted in the tumor versus the reference cell population, indicating the associated shRNAs conferred a selective growth disadvantage (Fig. 1C and Supplementary Fig. 1C). In all models, positive (PSMA1 and RPL30) and negative (luciferase) controls displayed significant separation, confirming the high quality of the screens (Supplementary Fig. 1D).
Next, we leveraged a modified redundant shRNA activity (RSA) analysis to identify “hits” (top-scoring genes)26 (Fig. 1D and Supplementary Table 3). EGFR emerged as a top hit in the wildtype KRAS B1003 model, but not in mutant-KRAS models (C0999, B11011, C1047), consistent with clinical findings where KRAS mutation is a negative predictor of response to EGFR blockade in CRCs. PIK3CA, AKT1, and mTOR emerged as top hits only in the PIK3CA-mutant model (B1003), but not in the PIK3CA-wild type context. Our results demonstrate strong correlation between model genotypes and functional phenotypes, supporting the robustness of in vivo screening to uncover genetic drivers.
To prioritize the genetic results with the highest chances of clinical impact, we conducted a high-throughput in vitro screening of drug compounds (Fig. 1E). Tumor model response to drug exposure was classified into 4 groups defined by the extension of the area under the curve (AUCn; Class1: AUCn≥0.7; Class2: AUCn≥0.4; Class3: AUCn>0.1; Class4: AUCn<0.1). Unsurprisingly, the most effective drugs (Class1) were classical cytotoxic agents, which were similarly efficacious across all models (Fig. 1E and Supplementary Table 4). Among targeted therapies, inhibitors targeting the proteasome, PLK1, HDAC, XPO1, CDK4/6 and mTOR scored as the most effective. In vitro drug screening results correlated well with the genetic screening, in which PSMD1, PLK1, HDAC2.3, XPO1, CDK4/6 and mTOR were identified as top-scoring hits in at least two of the four CRC models (Fig. 1F). Excluding XPO1, each of these is a well-known target in CRC currently under clinical investigation29–33, demonstrating that our dual screening strategy successfully captured essential, clinically relevant targets in CRC. XPO1 was prioritized for further study as a potentially novel therapeutic target in CRC.
Selective XPO1 inhibition induces DNA damage-dependent lethality in CRC
Exportin 1 (XPO1/CRM1) is a major nuclear-cytoplasmic exporter in eukaryotes that transports proteins and RNAs from the nucleus to the cytoplasm. Upregulation of XPO1 expression has been identified in some cancers, resulting in dysregulation of cargo proteins in the nuclear and cytoplasmic compartments.13, 34 In the TCGA dataset, XPO1 expression is upregulated in CRC versus normal tissue, and high expression of XPO1 correlated with poor prognosis (Supplementary Fig. 2A-B; GSE17536).3, 35 Consistently, expression of XPO1 in all four CRC PDX models used in our screens and in established human CRC cell lines from ATCC was elevated compared to normal colon epithelial cells (Fig. 2A).
Figure 2.

Selective XPO1 inhibition drives DNA damage-dependent lethality in CRC (A) XPO1 expression level across CRC models (black, cell lines and PDX-derived primary cultures) and colon epithelial cells (light blue). (B) Knockdown efficiency of XPO1 using 2 independent shRNAs in B1011 PDX-derived cell line compared to 2 shNT (Non-targeting) controls. (C) Colony formation assay in C0999, B1003 and B1011 cells expressing shNT or XPO1-targeting shRNA. (D) Sensitivity to KPT-330 across a panel of CRC models (cell lines and PDX-derived primary cultures) and colon epithelial cells (light blue) based on ATP viability assay (96h). (E) Expression of indicated proteins in CRC (black) or normal colon epithelial (light blue) cells treated with KPT-330 at indicated doses for 24h. (F) Nuclear fraction of protein expression in B1011 PDX-derived cells treated with KPT-330 at indicated dose for 24h. 5-FU serves as positive control. (G) Immunofluorescence staining of phospho-H2A.X and DAPI in B1011 PDX-derived primary cells treated with DMSO or KPT-330 at indicated doses for 24h. (H) FACS analysis for cell-cycle (BrdU incorporation) and DNA damage accumulation (phospho-H2A.X). β-actin and Histone H3 serve as loading controls in Western analyses. Representative image from 3 independent experiments is shown. All data are mean ± S.D. of biological replicates (n=3 each). All the experiments were repeated 3 times.
KPT-330 (selinexor) and KPT-8602 (eltanexor) are potent and specific XPO1 inhibitors that induce apoptosis upon accumulation of cargo proteins in the nucleus.13, 14, 17 In a recent early phase clinical trial of KPT-330 in advanced solid tumors, biological response was observed at tolerated doses.19 Here we aim to evaluate the efficacy of XPO1 inhibitor therapy in CRC and identify strategies to optimize clinical benefit.
Based on the Project DRIVE dataset, XPO1 is an essential gene in ~80% of cancer cell lines (Supplementary Fig. 2C).36 We confirmed that genetic or pharmacologic XPO1 inhibition impaired cell growth in 2D, colony formation and 3D assays CRC PDX-derived cell cultures and established cell line models, and we observed markedly attenuated effects in normal colon epithelial cell lines (Fig. 2B-E and Supplementary Fig. 2D). In PDX-derived cultures, increasing KPT-330 concentration correlated with increased p53 protein levels in the nucleus, whereas XPO1 as well as DNA damage repair proteins RAD51 and RAD50 decreased (Fig. 2F-G and Supplementary Fig. 2E-G). Consistently, phospho-H2A.X levels, indicative of DNA damage, increased in a dose-dependent manner, leading to accumulation of cells in sub-G0/G1 and a reduced number of cells in G2/M (Fig. 2H and Supplementary Fig. 2H). The abundance of MSH and MLH, which are frequently dysregulated in CRC, was not affected by XPO1 inhibition, suggesting that XPO1 inhibition impacts only a subset of DNA damage response (DDR) genes (Supplementary Fig. 2E). These data indicate that XPO1 inhibition affects expression of specific DDR genes in CRC, leading to cell cycle arrest and DNA damage accumulation.
XPO1 inhibition induces TP53-independent DNA damage, while drug recovery depends on TP53 mutational status
TP53 mutations have been identified in 40% to 50% of sporadic CRCs, and TP53 mutational status is a relevant prognostic marker for patients with CRC.37–39 Based on our finding of increased DNA damage and cell cycle defects in XPO1-inhibited cell cultures, we hypothesized that dysregulation of p53 may sensitize CRC cells to XPO1 inhibition. To test this, we evaluated the response of TP53-null isogenic pairs of HCT11640 and RKO41 cell lines to KPT-330 treatment. KPT-330 induced apoptosis and DNA damage independent of TP53 status (Fig. 3A-C and Supplementary Fig. 3A-B). Similarly, all tested CRC PDX-derived models showed acute sensitivity to XPO1 inhibition irrespective of their TP53 status (Fig. 3D, Fig. 2D and Supplementary Fig. 3C). To study recovery post-KPT-330 treatment two cell lines carrying mutant TP53 R273H (HT29 and WiDr) were exposed to KPT-330 for 48h and then allowed to recover until they reached confluence. Live-cell analysis revealed delayed recovery in TP53 wild type models compared to vehicle-treated control cultures, whereas TP53-mutant cell lines exposed to KPT-330 recovered similarly to vehicle-treated controls (Fig. 4A). Similar results were obtained in two additional CRC TP53 R248W cell lines (COLO320DM and SNU-C5) and one TP53 R175H (SNU-61) cell line (Supplementary Fig. 4A).
Figure 3.
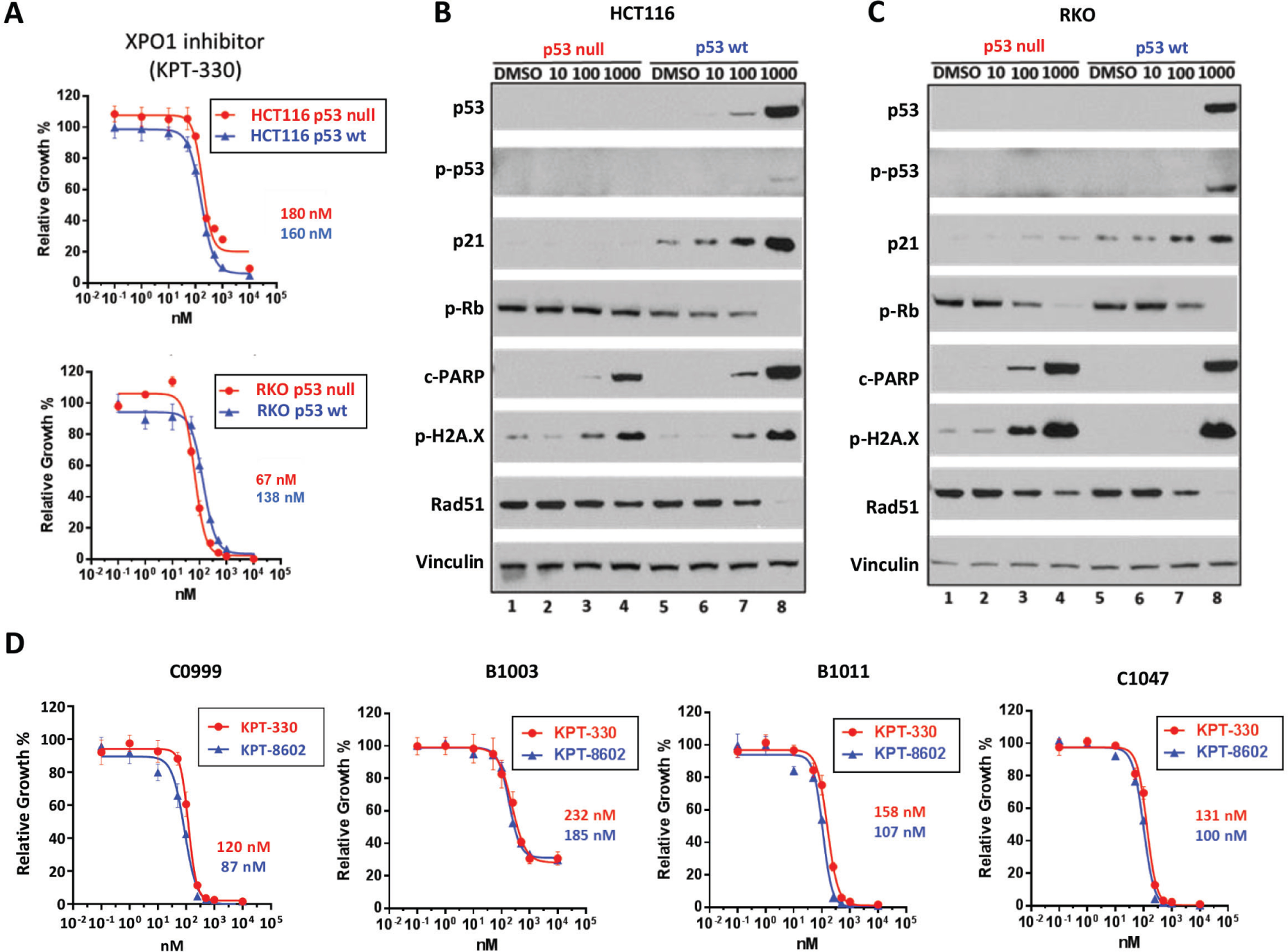
Selective XPO1 inhibition induces DNA damage and apoptosis independent of TP53 status in CRC (A) Sensitivity to KPT-330 in HCT116 (upper panel) and RKO (lower panel) TP53 isogenic pairs based on ATP viability assay (96h). (B, C) Protein expression in HCT116 (B) and RKO (C) TP53 isogenic pairs treated with KPT-330 at indicated dose for 24h. (D) Four CRC PDX-derived cell lines were treated with KPT-330 or KPT-8602 at indicated concentration for 96h, and viability was assessed based on ATP activity. IC50 values are shown on the side. Representative image from 3 independent experiments is shown. All data are mean ± S.D. of biological replicates (n=3 each). All the experiments were repeated 3 times.
Figure 4.

Differential TP53-dependent adaptation to XPO1 inhibition in CRC (A) TP53 wild type (RKO and SW48) and mutant (HT29 and WiDr) cells were treated with DMSO or KPT-330 (100nM) for 48h. Then, drugs were washed out and cells re-seeded onto 24-well plates. Confluence was monitored by IncuCyte to evaluate the recovery dynamics. (B) TP53 wild type (RKO and SW48) and mutant (HT29 and WiDr) cells were treated with DMSO or KPT-330 (100nM) for 48h. Then, drugs were washed out and protein expression analyzed by Western blotting at time indicated after washout (red box, DDR proteins; green box, Rb). (C) Sensitivity to KPT-330 followed by AZD-6738 or palbociclib in TP53 wild type (RKO and SW48) and mutant (HT29 and WiDr) cells (n=3). All the cells were treated with KPT-330 (100nM) for 48h followed by washout and reseeding onto 24-well plates. Cells were then treated with DMSO, AZD-6738 (1μM) or palbociclib (1μM). Confluence was monitored by IncuCyte to evaluate recovery dynamics during the second treatment. (D) Heatmap of normalized confluence (mean of three replicates) at 144h for TP53 wild type (RKO and SW48) and mutant (HT29, WiDr, COLO320DM, SNU-C5, SNU-61) CRC cells treated with KPT-330 (100nM) followed by either DMSO, AZD-6738 (1μM) or palbociclib (1μM). Representative image from 3 independent experiments is shown. All data are mean ± S.D. of biological replicates (n=3 each). All experiments were repeated 3 times.
RPPA analysis across a time course of recovery identified key cell cycle regulatory proteins, including PLK1, cyclin B1, CDK1, and WEE1, that were consistently upregulated in all cell lines after XPO1 inhibitor treatment (Supplementary Fig. 3D and Supplementary Table 5), suggesting that all re-entered the cell cycle. In contrast, DDR proteins were enriched more in TP53-mutant versus wild type cell lines after recovery (Supplementary Fig. 3D and Supplementary Table 5). Immunoblot analysis confirmed increased phosphorylation of ATM, ATR, and activation of their downstream substrates CHK1, CHK2 and BRCA1 during recovery from XPO1 inhibition exclusively in TP53-mutant cell lines, suggesting that these cells activated the G2/M checkpoint to compensate for the defective G1 checkpoint (Fig. 4B and Supplementary Fig. 4B). A similar dependency was previously observed in G1 checkpoint-defective neuroblastoma cells.42
To test whether the enhanced activation of the G2/M checkpoint in TP53-mutant cell lines may be therapeutically exploited, we treated cells with KPT-330 for 48h, followed by wash-out and treatment with the ATR inhibitor, AZD-6738. As anticipated, AZD-6738 profoundly inhibited recovery from KPT-330 treatment specifically in the TP53-mutant context, including in models harboring each of the three most frequent TP53 mutations in CRC (R273H, R248W, and R175H)43, with minimal impact observed in TP53 wild type models (Fig. 4C-D and Supplementary Fig. 4C). Consistent with our RPPA profiling data and mechanistic hypothesis, sequential treatment with KPT-330 followed by the CDK4/6 inhibitor, palbociclib, showed significant anti-tumor activity across all models, independent of TP53 status (Fig. 4C-D and Supplementary Fig. 4C).
Our findings support that adaptation to XPO1 inhibition requires re-start of the cell cycle machinery and that, exclusively in the TP53-mutant context, activation of ATM/ATR signaling is required to cope with accumulating DNA damage.
Pharmacological XPO1 inhibition in serial combination with a selective ATR inhibitor has potent anti-tumor activity in vivo
In anticipation of XPO1 inhibitor clinical studies in CRC, we selected KPT-8602 (eltanexor), a second-generation XPO1 inhibitor with diminished blood-brain barrier penetration and fewer side effects that might limit treatment at effective doses.44, 45 KPT-8602 behaved similarly to KPT-330 in CRC models and normal colon epithelial cells in vitro, with potent anti-proliferative activity observed solely in tumor cells (Fig. 3D and Supplementary Fig. 3C). In mice bearing tumors derived from B1011 cells (TP53-mutant CRC PDX) KPT-8602 15 mg/kg (mpk) dosed on a 5 days on/2 days off schedule17 for 2 weeks resulted in strong tumor growth inhibition (TGI), but the animals experienced >15% body weight loss (Supplementary Fig. 5A-B). The better-tolerated doses of 5 and 10 mpk both induced superior TGI compared to 5-Fluorouracil (5-FU), and with null or negligible body weight loss (Fig. 5A-B). XPO1 target engagement, inhibition of proliferation (Ki67), as well as induction of apoptosis (cleaved-PARP) and DNA damage (phospo-H2A.X) were confirmed by immunohistochemistry in KPT-8602- versus vehicle-treated tumors (Fig. 5C).
Figure 5.

Second-generation XPO1 inhibitor KPT-8602 shows potent anti-tumor activity in TP53-mutant CRC (A, B) Animals harboring tumors derived from TP53-mutant B1011 PDX model were randomized to treatment with vehicle, 25 mpk 5-FU, or 5 or 10 mpk KPT-8602. Arrows indicate days of oral dosing for KPT-8602. Tumor volumes (A) and body weight changes (B) are shown. (C) Representative images and signal quantification of IHC staining (Hematoxylin and Eosin (H&E), XPO1, Ki-67, cleaved-PARP, phospho-H2A.X) for B1011 treated with either vehicle or KPT-8602 at 10mpk. (D) Animals harboring B1011-derived tumors were randomized to vehicle or 10 mpk KPT-8602 for 12 days. Tumors were collected during drug recovery period at indicated time points. (E) Top ten enriched REACTOME pathways (Fisher’s exact test) for differentially expressed genes during drug recovery period at indicated time points. (F) Representative images of IHC staining (H&E, phospho-ATR/ATM, phospho-H2A.X) from tumors harvested from animals described in (D) at indicated time point during recovery. NS, not significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001 by unpaired two-tailed t-test. Bar = 100 μm.
To understand the molecular underpinnings of recovery from KPT-8602 treatment in vivo, we conducted RNA-sequencing on tumors excised at 24h and 6 days after the end of treatment (Fig. 5D). Interferon signaling, cell-cycle, and DNA replication emerged as the top transcriptionally deregulated pathways after KPT-8602 treatment, and they all reactivated during recovery, suggesting the existence of transcriptional programs that drive the entry of surviving cancer cells into the proliferative phase (Fig. 5E and Supplementary Table 6). Immunohistochemical analysis detected upregulation of phospho-ATR/ATM and phospho-H2A.X during recovery, indicating post-transcriptional regulation of DDR proteins also contribute to adaptation to XPO1 inhibition in vivo in TP53-mutant CRC (Fig. 5F).
We next evaluated sequential therapy combinations in vivo. Animals with tumors derived from B1011 were randomized to receive treatment with KPT-8602 followed by AZD-6738 or palbociclib, a Cdk4/6 inhibitor in clinical trials in CRC (ClinicalTrials.gov: NCT02223923, NCT02668666).46 Consistent with in vitro data, both sequential treatment regimens yielded robust TGI and prolonged survival compared to vehicle (Fig. 6A-D). As expected, TGI induced by sequential combinations was comparable to prolonged KPT-8602 treatment, but sequential therapy was associated with improved tolerability (Supplementary Fig. 5C-D). Moreover, combination therapy had superior TGI and survival benefit compared with either palbociclib or AZD-6738 alone (Fig. 6A-D and Supplementary Fig. 5E-F). Consistent with our mechanistic studies, the reverse therapy sequence was equally effective in the case of palbociclib, but inferior TGI was achieved when AZD-6738 was provided prior to KPT-8602 (Supplementary Fig. 5E-F). These data confirm exquisite dependency of TP53-mutant CRC tumors on cell cycle and DDR machineries during recovery from XPO1 inhibition.
Figure 6.

Sequential dosing with KPT-8602 and palbociclib or AZD-6738 prolongs treatment response and survival in TP53-mutant CRCs (A, B) Animals harboring tumors derived from TP53-mutant B1011 model were randomized to treatment with vehicle, palbociclib (100mpk) or KPT-8602 (10mpk) for 2wk followed by vehicle or palbociclib for 2wk. Tumor growth (A) and Kaplan-Meier survival (B) curves are shown. (C, D) Animals harboring tumors derived from B1011 were randomized to treatment with vehicle, AZD-6738 (50mpk) or KPT-8602 (10mpk) for 2wk followed by vehicle or AZD-6738 for 2wk. Tumor growth (C) and Kaplan-Meier survival (D) curves are shown. (E) End-point tumor growth comparison (%) between treated groups (K: KPT-8602, K+A: KPT-8602+AZD-6738) and vehicle in three TP53 wild type and three TP53-mutant CRC PDXs. Endpoint tumor volumes were defined as the last measurements for each tumor when vehicle-treated tumors reached the ethical limit (see Suppl. Fig. 6). (F) Illustration of TP53-dependent adaption to DNA damage induced by XPO1 inhibition and the informed sequential combinations for patient-stratified clinical trial designs with CDK4/6 or ATR inhibitors in CRC. Tumor growth analysis: NS = not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by unpaired two-tailed t-test. Survival analysis: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by Mantel-Cox test.
To validate TP53 as a patient stratification biomarker in CRC, we selected three wild type and three mutant TP53 CRC PDX models. As anticipated, sequential treatment with KPT-8602 and AZD-6738 resulted in statistically significantly greater TGI versus KPT-8602 alone only in TP53-mutant models, whereas KPT-8602 alone resulted in similar TGI as combination therapy with AZD-6738 in TP53 wild type models (Fig. 6E and Supplementary Fig. 6A-F). Our results validate TP53 mutational status as a valuable biomarker of ATM/ATR checkpoint dependency in CRC cells that survive XPO1 inhibition and support clinical evaluation of sequential administration of XPO1 and ATR inhibitor drugs in TP53-mutated CRC (Fig. 6F).
Discussion
Our study integrated in vivo functional genomics and high-throughput in vitro drug screening to identify rapidly translatable therapeutic strategies in CRC. Our screens were conducted in patient-derived CRC samples, which better recapitulate the genetic and functional heterogeneity of human tumors compared to established tumor cell lines. We selected shRNA-based gene suppression, rather than CRISPR-based inactivation, to better mimic the biological activity of targeted drugs, which do not usually completely suppress gene function. To our knowledge, this is the first attempt to systematically combine PDX-centric functional genomics with in vitro high-throughput drug screens, and our data support this as a viable alternative approach to labor- and resource-intensive PDX pre-clinical trials47 designed to inform drug repositioning.
Our approach was adequate to captured clinically relevant, genetic context-specific dependencies in CRC, as demonstrated by the emergence of EGFR as a positive screening hit solely in wild type KRAS models, consistent with published data and the established use of KRAS mutational status as a biomarker for EGFR inhibitor treatment in patients with CRC. One advantage of our strategy is that clinical safety and efficacy readouts are already available for the drugs/drug targets included in our screening platform. For XPO1, inhibitor drugs in clinical testing for other indications have generated promising data, and selinexor has been approved for treatment of advanced multiple myeloma and diffuse large B cell lymphoma.20, 46 However, XPO1 targeting in CRC has not been adequately evaluated. We show that XPO expression is higher in CRC cells compared to normal colon epithelial cells, and that XPO1 inhibition potently inhibits proliferation and induces apoptosis of malignant cells compared to normal cells. Mechanistically, our data suggest that XPO1 inhibition results in a toxic accumulation of DNA damage in CRC cells that is not detected in normal colon epithelial cells, indicating a tumor-specific functional dependency on XPO1.
Multiple tumor suppressor proteins (TSPs) and transcription factors in the nucleus, such as p53, Rb, p27, and FOXO3a, protect cells by regulating cell growth, apoptosis and DNA damage repair;13, 48–50 thus, cytoplasmic mislocalization of TSPs can lead to tumor progression. One strategy to prevent localization of essential TSPs in the cytoplasm is to inhibit their nuclear export. XPO1 is the sole nuclear export receptor for multiple TSPs. Overexpression of XPO1 is reported in different cancers types and can correlate with poor prognosis.51, 52 The XPO1 inhibitors, KPT-330 and KPT-8602, induce nuclear accumulation of TSPs and restore their tumor suppressor activity.15, 17, 18, 45 Thus, XPO1 inhibition represents a unique therapeutic concept.
A previous study indicated that p53 deficiency or loss significantly contributed to XPO1 inhibitor resistance in thymic epithelial tumors.16 In contrast, our data from CRC cell lines with varied TP53 mutational status uncovered a mechanism of DNA damage accumulation unrelated to p53 status upon XPO1 inhibition. In addition, we identified a direct association between TP53 mutational status and recovery dynamics following XPO1 inhibition. Our findings suggest that p53 inhibits adaptation to DNA damage induced by XPO1 inhibition and engage the G1/S checkpoint, while p53-defective cells lack this ability and are highly dependent on the ATM/ATR axis at the G2/M checkpoint. These results are consistent with previous studies showing that cancer cells harboring TP53 loss-of-function mutation have a dysfunctional G1/S checkpoint and primarily rely on the G2/M checkpoint to arrest the cell cycle and execute DNA repair.53, 54 Consistently, it has been demonstrated that TP53-deficient cells depend on ATM and ATR-mediated checkpoint signaling through the p38 MAPK/MK2 pathway to repair DNA damage.55
We show that TP53-mutant CRC cells displayed higher sensitivity to sequential XPO1 inhibitor treatment followed by ATR inhibition compared to wild type TP53 CRC cells in vitro and in vivo, which explicates a rational therapeutic approach. The consistency of these results across models carrying some of the most frequent TP53 mutations in CRC (R248W, R273H, and R175H)43 compels the clinical investigation of sequential XPO1-ATR inhibitor therapy in TP53mutant CRC. TP53 mutation occurs in approximately 40%−50% of sporadic CRCs37–39, and KPT-330 and AZD6738 are in clinical trials in solid tumors, with data from the former showing clinical activity with an acceptable safety profile.19 We also demonstrated that palbociclib, which is approved for treatment of estrogen receptor-positive metastatic breast cancer56 and is in clinical development for additional indications, including CRC,27 can retard recovery from XPO1 inhibitor treatment in CRC models with functional p53, illuminating another actionable clinical opportunity to evaluate targeted therapies in biomarker-defined CRC.
The accumulation of DNA damage induced by XPO1 inhibition suggests that patients with CRC who receive this treatment may also benefit from immune checkpoint blockade (ICB). Recently, anti-PD-1 (nivolumab and pembrolizumab) and anti-CTLA4 (ipilimumab) therapies have been approved for a subset of patients with advanced CRC characterized by deficient DNA mismatch repair (dMMR) or microsatellite instability-high (MSI-H).57, 58 MMR is essential for DNA repair, and dMMR tumors harbor high mutational burdens associated with high neoantigen loads and T-cell infiltration, which in turn should elicit profound immunogenic responses by the host and in response to ICB therapy.58–61 However, only 3–6% of patients with advanced-staged CRC have dMMR or MSI-H tumors that are likely to respond to ICB therapy.62 To expand the application of ICB therapy, several clinical studies are evaluating ICB therapy combined with DNA damaging agents or DDR inhibitors, such as PARP inhibitors.63, 64 Based on our finding that coupling XPO1 with ATR inhibitor treatment results in massive accumulation of DNA damage, combining XPO1 and ATR inhibitors followed by ICB therapy to treat TP53-mutant CRC may be beneficial. Further studies are needed to provide mechanistic insights regarding this triple combination, as well as identify the combination sequence strategy and drug doses to optimize clinical benefit.
Taken together, our findings suggest that administration of XPO1 inhibitors, especially in combination with ATR inhibitors, may be a novel therapeutic approach to treat patients with CRC. This mechanism-based combination strategy may prevent treatment adaptation to produce more durable responses in patients with this disease.
Supplementary Material
“What You Need to Know” Summary.
Background and context:
Approved targeted therapies to achieve enduring therapeutic responses in patients with CRC are limited (e.g., EGFR inhibitors in KRAS wild type tumors).
New findings:
Sequential treatment with an XPO1 inhibitor followed by an ATR inhibitor induces massive DNA damage in CRC cells harboring TP53 mutations, thereby shrinking tumor burden and prolonging survival in preclinical models.
Limitations:
Lack of experiments assessing the effects of XPO1-ATR combination treatment in an immunocompetent system limits our characterization of the therapeutic response as well as the evaluation of potential interactive effects with immunotherapy.
Impact:
Our successful results with XPO1 and ATR inhibitors, which have known safety profiles, should provide the foundation for initiating clinical trials for patients with CRC with genomically defined TP53 mutations.
Lay Summary:
Developing an approach that combines functional genomics, drug screening and patient-derived xenograft models, we uncovered novel rational drug combinations in colorectal cancer based on TP53 mutational status.
Acknowledgments:
We wish to thank the members of Viale, Draetta, Genovese and Carugo labs for discussions and reagents. Special thanks to Dr. Maria Emilia Di Francesco, Dr. Christopher Carroll and the IACS platform for advice and reagents. We thank the UTMDACC Department of Veterinary Medicine, the UTMDACC Sequencing & Non-coding RNA Program and the UTMDACC Flow Facility. The GCC High Throughput Screening Program was supported by CPRIT Grant RP150578. G.F.D. was supported by the Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Grant, Pancreatic Cancer Action Network Translational Research Grant 17-65-DRAE, and the Sewell Family Chair in Genomic Medicine. A.C. was supported by the FIRC-AIRC fellowship. G.G. was supported by the Barbara Massie Memorial Fund and the CPRIT Grant RP170722.
Abbreviations:
- ATR
Ataxia telangiectasia and Rad3-related
- CDK4/6
Cyclin Dependent Kinase 4/6
- CRC
Colorectal Cancer
- PDX
Patient-Derived Xenograft
- shNT
Short hairpin Non Targeting
- RNA shRNA
Short hairpin RNA
- XPO1
Exportin 1
Footnotes
Competing interests: G.F.D. is scientific advisor of Karyopharm Therapeutics. All other authors have no conflicts.
Data and materials availability: Transcriptomic analysis upon XPO1 inhibition will be deposited to archives.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes:
- 1.Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77. [DOI] [PubMed] [Google Scholar]
- 5.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pauli C, Hopkins BD, Prandi D, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 2017;7:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopetz S, Desai J, Chan E, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol 2015;33:4032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bossi D, Cicalese A, Dellino GI, et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov 2016;6:650–63. [DOI] [PubMed] [Google Scholar]
- 10.Dietlein F, Thelen L, Jokic M, et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov 2014;4:592–605. [DOI] [PubMed] [Google Scholar]
- 11.Carugo A, Genovese G, Seth S, et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep 2016;16:133–147. [DOI] [PubMed] [Google Scholar]
- 12.Kodama M, Kodama T, Newberg JY, et al. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc Natl Acad Sci U S A 2017;114:E7301–E7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer 2004;4:106–17. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, McMillan E, Kim HS, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016;538:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012;120:4621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conforti F, Zhang X, Rao G, et al. Therapeutic Effects of XPO1 Inhibition in Thymic Epithelial Tumors. Cancer Res 2017;77:5614–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vercruysse T, De Bie J, Neggers JE, et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin Cancer Res 2017;23:2528–2541. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Camacho SC, Silvers TR, et al. Inhibition of the Nuclear Export Receptor XPO1 as a Therapeutic Target for Platinum-Resistant Ovarian Cancer. Clin Cancer Res 2017;23:1552–1563. [DOI] [PubMed] [Google Scholar]
- 19.Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients With Advanced Solid Tumors. J Clin Oncol 2016;34:4142–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.XPO1 Inhibitor Approved for Multiple Myeloma. Cancer Discov 2019;9:1150–1151. [DOI] [PubMed] [Google Scholar]
- 21.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet 2012;13:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401–9. [DOI] [PubMed] [Google Scholar]
- 23.Di Nicolantonio F, Mercer SJ, Knight LA, et al. Cancer cell adaptation to chemotherapy. BMC Cancer 2005;5:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fodale V, Pierobon M, Liotta L, et al. Mechanism of cell adaptation: when and how do cancer cells develop chemoresistance? Cancer J 2011;17:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandarlapaty S Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov 2012;2:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birmingham A, Selfors LM, Forster T, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 2009;6:569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Texas A & M University System. Health Science Center. Institute of Biosciences and Technology. The Institute of Biosciences and Technology, Texas A&M University: [Google Scholar]
- 28.Katsiampoura A, Raghav K, Jiang ZQ, et al. Modeling of Patient-Derived Xenografts in Colorectal Cancer. Mol Cancer Ther 2017;16:1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 2008;26:1603–10. [DOI] [PubMed] [Google Scholar]
- 30.O’Hara MH, Edmonds C, Farwell M, et al. Phase II pharmacodynamic trial of palbociclib in patients with KRAS mutant colorectal cancer. Journal of Clinical Oncology 2015;33:626–626. [Google Scholar]
- 31.Schoffski P, Awada A, Dumez H, et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer 2012;48:179–86. [DOI] [PubMed] [Google Scholar]
- 32.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 2017;14:417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tampakis A, Tampaki EC, Nebiker CA, et al. Histone deacetylase inhibitors and colorectal cancer: what is new? Anticancer Agents Med Chem 2014;14:1220–7. [DOI] [PubMed] [Google Scholar]
- 34.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 2012;83:1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald ER 3rd, de Weck A, Schlabach MR, et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017;170:577–592.e10. [DOI] [PubMed] [Google Scholar]
- 37.Lopez I, PO L, Tucci P, et al. Different mutation profiles associated to P53 accumulation in colorectal cancer. Gene 2012;499:81–7. [DOI] [PubMed] [Google Scholar]
- 38.Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989;244:217–21. [DOI] [PubMed] [Google Scholar]
- 39.Li XL, Zhou J, Chen ZR, et al. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol 2015;21:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jallepalli PV, Lengauer C, Vogelstein B, et al. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 2003;278:20475–9. [DOI] [PubMed] [Google Scholar]
- 41.Sur S, Pagliarini R, Bunz F, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009;106:3964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu H, Cheung IY, Wei XX, et al. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G1 checkpoint-defective neuroblastoma. Int J Cancer 2011;129:1953–62. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Zhang J, Tong JHM, et al. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hing ZA, Fung HY, Ranganathan P, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016;30:2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Etchin J, Berezovskaya A, Conway AS, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017;31:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guichard SM, Brown E, Odedra R, et al. Abstract 3343: The pre-clinical in vitro and in vivo activity of AZD6738: A potent and selective inhibitor of ATR kinase. Cancer Research 2013;73:3343–3343. [Google Scholar]
- 47.Gao H, Korn JM, Ferretti S, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25. [DOI] [PubMed] [Google Scholar]
- 48.Takayama T, Miyanishi K, Hayashi T, et al. Colorectal cancer: genetics of development and metastasis. J Gastroenterol 2006;41:185–92. [DOI] [PubMed] [Google Scholar]
- 49.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005;24:2899–908. [DOI] [PubMed] [Google Scholar]
- 50.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000;408:307–10. [DOI] [PubMed] [Google Scholar]
- 51.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell 2012;23:3673–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao Y, Dong Y, Lin F, et al. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep 2009;21:229–35. [PubMed] [Google Scholar]
- 53.Koniaras K, Cuddihy AR, Christopoulos H, et al. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 2001;20:7453–63. [DOI] [PubMed] [Google Scholar]
- 54.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reinhardt HC, Aslanian AS, Lees JA, et al. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007;11:175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhillon S Palbociclib: first global approval. Drugs 2015;75:543–51. [DOI] [PubMed] [Google Scholar]
- 57.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008;134:988–97. [DOI] [PubMed] [Google Scholar]
- 62.Nosho K, Baba Y, Tanaka N, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol 2010;222:350–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li A, Yi M, Qin S, et al. Prospects for combining immune checkpoint blockade with PARP inhibition. J Hematol Oncol 2019;12:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol 2013;24:75–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.