

Abstract
Under-expression or over-expression of protein kinases has been shown to be associated with unregulated cell signal transduction in cancer cells. Therefore, there is a major interest in designing protein kinase inhibitors as anticancer agents. We have previously reported [WR]5, a peptide containing alternative arginine (R) and tryptophan (W) residues as a non-competitive c-Src tyrosine kinase inhibitor. A number of larger cyclic peptides containing alternative hydrophobic and positively charged residues [WR]x (x = 6–9), and hybrid cyclic-linear peptides, [R6K]W6 and [R5K]W7, containing R and W residues were evaluated for their protein kinase inhibitory potency. Among all the peptides, cyclic peptide [WR]9 was found to be the most potent tyrosine kinase inhibitor. [WR]9 showed higher inhibitory activity (IC50= 0.21 μM) than [WR]5, [WR]6, [WR]7, and [WR]8 with IC50 values of 0.81, 0.57, 0.35, and 0.33 μM, respectively, against Src kinase as determined by a radioactive assay using [γ-33P]ATP. Consistent with the result above, [WR]9 inhibited other protein kinases such as Abl kinase activity with an IC50 value of 0.35 μM, showing 2.2-fold higher inhibition than [WR]5 (IC50= 0.79 μM). [WR]9 also inhibited PKCa kinase activity with an IC50 value of 2.86 μM, approximately 3-fold higher inhibition than [WR]5 (IC50=8.52 μM). A similar pattern was observed against Braf, c-Src, Cdk2/Cyclin A1, and Lck. [WR]9 exhibited IC50 values of <0.25 μM against Akt1, Alk, and Btk. These data suggest that [WR]9 is consistently more potent than other cyclic peptides with smaller ring size and hybrid cyclic-linear peptides [R6K]W6, and [R5K]W7 against selected protein kinases. Thus, the presence of R and W residues in the ring, ring size, and the number of amino acids in the structure of the cyclic peptide were found to be critical in protein kinase inhibitory potency. We identified three putative binding pockets through automated blind docking of cyclic peptides [WR](5–9). The most populated pocket is located between the SH2, SH3, and N-lobe domains, on the opposite side of the ATP binding site. The second putative pocket is formed by the same domains and located on the ATP binding site side of the protein. Finally, a third pocket was identified between the SH2 and SH3 domains. These results are consistent with the non-competitive nature of the inhibition displayed by these molecules. Molecular dynamics simulations of the protein-peptide complexes indicate that the presence of either of [WR]5 or [WR]9 affects the plasticity of the protein, and in particular the volume of the ATP binding site pocket, in different ways. These results suggest that the second pocket is most likely the site where these peptides bind and offer a plausible rationale for the increased affinity of [WR]9.
Graphical Abstract
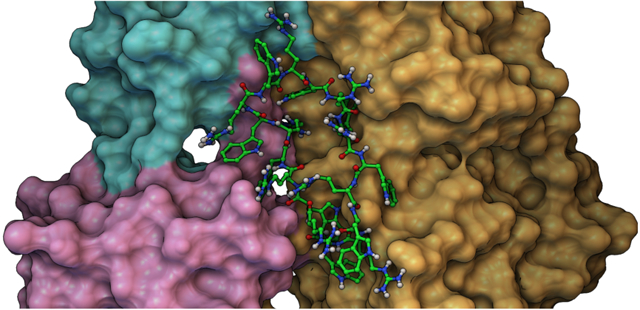
INTRODUCTION
Protein kinases play a major role in the cellular signaling pathways. Studies have also suggested that protein kinases are associated with enhancing unregulated cell signal transduction in cancer cells.1, 2 For example, among protein-tyrosine kinases (PTKs), overexpression of c-Src kinase has been observed in cancer cells.3 c-Src kinase phosphorylates the side chain of tyrosine residues in protein substrates, leading to the protein substrate1 conformational change, thereby changing the activity of the protein. Under-expressed or overly expressed kinase can lead to a change of signaling and catastrophic results for the body.4, 5 Thus, researchers have delved into characterizing the PTKs that potentiate such pathways and developing strategies to disrupt the unregulated proliferating cellular signaling.
The sequence of amino acids in the structure of the protein substrate can be used to develop peptide mimics that target non-conserved regions of PTK. Furthermore, designing peptides specific to the conserved regions of the PTK that block the binding of ATP has become a subject of major interest.6,7
As a representative example of the Src family kinases, the c-Src protein is composed of three major domains, SH2, SH3, and the kinase catalytic domain. The SH2 and SH3 are vital in protein-protein interactions, while the kinase catalytic domain contains the kinase active site that includes ATP binding and substrate-binding sites.3 There is still interest in developing different kinase inhibitors for a variety of diseases.8 The use of different sequences and configurations of amino acids can create a variety of structures.
In continuation of our efforts in designing protein kinase inhibitors using cyclic peptides or those that interact with binding pockets outside the catalytic site,9–15 here we investigated cyclic peptides that potentially act as kinase inhibitors.
Cyclic peptides have been widely used as therapeutic agents and biochemical tools.16 Cyclic peptides are made up of amino acids linked together by a peptide bond through the head to tail cyclization. Linear peptides are less stable and have lower bioavailability than cyclic peptides because they are more prone to undergo proteolytic degradation in the body. On the other hand, cyclic peptides have been shown to be more stable and potent in comparison to linear peptides and are not readily degraded in the body via hydrolysis.16
A linear peptide is a flexible structure that could lead to weak interactions with the target site. The cyclic structure reduces the conformation freedom of the peptides. The constrained conformation of cyclic peptides allows high-affinity binding to the target receptors due to the decrease in entropy term of the Gibbs free energy.17
We have previously reported that a cyclic peptide [WR]5 showed a c-Src kinase inhibitory activity with an IC50 value of 2.8 μM using a Transcreener® ADP2 fluorescence intensity assay through a non-competitive inhibition pattern against ATP for both Csk and c-Src.18 A corresponding linear peptide [WR]5 showed an IC50 of 7.1 μM, suggesting the importance of cyclic nature in improving binding affinity. More structure-activity relationships were required to determine the importance of arginine and tryptophan in generating c-Src kinase inhibitory activity. Furthermore, more investigation was needed to explore the selectivity of this class of cyclic peptides against other protein kinases.
Thus, cyclic peptides with a higher number of positively charged and hydrophobic peptides [WR]x (x = 6–9) (Figure 1) and hybrid cyclic-linear peptides, [R6K]W6, and [R5K]W7 (Figure 2), were synthesized to be evaluated as c-Src kinase inhibitors. The previous data18 suggested that cyclic [WR]5 is using other potential sites in the catalytic domain for generating kinase inhibitory activity, possibly by binding to adjacent domains. Herein we studied the roles of the ring size, hydrophobicity, and charge on their c-Src inhibitory potency in comparison to [WR]5. We also explored their effects on protein kinase selectivity. Thus, the inhibitory activity of all the cyclic peptides was determined against Abl1, Akt1, Alk, Braf, Btk, Cdk2/Cylin A1, Lck, and PKCa. The inhibitory activity of cyclic peptides against protein kinases was compared with hybrid cyclic-linear peptides [R5K]W7 and [R6K]W6. Finally, we used molecular modeling to delineate a plausible binding pocket for the interaction of [WR]9 with c-Src kinase.
Figure 1.
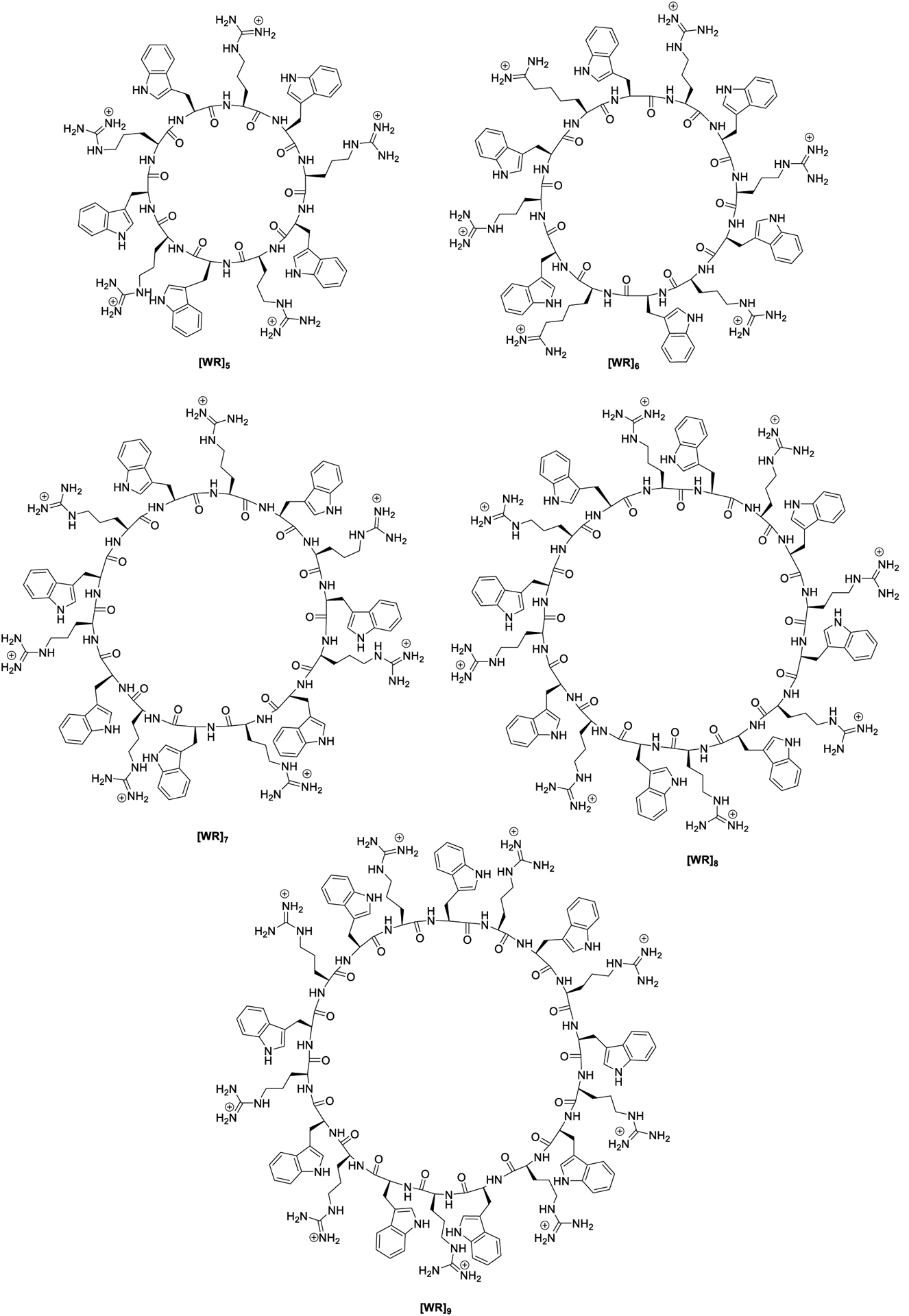
Chemical structures of synthesized cyclic peptides.
Figure 2.
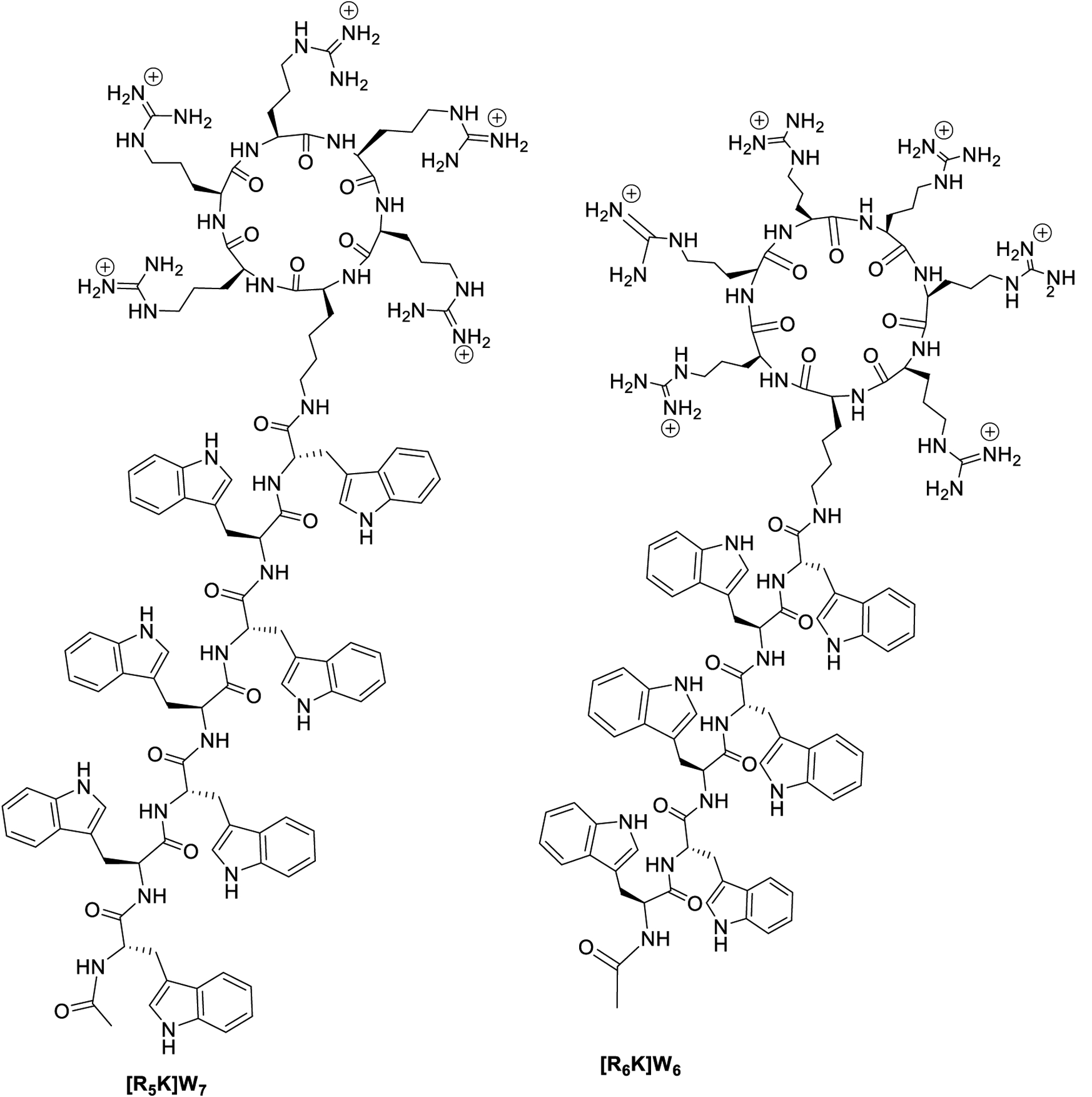
Hybrid cyclic-linear peptides containing positively-charged arginine residues on the ring attached to a hydrophobic tryptophan chain.
The goal here was to determine whether increasing the hydrophobicity, charge, and ring size will affect the tyrosine kinase inhibitory activities in comparison to [WR]5, to establish structure-tyrosine kinase inhibitory relationships, and find the most potent lead compound for further mechanistic studies and X-ray crystallography.
RESULTS AND DISCUSSION
Chemistry
Fmoc solid-phase peptide synthesis followed by solution-phase chemistry was used to synthesize all the cyclic peptides [WR]x (x = 5–9) (Figure 1) as described previously.19 The linear peptides were first assembled on H-Trp(Boc)-2-chlorotrityl resin by step-by-step coupling and deprotection. The cleavage of the side-chain linear peptides from the resin was accomplished in the presence of a cleavage cocktail of dichloromethane (DCM):trifluoroethanol (TFE):acetic acid (AcOH). The N- to C-terminal cyclization was conducted in the presence of coupling reagent, N,N′-diisopropylcarbodiimide (DIC), 1-hydroxy-7-azabenzotriazole (HOAt), and a base, N,N-diisopropylethylamine (DIPEA). Final deprotection was carried out by freshly prepared cleavage cocktail reagent R (trifluoroacetic acid (TFA):thioanisole:1,2-ethanedithiol:anisole). The cyclic peptides were purified using reverse-phase HPLC and characterized by using high-resolution matrix-assisted laser desorption/ionization (MALDI). As a representative example of these cyclic peptides, the synthesis of [WR]7 is shown in Scheme 1 according to the previously reported procedure by us.19
Scheme 1.
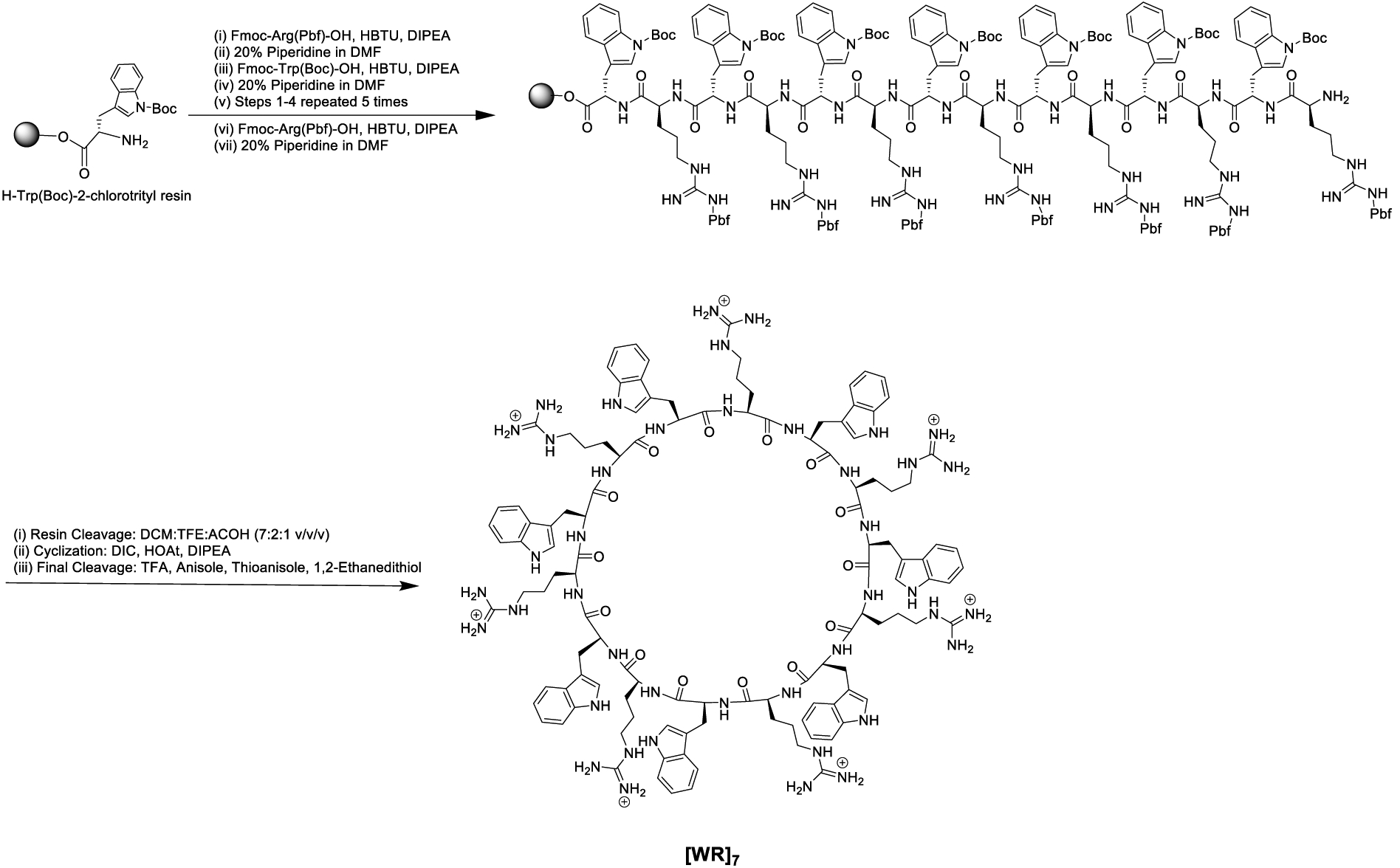
Synthesis of cyclic peptide [WR]7.
The synthesis of hybrid cyclic-linear peptides, [R6K]W6 and novel peptide [R5K]W7, was accomplished according to the previously reported procedure.20 As a representative example, the synthesis of [R6K]W6 is depicted in Scheme 2. After assembly of building block amino acids, Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, and Dde-Lys(Fmoc)-OH on H-Arg(Pbf)-2-chlorotrityl resin, the Fmoc of the lysine was removed in the presence of piperidine followed by continuing the coupling of Fmoc-Trp(Boc)-OH on the side chain of lysine. After Fmoc deprotection and capping in the presence of acetic anhydride, the Dde group of lysine residue was removed in the presence of hydrazine in DMF (2% v/v). After resin cleavage in the presence of DCM:TFE:AcOH, the cyclization was accomplished in the presence of DIC and HOAt. The final cleavage was achieved in the presence of TFA, anisole, thioanisole, dithiothreitol (DTT) (Scheme 2).
Scheme 2.
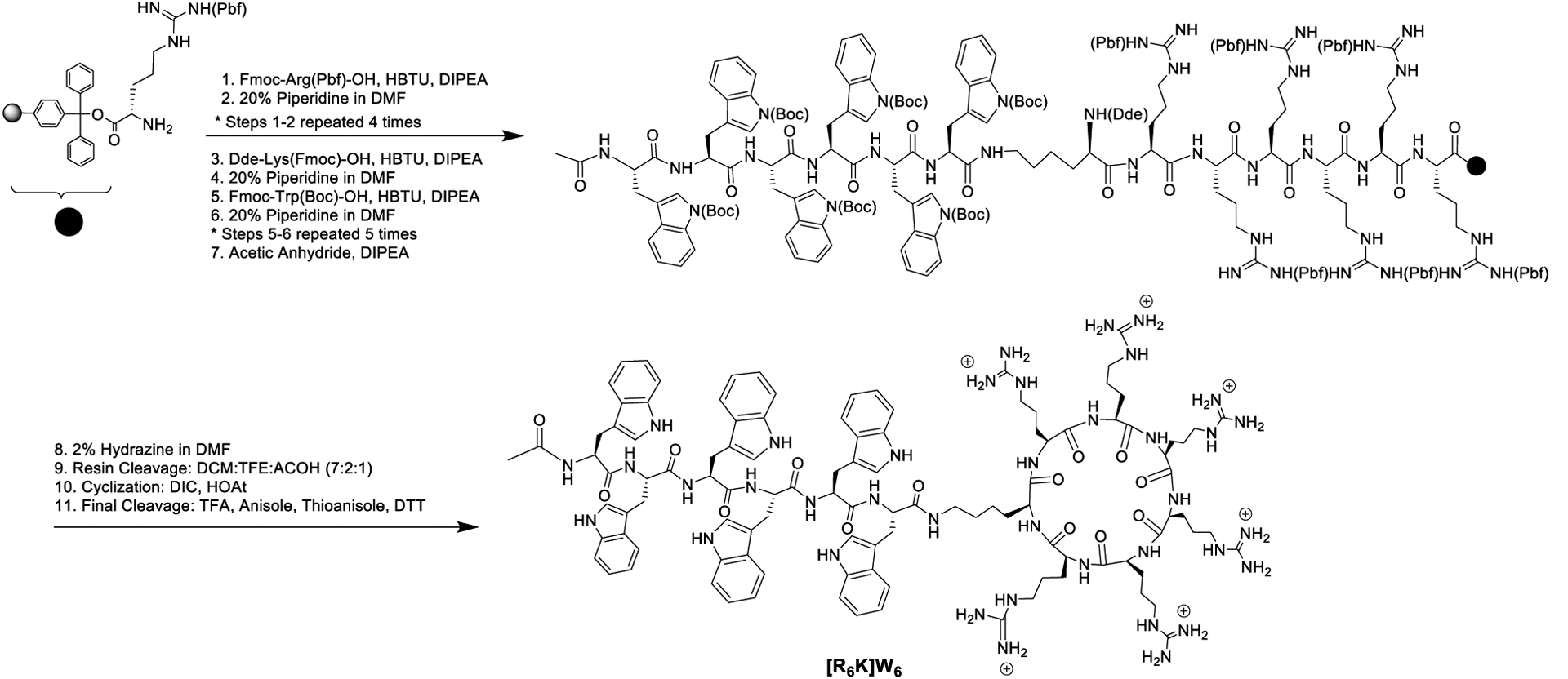
Synthesis of hybrid cyclic-linear peptide [R6K]W6.
Protein Kinase Inhibitory Activity
Four compounds, [WR]5, [WR]9, [R6K]W6, and [R5K]W7 were selected for initial screening against Abl1, c-Kit, c-Src, Cdk2/Cyclin A1, Csk, EGFR, Lck, mTor/Frap1, P38a/MAPK12, and PKCa at a single dose duplicate at a concentration of 5 μM (Table 1). The compounds did not significantly inhibit the kinase activity of c-Kit, EGFR, mTor/Frap1, and P38a/MAPK14 at this concentration. The slightly higher percentage than 100% enzyme activity in the screening assay is due to the experimental error. Significantly higher than 100% enzyme activity is presumed to be due to some activation of the enzyme in the presence of the peptide. However, our goal was to design and evaluate the kinase inhibitors. Therefore, these kinases were not selected for further studies. However, there was some significant inhibition of Abl1 (93.0%), c-Src (98.6%), Cdk2/Cyclin A1 (94.4%), Csk (92.3%), and PKCa (95.0%) in the presence of [WR]9 (Table 1).
Table 1.
Initial screening of peptides at a concentration of 5 μM (single dose duplicate).
| % Enzyme Activity (relative to DMSO controls) | ||||
|---|---|---|---|---|
| [WR]5 | [WR]9 | [R6K]W6 | [R5K]W7 | |
| Abl1 | 49.2 | 7.0 | 18.4 | 17.1 |
| c-Kit | 41.0 | 38.8 | 83.1 | 88.0 |
| c-Src | 6.2 | 1.4 | 141.2 | 95.1 |
| Cdk2/cyclin A1 | 33.9 | 5.6 | 5.9 | 7.0 |
| Csk | 11.3 | 7.7 | 91.8 | 69.7 |
| EGFR | 69.1 | 72.6 | 126.6 | 119.0 |
| Lck | 32.2 | 18.1 | 88.0 | 89.7 |
| mTOR/FRAP 1 | 146.9 | 133.3 | 152.9 | 111.9 |
| P38a/MAPK14 | 92.0 | 61.0 | 51.4 | 64.2 |
| PKCa | 8.9 | 5.0 | 6.8 | 15.8 |
The data suggested that [WR]9 is selective towards specific kinases. Thus, the protein kinase inhibitory activity of the cyclic peptides analogs, [WR]5, [WR]6, [WR]7, [WR]8, and [WR]9, and hybrid cyclic peptides, [R6K]W6, and [R5K]W7, were compared for the kinase inhibition activity of in 10 selected kinases shown in Table 2. Abl1, Atk1, Alk, Braf, Btk, Cdk2/Cyclin A1, Csk, c-Src, Lck, and PKCa were selected because of their importance in cancer research. There are also commercially available drugs against a number of these protein kinases. For example, Abl is a non-receptor kinase involved in the regulation of cell growth, differentiation, migration, and immune response.21 Ponatinib, bosutinib, and dasatinib are being used against Abl. Talfinavir, Ibrutinib, Ceritinib are small molecule inhibitors that are commonly used against Braf, BTK, and ALK, respectively.22 We also explored the selectivity of compounds towards the Src family of non-receptor kinases (e.g., c-Src, Lck).
Table 2.
Protein kinase inhibitory activity of peptides (IC50, μM)1.
| Kinase | [WR]5 | [WR]6 | [WR]7 | [WR]8 | [WR]9 | [R6K]W6 | [R5K]W7 |
|---|---|---|---|---|---|---|---|
| Abl1 | 0.79 | 0.91 | 0.35 | 0.41 | 0.35 | 1.02 | 1.49 |
| Akt1 | 1.31 | 1.26 | 0.76 | 0.42 | <0.25 | 1.25 | 1.36 |
| Alk | 0.41 | 0.28 | <0.25 | <0.25 | <0.25 | ND1 | ND |
| Braf | 3.72 | 1.45 | 0.77 | 0.68 | 0.48 | 1.04 | 2.34 |
| Btk | 0.57 | <0.25 | <0.25 | <0.25 | <0.25 | ND | ND |
| Cdk2/Cyclin A1 | 2.97 | 1.45 | 1.17 | 0.92 | 0.57 | 1.27 | 2.63 |
| Csk | ND | ND | ND | ND | 0.34 | ND | ND |
| c-Src | 0.81 | 0.57 | 0.35 | 0.33 | 0.21 | ND | ND |
| Lck | 3.07 | 2.74 | 2.91 | 2.76 | 2.52 | ND | ND |
| PKCa | 8.52 | 6.66 | 4.88 | 2.88 | 2.86 | 7.49 | 12.80 |
Average of triplicate;
ND = Not determined
As reported before18, [WR]5 is a c-Src kinase inhibitor. Among all the peptides, [WR]9 containing alternative tryptophan (W) and arginine (R) residues was shown to be the most potent c-Src kinase inhibitor with an IC50 value of 0.21 μM higher than [WR]5, [WR]6, [WR]7, [WR]8, with IC50 values of 0.81, 0.57, 0.35, and 0.33 μM, respectively. The order of inhibition was [WR]9 > [WR]8 > [WR]7 > [WR]6 > [WR]5. These data demonstrate that there is a direct correlation between the number of tryptophan and arginine residues in the cyclic peptides and kinase inhibition, suggesting that these residues contribute to kinase inhibitory activity.
To study the effect of the ring size on the kinase inhibitory activity, the IC50 of the synthesized cyclic peptides [WR]x (x=5–9) against other protein kinases were also obtained as shown in Table 2. A similar pattern was observed for Atk1, Alk, Braf, Cdk2/Cyclin A1, Lck, and PKCa. [WR]9 showed significant inhibitory activity against Abl, Cdk2/Cyclin A1, Csk, and PKCa and found to be consistently more potent than other peptides with smaller size ring. [WR]9 inhibited Abl1 kinase activity with an IC50 value of 0.35 μM, showing 2.2-fold higher inhibitory activity than [WR]5 (IC50= 0.79 μM). [WR]9 also inhibited PKCa kinase activity with an IC50 value of 2.86 μM, about 3-fold higher inhibitory activity than [WR]5 (IC50 =8.52 μM). Consistently, [WR]9 inhibited Cdk2/Cyclin A1 kinase activity with an IC50 of 0.57 μM that is 5.2 fold higher inhibitory activity when compared to [WR]5 (IC50 = 2.97 μM). The data suggest that [WR]9 is more potent than [WR]5 against kinases such as c-Src, Abl, and PKCa (Table 2).
It was found that [WR]9 was more selective towards Akt1, Alk, and Btk with IC50 values of less than 0.25 μM. The lowest concentration used in the assay was 0.25 μM at which [WR]9 showed at least 50% inhibition against these enzymes. Therefore, IC50 values of less than 0.25 μM were reported for some compounds. The peptide was 12 and 14 times more selective against c-Src (IC50 = 0.21 μM) versus Lck (IC50 = 2.52 μM) and PKCa (IC50 = 2.86 μM), respectively. A similar selectivity was found for all the other peptides with smaller ring size towards c-Src versus Lck and PKCa. However, [WR]9 and all the other compounds were more potent against Btk and Alk. None of the peptides exhibited any kinase inhibitory activity against GSK3a.
To determine the importance of the presence of hydrophobic and positively-charged residues on the ring, two other peptides with arginine residues on the ring and tryptophan residues outside the ring were evaluated for protein kinase activity and compared with [WR]9. It was found that [R6K]W6 and [R5K]W7 were generally less potent than [WR]9, [WR]8, [WR]7, and [WR]6 against Abl1, Alk, Braf, Cdk1/Cyclin A1, and PKCa. These data indicate that the presence of both arginine and tryptophan residues on the ring will provide a more effective strategy for generating more potent kinase inhibitors in this class.
Molecular Modeling
Kinase inhibitory assay revealed that [WR]9 exhibits the highest activity against different kinase families. Although the full structure for the majority of kinases tested here is not yet determined, the structure of two protein kinases c-Src (PDB ID: 2SRC) and Abl1 (PDB ID: 2FO0) have been extensively studied, and their high-resolution crystal structures are available in the protein data bank (PDB). Molecular modeling was performed using c-Src, which was solved at a higher resolution (1.5Å) than Ab11 (2.27Å).
We performed blind docking by placing a docking box on the c-Src with 20Å padding on every side to accommodate the binding of the largest peptides under consideration, anywhere on the receptor. We performed docking of the [WR]x cyclic peptide with AutoDock CrankPep, or ADCP in short24, 25 for x varying from 5 to 9. Each peptide was docked using 400 independent searches, yielding 77818, 70469, 65808, 64110, and 61131 docked poses for [WR][9,8,7,6,5], respectively. We clustered these solutions using an 80% threshold on residue contacts similarity and obtained 42, 223, 144, >300, and >300 clusters for [WR][9,8,7,6,5], respectively. The calculation of contacts similarity is identical to the calculation of the fraction of native contacts but substituting the native complex with a cluster seeding structure. The cutoff for this calculation was set to 5Å. The improved clustering observed for [WR]9 (i.e., poses clustering into fewer clusters) provides a first indication that the interaction energy landscape is better defined for this peptide, with fewer poses clearly outperforming the rest of the docked poses.
This clustering performed by ADCP is based on residue identity in the peptide (i.e., residue number) and therefore does not account for the cyclic symmetry created by the repeats of the [WR] pattern. We performed a second clustering with the DBSCAN26 density-based clustering algorithm, using the docked peptides’ geometric centers. The 42 clusters reported for [WR]9 coalesced to 4 clusters while we obtained 14, 10, 9, and 5 clusters for [WR][8,7,6,5], respectively. ADCP initially builds a linear extended 3D conformation of the peptide from its sequence. It then uses a potential to cyclize the peptide backbone during the docking. For the longer peptides (x = 7, 8, 9), some docked poses ended up wrapping the peptide around the extended loop connecting the SH2 and N-lobe. These 27 out of 144, 60 out of 233, and 28 out of 42 topologically impossible solutions for [WR]7, [WR]8, and [WR]9 respectively, were discarded. The remaining solutions were visually inspected and found to aggregate in 3 pockets depicted in Figure 3. The first pocket (pocket 1) is located between the SH2, SH3, and N-lobe domains, on the opposite side of the ATP binding site. This pocket held the docking solution with the best docking score for every peptide we docked (Figure 4).
Figure 3.
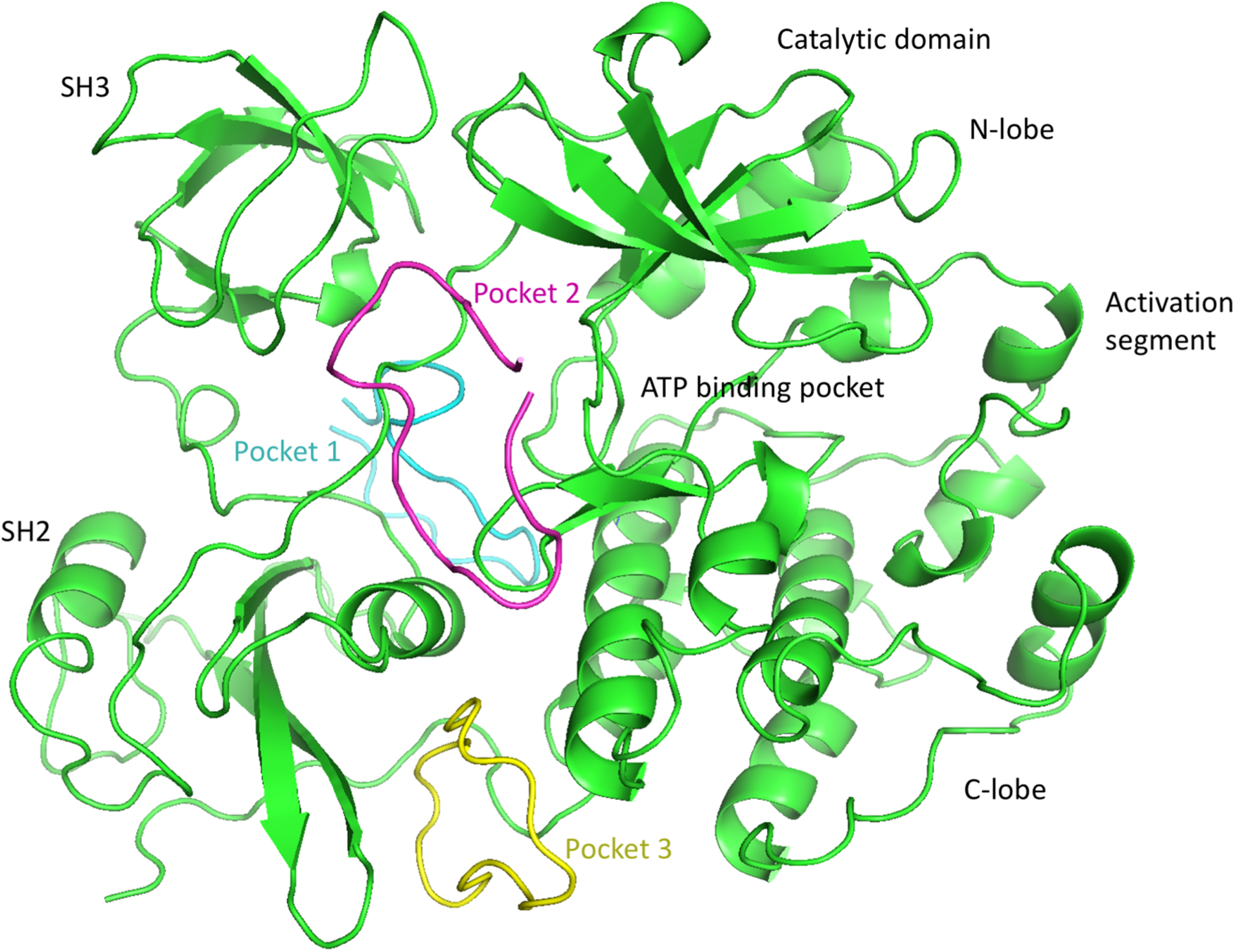
Three Binding sites identified by blind-docking cyclic [WR][5–9] peptides with AutoDock CrankPep. A cartoon representation of Protein kinases c-Src (PDB ID: 2SRC) is shown in green with its domains labeled. Representative of docked poses of [WR]9 for pockets 1, 2 and 3 are depicted as backbone tubes colored cyan, magenta and yellow respectively. All peptides from [WR]5 through [WR]9 docked primarily in these 3 pockets.
Figure 4.
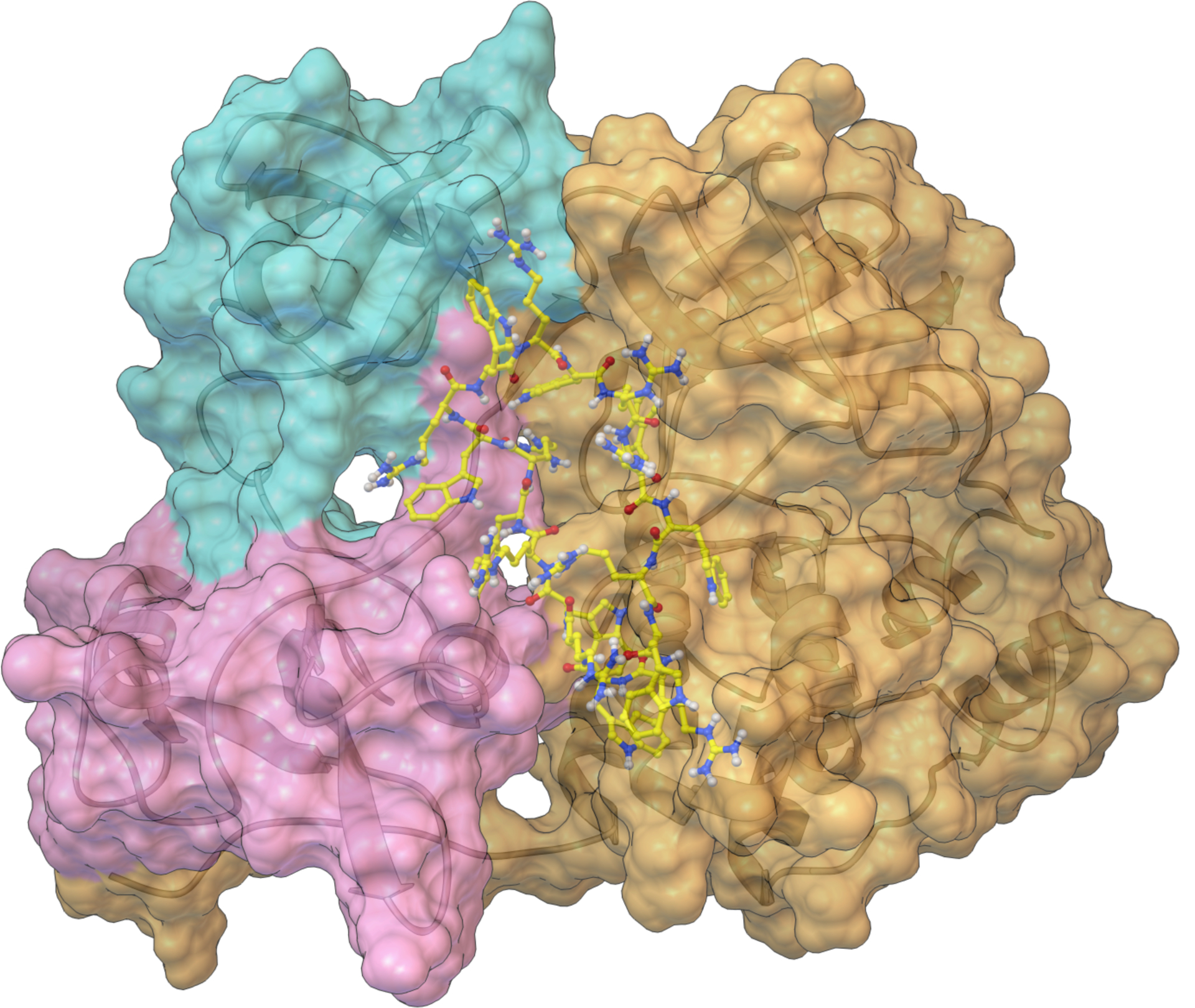
The interaction of [WR]9 with c-Src. SH3 SH2 and SH1 domains are colored cyan, magenta, and gold, respectively. This Figure was produced with the Python Molecular Viewer35 and the molecular surface computed with MSMS36.
The second putative pocket (pocket 2) is delineated by the same domains but located on the side of the ATP binding site. Finally, a third pocket (pocket 3) was identified between the SH2 and SH3 domains. We selected the docked pose with the best docking score as a representative for each one of the 3 pockets for [WR]5 and [WR]9 for further analysis. The docking scores of docked poses improve with peptide length. i.e. the longer peptides have lower negative docking scores. This is to be expected given the largely additive nature of the AutoDock27 scoring functions. Larger ligands generate larger scores as more atoms can interact with receptor atoms, adding to the score. In fact, the correlation between the docking score and the number of atoms for the best docked poses of [WR]5, [WR]6, [WR]7, [WR]8, and [WR]9 is 0.978, indicating that these molecules varying in size from 125 to 225 atoms achieve the same ligand efficiency. Hence, docking scores cannot be used directly to rank ligands for binding affinity. Moreover, the full-atoms docked poses produced by ADCP are rather crude and unoptimized models, built from the coarse representation used internally by ADCP. To improve the geometry of these docked complexes, allow the receptor to accommodate the bound peptides, and investigate the dynamic behavior of these complexes, we performed 100 nanoseconds of Molecular Dynamics (MD) simulations in implicit solvent saving snapshots every 2500 steps of 2 femtoseconds each (i.e., every 5 pico seconds). MD was performed for the representative docked pose of [WR]5 and [WR]9 in each of the 3 pockets, as well as with the c-Src apo structure (i.e., with no peptide bound and no ligand in the ATP binding site). We aligned the MD trajectories using mdlovofit28 and computed RMSD and RMSF values with this program, using threshold of 1Å to select the subset of atoms used for aligning the MD frames. The RMSD of the peptide in all 6 MD runs of the protein-peptide complexes remained low, indicating that none of the docked peptide dissociated from its receptor. The RMSF values of the receptor’s C-alpha atoms indicated that the presence of the peptide affects the plasticity of the receptor in various ways (Figure 5A). The blue line depicts the RMSF value of residues of the Apo s-Src structure, while the green and gold lines show the RMSF values of the protein with [WR]5 and [WR]9 bound, respectively. Dashed black vertical lines delineate domain boundaries and dashed silver lines identify two particular regions of interest in the catalytic domain, namely the lid of the ATP-binding pocket (LID) and activation loop (LOOP). Shifts between the blue and the other lines (green and gold) indicate a change of plasticity of the receptor. Figure 5B shows the difference in RMSF values between the Apo structure and the complexes formed with [WR]5 (green) and [WR]9 (gold).
Figure 5.
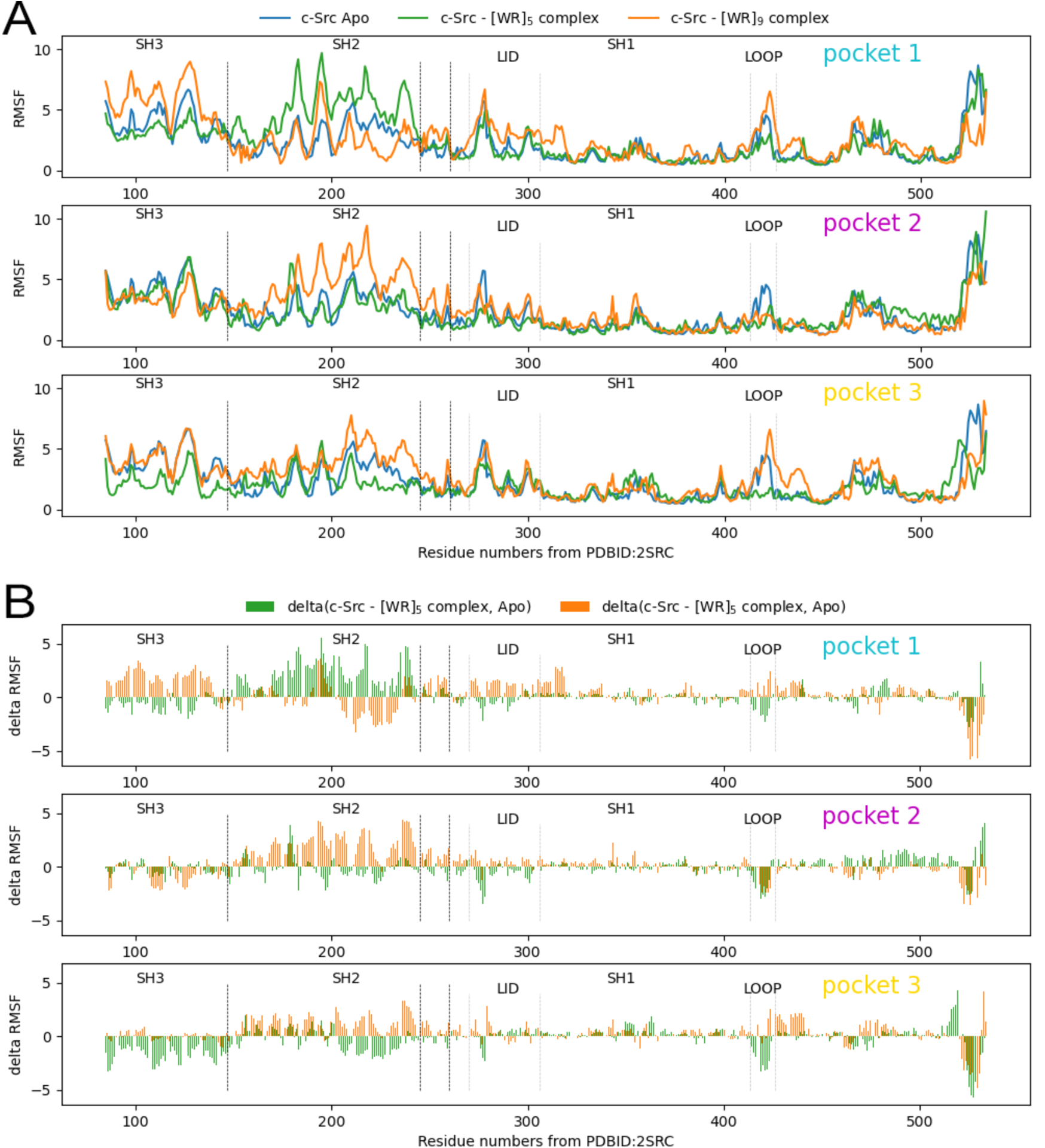
A) RMSF values for Cα atoms the over a 100ns MD simulation. The 3 plots correspond to results for the 3 putative binding pockets. In each plot, curves show the RMSF values: for the Apo structure (blue), for [WR]5 bound to c-Src (green), and [WR]9 bound to c-Src (gold) when bound in pocket 1 (top), pocket 2 (middle) and pocket 3 (bottom). Vertical dashed lines delineate the domains and regions of interest. B) RMSD delta values. The plots depict the difference in RMSF values between the Apo structure and the protein complexed with [WR]5 (green) and [WR]9 (gold).
The presence of the peptides affects the plasticity of the protein, in particular in the SH3 and SH2 domains. While the peptides bound in pockets 1 and 3 seem to have opposite effects on the flexibility of the activation loop, their presence in in pocket 2 consistently rigidify the activation loop (Fig 5B, pocket 2, LOOP). This provides a possible explanation of the inhibitory activity through allosteric modulation of the activation loop. Assuming that pocket 2 is the actual binding site for these peptides, we can also see that these two peptides seem to have different effects on the lid region of the ATP binding site (LID).To further investigate how the presence of the peptide in pocket 2 affects c-Src activation, we computed the volume of the ATP binding site using every 100th frame (i.e., every 50 pico-seconds) of the trajectories [Figure 6] using the POVME 29 software program. Figure 6 shows that the volume of the ATP-binding pocket is modulated pocket by the peptide binding in pocket 2. The volume of the ATP-binding pocket favored when [WR]9 binds in pocket 2 is closer to the volume of the Apo structure and might be less favorable to ATP binding leading to an increased inhibitory activity.
Figure 6.
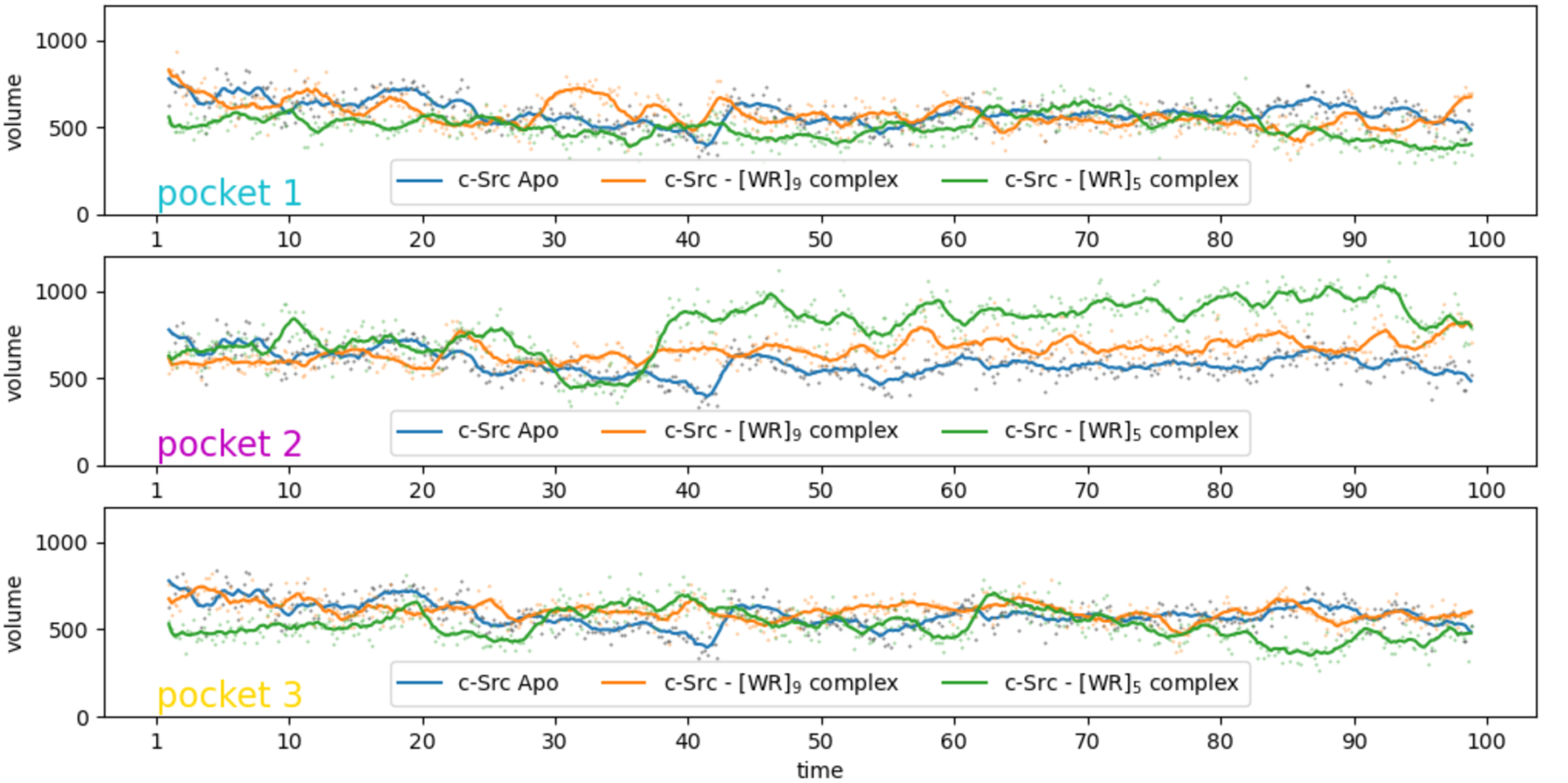
Volume of the ATP-binding pocket during the MD simulations of the c-Src Apo structure (blue) and with [WR]5 and [WR]9 bound to c-Src (green and gold, respectively) in the 3 identified binding pockets. The volume values are depicted as dots and the lines provide a moving average using a window of 10 points. The peptide bound in pocket 2 modulates the volume of the ATP binding pocket.
We hypothesize that pocket 2 is the actual binding site for the peptides and that the difference in affinity could be related to the way these peptides affect the plasticity of the ATP binding site pocket. To further explore this hypothesis, we computed average structures for the 6 complexes using the second half of the trajectories and evaluated the complexes with a newly developed random forest-based classifier, trained to segregate proper protein-peptide complexes from decoys [manuscript submitted]. This classifier relies on AutoDock4 energy terms, along with various peptide descriptors and buried surface areas, to assign a probability of correctness to a putative protein-peptide complex obtained by docking. The poses in pocket 2 all yielded a higher probability of being correct than the complexes where the peptide is located in pockets 1 and 3. This result further bolsters our confidence that pocket 2 is the actual binding site.
We have previously shown that the synthesized peptides were not cytotoxic to normal (LLCPK) cells at a concentration of 10 μM.19 These compounds have the potential to be used as protein kinase inhibitors. In the future, the co-crystallization of the most potent and selective compounds with the enzyme will be used to understand the exact mode of interaction. The crystal structure will enable us to test our hypothesis and further understand the structural requirements for designing the next generation of compounds with optimal protein kinase inhibitory activity.
CONCLUSION
The synthesized peptides [WR]x (x = 5–9) and hybrid cyclic-linear peptides [R6K]W6 and [R5K]W7 were evaluated for their inhibitory activity on different protein kinases. [WR]5 was previously shown to inhibit c-Src kinase activity. Herein, we have shown that [WR]9 is a more potent c-Src kinase inhibitor than other cyclic peptides with a smaller size in the order of [WR]9 > [WR]8 > [WR]7 > [WR]6 > [WR]5. A similar pattern was also observed for the inhibitory activity against Atk1, Alk, Braf, Cdk2/Cyclin A1, Lck, and PKCa (Table 3). [WR]9 was found to be more selective toward Btk and Alk and showed selectivity against c-Src versus Lck in the Src family of kinases. Docking studies revealed three putative binding pockets for the [WR][5–9] peptides. Additional modeling based on molecular dynamics of the docked complexes point toward pocket 2, located between the SH2, SH3, and N-lobe domains of c-Src, as the actual binding site and provides a plausible rationalization for the difference in binding affinity between [WR]5 and [WR]9. Additional experimental and computational studies will shed more light on the mode of inhibition and interactions of positively charged and hydrophobic amino acids of the cyclic peptide with c-Src, Btk, and Alk.
Table 3.
Substrates used for protein kinase assays.
| Kinases | Kinase (Invitrogen) Cat# | Kinase Conc. in Reaction (nM) | Substrate | Substrate Conc. In Reaction |
|---|---|---|---|---|
| Abl1 | PR4348B | 0.25 | ABLtide | 20 μM |
| Csk | PR3080B | 1.5 | pEY | 0.2 mg/ml |
| c-Src | P3044 | 0.6 | pEY | 0.2 mg/ml |
| Akt1 | PR3878D | 8 | Crosstide | 20 μM |
| Gsk3a | PR9188A | 3 | Phospho- | 20 μM |
| Cdk2/Cyclin A1 | C29–10BG | 15 | Histone | 20 μM |
| PKCa | PR1455C | 0.5 | Histone | 20 μM |
| Braf | PR6995A | 30 | MEK1 | 5 μM |
| Lck | P3043 | 8 | pEY + | 0.2 mg/ml |
| Btk | PV3363 | 8 | pEY | 0.2 mg/ml |
| Alk | PV3867 | 1.5 | pEY | 0.2 mg/ml |
| c-Kit | PV3589 | 200 | pEY + | 0.2 mg/ml |
| EGFR | PV3872 | 3 | pEY + | 0.2 mg/ml |
| mTOR/FR AP1 | PV4754 | 300 | PEBP1/E | 10 μM |
| P38a/MAP K14 | PV3304 | 20 | MBP | 20 μM |
EXPERIMENTAL SECTION
Materials
All Fmoc-protected amino acids and resins were purchased from AAPPTEC (Louisville, KY, USA). All the other chemicals and reagents were purchased from MilliporeSigma (Milwaukee, WI, USA). The chemical structures of the final purified peptides were confirmed by high-resolution MALDI-TOF (GT 0264) from Bruker Inc (Fremont, CA, USA). Final peptides were purified by a reversed-phase HPLC (LC-20AP) from Shimadzu (Canby, OR, USA) using a gradient system of water and acetonitrile and a reversed-phase preparative column (Waters, XBridge BEH130 Prep C18).
Solid Phase Peptide Synthesis.
The synthesis of the cyclic peptides was accomplished according to the previously reported procedure by us.19 In brief, Fmoc-Arg(Pbf)-OH and Fmoc-Trp(Boc)-OH were used as building block amino acids in the synthesis. The linear protected peptides were synthesized on trityl resin using 0.30 mmol scale. The square bracket represents cyclic peptide, and parentheses represent linear peptide. The tryptophan preloaded resin H-Trp(Boc)-2-chlorotrityl was swelled in N,N-dimethylformamide (DMF) under dry nitrogen for 30 min. The solvent was filtered. The Fmoc protected amino acid was coupled to the N-terminal with the use of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), N,N-diisopropylethylamine (DIPEA), and DMF mixing for 1 h under inert nitrogen gas. The reaction solution was filtered off after the coupling reaction was completed. The resin was washed twice with 15 mL of DMF for 5 min. Fmoc deprotection was conducted by using piperidine in DMF (20% v/v, 10 mL, 2 × 15 min). The reaction solution was filtered off after the deprotection. The resin was washed twice with DMF (15 mL, 2 × 5 min). The remaining amino acids were coupled using the same protocol. Fmoc deprotection was performed on the final amino acid by using piperidine in DMF (20% v/v, 10 mL, 2 × 15 min). The resin was washed twice with DMF (15 mL, 2 × 5 min) and dried using methanol (15 mL, 5 min). To cleave the resin, a freshly prepared cocktail of dichloromethane:trifluoroethanol:acetic acid (DCM:TFE:AcOH; 35 mL:10 mL: 5 mL) was used. The cleavage cocktail was added to the resin and mixed for 3 h. The solvents were evaporated under vacuum using hexane (2 × 20 mL) and DCM (2 ×15 mL) to remove the acetic acid from the mixture. The crude material was solidified as a white fluffy solid. MALDI analysis was used to confirm the molecular weight of the linear side-chain protected peptides. The protected linear peptides were cyclized in the presence of anhydrous DMF (100 mL), anhydrous DCM (25 mL), N,N’-diisopropylcarbodiimide (DIC; 188 μL), 1-hydroxy-7-azabenzotriazole (HOAt; 122.5 mg). The cyclization reaction occurred overnight under dry nitrogen. The monitoring of the reaction was done by MALDI analysis was to detect the completion of cyclization by checking the molecular weight of the cyclized peptide after deprotection of side-chain protecting groups. The solvents were removed under low pressure. A total cleavage cocktail was then prepared using TFA:thioanisole:1,2 ethanedithiol:anisole (6300 μL:350 μL:210 μl:140 μL). The mixture was stirred for 3 h. The cold diethyl ether was added to the reaction mixture to precipitate the crude peptides. MALDI analysis was used to confirm the M. Wt of the cyclic peptides without any protecting groups as described previously.19 All the peptides were purified using reversed-phase HPLC and lyophilized. The purity of peptides was >95%, according to the HPLC analysis.
The synthesis of hybrid cyclic-linear peptides, [R6K]W6 and [R5K]W7 was accomplished according to the previously reported procedure.20 Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, and Dde-Lys(Fmoc)-OH, were assembled on H-Arg(Pbf)-2-chlorotrityl resin. After conjugation of lysine residue containing Dde, the Fmoc of the lysine was removed in the presence of piperidine in DMF (20% v/v). The peptide synthesis was continued by the coupling of Fmoc-Trp(Boc)-OH on the side chain of lysine. After Fmoc deprotection on the last amino acid in the peptide chain, the free amino group was capped in the presence of acetic anhydride (3 equiv.) and DIPEA (6 equiv.). The removal of Dde group of lysine residue was accomplished in the presence of hydrazine in DMF (2% v/v). After washing, the resin cleavage was accomplished in the presence of DCM:TFE:AcOH for 3 h. The solvent was removed under vacuum. The crude peptide was used for cyclization. The cyclization was accomplished in the presence of DIC, and HOAt in anhydrous DMF and DCM. Cyclization was confirmed by MALDI. The solvent was removed under a rotatory evaporator and the final cleavage was performed by using a freshly prepared cleavage cocktail, TFA, anisole, and thioanisole for 3 h to afford the crude peptides that were purified by HPLC. MALDI-TOF data for [R6K]W6 has been previously reported and matched.20
Protein Kinase Assays.
The compounds were tested against c-Src, Abl1, c-Kit, Csk, EGFR, mTOR/FRAP1, P38a/MAPK14, PKCaAkt1, Alk, Braf, Btk, Cdk2/Cyclin A1, Lck, GSK3a, and PKC, in duplicate by Reaction Biology Corporation. Compounds were tested in a 5-dose IC50 mode with 3-fold serial dilution starting at 20 μM or single-dose triplicate at 5 μM. Staurosporine and GW5074 were used as the control compounds and tested in a 10-dose IC50 starting at 20 μM with a 4-fold or 3-fold serial dilution, respectively. All protein kinase reactions were performed using 200 μM of ATP. The peptides were first pre-incubated with the kinase and substrate (Table 3) mixture for about 20 min. ATP was then added to initiate the reaction. The reaction was continued for 2 h at room temperature. Curve fittings were conducted when the activities at the maximum concentration of the peptides were less than 65%. The detailed experimental procedure has been reported by us previously.13 An IC50 value < 0.04 nM or >1 μM is estimated based on the best curve fitting available. The substrates for kinases are depicted in Table 3.
Molecular Modeling
Docking.
Blind docking of the cyclic peptides was performed using ADCP v 1.0. We relied on the AGFRgui software program30 to prepare the c-Src (PDB ID: 2SRC), define the docking box as the bounding box of the receptor with 20Å padding, and computing the AutoDock affinity maps. We performed automated docking of the [WR](5–9) peptides into this docking box, performing 400 independent searches, allowing each search to consume 54 million evaluations of the scoring function in order to have at least 3 million evaluations per amino acids for the longest peptide. The results of each docking were clustered using a clustering cutoff of 80% identity in residue contacts calculated in a way similar to native contacts. The resulting docking poses were further clustered by applying the DBSCAN density-based clustering algorithm to the geometric center of the peptides to account for the symmetries created by the cyclic repeats in the peptides. The DBSCAN epsilon parameter was set to 2.0 and the minimum size of a cluster was set to 2.
Molecular Dynamics.
We prepared the receptor and peptide, parametrized them for Amber forcefield, and performed molecular dynamics in explicit solvent (MD) using PDBfixer31 and OpenMM32. The solvent was added using the OpenMM Modeller. We performed an initial minimization of 500 steps of 2 femtoseconds each by 100 nanoseconds of simulation with explicit solvent.
Analysis.
The solvent was stripped from the trajectories using catDCD.33,34 We used MDlovofit27 with the -mapfrac option to get the RMSD of the alignment of a subset of the atoms, as a function of the size of this subset. For each trajectory, we picked the fraction for which the subset of atoms used for the alignment yields and RMSD closest to 1.0 and >= 1.0. These values are 0.59 for the apo receptor; 0.37, 0.61, and 0.5 for [WR]5 bound in pockets 1, 2, and 3; and 0.5, 0.57, 0.36 for [WR]9 bound in pockets 1, 2 and 3, respectively. We aligned the trajectories frames using these fractional values and computed RMSD and RMSF values with MDlovofit28.We used POVME29 to calculate the volume of the ATP binding pocket over the MD trajectory by placing spheres of radius 6.0Å on each atom of the ANP molecule in the c-Src crystal structure (PDBID:2SRC) and using them as inclusion spheres.
Acknowledgments
The authors also acknowledge the fellowship support to K. Z. from Jazan University, Saudi Arabia, and Chapman University School of Pharmacy core facility. The Table of Content graphics and Figures 2 and 4 were produced with the Python Molecular Viewer35 and the molecular surface computed with MSMS36.
Funding
The authors acknowledge financial support from the Chapman University School, Irvine, CA and Kay Family Foundation. The docking and modeling work performed by MSF was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM096888 to M.F.S.
Footnotes
Data and Software Availability
Input data for the work presented in this work and relevant output files here are available on GitHub at https://github.com/sannerlab/WR5-9paper2021 under the LGPL v2.1 OpenSource license.
Specifically, we provide SMILE strings for all the peptides along with IC50 values for the tyrosine kinases considered in this work. The repository also provides the input data for docking. i.e. the receptor PDBQT, along with the receptor maps generated for this receptor. It also provides the set of translational and the constrains file used as input by ADCP to perform docking. As the docking trajectories from ADCP runs were clustered with a modified version of the clusterADCP.py available in the ADFRsuite v1.0, the modified version of this file is provided in the scripts folder, along with pose clustering script (clusterCG.py and associate DBSCAN.py) using the DBSCAN algorithm to cluster poses based on their center of gravity. We also provide the input files for the molecular dynamics simulations (i.e. .parm7, .rst7 and corresponding .pdb files) for the [WR]5 and [WR]9 peptides docked in pockets 1, 2 and 3. These docked poses were used to run the MD simulation from which RMSF values reported in Figure 5 were obtained.
For the docking calculation we used the ADCP and AGFRgui software programs from the ADFRsuite v1.0 freely available for download from https://ccsb.scripps.edu/adfr. The box used for generating AutoDock affinity maps with AGFRgui and dock the peptides with ADCP was centered on (20.279, 34.208, 65.721) and its size was set to (278,292,272) grid points with a grid spacing of 0.374Å. These maps are available in the GitHub repository for convenience.
The molecular dynamics simulations were performed using the OpenMM software package from a miniconda3 installation performed following the steps provided at https://docs.conda.io.projects/vonda/en/latest/uesr-guide. The input files available in the GitHub repository for convenience.
The MDlovofit software (v 16.264) was downloaded from http://leandro.iqm.unicamp.br/mdlovofit/download.shtml.
The POVME software v3 was downloaded from https://github.com/POVME/POVME. The input files for POVME are available in the GitHub repository for convenience.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Thomas SM; Brugge JS Cellular Functions Regulated By Src Family Kinases. Annu. Rev. Cell Dev. Biol 1997, 13, 513–609. [DOI] [PubMed] [Google Scholar]
- 2.Warmuth M; Damoiseaux R; Liu Y; Fabbro D; Gray N SRC Family Kinases: Potential Targets For The Treatment Of Human Cancer And Leukemia. Curr. Pharm. Des 2003, 9, 2043–2059. [DOI] [PubMed] [Google Scholar]
- 3.Machida K; Mayer BJ The SH2 Domain: Versatile Signaling Module And Pharmaceutical Target. Biochim. Biophys. Acta 2005, 1747, 1–25. [DOI] [PubMed] [Google Scholar]
- 4.Shukla D; Meng Y; Roux B; Pande VS Activation Pathway Of Src Kinase Reveals Intermediate States As Targets For Drug Design. Nat. Commun 2014, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blume-Jensen P; Hunter T Oncogenic Kinase Signalling. Nat. 2001, 411, 355–365. [DOI] [PubMed] [Google Scholar]
- 6.Toledo LM; Lydon NB; Elbaum D The Structure-Based Design Of ATP-Site Directed Protein Kinase Inhibitors. Curr. Med. Chem 1999, 6, 775–805. [PubMed] [Google Scholar]
- 7.Schenone S; Bruno O; Ranise A; Bondavalli F; Brullo C; Fossa P; Mosti L; Menozzi G; Carraro F; Naldini A; Bernini C; Manetti F; Botta M New Pyrazolo[3,4-d]Pyrimidines Endowed With A431 Antiproliferative Activity And Inhibitory Properties Of Src Phosphorylation. Bioorg. Med. Chem. Lett 2004, 14, 2511–2517. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J; Yang PL; Gray NS Targeting Cancer With Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- 9.Parang K; Till JH; Ablooglu AJ; Kohanski RA; Hubbard SR; Cole PA Mechanism-Based Design Of A Protein Kinase Inhibitor. Nat. Struct. Biol 2001, 8, 37–41. [DOI] [PubMed] [Google Scholar]
- 10.Lee S; Lin X; Nam NH; Parang K; Sun G Determination Of The Substrate-Docking Site Of Protein Tyrosine Kinase C-Terminal Src Kinase. Proc. Natl. Acad. Sci 2003, 100, 14707–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S; Ayrapetov MK; Kemble DJ; Parang K; Sun G Docking-Based Substrate Recognition By The Catalytic Domain Of A Protein Tyrosine Kinase, C-Terminal Src Kinase (Csk). J. Biol. Chem 2006, 281, 8183–8189. [DOI] [PubMed] [Google Scholar]
- 12.Kumar A; Ye G; Wang Y; Lin X; Sun G; Parang K Synthesis And Structure-Activity Relationships Of Linear And Conformationally Constrained Peptide Analogues Of CIYKYY As Src Tyrosine Kinase Inhibitors. J. Med. Chem 2006, 49, 3395–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chhikara BS; Shraf S; Mozaffari S; St. Jeans N; Mandal D; Tiwari RK; Ul-Haq Z; Parang K Phenylpyrazalopyrimidines As Tyrosine Kinase Inhibitors: Synthesis, Antiproliferative Activity, And Molecular Simulations. Molecules 2020, 25, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiwari RK; Brown A; Sadeghiani N; Shirazi AN; Bolton J; Tse A; Verkhivker G; Parang K; Sun G Design, Synthesis, And Evaluation Of Dasatinib-Amino Acid And Dasatinib-Fatty Acid Conjugates As Protein Tyrosine Kinase Inhibitors. Chem. Med. Chem 2017, 12, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nam NH; Ye G; Sun G; Parang K Conformationally Constrained Peptide Analogues Of Ptyr-Glu-Glu-Ile As Inhibitors Of The Src SH2 Domain Binding. J. Med. Chem 2004, 47, 3131–3141. [DOI] [PubMed] [Google Scholar]
- 16.Lattig-Tunnemann G; Prinz M; Hoffmann D; Behlke J; Palm-Apergi C; Morano I; Herce HD; Cardoso MC Backbone Rigidity And Static Presentation Of Guanidinium Groups Increases Cellular Uptake Of Arginine-Rich Cell-Penetrating Peptides. Nat. Commun 2011, 2, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joo SH Cyclic Peptides As Therapeutic Agents And Biochemical Tools. Biomol. Ther 2012,20, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasrolahi Shirazi A; Tiwari RK; Brown A; Mandal D; Sun G; Parang K Cyclic Peptides Containing Tryptophan And Arginine As Src Kinase Inhibitors. Bioorg. Med. Chem. Lett 2013, 23, 3230–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanna SE; Mozaffari S; Tiwari RK; Parang K Comparative Molecular Transporter Efficiency Of Cyclic Peptides Containing Tryptophan And Arginine Residues. ACS. Omega 2018, 3, 16281–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mozaffari S; Bousoik E; Amirrad F; Lamboy R; Coyle M; Hall R; Alasmari A; Mahdipoor P; Parang K; Montazeri Aliabadi H Amphiphilic Peptides For Efficient siRNA Delivery. Polymers 2019, 11. 10.3390/polym11040703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wessler S; Backert S Abl Family Of Tyrosine Kinases And Microbial Pathogenesis. Int. Rev. Cell. Mol. Biol 2011, 286, 271–300. [DOI] [PubMed] [Google Scholar]
- 22.Redaelli S; Piazza R; Rostagno R; Magistroni V; Perini P; Marega M; Gambacorti-Passerini C; Boschelli F Activity Of Bosutinib, Dasatinib, And Nilotinib Against 18 Imatinib-Resistant BCR/ABL Mutants. J. Clin. Oncol 2009, 27, 469–471. [DOI] [PubMed] [Google Scholar]
- 23.Tiwari R; Brown A; Narramaneni S; Sun G; Parang K Synthesis and evaluation of conformationally constrained peptide analogues as the Src SH3 domain binding ligands. Biochimie. 2010, 92, 1153–1163. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Yuqi, Sanner Michel F.. Autodock Crankpep: Combining Folding And Docking To Predict Protein–Peptide Complexes. Bioinformatics. Vol. 35, Issue 24, 2019, 5121–5127, 10.1093/bioinformatics/btz459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Yuqi, Sanner Michel F. Docking Flexible Cyclic Peptides With Autodock Crankpep. J. Chem. Theory. Comput 2019, 15, 5161–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schubert E, Sander J, Ester M, Kriegel HP, Xu X. DBSCAN Revisited, Revisited: Why And How You Should (Still) Use DBSCAN. ACM. Trans. on Data. Sys. (TODS) 2017, 42,1–21. [Google Scholar]
- 27.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS and Olson AJ Autodock4 And Autodocktools4: Automated Docking With Selective Receptor Flexibility. J. Comput. Chem, 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martínez L Automatic Identification Of Mobile And Rigid Substructures In Molecular Dynamics Simulations And Fractional Structural Fluctuation Analysis. PloS one, 2015,10, 10.1371/journal.pone.0119264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durrant JD, Votapka L, Sørensen J, Amaro RE. POVME 2.0: An Enhanced Tool For Determining Pocket Shape And Volume Characteristics. J. Chem. Theory Comput. 2014; 10:5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Forli S, Omelchenko A, Sanner MF AutoGridFR: Improvements on AutoDock Affinity Maps and Associated Software Tools. J. Comput. Chem 2019, 40, 2882–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peter Eastman, PDBFixer, https://github.com/pandegroup/pdbfixer (2013).
- 32.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD and Wiewiora RP, Openmm 7: Rapid Development Of High Performance Algorithms For Molecular Dynamics. PLoS Comput. Bio, 2017. 13, 10.1371/journal.pcbi.1005659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godwin R, and Salsbury FR. Catdcd. Interface Code 2015.
- 34.Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 35.Sanner MF Python: A Programming Language for Software Integration and Development. J. Mol. Graphics Mod, 1999, 17, 57–61. [PubMed] [Google Scholar]
- 36.Sanner MF, Spehner J-C, and Olson AJ Reduced Surface: An Efficient Way To Compute Molecular Surfaces. Biopolymers, 1996, 38, 305–320. [DOI] [PubMed] [Google Scholar]