

Abstract
The severe acute respiratory syndrome coronavirus (SARS‐CoV) emerged in humans in 2002. Despite reports showing Chiroptera as the original animal reservoir of SARS‐CoV, many argue that Carnivora‐hosted viruses are the most likely origin. The emergence of the Middle East respiratory syndrome coronavirus (MERS‐CoV) in 2012 also involves Chiroptera‐hosted lineages. However, factors such as the lack of comprehensive phylogenies hamper our understanding of host shifts once MERS‐CoV emerged in humans and Artiodactyla. Since 2019, the origin of SARS‐CoV‐2, causative agent of coronavirus disease 2019 (COVID‐19), added to this episodic history of zoonotic transmission events. Here we introduce a phylogenetic analysis of 2006 unique and complete genomes of different lineages of Orthocoronavirinae. We used gene annotations to align orthologous sequences for total evidence analysis under the parsimony optimality criterion. Deltacoronavirus and Gammacoronavirus were set as outgroups to understand spillovers of Alphacoronavirus and Betacoronavirus among ten orders of animals. We corroborated that Chiroptera‐hosted viruses are the sister group of SARS‐CoV, SARS‐CoV‐2 and MERS‐related viruses. Other zoonotic events were qualified and quantified to provide a comprehensive picture of the risk of coronavirus emergence among humans. Finally, we used a 250 SARS‐CoV‐2 genomes dataset to elucidate the phylogenetic relationship between SARS‐CoV‐2 and Chiroptera‐hosted coronaviruses.
Introduction
Coronaviruses (CoVs) are members of the subfamily Orthocoronavirinae (formally known as Coronavirinae) in the family Coronaviridae, order Nidovirales, following the current classification of the International Committee on Taxonomy of Viruses (ICTV). The virion of a coronavirus is relatively large among viruses and is enveloped, spherical and c.120 nm in diameter. Moreover, coronavirus genomes are the longest of all characterized RNA viruses. Coronaviruses are positive‐sense single‐stranded RNA viruses with monopartite and linear genomes of 27–32 kb in length (Woo et al., 2010) and complex gene expression (Luytjes, 1995; Irigoyen et al., 2016).
Orthocoronavirinae consists of four genera. Gammacoronavirus (GammaCoVs) and Deltacoronavirus (DeltaCoVs) are coronaviruses that originated from Aves (birds), with only a few known lineages that infect mammals (Woo et al., 2014; Duraes‐Carvalho et al., 2015). Alphacoronavirus (AlphaCoVs) and Betacoronavirus (BetaCovS) originated from Chiroptera (bats) and infect different mammals, including humans (Woo et al., 2012). Coronavirus infections in domestic animals can lead to significant economic losses (Li et al., 2007; Boileau and Kapil, 2010; Hansa et al., 2012; Mandelik et al., 2018). The episodic emergence of Alphacoronavirus and Betacoronavirus in the human population are even greater concerns.
Coronaviruses cause respiratory or enteric diseases in most cases. Neurological illness or hepatitis (Lai and Cavanagh, 1997) occur less frequently. Currently, the United States Centers for Disease Control (CDC) website (Centers for Disease Control and Prevention, 2020) lists seven common human coronaviruses (HCoVs): two Alphacoronavirus (HCoV‐229E and HCoV‐NL63) and five Betacoronavirus (HCoV‐OC43, HCoV‐HKU1, SARS‐CoV, MERS‐CoV and SARS‐COV‐2). We add the human enteric coronavirus 4408 (HECV‐4408) to that list. HECV‐4408 was first isolated from a child with acute gastroenteritis. HECV‐4408 was reported to be antigenically and genetically more closely related to bovine coronavirus (BCoV) than to HCoV‐OC43 (Zhang et al., 1994). The HCoVs mentioned above cause infections in infants, young children and elderly individuals (Su et al., 2016).
Despite many disease phenotypes, coronaviruses were not deemed highly pathogenic to humans until the outbreak of the severe acute respiratory syndrome (SARS) caused by SARS‐CoV, which emerged in humans in 2002 in Guangdong province, China (Ksiazek et al., 2003; Zhong et al., 2003). The dangers of emerging coronaviruses infections in humans were made even more evident by the recent outbreaks of coronavirus disease including: (i) the Middle East respiratory syndrome (MERS) caused by MERS‐CoV, which emerged in humans and camels in the Middle East in 2012 (Zumla et al., 2015) and (ii) the pandemic coronavirus disease 2019 (COVID‐19) caused by SARS‐CoV‐2, which was recognized in humans in Hunan province, China, in 2019 (World Health Organization, 2020g) but may have emerged earlier in rural Yunnan, China.
A recent literature review of the zoonotic origins of HCoVs (Ye et al., 2020) describes that the fundamental hosts of HCoVs can be Rodentia (for HCoV‐OC43 and HCoV‐HKU1) or Chiroptera (for HCoV‐NL63, HCoV‐229E, SARS‐CoV, MERS‐CoV and SARS‐CoV‐2). According to Ye et al. (2020), data on intermediate hosts of HCoV‐NL63 and HCoV‐HKU1 are absent. Furthermore, Ye et al. (2020) also point out that there is an open debate about the existence of intermediate hosts of HCoV‐229E and SARS‐CoV‐2. In this paper, we challenge all assumptions on the origins of coronaviruses using large‐scale phylogenetic analysis based on as much publicly available information as possible. Although we test the origins of different viruses of the subfamily Orthocoronavirinae, we focus mainly on SARS‐CoV, MERS‐CoV and SARS‐CoV‐2, which cause severe disease in humans.
Severe acute respiratory syndrome coronavirus (SARS‐CoV)
In 2002–03, SARS‐CoV infected 8098 people and caused 774 deaths. The first case of SARS‐CoV was confirmed in November 2002 and the last case was confirmed in May 2003. However, travel restrictions associated with the disease were not released until June and July 2003 (Centers for Disease Control and Prevention, 2005; World Health Organization, 2015b).
SARS‐CoV evolved from Chiroptera hosts and spread to humans hosts. Small carnivores such as Paguma larvata (commonly known as masked palm civet or gem‐faced civet) were infected post‐human infection and are thus irrelevant to the lineage of the virus that spread, human to human, and around the world including Asia, Europe, South Africa and North America (Janies et al., 2008). We publicized this result in 2008 (Caldwell, 2008), but the idea of intermediate hosts between bats and humans persists in some public health discourse (Roos, 2004; World Health Organization, 2020f) but not universally (Yip et al., 2009; Bolles et al., 2011). As a consequence of such discourse, tens of thousands of small carnivores were culled in Guangdong in a futile attempt to contain SARS‐CoV (Normile, 2004).
Genomic sequences of a Chiroptera‐hosted virus that shared common ancestry with human‐hosted SARS‐CoV clade were published in 2016 (e.g. SARS‐like coronavirus WIV16; NCBI GenBank accession number KT444582, collected 21 July 2013) (Yang et al., 2016). These data indicate that close relatives of SARS‐CoV continued to circulate in nature in Chiroptera long after the SARS‐CoV lineage was considered extinct in humans. Others have found additional SARS‐like viruses in nature (Hu et al., 2017) and verified their potential for human infection in the lab (Menachery et al., 2015).
Middle East respiratory syndrome coronavirus (MERS‐CoV)
The Middle East respiratory syndrome coronavirus (MERS‐CoV) was discovered in 2012 in the Middle East. As of November 2019, MERS‐CoV infected 2494 people and resulted in 858 associated deaths (World Health Organization, 2020c). Since the discovery of MERS‐CoV, there has been careful tracking and reporting of human cases by the World Health Organization (World Health Organization, 2020c). MERS‐CoV is a novel Betacoronavirus closely related to the Neoromicia capensis coronavirus (NeoCoV), a bat coronavirus (see GenBank accession number KC869678) that was discovered in the South African bat species Neoromicia capensis (commonly known as Cape serotine bat; see GenBank accession numbers KJ756000 and KJ756001) (Corman et al., 2014). MERS‐CoV infects humans and Camelus dromedarius (commonly known as dromedary, Somali camel or Arabian camel). The zoonosis between bats, humans and camels is complex. In 2015, MERS‐CoV spread from the Middle East to South Korea where it led to 186 cases (185 in South Korea, one in China) and 38 deaths (World Health Organization, 2015a).
Thus, it is clear that MERS‐CoV spread among humans in areas where camels are not husbanded. There were few cases of travellers carrying MERS‐CoV outside the Middle East and South Korea. However, no sustained transmission occurred in these traveller cases in Europe, China, Southeast Asia or the Americas. Camels are seropositive for antibodies in response to a MERS‐CoV‐like virus in the Canary Islands (Gutiérrez et al., 2015), Nigeria, Tunisia and Ethiopia (Reusken et al., 2014). Recently, humans who are handlers of Artiodactyla, exclusive of camels, in Kenya were found to show seropositivity to MERS‐CoV (Liljander et al., 2016).
There was an early bat–human MERS‐CoV and a recent bat–hedgehog transmission, but these events are peripheral to the main epidemic lineage that led to the MERS‐CoV outbreak in 2012. In 2018, there also was a travel case of MERS‐CoV from a South Korean man who returned from Kuwait (World Health Organization, 2020b). In 2019–20, MERS‐CoV still occurred in humans in Saudi Arabia (World Health Organization, 2020d). Thus, unlike SARS‐CoV, MERS‐CoV still circulates in humans and may persist in camels and other Artiodactyla.
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2)
In November 2019, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), which causes COVID‐19, emerged in the human population in Wuhan, Hubei Province, China. As of end March 2021, cases of COVID‐19 exceeded 127 million and deaths exceeded 2.78 million worldwide according to https://covid19.who.int/, accessed on 30/3/2021 (World Health Organization, 2021).
Close relatives of SARS‐CoV‐2 virus were circulating in Chiroptera in 2013 (e.g. hCoV‐19/bat/Yunnan/RaTG13/2013 collected in Pu'er, Yunnan, China; GISAID's EpiCoV accession number EPI_ISL_402131). Sequence data from the RaTG13 SARS‐like coronavirus were submitted to a public database 6.5 years post‐isolation on 24 January 2020.
Pu'er is ˜2000 km southwest of Wuhan. Pu'er is nestled in the region where China has borders with Vietnam, Laos, Cambodia, Myanmar, and is not far from Thailand, India and Bangladesh. There are also reports that SARS‐like viruses have infecting rural human populations in 2015 in Jinning County, Yunnan province, China (Wang et al., 2018). On 11 March 2020, the World Health Organization declared a pandemic due to the spread of SARS‐CoV‐2 (World Health Organization, 2020g).
Current public data indicate that the key epidemiological event in the history of SARS‐CoV‐2 was that a Chiroptera‐hosted lineage of viruses infected an urban human population in Wuhan, China (Zhao et al., 2020) and this is perhaps linked to earlier infections in rural populations (Wang et al., 2018).
The fundamental role of Chiroptera hosts for SARS‐CoV‐2 is also consistent with the history of the SARS/MERS clade. Chiroptera of various species as the original host for the three most medically important coronaviruses (SARS‐CoV, MERS‐CoV and SARS‐CoV‐2) is no longer disputed by most public health experts (Menachery et al., 2015; Yang et al., 2016; Cyranoski, 2017; Hu et al., 2017; Han et al., 2019; Zhou P. et al., 2020). However, there remains a contingent of groups that support the idea that there is a yet to be discovered intermediate host species other than Chiroptera (Andersen et al., 2020; Sun et al., 2020b; Zhang and Holmes, 2020; Zhang et al., 2020). In an event reminiscent of small carnivores being infected with SARS‐CoV derived from humans followed by additional exchanges between the two hosts (Janies et al., 2008), in late 2020 farmed minks in Denmark (host species Neovison vison), Netherlands (host species Mustela lutreola) and elsewhere were infected with SARS‐CoV‐2 from humans and passed the virus back to humans (Oreshkova et al., 2020; Oude Munnik et al., 2021; World Health Organization, 2020e).
In separate or in combination, several strategies were used to infer the intermediate host involved in human infection by SARS‐CoV‐2. Ji et al. (2020) used the relative synonymous codon usage (RSCU) bias and showed that the RSCU bias of SARS‐CoV‐2 is closer to that of snakes. However, betacoronaviruses have never been detected in snakes (King et al., 2011). Moreover, possible new betacoronaviruses are unlikely to cross over mammals to infect reptiles (Liu et al., 2020b), and wild snakes were less active in winter when human infection most likely occurred.
Ji et al. (2020) also pointed out that, among mammals, the RSCU bias of SARS‐CoV‐2 is closer to that of a marmot. Yuan et al. (2020) believe that this is an indication that rodents are the intermediate hosts of SARS‐CoV‐2. Yuan et al. (2020) presented other arguments in favour of rodents, especially squirrels, as an intermediate host but empirical evidence is absent. Moreover, Yuan et al. (2020) admitted that RSCU comparisons may be inappropriate when there is a large difference in the number of codons among the sequences that are being compared.
Sequence similarity has also been used to identify the putative intermediate host of SARS‐CoV‐2. For example, Xiao et al. (2020) and Lam et al. (2020) showed a high similarity of 97.4% within the receptor‐binding domain of SARS‐CoV‐2 and some coronaviruses isolated from Manis javanica (also known as Malayian pangolins; one of the eight species of the genus Manis of the family Manidae, order Pholidota, that are commonly named pangolins). Still, the full‐length genome similarities between the pangolin coronaviruses and SARS‐CoV‐2 (85.5–92.4%) are much lower than that between bat (Chiroptera) coronaviruses (specifically BatCoV RaTG13) and SARS‐CoV‐2 (96.2%). Furthermore, China's pangolins are endangered to the point of near extinction. Their low population density makes it nearly impossible that pangolins are an intermediate host. They also have long been banned from sale and have not been seen recently in Wuhan's wet markets (Yuan et al., 2020).
Other strategies, more speculative than those listed above, have been used to suggest that SARS‐CoV‐2 came from a laboratory accident at the Wuhan Institute of Virology (Rogin, 2020). The evidence indicates that SARS‐CoV‐2 was not purposefully manipulated (Andersen et al., 2020). Moreover, the notion that the SARS‐CoV‐2 pandemic resulted from a laboratory accident at the Wuhan Institute of Virology (Rogin, 2020) is not necessary to explain the pandemic. Based on serology evidence collected in October 2015, it is possible that members of the human population in Yunnan, or close contacts, carried SARS‐CoV‐like viruses in themselves to Wuhan (Wang et al., 2018). There also is evidence supporting that SARS‐CoV‐2's spike protein is derived from natural selection and is not the product of purposeful manipulation. For example, although the receptor‐binding domain (RBD) of the first variant of SARS‐CoV‐2 (Wuhan‐Hu‐1) binds to human ACE2, computational analysis suggests that the interaction is not ideal and that the Wuhan‐Hu‐1 RBD differs from the SARS‐CoV RBD that was shown to be optimal for receptor binding (Sheahan et al., 2008; Wan et al., 2020).
The natural emergence of new SARS‐COV‐2 variants in the UK, South Africa and elsewhere independently acquiring mutations that improve fitness, interaction with the host cell or transmissibility (Plante et al., 2021; World Health Organization, 2020a; Galloway et al., 2021) strengthens the hypothesis that the Wuhan‐Hu‐1 RBD is suboptimal for receptor binding and demonstrates that it could have been improved with minimal modifications if it had been purposefully manipulated.
Finally, many authors relied on phylogenetic approaches to estimate the proximal origin of SARS‐CoV‐2. So far, phylogenetic analyses by these authors indicate either bat (e.g. Lu et al., 2020; Sun et al., 2020a; Zhou P. et al., 2020) or pangolin (e.g. Lam et al., 2020; Xiao et al., 2020; Zhang et al., 2020) coronaviruses as the sister group of SARS‐COV‐2. These papers often suffer from the lack of diversity of genes and hosts sampled, both of which can impact the estimation of ingroup relationships (Schneider et al., 2020; Wenzel, 2020). In our manuscript, we overcome these shortcomings in phylogenetic analyses of coronaviruses by including as much data from the coronaviruses as possible to determine orthology and performing simultaneous analysis of a comprehensive sample of the Orthocoronavirinae subfamily including its four genera (i.e. AlphaCoVs, BetaCoVs, DeltaCovs and GammaCoVs).
Aims
In this paper, we present a comprehensive phylogeny based on 2006 Orthocoronavirinae genomes and metadata. The phylogeny serves to challenge persistent assumptions, and re‐evaluate hypotheses with new data concerning host shifts in highly pathogenic lineages (SARS‐CoV, MERS‐CoV and SARS‐CoV‐2), lineages of relatively benign human coronaviruses (HCoV‐NL63, HCoV‐229E, HCoV‐HKU1, HCoV‐OC43 and HECV‐4408), and coronaviruses that infect wildlife and livestock (e.g. mouse hepatitis virus (MHV), porcine epidemic diarrhoea virus (PEDV), transmissible gastroenteritis virus (TGEV). We aim to put the full record of coronavirus host shifts in a single comprehensive phylogenetic framework based on genomes. This phylogeny and the analyses of the host evolution provide vital information for the assessment of zoonotic episodes (Bolles et al., 2011). Moreover the comparison of sister‐group and orthology relationships among Orthocoronavirinae will allow for selection of viruses that can be used as proxies in research under standard laboratory conditions (Biosafety Level 2, BSL‐2) when higher Biosafety level labs (e.g. BSL‐3) are scarce.
Methods
Computation resources
All analyses were performed using UNC Charlotte's Linux clusters operated by University Research Computing (https://urc.uncc.edu).
Sample selection
Terminal selection includes unique sequences of Orthocoronavivinae from NCBI's RefSeq or GenBank, plus all complete SARS‐CoV‐2 sequences (i.e. ≥26 000 bp) from GISAID that were available on 17 February 2020. The final dataset comprises 2006 terminals. The outgroup is composed of three of the four genera of the subfamily Orthocoronavirinae, including 630 Alphacoronavirus, 265 Gammacoronavirus and 12 Deltacoronavirus. The ingroup comprises 1099 Betacoronavirus and includes 170 different samples of SARS‐CoV‐2. A complete list of selected terminals and accession numbers is provided in Appendix S1a.
Although our dataset is adequate to infer the origins of the different coronaviruses, we note that it is insufficient to discuss the epidemiology of SARS‐CoV‐2 during the COVID‐19 pandemic. Since 17 February 2020, there has been an accelerating increase in SARS‐CoV‐2 genomic information. For example, 559 218 complete or nearly complete genomes of SARS‐COV‐2 and related viruses were submitted to the GISAID's EpiCov database (https://www.epicov.org/) between 17 February 2020 and 17 February 2021. In that period, many cases of anthroponotic transmission (namely, transmission of a pathogen from humans to animals under natural conditions) of SARS‐CoV‐2 have been reported in cats, dogs, tigers, lions and minks (Abdel‐Moneim and Abdelwhab, 2020). These events are related to anthroponotic transmission during the pandemic rather than the fundamental emergence of SARS‐CoV‐2 from animals to humans.
The final nucleotide matrix (Appendix S1b) comprises 38 274 characters divided into four partitions, representing the genes ORF1ab (translated by ribosomal frameshifting), S (spike glycoprotein trimer), M (membrane protein) and N (nucleoprotein).
Since 11 November 2020, until this version of our manuscript, the website for Xiao et al. (2020) bore a warning reading “11 November 2020 Editor's Note: Readers are alerted that concerns have been raised about the identity of the pangolin samples reported in this paper and their relationship to previously published pangolin samples. Appropriate editorial action will be taken once this matter is resolved.” The sequence produced and described by Xiao et al. (2020) (of a Pan_SL‐CoV_GD virus, BioProject accession number PRJNA607174, GISAID accession number EPI_ISL_410721) is included in this manuscript. However, because we do not implicate pangolins as important in the emergence of SARS‐CoV‐2, removal of these data would not change the conclusions of our paper.
Gene annotations
The gene composition of the different genera of the subfamily Orthocoronavirinae vary, and not all genome sequences are entirely annotated. We selected four genes (ORF1ab, S, M and N) that are shared among all four genera of Orthocoronavirinae to partition the genome sequences of these viruses. To annotate these genes, we created a query database that contains nucleotide sequences of 194 gene sequences (44 ORF1ab, 49 M, 50 S and 51 N) from NCBI's RefSeq and Genbank. The results of nucleotide‐to‐nucleotide Blast (blastn) v2.4.0+ (Altschul et al., 1990; Camacho et al., 2009) were parsed using a python v3 script (parseOutfmt6.py, available at https://gitlab.com/phyloinformatics/parseoutfmt6https://gitlab.com/phyloinformatics/parseoutfmt6). Only terminals for which we could unambiguously annotate all four partitions were kept.
Multiple sequence alignment
Sequences of ORF1ab were aligned using Mafft v7.453 (Katoh et al., 2002; Katoh and Standley, 2013, available at mafft.cbrc.jp/alignment/server) with the iterative strategy “FFT‐NS‐i” (accurate but slow). The command line used was “mafft –reorder –anysymbol –maxiterate 1000 –6merpair input.”' Sequences of the other three partitions (M, S and N) were aligned using a translation‐based method with Mafft at the TranslatorX server (available at http://translatorx.co.uk/, accessed February 2020).
Parsimony analysis
Phylogenetic analysis under the parsimony optimality criterion was performed following the framework of a total evidence analysis (Kluge, 1989, 2004). We applied equal weights to all classes of character state transformation events. This approach considers that evolutionary cladogenesis and transformation series are unique and idiographic events. Historical inference under parsimony is idiographic in that the method aims to infer particular events rather than universal trends or laws. Such parsimony treats all hypothesized homologues and evolutionary transformations as ontological individuals that are unique, concrete and singular (Grant and Kluge, 2004; Kluge and Grant, 2006; Grant and Kluge, 2009). The parsimony approach differs from the alternative statistical and parametric optimality criteria that treat cladogenesis and transformation series as probabilistic events rather than heritable properties (Siddall and Kluge, 1997; Grant and Kluge, 2003; Grant et al., 2006).
Tree search was performed using TNT v1.1 (Goloboff et al., 2008). First, ten individual iterations (see Appendix S1c) were performed using a new technologies search employing the ratchet, Tree Bisection and Re‐connection (TBR) and Sub‐tree Pruning and Re‐grafting (SPR) tree searching strategies. A total of 100 rounds of tree fusing (command line “tfuse = rounds 100”) were executed using all trees found this way. A strict consensus of the most parsimonious trees was taken (with command “nelsen”) and used as our final parsimony tree but all character optimization (including the inference of host transformation events) was performed on binary trees. Bootstrap clade frequencies were calculated using 1000 pseudo‐replicates (command “resample boot replications 1000”). Goodman–Bremer support values were calculated using the “Bremer.RUN” macro (Appendix S1c). Goodman–Bremer values calculated this way were used to obtain the ratio of explanatory power (REP), as defined in Grant and Kluge (2007).
We used the 1031 shortest trees from the parsimony analyses for host character optimization. We normalized host information across all terminals to the ordinal level. The complete dataset comprised ten orders of animals: Artiodactyla (cloven‐hooved mammals, such as camels, pigs and cows), Aves (birds), Carnivora (eutherian mammals such as civet cats), Chiroptera (bats), Eulipotyphla (including hedgehogs), Lagomorpha (including hares and rabbits), Perissodactyla (odd‐toed ungulates, such as horses), Pholidota (pangolins), Primates (eutherian mammals such as moor macaque and chimpanze) and Rodentia (rodents such as rats). Due to the medical and societal importance of SARS‐CoV, MERS and SARS‐CoV‐2, humans were treated as a group apart from Primates. To investigate the different host shift events and their frequency with Orthocoronavirinae, we employed the commands CHANGE and APO from TNT as well as functions from YBYRÁ (Machado, 2015) and retrieved host shift information across different clades of Orthocoronavirinae.
Maximum‐likelihood analysis
We used IQ‐tree v1.6.12 (Nguyen et al., 2015; Chernomor et al., 2016; Kalyaanamoorthy et al., 2017; Hoang et al., 2018) for maximum‐likelihood (ML) analysis. Model selection implemented a greedy strategy (Lanfear et al., 2012) that starts with the full partition model and sequentially merges two genes until the model fit does not increase any further (argument: “‐m TESTMERGE”). Tree‐search started with 1000 initial parsimony trees (argument: “‐ninit 1000”). Bootstrap values were calculated using 1000 pseudoreplicates (argument: “‐bb 1000”). SH‐like approximate likelihood ratio test (SH‐aLRT; Guindon et al., 2010) using 1000 replicates (argument: “‐alrt 1000”).
IQ‐tree results were used as a constraint to recalculate branch lengths for our complete alignment matrix and each of our four alignment partitions. This way, we obtained five ML trees for (I) all of the partitions, (II) gene S, (III) gene M and (IV) gene N. We employed those five ML trees and two different outgroups (Deltacoronavirus or Deltacoronavirus + Gammacoronavirus) and their respective alignment matrices to calculate mutation rates using treetime v0.7.5 (Sagulenko et al., 2018) and following instructions in the documentation (revision f1c83c30, available at https://treetime.readthedocs.io). According to Sagulenko et al. (2018), treetime is known to underestimate evolutionary rates when branch lengths are long but it returns accurate estimates for low‐diversity samples. treetime strikes a useful compromise between inflexible but fast heuristics and computationally expensive Bayesian approaches that are not feasible in datasets the size of ours.
We also employed TreeTime to estimate the date of host shift along branches for the complete ML tree (I) using the “mugration” option. The mugration analysis in treetime assumes that host shifts can be modelled as a time‐reversible process with comparable sampling probabilities of the different states, treating host shifts as if they were mutations between different sequence states. As a result of this analysis, treetime generates a generalized time‐reversible (GTR) model for transformations between the different states and reports all of the most likely character state transformations. Unlike host shift optimization under the parsimony criterion (described above), which reports nonambiguous transformations only, host shift analysis in treetime reports a complete list of character state transformation events.
Finally, we calculated individual trees per data partition using the same unconstrained strategy described for the total evidence analysis above. We used YBYRÁ and msdist (Bogdanowicz and Giaro, 2011) to estimate the clade variation and match split (MS) distances among all trees, respectively.
Sensitivity to the removal of putative recombinant genomes
We tested the effect of excluding putative recombinant genomes in tree search experiments. We selected an initial subset of 505 terminals representing at least one viral strain from our complete dataset of 2006 terminals. We identified putative recombinants using RDP v5 (Martin et al., 2015) with the RND and GENECOV algorithms. We removed all putative recombinant sequences found this way. Parsimony analyses of the datasets with and without recombinants (i.e. with 505 terminals and 505 terminals minus 190 putative recombinants, respectively) followed the same tree search procedures applied for the complete dataset, described above. The trees resulting from the best heuristic searches of the smaller datasets were compared using YBYRÁ.
Contribution of genome recombination to the emergence of SARS‐CoV‐2
Recently, different authors (e.g. Li et al., 2020; Shang et al., 2020b) presented recombination detection analyses among bat‐hosted SARS‐like CoVs and pangolin‐hosted SARS‐like CoVs and the sequences of SARS‐CoV‐2 including its reference sequence (the human derived Wuhan‐Hu‐1, RefSeq accession number NC_045512, GenBank accession number MN908947.3). The bat‐hosted SARS‐like CoV RaTG13 (GISAID accession number EPI_ISL_402131) is the virus that shares the highest observed level of raw genetic similarity (96.3%) with the Wuhan‐Hu‐1. However, the evidence presented by Li et al. (2020) and Shang et al. (2020b) suggests that pangolin‐hosted CoVs sampled from Guangdong, China (i.e. Pan_SL‐CoV_GD viruses such as GISAID accession number EPI_ISL_410721) and bat‐hosted viruses represent samples of the ancestors that contributed to the RBD of SARS‐CoV‐2.
Referring to betacoronaviruses in general and SARS‐CoV‐2‐related viruses, Li et al. (2020) argue that “there is extensive recombination among all of these viruses.” Therefore, although we are interested in evaluating host transitions, including the one that led to the human infection by SARS‐CoV‐2, we must quantify the extent to which putative recombination events changed the genetic content of the lineage of viruses that led to SARS‐CoV‐2 and how putative recombination impacts our ability to infer the phylogeny and host transitions in the history of SARS‐CoV‐2.
In order to perform these analyses, we aligned the complete genomes of SARS‐CoV‐2 Wuhan‐Hu‐1 to two of the nonhuman‐hosted SARS‐like viral genomes that are most genetically similar to it (bat‐hosted COV RaTG13 and a representative of the Pan_SL‐CoV_GD clade; GISAID accession number EPI_ISL_410721), as well as to two other related bat‐hosted SARS‐like viruses (bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21; GenBank accession numbers MG772933.1 and MG772934.1, respectively). Genome sequences were aligned using the Mafft v7.450 (Katoh et al., 2002; Katoh and Standley, 2013) plugin in geneious prime® v2020.1.2 (https://www.geneious.com). We employed the alignment described above for recombination detection analysis utilizing the geneious plugin dualbrothers (Suchard et al., 2002, 2003; Minin et al., 2005) and RIP v3.0 (Siepel et al., 1995, web version available from https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). See additional detail in Appendix S1d.
Independent analysis of the SARS‐CoV‐2‐related clade
The phylogenetic relationships within the SARS‐CoV‐2‐related clade were further examined based on the results of the phylogenetic analyses of 2006 complete genomes of Orthocoronavirinae viruses, described above.
We selected complete genomes >26 kbp in length of SARS‐CoV‐2 from NCBI (100 samples) and GISAID (241) available as of 3 February 2020. We also selected nine genomes from pangolin‐hosted SARS‐CoV‐2 viruses (GenBank accession number MT084071 and GISAID accession numbers EPI_ISL_410538, EPI_ISL_410539, EPI_ISL_410540, EPI_ISL_410541, EPI_ISL_410542, EPI_ISL_410543, EPI_ISL_410721 and EPI_ISL_412860) and one genome of the bat‐hosted SARS‐like CoV RaTG13 (GISAID accession number EPI_ISL_402131). To remove redundancy, we used only one representative of each group of human‐hosted viruses with identical sequences. We used reference SARS‐CoV‐2 sequence from NCBI's RefSeq (accession number NC_045512) to create gene databases and annotate 11 genes (ORF1ab, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N and ORF10) using Blast and parseoutfmt6m and following the same guidelines described for the gene annotation of the 2006 genomes of Orthocoronavirinae (see above). We discarded all terminals for which we could not unambiguously annotate these 11 genes. The final SARS‐CoV‐2 dataset included 250 genome sequences. We conducted phylogenetic analysis using TNT and IQ‐tree followed the same procedures described above for the analyses of the 2006 terminals dataset.
We leveraged the alignment matrices and phylogenies obtained using the methodology described above to classify mutations, including insertions or deletions (indels) in the SARS‐CoV‐2‐related clade that could help to identify unique characteristics of the clade. Additional sequence comparisons were performed using the plotindels software that was developed in‐house and is available at GitLab (https://gitlab.com/phyloinformatics/plotindels).
In this analysis, we focused on the mutations that formed the SARS‐CoV‐2 S gene and its receptor‐binding motif (RBM), which constitutes the part of the RBD that anchors the receptor‐binding loop to the core of the RBD (Li et al., 2005; Tai et al., 2020). The viral spike glycoprotein mediates virus entry into the host cell. The efficiency of the spike's RBD interaction with the host receptors is a key factor determining the host range of many coronaviruses.
Distinct features of the SARS‐CoV‐2 virus over its relatives SARS‐CoV and MERS include the high efficiency of SARS‐CoV‐2 binding to the receptor on human cells, the angiotensin‐converting enzyme 2 (ACE2) and SARS‐CoV‐2's use of the host enzyme furin to preactivate the spike protein and facilitate cell entry. These features enable SARS‐CoV‐2 to be more infectious and transmissible than SARS‐CoV and HCoV‐NL63 which bind ACE2, albeit inefficiently (Brielle et al., 2020; Shang et al., 2020a). Note that not all coronaviruses bind to the same receptors. For example, MERS‐CoV binds to a distinct receptor, dipeptidyl peptidase 4 (DPP4) (Mou et al., 2013).
The evolution of sequences and structures that mediate virus–cell interaction frequently is invoked in discussions of the zoonotic events (Janies et al., 2008; Li, 2016; Liu et al., 2020b). Thus we account for these mutations and indels in all lineages in our dataset.
Results
A graphical abstract of the main results shown here is available at Appendix S1e.
Parsimony tree search results for Orthocoronavirinae
A total of six equally most heuristically parsimonious trees were found for the dataset of 2006 terminals, each with 560 229 steps. A NEXUS file with the best heuristic results, the strict consensus, and trees with Goodman–Bremen, REP and bootstrap values are provided in Appendix S1f. The average bootstrap value on the consensus tree was 75.17% (median = 90%, mode = 100%; see Appendix S1g). The strict consensus tree is divided into three parts to allow visualization (Figs 1, 2 and 3). Goodman–Bremer support values were calculated for 473 different nodes, ranging from 1 to 1 000 000 (median = 2266.18, mode = 39). The REP varied from 5.048 × 10−6% to 5.048%.
Fig. 1.
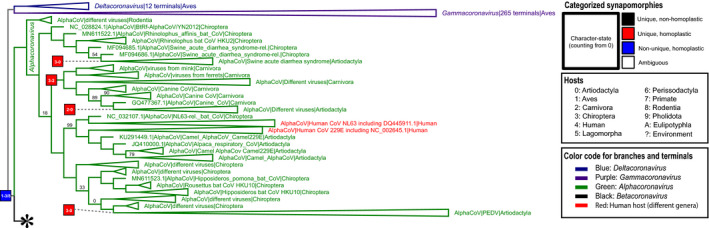
The figure shows the strict consensus of the best six heuristic solutions from the phylogenetic tree search performed under the parsimony criterion (part 1 of 3). The tree is rooted on Deltacoronavirus and Gammacoronavirus and the figure focuses on Alphacoronavirus. Clade frequencies calculated using the bootstrap strategy are at the top of each branch except if equal to 100%. Some clades are collapsed to improve visualization. See Appendix S1h for a complete version of this tree with branch lengths. The asterisk indicates the branch that continues on part 2. [Colour figure can be viewed at wileyonlinelibrary.com]
Fig. 2.
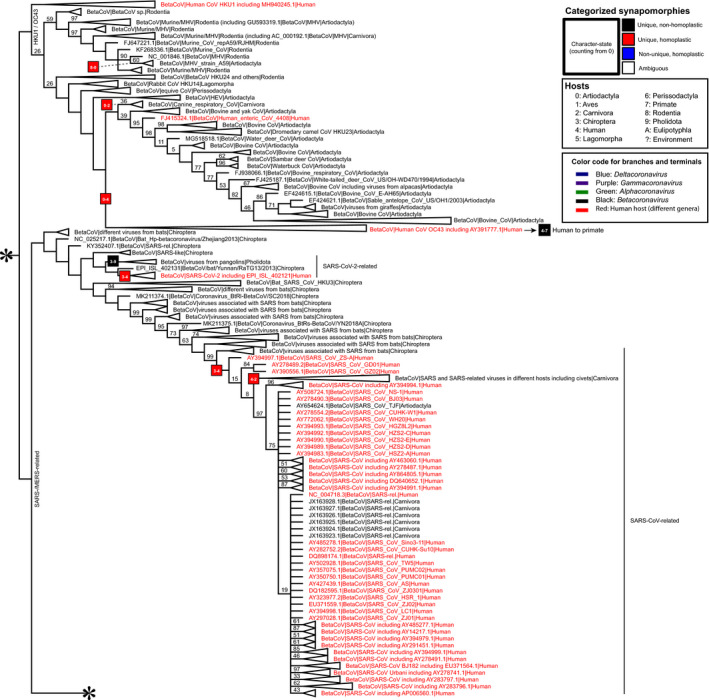
Strict consensus from parsimony analyses (part 2 of 3). The asterisks on the top and bottom indicate the continuity with Figs 1 and 3, respectively. See Appendix S1h for a complete version of this tree with branch lengths. The figure focuses on clades related to SARS‐CoV and SARS‐CoV‐2. Clade frequencies calculated using the bootstrap strategy are at the top of each branch except if equal to 100. Some clades are collapsed to improve visualization. [Colour figure can be viewed at wileyonlinelibrary.com]
Fig. 3.
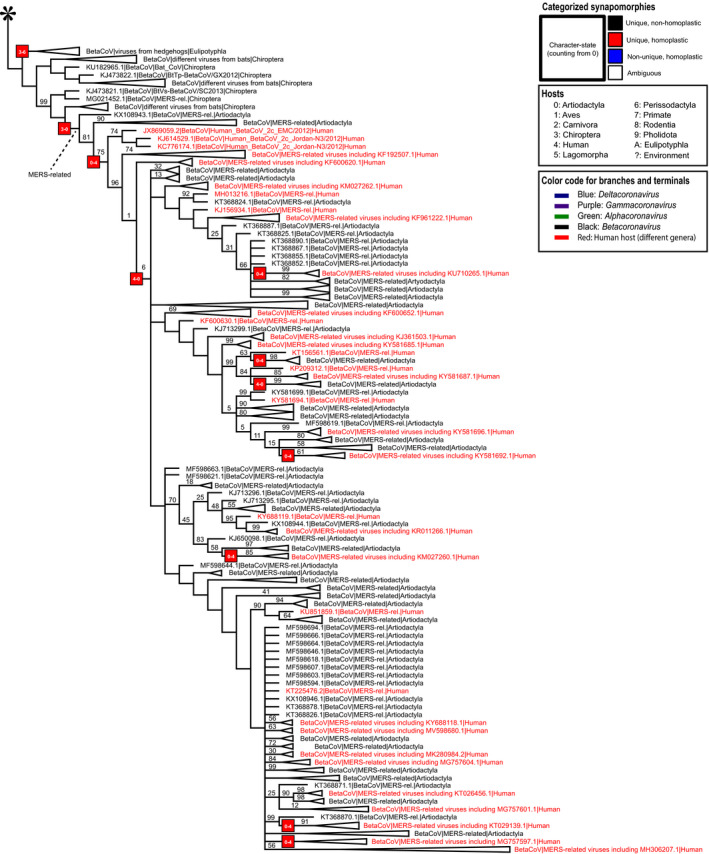
Strict consensus from parsimony analyses (part 2 of 3). The asterisk on the top indicates the continuity with Fig. 2. See Appendix S1h for a complete version of this tree with branch lengths. The figure focuses on MERS‐related clades. Clade frequencies calculated using the bootstrap strategy are at the top of each branch except if equal to 100. Some clades are collapsed to improve visualization. [Colour figure can be viewed at wileyonlinelibrary.com]
Parsimony analysis recovered the monophyly of all four genera. Only one sequence, a camel‐hosted coronavirus HKU23 (GenBank accession number KT368891.1), was found outside its originally assigned genus. The authors who submitted this sequence of the camel‐hosted coronavirus HKU23 to NCBI's GenBank classified it as an Alphacoronavirus. However, our phylogeny placed it within a clade that includes two betacoraviruses (two dromedary camel‐hosted coronavirus HKU23; GenBank accession numbers KF906251.1 and KF906251.1). In parenthetical notation, this clade can be described as (KF906251.1, KF906251.1, KT368891.1). Therefore, the sequence KT368891.1 could have been mistakenly assigned to the wrong genus by its submitters.
We measured the similarity of these sequences using two different strategies. First, we aligned these three genomes using the mauve v2.4.0 (Darling et al., 2010) aligner and observed a single LCB block of 31 041 bp. The sum of the lengths of matches in this LCB is 31 036 bp (99.83%), indicating that these sequences have a remarkable similarity throughout their contiguous length. Second, we measured the similarity among these sequences using the program distmat (available with EMBOSS v6.6.0; Rice et al., 2000). In a distance matrix calculated using the Tajima–Nei correction method (base positions: “123”, gap weighting: 0.0), the distance between the two HKU23 dromedary camel coronaviruses (KF906251.1 and KF906251.1) was 0.18. The distance between the camel coronavirus (KT368891.1) and the most distant dromedary coronavirus (KF906251.1) was 0.26. However, the distance between the camel coronavirus HKU23 (KT368891.1) and its sister taxon (KF906251.1) was 0.17, the smallest in this distance matrix. Based on these results, we classify the camel coronavirus HKU23 (KF906251.1) as a Betacoronavirus.
Wildcard taxa search with YBYRÁ does not indicate that there are any sequences that behave as a rogue terminal. According to our work with YBYRÁ, the terminals that are responsible for the most polytomies are representatives of SARS‐CoV‐2, which are similar to each other and comprise an unresolved clade (i.e. a collapsed branch of the cladogram). Although parsimony analysis can, at least on some occasions, be affected by long‐branch attraction (LBA; see review in Bergsten, 2005), we did not observe any two long nonsister branches within clades composed of otherwise short branches. We also did not observe any event in which outgroup sequences were attracted to long ingroup branches.
The host shift analysis in TNT was performed on the pool of the shortest binary trees. This analysis shows the minimum and the maximum number of host shifts across the best heuristic tree solutions (Appendix S1i). Throughout all Orthocoronavirinae (Table 1A) humans were infected with coronaviruses from Artiodactyla and Chiroptera at least 17 and six times, respectively. Carnivora and Rodentia hosts could be unambiguously assigned as sources of human coronavirus infection no more than one time each.
Table 1.
Minimum and maximum number of host shifts thought different clades of Orthocoronavirinae. Clades listed here are indicated in Figs 1, 2 and 3, which also include transformations that are found in all the best heuristic solutions from parsimony analysis. Empty cells indicate zero transformations. When the number of transformations is sensitive to tree topology and optimization strategy, the cells indicate the minimum and the maximum number of expected transformations. This table is divided into eight parts: (A) Complete phylogeny of Orthocoronavirinae; (B) Alphacoronavirus; (C) Betacoronavirus; (D) The clade that includes both HCoV‐HKU1 and HCoV‐OC43 betacoronaviruses; (E) Viruses related to SARS‐CoV, SARS‐CoV‐2 and MERS‐CoV; (F) SARS‐CoV and related viruses; (G) SARS‐CoV‐2 and related viruses; and (H) MERS‐CoV and related viruses
| To | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Ar) | (Av) | (Ca) | (Ch) | (Eu) | (Hu) | (La) | (Pe) | (Ph) | (Pr) | (Ro) | ||
| (A) Orthocoronavirinae | ||||||||||||
| From | (Ar)tiodactyla | — | 2 | 17–22 | 0–1 | 0–1 | ||||||
| (Av)es | — | 0–1 | 0–1 | |||||||||
| (Ca)rnivora | 1 | — | 0–1 | |||||||||
| (Ch)iroptera | 3–4 | 1 | — | 1 | 3–5 | 1 | 0–2 | |||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 9–14 | 6–7 | 0–1 | — | 1 | 0–1 | ||||||
| (La)gomorpha | 0–1 | — | 0–1 | |||||||||
| (Pe)rissodactyla | 0–1 | 0–1 | — | |||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | 4–5 | 1 | 0–2 | 0–1 | 0–1 | 0–1 | — | |||||
| (B) Alphacoronavirus | ||||||||||||
| From | (Ar)tiodactyla | — | 1 | 0–1 | ||||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | 1 | — | ||||||||||
| (Ch)iroptera | 2–3 | 1 | — | 1–2 | ||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 0–1 | 0–1 | — | |||||||||
| (La)gomorpha | — | |||||||||||
| (Pe)rissodactyla | — | |||||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | 1 | — | ||||||||||
| (C) Betacoronavirus | ||||||||||||
| From | (Ar)tiodactyla | — | 1 | 17–21 | 0–1 | 0–1 | ||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | 0–1 | ||||||||||
| (Ch)iroptera | 1 | — | 1 | 2 | 1 | 0–1 | ||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 9–13 | 6–7 | 0–1 | — | 1 | 0–1 | ||||||
| (La)gomorpha | 0–1 | — | 0–1 | |||||||||
| (Pe)rissodactyla | 0–1 | 0–1 | — | |||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | 4–5 | 1 | 0–1 | 0–1 | 0–1 | — | ||||||
| (D) HKU1/OC43 | ||||||||||||
| From | (Ar)tiodactyla | — | 1 | 2 | 0–1 | 0–1 | ||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | |||||||||||
| (Ch)iroptera | — | |||||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | — | 1 | 1 | |||||||||
| (La)gomorpha | 0–1 | — | 0–1 | |||||||||
| (Pe)rissodactyla | 0–1 | 0–1 | — | |||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | 4–5 | 1 | 0–1 | 0–1 | — | |||||||
| (E) SARS‐/MERS‐related | ||||||||||||
| From | (Ar)tiodactyla | — | 15–19 | |||||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | 0–1 | ||||||||||
| (Ch)iroptera | 1 | — | 2 | |||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 9–13 | 6–7 | — | |||||||||
| (La)gomorpha | — | |||||||||||
| (Pe)rissodactyla | — | |||||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | — | |||||||||||
| (F) SARS‐CoV‐related | ||||||||||||
| From | (Ar)tiodactyla | — | ||||||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | 0–1 | ||||||||||
| (Ch)iroptera | — | 1 | ||||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 1 | 6–7 | — | |||||||||
| (La)gomorpha | — | |||||||||||
| (Pe)rissodactyla | — | |||||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | — | |||||||||||
| (G) SARS‐CoV‐2‐related | ||||||||||||
| From | (Ar)tiodactyla | — | ||||||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | |||||||||||
| (Ch)iroptera | — | 1 | 1 | |||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | — | |||||||||||
| (La)gomorpha | — | |||||||||||
| (Pe)rissodactyla | — | |||||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | — | |||||||||||
| (H) MERS‐related | ||||||||||||
| From | (Ar)tiodactyla | — | 15–19 | |||||||||
| (Av)es | — | |||||||||||
| (Ca)rnivora | — | |||||||||||
| (Ch)iroptera | 1 | — | ||||||||||
| (Eu)lipotyphla | — | |||||||||||
| (Hu)mans | 8–12 | — | ||||||||||
| (La)gomorpha | — | |||||||||||
| (Pe)rissodactyla | — | |||||||||||
| (Ph)olidota | — | |||||||||||
| (Pr)imates | — | |||||||||||
| (Ro)dentia | — | |||||||||||
Humans were the source of coronaviruses infecting other hosts a minimum of 16 times (nine times to Artiodactyla, six times to Carnivora, and once to other Primates). Half of the remaining host shifts are from Rodentia to Artiodactyla (at least four times) and Chiroptera to Artiodactyla (at least three times).
In Alphacoronavirus (Table 1B), the most frequent type of host shift is from Chiroptera to Artiodactyla (two to three times). All alphacoronaviruses infecting humans originated from a Chiroptera or Artiodactyla host. Chiroptera to human transmissions occurred one to two times. Artiodactyla to human transmissions occurred zero to one time, depending on the tree. Likewise, when considering just the betacoronavirus clade (Table 1C), all human infections originated from Artiodactyla and Chiroptera (≥17 and two times, respectively) or from Carnivora (one to three times).
The clade that includes HCoV‐HKU1 and HCoV‐OC34 (Table 1D) has two host shifts from Artiodactyla to humans. There are no other host shifts from an animal species to humans in this clade.
Table 1E–H makes clear that most host shifts from Artiodactyla to humans (15–19 times) are in the clade associated with MERS‐CoV (Table 1E and H). The clade of SARS‐CoV‐related viruses (Table 1F) shows one host shift from Chiroptera to humans and zero or one host shift from Carnivora to humans, depending on the tree. The clade of SARS‐CoV‐2‐related viruses (Table 1G) has only two types of transmissions, which are independent: once from Chiroptera to humans, and once from Chiroptera to Pholidota. Finally, the MERS‐related clade (Table 1H) harbours 15 to 19 host shifts from Artyodactyla to humans, eight to 12 host shifts from humans to Artiodactyla, and one host shift from Chiroptera to Artiodactyla.
Characterization of different types of character state transformations in YBYRÁ allowed us to separate ambiguous and nonambiguous transformations, and classify host shifts into homoplastic and nonhomoplastic events, including reversions. Please note that a glossary of terms is included (Appendix S3). The categorized character‐state changes represent a consensus among all possible optimization schemes considering all best heuristic solutions from the parsimony analysis. For example, we highlight the transformation from Artiodactyla to humans that leads to the HCoV‐OC43 clade. This transformation is characterized as unique and homoplastic (Fig. 1). We also call the readers' attention to transformations in viral lineages that exhibit Chiroptera‐hosted to human‐hosted history in SARS‐CoV and SARS‐CoV‐2 clades (Fig. 2). In the clade of SARS‐CoV, there is a unique and homoplastic transformation from Chiroptera hosts to humans hosts before humans infected Carnivora hosts (Janies et al., 2008) Likewise, in the clade of SARS‐CoV‐2 related viruses, there is a unique and homoplastic transformation from Chiroptera hosts to human hosts. In lineages leading to the clade of SARS‐CoV‐2 in humans, there is a unique and nonhomoplastic transformation from Chiroptera to Pholidota that is independent of the human infection by SARS‐CoV‐2.
Chiroptera hosts play a fundamental role in the history of human infections by MERS‐CoV. In the clade of MERS‐related betacoronaviruses (Fig. 3), a unique and homoplastic change from Chiroptera hosts to Artiodactyla hosts occurs before another unique and homoplastic change from Artiodactyla to humans, which is followed by numerous host shifts between Artiodactyla and humans.
Maximum‐likelihood tree search results for Orthocoronavirinae
The ML tree (Fig. 4) also recovered the monophyly of the four genera of Orthocoronavirinae. The log‐likelihood of the consensus tree is . Node labels show the support values formatted as SH‐aLRT support (%)/bootstrap values (%). The branch lengths are proportional to the number of nucleotide substitutions per nucleotide site. As for the results from parsimony analysis, we did not detect any branch distortions.
Fig. 4.
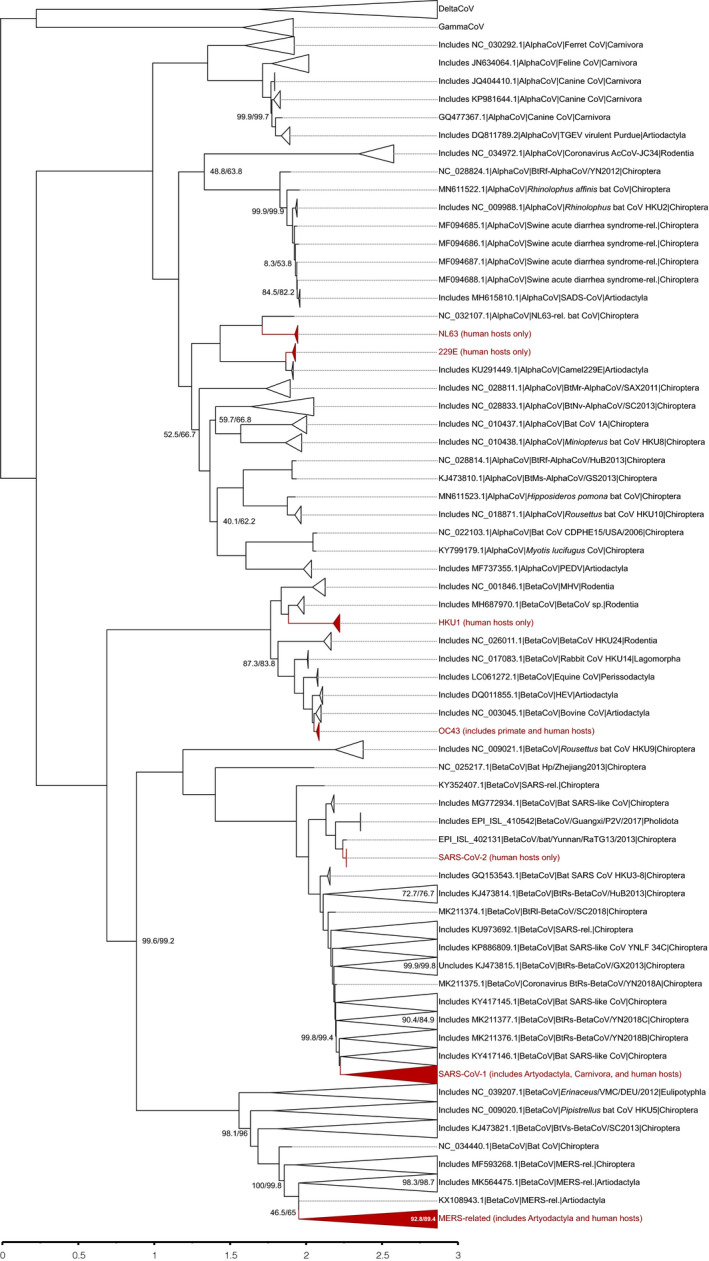
Maximum‐likelihood tree (log‐likelihood: ). Node labels show the support values formatted as SH‐aLRT support (%)/bootstrap values (%) except when values are both equal to 100. The branch lengths are proportional to the average number of nucleotide substitutions per aligned position. Branches of coronaviruses that infect humans are shown in grey and marked with an asterisk (*). Some clades were collapsed and named according to representative sequences to facilitate visualization. A complete version of this tree is available in Appendix S1. [Colour figure can be viewed at wileyonlinelibrary.com]
The ML tree is similar to the best heuristic results from parsimony analysis. There are no clades in the ML tree that are not found in at least one of the best heuristic results from parsimony analysis. In the histogram on Fig. 5 we show that only a fraction of all the clades from ML could not be found in all of the parsimony trees, and that the majority of the clades were found in all trees. Also, as shown in Fig. 5, the vast majority of the clades from the ML tree that are not found in the set of parsimony trees were <10 terminals.
Fig. 5.
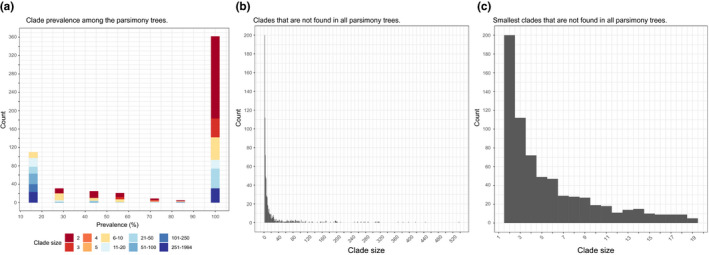
Histograms comparing the clades in the ML tree with the clades in all of the six best heuristic results from the parsimony analysis: (a) prevalence of clades from the ML tree among parsimony analysis; (b) all the clades that are not present in all parsimony trees; and (c) clades smaller than 20 terminals that are not present in all parsimony trees. [Colour figure can be viewed at wileyonlinelibrary.com]
The ML analysis served to inform model‐based analyses with the treetime program (Sagulenko et al., 2018). Complete results including evolutionary rates estimates and host shift analysis using “mugration” models are in Appendix S1j. Table 2 summarizes treetime results for the clades of alphacoronaviruses and betacoronaviruses known to infect humans. Table 2 also gives each of the viruses' earliest publications, which we can use as a conservative threshold for that clade's earliest expected date. A complete version of this table is available in Appendix S1j, including the publications' digital object identifiers and the details about the earliest genetic sequences submitted to NCBI's databases for each virus.
Table 2.
Main results from different treetime experiments, including branch length and date estimates for selected viruses. Minimum and maximum dates correspond to treetime estimates of the dates of emergence of the lineage. In MERS‐CoV, values that could not be computed for partitions M, N and S are not shown. A complete version of this table is available in Appendix S1j
| Genus | Virus | Partition | Branch length | Min. date | Max. date | Earliest paper's date |
|---|---|---|---|---|---|---|
| AlphaCoV | HCoV‐229E | All | 0.277057 | 16‐May‐1979 | 14‐Nov‐2001 | 28‐Nov‐1966 |
| M | 0.387593 | 6‐Jul‐1969 | 15‐Jan‐1984 | |||
| N | 0.360389 | 18‐Mar‐1974 | 17‐Jun‐1977 | |||
| ORF1ab | 0.293851 | 1‐Jan‐1982 | 4‐Jan‐1990 | |||
| S | 0.01769 | 11‐Mar‐1971 | 11‐Mar‐1971 | |||
| AlphaCoV | HCoV‐NL63 | All | 0.214784 | 2‐Apr‐1983 | 1‐Jan‐2010 | 21‐Mar‐2004 |
| M | 0.300646 | 13‐Jul‐1983 | 17‐Dec‐1987 | |||
| N | 0.319683 | 11‐Aug‐1983 | 11‐Aug‐1983 | |||
| ORF1ab | 0.169852 | 28‐Feb‐1983 | 19‐Jan‐2000 | |||
| S | 0.469001 | 31‐Jul‐1983 | 31‐Jul‐1983 | |||
| BetaCoV | HCoV‐OC43 | All | 0.016838 | 7‐Oct‐1986 | 1‐Aug‐2014 | 1‐Dec‐1967 |
| M | 0.03109 | 0‐Jul‐1984 | 28‐Nov‐1984 | |||
| N | 0.012209 | 4‐Nov‐1984 | 28‐Nov‐1984 | |||
| ORF1ab | 0.014887 | 1‐Oct‐1984 | 9‐Sep‐1998 | |||
| S | 0.02301 | 3‐Sep‐1984 | 23‐Sep‐1984 | |||
| BetaCoV | HECV‐4408 | All | 0.005943 | 1‐Jan‐1989 | 19‐Jan‐2014 | 28‐Jan‐1994 |
| M | 0.004738 | 8‐Jan‐1988 | 27‐Jun‐1988 | |||
| N | 0.003355 | 1‐Jan‐1988 | 1‐Jan‐1988 | |||
| ORF1ab | 0.005562 | 1‐Jan‐1988 | 2‐Jun‐2000 | |||
| S | 0.007408 | 1‐Jan‐1988 | 1‐Jan‐1988 | |||
| BetaCoV | SARS‐CoV | All | 0.028383 | 15‐Jan‐2010 | 24‐Feb‐2019 | 15‐May‐2003 |
| M | 0.026576 | 5‐Apr‐2003 | 20‐Apr‐2003 | |||
| N | 0.010167 | 20‐Apr‐2003 | 20‐Apr‐2003 | |||
| ORF1ab | 0.02828 | 13‐Apr‐2003 | 13‐Apr‐2003 | |||
| S | 0.032441 | 9‐Apr‐2003 | 9‐Apr‐2003 | |||
| BetaCoV | HCoV‐HKU1 | All | 0.294901 | 20‐Jul‐2003 | 15‐Jan‐2016 | 3‐Sep‐2004 |
| M | 0.422428 | 14‐Nov‐2002 | 19‐Apr‐2004 | |||
| N | 0.348589 | 12‐Sep‐2004 | 15‐Oct‐2004 | |||
| ORF1ab | 0.287167 | 28‐Nov‐2004 | 28‐Nov‐2004 | |||
| S | 0.256345 | 3‐Nov‐1998 | 3‐Nov‐1998 | |||
| BetaCoV | MERS‐CoV | All | 0.000394 | 28‐Mar‐2012 | 23‐Apr‐2012 | 17‐Oct‐2012 |
| ORF1ab | 0.000485 | 28‐Mar‐2012 | 28‐Mar‐2012 | |||
| BetaCoV | SARS‐CoV‐2 | All | 0.021728 | 21‐Dec‐2019 | 24‐Dec‐2019 | 23‐Jan‐2020 |
| M | 0.025206 | 28‐Sep‐2019 | 24‐Dec‐2019 | |||
| N | 0.020273 | 24‐Dec‐2019 | 24‐Dec‐2019 | |||
| ORF1ab | 0.017981 | 17‐Dec‐2019 | 17‐Dec‐2019 | |||
| S | 0.037718 | 24‐Dec‐2019 | 24‐Dec‐2019 |
We retrieved the oldest and more recent dates for each virus from the different treetime experiments (Table 2). In some cases, the variation is negligible; for example, from 28 September to 21 December 2019, for SARS‐CoV‐2. However, depending on the virus, data partitioning and treetime parameters, there were more pronounced variations; for example, from 6 July 1969 to 14 November 2001, for HCoV‐229E. The date estimates also varied within the same clade depending on the partition; for example, the minimum dates for the SARS‐CoV date varied from 9 April 2003, to 15 January 2010.
In a similar way to date estimations, Table 2 also shows that branch length estimations (which correspond to the number of expected mutations per site in each branch) varied depending on the clade and data partitioning. The ratio between the maximum and minimum branch length varied from c.1.64 in HCoV‐HKU1 to c.21.91 in HCoV‐229E.
Moving from treetime analyses to the results of phylogenetic analsyses using different partition schemes, we observed that unconstrained gene trees based on partitions (ORF1ab, S, M and N) were topologically distant from the tree based on all datasets combined according to the MS distances for unrooted binary trees calculated with the msdist program (Table 3). The distance between the tree from all datasets to the trees from each partition increases as each partition size decreases. We used YBYRÁ to compare all clades among these trees. Table 4 illustrates the frequency in which a few selected clades were recovered in trees from different partitions. The unconstrained gene trees are provided in Appendix S1l.
Table 3.
Matching split distances for unrooted binary phylogenetic trees from ML analyses
| Total evidence | ORF1ab | S | M | N | |
|---|---|---|---|---|---|
| Total evidence | 0 | ||||
| ORF1ab | 13.775 | 0 | |||
| S | 26.323 | 28.544 | 0 | ||
| M | 60.817 | 61.578 | 68.542 | 0 | |
| N | 32.056 | 34.453 | 38.365 | 65.285 | 0 |
Table 4.
Percentage of trees containing the selected clades (i.e. clade frequency) among ML trees resulting from the analyses of different partitions independently (for genes ORF1ab, S, M and N) or combined (all)
| Clade | Clade frequency (%) | All | ORF1ab | S | M | N |
|---|---|---|---|---|---|---|
| DeltaCoV | 100 | Yes | Yes | Yes | Yes | Yes |
| GammCoV | 80 | Yes | Yes | No | Yes | Yes |
| AlphaCoV | 80 | Yes | Yes | No | Yes | Yes |
| HCoV‐NL63 | 100 | Yes | Yes | Yes | Yes | Yes |
| HCoV‐229E | 100 | Yes | Yes | Yes | Yes | Yes |
| BetaCoV | 40 | Yes | Yes | No | No | No |
| HCoV‐OC43 | 100 | Yes | Yes | Yes | Yes | Yes |
| HCoV‐HKU1 | 80 | Yes | Yes | Yes | No | Yes |
| SARS‐CoV‐2 | 100 | Yes | Yes | Yes | Yes | Yes |
| SARS‐CoV‐2 + RaGT13 | 100 | Yes | Yes | Yes | Yes | Yes |
| SARS‐CoV‐2 + pangolin CoVs | 0 | No | No | No | No | No |
| SARS‐CoV‐related | 80 | Yes | Yes | Yes | Yes | No |
| MERS‐related | 100 | Yes | Yes | Yes | Yes | Yes |
Sensitivity to removal of putative recombinant genomes
We used RDP v5 to search for putative recombinant events on a subset of 505 terminals selected to represent the main lineages of coronaviruses found in the complete dataset of 2006 terminals. The RDP and GENECOV algorithms indicated that 190 terminals were possible recombinant genomes (see Appendix S1k). We removed those 190 putative recombinants from the initial subset to generate a new matrix of 315 terminals.
The subset of 505 terminals resulted in 50 most parsimonious trees of 527 991 steps. The subset of 315 terminals resulted in nine most parsimonious trees, with 366 045 steps. Alignment matrices and trees are available in Appendix S1m. We used YBYRÁ to compare the strict consensus trees generated from both datasets. The data plot in Fig. 6 illustrates how the most parsimonious trees differ among datasets using the percentage of shared branches among trees. The distance between trees from each dataset is higher than the distance of the trees within each dataset. However, the histograms on Fig. 7 indicate that strict consensus topologies are mostly congruent, with most of the differences being of clades with <10 terminals. We found that, in this particular case, the distances between these trees do not affect downstream analysis of character evolution.
Fig. 6.
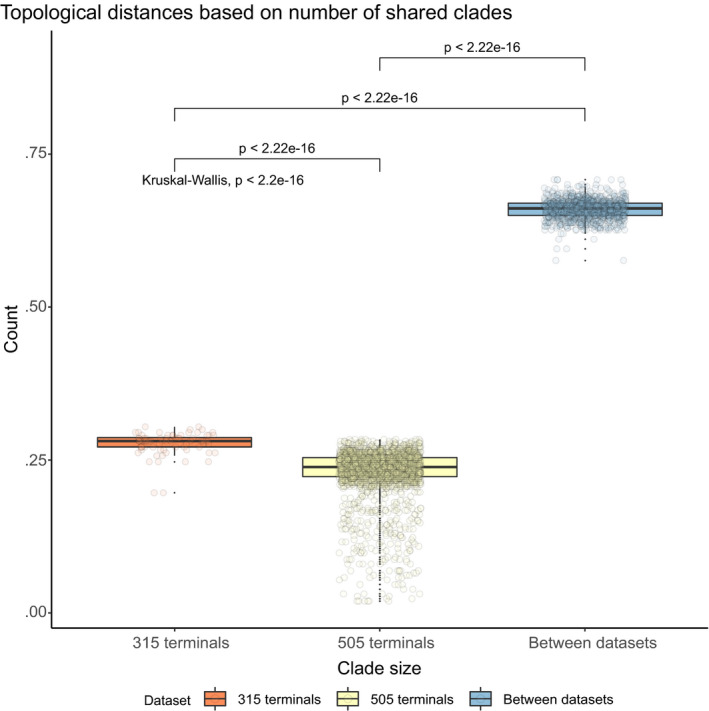
Data plot illustrating how the most parsimonious trees differ among datasets using the percentage of shared branches among trees. This is calculated using local distances (LD) calculated as one minus the ratio between the number of shared branches between trees (S) and the different branches in both trees (U): . Orange, distance among best heuristic results for the 315 terminals dataset; yellow: distance among best heuristic results for the 505 terminals dataset; blue: distance between trees from each dataset. [Colour figure can be viewed at wileyonlinelibrary.com]
Fig. 7.
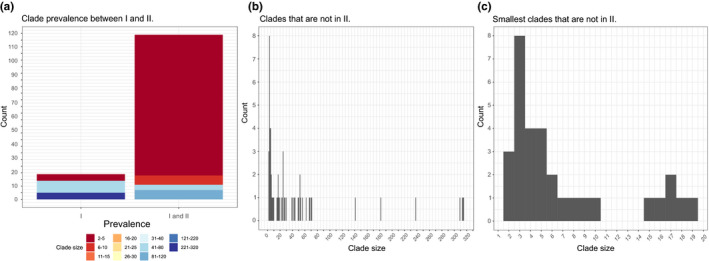
Clade prevalence on the strict consensus trees from the analyses of the 505 terminal dataset (I) and the subset of 315 terminals from which we removed putative recombinants (II). (a) Clade prevalence between I and II. (b) All clades that are not in II. (c) The smallest clades (<20 terminals) that are not in II. [Colour figure can be viewed at wileyonlinelibrary.com]
Upon close examination, we noticed that topological modifications in the consensus tree from the smallest dataset make groups expected to be monophyletic (and that were retrieved as such in our analysis of the complete dataset) become paraphyletic or polyphyletic. For example, depending on the root position, with smaller datasets Deltacoronavirus would be split into a minimum of two clades that comprised either a paraphyletic assemblage in the trees from the larger dataset or a polyphyletic assemblage in trees from the smaller dataset. Although Alphacoronavirus is a polyphyletic group in both cases, the consensus tree from the larger dataset organizes alphacoronaviruses in a minimum of five clades, whereas the consensus tree from the smaller dataset organizes alphacoronaviruses in a minimum of six clades.
Likewise, Betacoronavirus comprises at least three and at most nine clades in the consensus trees from the larger and smaller datasets, respectively. The only exception to this pattern seems to be the group of SARS‐CoV‐related viruses that form a polyphyletic group composed of three (dataset of 515 terminals) or two (dataset of 315 terminals) clades. The comparison of selected clades of interest (summarized in Table 5) between the two strict consensus trees indicates that the monophyly of these clades is recovered when more data are analyzed. This result is further supported from the phylogenetic analyses of the complete dataset (2006 terminals), described above.
Table 5.
Status (monophyletic, paraphyletic or polyphyletic) of different groups and the minimum number of clades they form (*) in the strict consensus tree of the phylogenetic analyses of two datasets: a large dataset of 505 terminals and a smaller dataset of 315 terminals from which we removed 190 putative recombinant genomes. All datasets include four partitions (ORF1ab, M, S and N) and represent subsets of the complete dataset of 2006 terminals. SARS‐CoV‐2‐related clade indicates SARS‐CoV‐2 plus associated Chiroptera‐hosted viruses
| Dataset | Clade | Status | No. of clades (*) |
|---|---|---|---|
| 505 terminals | Deltacoronavirus | Paraphyletic | 2 |
| 315 terminals | Deltacoronavirus | Polyphyletic | 2 |
| 505 terminals | Gammacoronavirus | Monophyletic (single terminal) | 1 |
| 315 terminals | Gammacoronavirus | Monophyletic (single terminal) | 1 |
| 505 terminals | Alphacoronavirus | Polyphyletic | 5 |
| 315 terminals | Alphacoronavirus | Polyphyletic | 6 |
| 505 terminals | Betacoronavirus | Polyphyletic | 3 |
| 315 terminals | Betacoronavirus | Polyphyletic | 9 |
| 505 terminals | MERS‐related | Monophyletic | 1 |
| 315 terminals | MERS‐related | Monophyletic | 1 |
| 505 terminals | SARS‐CoV‐related | Polyphyletic | 3 |
| 315 terminals | SARS‐CoV‐related | Polyphyletic | 2 |
| 505 terminals | SARS‐CoV‐2‐related | Monophyletic | 1 |
| 315 terminals | SARS‐CoV‐2‐related | Monophyletic | 1 |
Putative recombination involving SARS‐CoV‐2 and pangolin‐hosted CoVs
The multiple sequence alignment of the reference sequence of SARS‐CoV‐2 (Wuhan‐Hu‐1), bat coronavirus RaTG13, bat‐SL‐CoVZC45 (GenBank accession number MG772933), bat‐SL‐CoVZXC21 (GenBank accession number MG772934.1), and a representative of the Pan_SL‐CoV_GD clade (GISAID accession number EPI_ISL_410721) had a total length of 29 927 (with 24 175 nucleotide positions that are identical among all sequences).
Recombination detection using dualbrothers found three different topologies that were favoured depending on the position sliding window. The first topology places Wuhan‐Hu‐1 as the sister group of RaTG13. This topology is preferred in the majority (97.34%) of the positions of the multiple sequence alignment (a total of 29 132 nucleotides in positions 210–2177; 2556–22 900; and 23 108–29 927). The second topology places Wuhan‐Hu‐1 as the sister group of the pangolin‐hosted CoV. This topology is favoured from positions 1 to 210 (210 nucleotides of the 5′UTR) and from position 22 901 to 23 107 (207 nucleotides of the spike glycoprotein coding gene that includes the ACE2 receptor binding site), which represents 1.39% of all positions. Finally, the third topology places RaTG13 as the sister of the pangolin‐hosted CoV. We found this last topology from position 2178 to 2555 (378 nucleotides of the nsp2 gene in ORF1a), corresponding to 1.26% of the total alignment. No other trees are highly supported by the data.
The results from dualbrothers analysis are consistent with those from RIP. However, RIP alignment analysis indicates that Wuhan‐Hu‐1 only significantly matches the pangolin‐hosted CoV in the ACE2 receptor binding site. RIP ignores gaps in the Wuhan‐Hu‐1 sequence and therefore considers only 29 903 positions of the original alignment. A significant best match between Wuhan‐Hu‐1 and Pan_SL‐CoV_GD is observed in only 417 positions (1.39% of the alignment). There are no significant matches to any other sequence besides RaTG13.
We provide additional details of the dualbrothers and RIP analyses in Appendix S1d.
Topological and sequence similarity in the SARS‐CoV‐2‐related clade
Trees resulting from parsimony and ML analyses of the SARS‐CoV‐2‐related clade were topologically similar. A visual representation of this tree topology is available at Appendix S1e, panel D. The files containing alignments, partition scheme and phylogenetic trees are described in Appendix S1n. The genomes of SARS‐CoV‐2 viruses infecting humans and SARS‐related viruses isolated from pangolins form two reciprocally monophyletic clades. Rooting the tree according to the phylogenetic analyses of the 2006 terminals dataset places pangolin‐hosted viruses as the sister group to a clade comprising human‐hosted SARS‐CoV‐2 and the CoV RaTG13 that was isolated from Rhinolophus affinis collected in Yunnan, China (GISAID accession number EPI_ISL_402131). The 240 unique sequences from SARS‐CoV‐2 (out of 341 SARS‐CoV‐2 samples), although not identical to each other, do not contain any mutations that resolve the polytomy of the SARS‐CoV‐2 clade.
A table summarizing alignment comparisons between the human‐hosted SARS‐CoV‐2 reference sequence (Wuhan‐Hu‐1) and related viruses found in humans, bats and pangolin hosts is available in Appendix S1o. The most common single nucleotide polymorphisms (SNPs) throughout all partitions are synonymous SNPs. Normalizing the number of SNPs by both the number of terminals per host group (bat, pangolin and human) and the length of each partition, SNPs are more frequent between the human‐hosted reference and ORF7a, S and ORF3a sequences from pangolin hosts, and less frequent in the E, ORF7a and ORF7b sequences from human hosts.
Following the same normalization strategy, we find most amino acid variation in ORF7b, ORF10 and ORF1ab from pangolin‐hosted viruses. There were no such variations in E from bat‐hosted viruses and pangolin‐hosted viruses, M and ORF6 from bat‐hosted virus, and ORF7b from human‐hosted viruses. See details in Appendix S1o.
Turing our attention to alignments rather than trees, we found more indels when comparing the SARS‐CoV‐2 sequences from human hosts (e.g. Wuhan‐Hu‐1) with related viral sequences from pangolin hosts (EPI_ISL_410538, EPI_ISL_410539, EPI_ISL_410540, EPI_ISL_410541, EPI_ISL_410542, EPI_ISL_410543, EPI_ISL_410544, EPI_ISL_410721, EPI_ISL_412860 and MT084071.1) than when comparing the reference to viral sequences from bats (EPI_ISL_402131). Considering the number of terminals per group of hosts and the length of each partition, we observed that the partitions ORF7a, ORF7b and ORF1ab have more indels than all other partitions.
These observations suggest that, although less common than other substitutions, indels are an essential part of the evolution of coronaviruses genomes in these zoonotic events. Additionally, indels among coronaviruses infecting pangolins, bats and humans in the SARS‐CoV‐2‐related clade help demonstrate that the sequence similarity between human‐hosted SARS‐CoV‐2 and bat‐hosted CoV RaTG13 is greater than that between human‐hosted SARS‐CoV‐2 and any other coronavirus from pangolin hosts in our dataset, thus further diminishing the hypothesis for pangolin–human host zoonosis in the evolution of SARS‐CoV‐2.
Turning our attention away from trees and alignments, the comparisons of k‐mers (5, 12, 31 and 100) from gene S among different groups of coronaviruses organized according to genera and host order or different lineages that infect humans show higher similarity between the human‐hosted SARS‐CoV‐2 reference sequence (Wuhan‐Hu‐1) and sequences from betacoronaviruses that infect bats (Fig. 8) in comparison to any other group. Shared k‐mer content is smaller between SARS‐CoV‐2 and betacoronaviruses infecting pangolins and humans than between SARS‐CoV‐2 and betacoronaviruses infecting bats and humans, but the difference is less pronounced in amino acid sequences than in nucleotide sequences, which is expected owing to the degenerate nature of the genetic code.
Fig. 8.
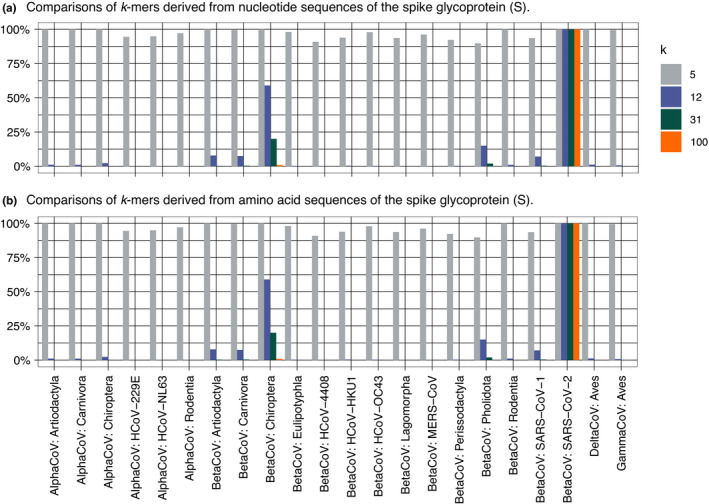
Comparisons between k‐mer content of the spike glycoprotein (S) of the SARS‐CoV‐2 reference sequence (Wuhan‐Hu‐1) and other groups of coronaviruses organized by genera and host order of specific lineage that infects humans. (a) Similarity between nucleotide sequences. (b) Similarity between amino acids sequences. [Colour figure can be viewed at wileyonlinelibrary.com]
We also inspected sequence similarity at the level of the RBM in gene S of SARS‐CoV‐2 (Wuhan‐Hu‐1), which interacts with the ACE2 receptors in humans. In the SARS‐CoV‐2 reference genome (Wuhan‐Hu‐1), the RBM is located between positions 22874 and 23080 of the RNA sequence (Lan et al., 2020). Distributed along a sequence of 69 amino acids, the RBM of the SARS‐CoV‐2 spike glycoprotein includes five key amino acid residues involved in ACE2 receptor binding. Numbered according to their position in the amino acid sequence of the surface glycoprotein of SARS‐COV‐2 (NCBI protein accession number YP_009724390.1), these positions are 455L, 486F, 493Q, 501N and 505Y.
Note that SARS‐CoV‐2 has a unique furin cleavage site insertion (PRRA) that is not found in any other CoVs in the Sarbecovirus group. The PRRA motif is in positions 681 to 684 of the surface glycoprotein (YP_009724390.1). This motif is absent in all other coronavirus genomes except SARS‐CoV‐2. However, the recently discovered bat‐hosted coronavirus RmYN02 contains a different insertion of three amino acids (PAA) in the same polybasic cleavage site, showcasing how such insertions can occur naturally in animal betacoronaviruses (Zhou H. et al., 2020). Unlike the RBM, the PRRA motif is a new insertion to the SARS‐CoV‐2 genome. Therefore, we do not report the sequence similarity at that specific position.
At both the nucleotide and the amino acid levels, the region of the five key amino acid residues of the RBM of Wuhan‐Hu‐1 is more similar to sequences from betacoronaviruses infecting bats (particularly those related to SARS‐CoV‐like viruses) than to any other viruses. For example, we found only two of the five key residues of the RBM of SARS‐CoV‐2 in the five different sequences of betacoronaviruses infecting pangolins that we examined (GISAID accession numbers EPI_ISL_410538, EPI_ISL_410539, EPI_ISL_410540, EPI_ISL_410541 and EPI_ISL_410542). However, at least three of these five residues are found in betaconaviruses infecting bats in the SARS‐CoV‐2 lineage (GenBank accession numbers KC881006, KF367457, KF569996, KT444582, KY417150, KY417151, KY417152, MK211376 and AB257344). Additional data from RBM sequence comparisons are available in Appendix S1p.
Discussion
In this paper we performed a comprehensive analysis across lineages of Orthocoronavirinae in terms of sampling of genomes, viral taxa and host source data.
We use the strict consensus tree from parsimony analysis as our main phylogenetic hypothesis as it is unencumbered by ad hoc assumptions that sap the explanatory power of the evidence (see discussion in Rindal and Brower, 2011).
We also examined hypotheses under the ML criterion and found that phylogenies based on ML analyses are largely congruent with phylogenies from parsimony. Thus, in the case of this Orthocoronavirinae dataset, the choice of optimality criterion does not significantly impact the results of downstream analysis, such as the discovery of host origins of viral lineages.
A primary objective of this paper is to challenge all assumptions on host and recombinant origins of coronaviruses, with special focus on lineages of Betacoronavirus that cause severe disease in humans: SARS‐CoV, MERS‐CoV and SARS‐CoV‐2. This challenge requires a comprehensive taxonomic sample of viruses such that many scenarios can be examined including origins of viral lineages from more than one ancestral host through recombination.
In our results, we find evidence for Chiroptera host origins of SARS‐CoV, SARS‐CoV‐2 and MERS‐CoV. In the case of SARS‐CoV, the Chiroptera are the ancestral host to viruses that infected humans and led to the outbreak that occurred in 2002–03. Even though Chiroptera have been reported as the ancestral hosts of SARS‐CoV in humans in the literature (Lau et al., 2005; Janies et al., 2008; Bolles et al., 2011), the misconception that Carnivora are the key hosts remains. Some argue that small carnivores such as civet cats Parguma larvata and racoon dogs Nyctereutes procyonoides were important amplifying hosts (Bolles et al., 2011). These were anthroponotic events local in Shenzen markets in the 2002–03 timeframe and do not explain the fundamental origins of SARS‐CoV (Janies et al., 2008). In the wake of the SARS‐CoV crisis, small carnivores to be sold in markets were widely culled (Normile, 2004; Watts, 2004). Most importantly, the discovery of SARS‐CoV in small carnivores in Shenzen markets in 2003 cannot explain the long‐term maintenance of SARS‐like viruses in Chiroptera that led to the emergence of SARS‐CoV‐2.
The persistence of the notion of the necessity for reservoir species for SARS‐like viruses that are not Chiroptera is an example of group‐think that has misled the field and distracts from understanding the source of the current COVID‐19 pandemic. This idea is manifest in the seemingly relentless pursuit of some market‐based animal whether be it snakes, pigs, minks, domestic cats, zoo tigers or pangolins as an intermediate host between bats and humans (King, 2020). Similar to minks, there have been reports of zoo tigers and domestic animals that have been infected with SARS‐CoV‐2 from humans.
MERS‐CoV may have led to some of this thinking. We have shown that the fundamental host origins of MERS‐CoV also are in Chiroptera but this lineage infected Artiodactyla (specifically dromedary camels) first then infected humans. Subsequent to these originating events MERS‐CoV continued to be exchanged among human and Artiodactyla.
Overview of host‐shifts in coronaviruses hosted by humans
Transmission of coronaviruses from animals to humans occur episodically. From 1966 until 2020, the scientific community described eight HCoVs (Table 2). On average, that is one new HCoV every 6.75 years. In the 1960s, two HCoVs were described approximately one year apart: HCoV‐229E (November 1966) and HCoV‐OC43 (December 1967). The first paper on HECV‐4408 would be published only 27 years later (January 1994), separated by nine years from the SARS‐COV outbreak in 2003. HCoV‐HKU1 and HCoV‐NL63 were described in the following year, 2004. It would take another eight years for the MERS‐COV outbreak in 2012, followed by the SARS‐CoV‐2 outbreak seven years later, in 2019.
Coronavirus transmission from animal to human host occur episodically at unpredictable intervals, so it is not wise to attempt to time when scientists will describe the next HCoV. However, it is safe to assume it will happen again based on the number of species in genera Alphacoronavirus and Betacoronavirus, the diversity of hosts, and ample contact between humans and those hosts.
Understanding the numerous host transformation events in Orthocoronavirinae that have and will occur is critical to design systems of preparedness and response for future outbreaks.
The optimization of hosts as discrete character states under parsimony (character categorization using YBYRÁ and TNT) and model‐based (mugration analysis in treetime) approaches resulted in largely congruent results except for ambiguities from traditional character optimization in TNT that are presented as a single most likely solution by treetime.
Our results shed light on the debate of the origins of different HCoVs (see Ye et al., 2020, for a recent review). HCoV‐NL63 is the sister group of a bat‐hosted CoV, and their ancestry is a lineage of bat‐hosted CoVs. This conclusion is in agreement with previous evidence supporting the zoonotic origin of HCoV‐NL63 (Huynh et al., 2012).
HCoV‐229E is the sister group of a camel‐hosted clade, and their ancestry is from an Artiodactyla host. Therefore, our results corroborate the previous hypothesis of the order of host transformations of HCoV‐229E from bats to camels and then to humans (Corman et al., 2015, 2016).
Our phylogenetic data confirm HCoV‐HKU1 as a lineage that originated from a rodent‐hosted virus (Su et al., 2016; Forni et al., 2017). Additionally, we confirmed that the HCoV‐OC43 came from bovids (Cui et al., 2019).
The evidence presented here corroborates the origination of SARS‐CoV and MERS‐CoV from Chiroptera‐hosted lineages. We agree with Janies et al. (2008) that SARS‐CoV was transmitted to small carnivores and artiodactyls after the emergence of SARS‐CoV in humans.
Our work also supports hypotheses for MERS‐CoV's evolutionary origin in bat‐hosted viral lineages followed by interspecies viral transmission involving human and dromedary camels as hosts (van Boheemen et al., 2012; Annan et al., 2013; Cotten et al., 2013). Considering the timescale and the number of host shifts between humans and artiodactyls in the MERS‐CoV‐related clade (≥23 in both directions), it seems that the virus that originated from bats was capable of infecting both humans and artiodactyls as soon as the first transmission event from a bat host occurred.
Finally, despite a confusing array of reports confirming (Lam et al., 2020; Xiao et al., 2020; Zhang et al., 2020) and denying (Liu et al., 2020a) the pangolin origin of SARS‐CoV‐2 on small datasets, we can clearly say that, based on our analyses of large datasets both in terms of taxonomic and genomic sampling, pangolin‐hosted (Manis javanica) CoVs are side events that are not part of the origins of SARS‐CoV‐2 infections in the human population.
The importance of gene annotation, data partitioning and outgroup sampling
A central goal of genetics is to understand how nucleotides encode complex biological functions. Genome annotation through bioinformatics is crucial to attaining this goal. In addition to the understanding of the genetic underpinnings of complex functions, an efficient process of gene annotation also is key to establishing the orthology required for comprehensive phylogenetic analysis (for example, see discussion in Gonçalves et al., 2020, p. 8).
Finally, the data partitioning that is made possible due to gene annotation is central to phylogenetic systematics under different optimality criteria (Buckley et al., 2001; Nylander et al., 2004; Brown and Lemmon, 2007; Kainer and Lanfear, 2015). Because there is variation of gene content among the genera of coronaviruses, data partitioning is needed to allow outgroup sampling in any analysis that considers genes that are not shared among outgroup and ingroup sequences. Outgroup comparison serves to root the topology and polarize character transformations (Farris, 1972, 1982). Rooting with the outgroup method is required to convert a network of abstract connections into a concrete evolutionary hypothesis (Lundberg, 1972).
Rooting with the midpoint method or other forced rooting, such as on viral lineages from small carnivores, has proven to be a major distraction in the study of coronavirus zoonosis that contributed to the pandemic of SARS‐CoV‐2 (Janies et al., 2008; Wenzel, 2020). For example, this forced rooting by Guan et al. (2003), perhaps in desperation for answers and responses to the outbreak of SARS‐CoV, forced the culling of civet cats that did not contribute to control the current or future HCoV outbreaks. Meanwhile, coronaviruses in bats began to develop the ability to directly interact with human cells (Menachery et al., 2015).
In the absence of outgroup sequences, many researchers interested in the phylogeny of viruses resort to strategies such as midpoint rooting (Liu et al., 2009; Moureau et al., 2015; Thézé et al., 2015; Kinene et al., 2016). Midpoint rooting is a fallback methodology, particularly for cases where a proper outgroup is unavailable, but can be less reliable the more inconsistent (for outgroup root consistency checks, see Maddison et al., 1984) the outgroup root is (Hess and De Moraes Russo, 2007).
There are other caveats for not performing outgroup sampling. Without outgroup comparison, we can test hypotheses of ingroup topology, but not its monophyly. We also can test the hypotheses of homology without reference to other taxas' character states, but we cannot evaluate the homologies of the entire ingroup. Finally, the inclusion of additional taxa increases the severity of the test of monophyly, and the inclusion of outgroup taxa can impact the phylogenetic relationships within the ingroup if the analysis is unconstrained (Grant, 2019).
Besides the motives delineated above, we argue that data partitioning in coronaviruses also is important owing to the variation of mutation rates. Our results unveil that mutation rates vary in different genes as well as within the same gene across different lineages. Information on the rate of mutations of viruses is required if we are to understand their mechanisms of evolution and combat them (for a discussion on the importance of estimating mutation rates in viruses, see Lynch, 2010; Sanjuán et al., 2010; Sanjuán and Domingo‐Calap, 2016; Peck and Lauring, 2018).
If we take the earliest publication dates in Table 2 as a small overestimation of each lineage's earliest emergence date, molecular clock analyses underestimated the age of HCoV‐229E and HCoV‐OC43, placing doubt on these analyses.
These results indicate the need for a thorough examination of why molecular clocks fail. These results further indicate that fields that depend on molecular clocks, such as phylodynamics, may overpromise pandemic forecasting.
The mixed results from treetime experiments underscore the importance of more research and development into gene annotation and data partitioning for phylogenetic systematics and translational work that depends on phylogenetics. For additional discussion on the importance of gene annotation and outgroup sampling in viral phylogenomics, see Schneider et al. (2020).
For example, Table 2 also shows that coronaviruses' mutation rates vary considerably depending on the clade and data partition. This variation of mutation rates impacts downstream analysis, such as inference of putative clocklike evolution, and pose challenges to translational research, such as developing new therapies and vaccines that viral lineages may rapidly escape.
Therefore, it is clear that mutation rates in Orthocoronavirinae should be calculated for each gene and each viral lineage individually. This may be challenging because bioinformatic tools for gene annotation in Orthocoronavirinae are lacking. The results presented herein justify the development of theory and practice for Orthocoronavirinae gene annotation and an effort to map mutation rates across its different lineages and genes.
Phylogenetic relationships in the SARS‐CoV‐2 clade
The lack of informative sites for the SARS‐CoV‐2 analysis results in a large polytomy in which only two sequences from patients from Finland appear to be consistently nested together. However, other authors have recently presented analyses of SARS‐CoV‐2 in humans in which they purport to find distinct clades (de Jesus et al., 2020; Fauver et al., 2020) These authors aimed to discuss the epidemiology of COVID‐19 itself. Our scope in this paper is much broader as we cover Orthocoronavirinae.
Our phylogenetic analyses consistently point to the bat‐hosted coronavirus RaTG13 (NCBI GenBank accession number MN996532) as the sister taxon to the SARS‐CoV‐2 clade. This bat‐hosted coronavirus was sequenced from a faecal swab taken from a Rhinolophus affinis bat located in China. That information by itself strongly suggests that SARS‐CoV‐2 originated from China. No new data have been released that changed this result.
On putative recombination events and the origins of SARS‐CoV‐2
Although we see no reservoir host in the history of SARS‐CoV‐2 there is a possibility that the viral lineage is a recombinant. In that case, SARS‐CoV‐2 could have had more different hosts for different parts for the genome. The specialized literature recognizes recombination between and within virus genomes as a major driver of virus evolution. “Viral recombination occurs when viruses of two different parent strains co‐infect the same host cell and interact during replication to generate virus progeny that have some genes from both parents. Recombination generally occurs between members of the same virus type (e.g. between two influenza viruses or between two herpes simplex viruses). Two mechanisms of recombination have been observed for viruses: independent assortment [in which viruses that have segmented genomes trade segments during replication] and incomplete linkage [between genes residing on the same piece of nucleic acid]. Either mechanism can produce new viral serotypes or viruses with altered virulence” (Fleischmann Jr., 1996). “In many different groups of viruses, genetic recombination is an important evolutionary process that generates much of the genetic diversity upon which natural selection acts” (Martin et al., 2015). Reviewing recombination in viruses is beyond our scope but is available elsewhere (Worobey and Holmes, 1999; Hu et al., 2003; Dolan et al., 2018).
Many recombination events were observed in different CoVs. Genetic recombination has been documented previously for animal CoVs, including MHV, TGEV, and feline and canine coronaviruses. Genetic recombination for HCoVs such as OC43, NL63, HKU1 and SARS‐CoV also has been observed. Some authors argue that the recombination of CoV in camels resulted in a dominant MERS‐CoV lineage that caused human outbreaks in 2015. For a review of epidemiology, genetic recombination and pathogenesis of CoVs, see Su et al. (2016).
Recently, Li et al. (2020) and Shang et al. (2020b) argued that there is molecular evidence indicating that the entire RBM in the spike glycoprotein of SARS‐CoV‐2 was introduced through recombination with coronaviruses from pangolins. According to the authors, this was a critical step in the evolution of SARS‐CoV‐2's ability to infect humans. It is imperative that we understand these claims from a phylogenetic perspective, taking into account all of the assumptions that are made during the inference of putative recombination events.
Li et al. (2020) compared the genomes of the SARS‐CoV‐2 virus (Wuhan‐Hu‐1, NC_045512.2) to six bat SARS‐like coronaviruses (Bat_SL‐CoVs; including the RaTG13 with GISAID accession number EPI_ISL_402131), one SARS‐CoV, and the two pangolin SARS‐like coronaviruses sampled from Guangdong (Pan_SL‐CoV_GD; including GISAID accession number EPI_ISL_410721) using SimPlot analysis (Lole et al., 1999) and the recombination detection tool RIP (Siepel and Korber, 1995). There are a number of algorithms such as these that can be used to infer putative recombination events between viral genomes (see Robertson and Feyertag, 2020). All of them use a sliding window of a certain length, frequently ranging from 100 to 500 bp, that is moved across a multiple genome alignment at steps that usually range from 2 to 10 bp. The sliding window is used to create a subset of data that will be used for the different similarity and phylogenetic analyses. Imagine, for example, a dataset of the cladogram (A, (B, (C, D))). When abrupt modifications in the similarity or phylogenetic patterns are observed, suggesting that D is more similar to B than C, or that the topology (A, (C, (B, D))) is more likely than (A, (B, (C, D))), this could be explained by random mutation, convergent evolution or recombination. Recombination is frequently considered the most likely explanation. However, it is important to note that the probability of convergence and the probability of recombination are not compared directly as both are not trivial to compute.
Among all known CoVs, SARS‐CoV‐2 shares the highest level of genetic similarity (96.3%) with a Bat_SL‐CoV named RaTG13 (GISAID accession number EPI_ISL_402131), sampled from a bat in Yunnan in 2013 (Zhou P. et al., 2020). That means that the distance between the human‐hosted SARS‐CoV‐2 and the bat‐hosted RaTG13 (3.7%) is c.2.3 times smaller than the distance between SARS‐CoV‐2 and the pangolin‐hosted Pan_SL‐CoV_GD‐like viruses (8.8%). However, when Li et al. (2020) used simplot and RIP to compare Wuhan‐Hu‐1 to Pan_SL‐CoV_GD and RaTG13, the authors found evidence for significant recombination breakpoints before and after the RBM (Li, 2016; Walls et al., 2020), as well as in other locations of the genome. About the findings, Li et al. (2020) wrote: “these observations suggest ancestral cross‐species recombination between pangolin and bat CoVs in the evolution of SARS‐CoV‐ 2 at the ORF1a and S genes. Furthermore, the discordant phylogenetic clustering at various regions of the genome among clade 2 CoVs also supports extensive recombination among these viruses isolated from bats and pangolins.”
Here, we show that the potential contribution from pangolin‐hosted CoVs to the genome of SARS‐CoV‐2 is likely only associated with the RBM in the S gene and correspond to as few as 207 nucleotides if we consider sliding windows from dualbrothers analysis overlapping regions in which Wuhan‐Hu‐1 significantly matches Pan_SL‐CoV_GD better than RaTG13, bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21. Those 207 nucleotides correspond to 0.69% of the complete Wuhan‐Hu‐1 genome. In this section of the alignment, there are 25 nucleotide differences between Wuhan‐Hu‐1 and Pan_SL‐CoV_GD, including a single nonsynonymous SNP. However, in this same region there are 57 SNPs between Wuhan‐Hu‐1 and RaTG13, with 15 nonsynonymous SNPs.
Considering the number of nucleotide substitutions and indels that separate the genomes of pangolin‐hosted‐CoVs from SARS‐CoV‐2, recombination events are not required to explain the observed sequence similarities because other processes, such as convergent evolution, cannot be ruled out. However, even if we explain the similarities between Wuhan‐Hu‐1 and Pan_SL‐CoV_GD as the product of ancestral recombination, the results presented by Li et al. (2020) as well as our inferences of recombination in the RBM of SARS‐CoV‐2 are not inconsistent with the phylogeny of Orthocoronavirinae presented herein.
We can explain the significant nucleotide similarity between bat coronavirus RaTG13 and SARS‐CoV‐2 by their proximal phylogenetic relationship despite possible ancestral recombination events. Historically, a recombination event involving a pangolin‐hosted virus sampled from Guangdong (Pan_SL‐CoV_GD) and unknown bat‐hosted viruses could have contributed to a tiny portion (<1.0%) of the genome of SARS‐CoV‐2. Nevertheless, this putative recombination event could have occurred even if RaTG13 and SARS‐CoV‐2 are in a clade that does not include pangolin‐hosted CoVs and the phylogeny presented here is not in itself sufficient to corroborate or falsify these hypotheses.
SARS‐CoV‐2 and the perceived need for an intermediate host
The known viral genomes that are most similar to SARS‐CoV‐2 include CoV RaTG13 (96.3%) and Pan_SL‐CoV_GD (91.2%). Ye et al. (2020) argued that the sequence divergence between SARS‐CoV‐2 and RaTG13 (3.7%) is too significant to assign ancestral relationships. Furthermore, Ye et al. (2020) advocated that an intermediate host between bats and humans would be necessary to explain the emergence of SARS‐CoV‐2 unless almost identical bat CoVs are found.
Serological evidence of humans from Jinning County, Yunnan Province, China being infected with viruses in 2015 that were also known from bat hosts is presented in Wang et al. (2018). Several of the infected people reported handling bats or seeing bats in their village. One of these infected people reported cross‐China travel to Shenzen.
According to Wassenaar and Zou (2020), “The use of bats in TCM [traditional Chinese medicine] is of great concern, and the use of the Greater horseshoe bat, Rhinolophus ferrumequinum, is of particular interest. The feces of this bat (Yè ming shǎ in Chinese [...]) is used to cure eye conditions, while body parts are dried and added to wine or ground into a powder for oral intake as a means to ‘detoxify’ the body.” (Greger, 2020) also mentioned Yè ming shǎ: “For only about thirty dollars a pound, anyone can go online and buy Chinese bat feces (Yè ming shǎ) to ‘treat… eye disorders.’ ” Also from Greger (2020): “While the drying of excrement would presumably inactivate coronavirus, the handling and trade of live and recently killed bats for use in traditional remedies could infect people directly or introduce opportunities for cross‐infection with other susceptible hosts. Even now, the Chinese government has been pushing traditional animal‐based remedies for the treatment of COVID‐19.”
Since the publication of Wassenaar and Zou (2020) and Greger (2020), the Chinese government has banned the eating and trading of wildlife due to the coronavirus crisis. Differently from previous efforts to regulate the management of wildlife in China, the current ban is expected to have permanent effects as it becomes law in the next few months. Nevertheless, the proposed legislation has loopholes for trade in wild animals for medicinal uses (Wildlife Conservation Society, 2020). Moreover, there are many ways besides traditional Chinese medicine that humans would come in contact with bats hosting coronaviruses or other potential human pathogens.
Bat‐hosted viruses of many taxa infect wild animals, domestic animals and humans (Plowright et al., 2015). A few examples include filoviruses (Ebola and Marburg virus), henipaviruses (Hendra and Nipah virus) and coronaviruses (SARS‐CoV), all of which cause severe disease in recipient hosts and have the potential to become pandemic (Chua et al., 2000; Leroy et al., 2005; Janies et al., 2008).
Therefore, if the divergence between CoV RaTG13 and SARS‐CoV‐2 is perceived as too high to place the former as the immediate ancestor of the latter, this does not mean that the immediate ancestor of SARS‐CoV‐2 has to be a virus hosted by an animal other than a bat.
It is reasonable to assume that we have not yet identified the CoVs that are more similar to SARS‐CoV‐2 owing to sampling bias. This realizeation leads to increased demand for screening wildlife for viruses immediately associated with transmission events leading to human infections. This realization also strengthens increasing claims for more rigorous wildlife disease surveillance as a strategy to abate future zoonotic disease outbreaks (Watsa and Wildlife Disease Surveillance Focus Group, 2020). However, given that bat‐hosted viruses are frequently associated with emerging zoonoses (Plowright et al., 2015) and that SARS‐CoV‐2 is phylogenetically closer to bat‐hosted CoVs than to CoVs hosted by any other animal, we can place bats as priority targets in efforts to increase our knowledge about the world's virome in general and the emergence of SARS‐CoV‐2 in particular.
Conflict of interest
None declared.
Supporting information
Appendix S1. List of supplementary digital materials with file descriptions. In all files containing molecular information from GISAID's EpiCoV database, we masked GISAID data; namely, each nucleotide was replaced by missing data (“?” or “N”), in compliance with that database's policies.
Appendix S1 a. Accession numbers of selected terminals.
Appendix S1 b. Data matrix and partition data.
Appendix S1 c. Scripts used for tree search.
Appendix S1 d. Protocol for recombination analyses.
Appendix S1 e. Graphical abstract.
Appendix S1 f. Best heuristic solutions from parsimony analyses.
Appendix S1 g. Scatter plots and histograms showing bootstrap values and clade sizes from parsimony analysis.
Appendix S1 h. Complete consensus tree from parsimony analyses.
Appendix S1 i. Table and tree describing host transformations.
Appendix S1 j. Digital files resulting from treetime analyses.
Appendix S1 k. Description of results from recombination detection analysis.
Appendix S1 l. Maximum‐likelihood trees.
Appendix S1 m. Subsets used for sensitivity analysis.
Appendix S1 n. Digital files resultong from the phylogenetic analyses of the SARS‐CoV‐2 related clade.
Appendix S1 o. Alignment comparisons in the SARS‐CoV‐2‐related clade.
Appendix S1 p. Alignment comparisons of the repeat binding motif of the spike glycoprotein.
Appendix S2. The complete GISAID acknowledgement table.
Appendix S3. Glossary.
Acknowledgements
Our methodology greatly benefits from comments from James Titus‐Mcquillan. We thank Ian J. Kitching for comments and suggestions in the final version of this manuscript. We gratefully acknowledge all of the scientists and submit laboratories listed (see “Supplementary Acknowledgement Table”, Appendix S2); without the aboratories involved in the collection, processing and deposition of SARS‐CoV‐2 sequences and the metadata in GISAID, we could not have completed this work. DJ and DM would like to acknowlege the support of the Belk family. We all would like to acknowledge UNC Charlotte and its various entities (The College of Computing and Informatics, the Department of Bioinformatics and Genomics, the University Professional Internship Program, the Bioinformatics Research Center and University Research Computing) for salary, space and computing support. The authors have no conflict of interest to declare.
References
- Abdel‐Moneim, A.S. and Abdelwhab, E.M. , 2020. Evidence for SARS‐CoV‐2 infection of animal hosts. Pathogens 9, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. , 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andersen, K.G. , Rambaut, A. , Lipkin, W.I. , Holmes, E.C. and Garry, R.F. , 2020. The proximal origin of SARS‐CoV‐2. Nat. Med. 26, 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annan, A. , Baldwin, H.J. , Corman, V.M. , Klose, S.M. , Owusu, M. , Nkrumah, E.E. , Badu, E.K. , Anti, P. , Agbenyega, O. , Meyer, B. et al., 2013. Human betacoronavirus 2c EMC/2012–related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten, J. , 2005. A review of long‐branch attraction. Cladistics 21, 163–193. [DOI] [PubMed] [Google Scholar]
- Bogdanowicz, D. and Giaro, K. , 2011. Matching split distance for unrooted binary phylogenetic trees. IEEE/ACM Trans. Comput. Biol. Bioinf. 9, 150–160. [DOI] [PubMed] [Google Scholar]
- van Boheemen, S. , de Graaf, M. , Lauber, C. , Bestebroer, T.M. , Raj, V.S. , Zaki, A.M. , Osterhaus, A.D. , Haagmans, B.L. , Gorbalenya, A.E. , Snijder, E.J. et al., 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3, e00473‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau, M.J. and Kapil, S. , 2010. Bovine coronavirus associated syndromes. Vet. Clin. Food Anim. Pract. 26, 123–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolles, M. , Donaldson, E. and Baric, R. , 2011. SARS‐CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr. Opin. Virol. 1, 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brielle, E.S. , Schneidman‐Duhovny, D. and Linial, M. , 2020. The SARS‐CoV‐2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses 12, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.M. and Lemmon, A.R. , 2007. The importance of data partitioning and the utility of Bayes factors in Bayesian phylogenetics. Syst. Biol. 56, 643–655. [DOI] [PubMed] [Google Scholar]
- Buckley, T.R. , Simon, C. and Chambers, G.K. , 2001. Exploring among‐site rate variation models in a maximum likelihood framework using empirical data: effects of model assumptions on estimates of topology, branch lengths, and bootstrap support. Syst. Biol. 50, 67–86. [PubMed] [Google Scholar]
- Caldwell, E. , 2008. Evolutionary history of SARS supports bats as virus source. Available at: https://news.osu.edu/evolutionary‐historyof‐sars‐supports‐bats‐as‐virus‐source/. Accessed 1 May 2020.
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. , 2009. BLAST+: architecture and applications. BMC Bioinform. 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention , 2005. SARS FAQ. Available at: https://www.cdc.gov/sars/about/faq.html. Accessed 1 May 2020.
- Centers for Disease Control and Prevention , 2020. Human coronavirus types. Available at: https://www.cdc.gov/coronavirus/types.html. Accessed August 2020.
- Chernomor, O. , Von Haeseler, A. and Minh, B.Q. , 2016. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K. , Bellini, W. , Rota, P. , Harcourt, B. , Tamin, A. , Lam, S. , Ksiazek, T. , Rollin, P. , Zaki, S. , Shieh, W.‐J. et al., 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288, 1432–1435. [DOI] [PubMed] [Google Scholar]
- Corman, V.M. , Baldwin, H.J. , Tateno, A.F. , Zerbinati, R.M. , Annan, A. , Owusu, M. , Nkrumah, E.E. , Maganga, G.D. , Oppong, S. , Adu‐Sarkodie, Y. et al., 2015. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 89, 11858–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V.M. , Eckerle, I. , Memish, Z.A. , Liljander, A.M. , Dijkman, R. , Jonsdottir, H. , Ngeiywa, K.J.J. , Kamau, E. , Younan, M. , Al Masri, M. et al., 2016. Link of a ubiquitous human coronavirus to dromedary camels. Proc. Natl. Acad. Sci. USA 113, 9864–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V.M. , Ithete, N.L. , Richards, L.R. , Schoeman, M.C. , Preiser, W. , Drosten, C. and Drexler, J.F. , 2014. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 88, 11297–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten, M. , Lam, T.T. , Watson, S.J. , Palser, A.L. , Petrova, V. , Grant, P. , Pybus, O.G. , Rambaut, A. , Guan, Y. , Pillay, D. et al., 2013. Full‐genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg. Infect. Dis. 19, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J. , Li, F. and Shi, Z.‐L. , 2019. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyranoski, D. , 2017. SARS outbreak linked to Chinese bat cave. Nature 552, 15–16. [DOI] [PubMed] [Google Scholar]
- Darling, A.E. , Mau, B. and Perna, N.T. , 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan, P.T. , Whitfield, Z.J. and Andino, R. , 2018. Mechanisms and concepts in RNA virus population dynamics and evolution. Annu. Rev. Virol. 5, 69–92. [DOI] [PubMed] [Google Scholar]
- Duraes‐Carvalho, R. , Caserta, L.C. , Barnabé, A.C. , Martini, M.C. , Ferreira, H.L. , Felippe, P.A. , Santos, M.B. and Arns, C.W. , 2015. Coronaviruses detected in Brazilian wild birds reveal close evolutionary relationships with Beta‐ and Deltacoronaviruses isolated from mammals. J. Mol. Evol. 81, 21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris, J.S. , 1972. Estimating phylogenetic trees from distance matrices. Am. Nat. 106, 645–668. [Google Scholar]
- Farris, J.S. , 1982. Outgroups and parsimony. Syst. Zool. 31, 328–334. [Google Scholar]
- Fauver, J.R. , Petrone, M.E. , Hodcroft, E.B. , Shioda, K. , Ehrlich, H.Y. , Watts, A.G. , Vogels, C.B. , Brito, A.F. , Alpert, T. , Muyombwe, A. et al., 2020. Coast‐to‐coast spread of SARS‐CoV‐2 during the early epidemic in the United States. Cell 181, 990–996.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann, W.R. Jr. , 1996. Viral Genetics, 4th edn, chapter Chapter 43. University of Texas Medical Branch at Galveston, Galveston, TX. Available at: https://www.ncbi.nlm.nih.gov/books/NBK8439/. Accessed 24 January 2021. [PubMed] [Google Scholar]
- Forni, D. , Cagliani, R. , Clerici, M. and Sironi, M. , 2017. Molecular evolution of human coronavirus genomes. Trends Microbiol. 25, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway, S.E. , Paul, P. , MacCannell, D.R. , Johansson, M.A. , Brooks, J.T. , MacNeil, A. , Slayton, R.B. , Tong, S. , Silk, B.J. , Armstrong, G.L. et al., 2021. Emergence of SARS‐CoV‐2 B.1.1.7 Lineage — United States, December 29, 2020–January 12, 2021. MMWR Morb. Mortal. Wkly Rep. 70, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloboff, P.A. , Farris, J.S. and Nixon, K.C. , 2008. TNT, a free program for phylogenetic analysis. Cladistics 24, 774–786. [Google Scholar]
- Gonçalves, C.C. , Bruce, T. , de Silva, C.O.G. , Fillho, E.X.F. , Noronha, E.F. , Carlquist, M. and Parachin, N.S. , 2020. Bioprospecting microbial diversity for lignin valorization: dry and wet screening methods. Front. Microbiol. 11, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, T. , 2019. Outgroup sampling in phylogenetics: Severity of test and successive outgroup expansion. J. Zool. Syst. Evol. Res. 57, 748–763. [Google Scholar]
- Grant, T. , Frost, D.R. , Caldwell, J.P. , Gagliardo, R. , Haddad, C.F. , Kok, P.J. , Means, D.B. , Noonan, B.P. , Schargel, W.E. and Wheeler, W.C. , 2006. Phylogenetic systematics of dart‐poison frogs and their relatives (Amphibia: Athesphatanura: Dendrobatidae). Bull. Am. Mus. Nat. Hist. 2006, 1–262. [Google Scholar]
- Grant, T. and Kluge, A.G. , 2003. Data exploration in phylogenetic inference: scientific, heuristic, or neither. Cladistics 19, 379–418. [DOI] [PubMed] [Google Scholar]
- Grant, T. and Kluge, A.G. , 2004. Transformation series as an ideographic character concept. Cladistics 20, 23–31. [DOI] [PubMed] [Google Scholar]
- Grant, T. and Kluge, A.G. , 2007. Ratio of explanatory power (REP): a new measure of group support. Mol. Phylogenet. Evol. 44, 483–487. [DOI] [PubMed] [Google Scholar]
- Grant, T. & Kluge, A.G. , 2009. Perspective: Parsimony, explanatory power, and dynamic homology testing. Syst. Biodivers. 7, 357–363. [Google Scholar]
- Greger, M. , 2020. How to Survive a Pandemic. Flatiron Books, New York, NY. [Google Scholar]
- Guan, Y. , Zheng, B. , He, Y. , Liu, X. , Zhuang, Z. , Cheung, C. , Luo, S. , Li, P. , Zhang, L. , Guan, Y. et al., 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.‐F. , Lefort, V. , Anisimova, M. , Hordijk, W. and Gascuel, O. , 2010. New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Gutiérrez, C. , Tejedor‐Junco, M.T. , González, M. , Lattwein, E. and Renneker, S. , 2015. Presence of antibodies but no evidence for circulation of MERS‐CoV in dromedaries on the Canary Islands, 2015. Eurosurveillance 20, 30019. [DOI] [PubMed] [Google Scholar]
- Han, Y. , Du, J. , Su, H. , Zhang, J. , Zhu, G. , Zhang, S. , Wu, Z. and Jin, Q. , 2019. Identification of diverse bat alphacoronaviruses and betacoronaviruses in China provides new insights into the evolution and origin of coronavirus‐related diseases. Front. Microbiol. 10, 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansa, A. , Rai, R. , Wani, M. and Dhama, K. , 2012. Pathology and diagnosis of corona virus infection in bovine. Indian J. Vet. Pathol. 36, 129–135. [Google Scholar]
- Hess, P.N. and De Moraes Russo, C.A. , 2007. An empirical test of the midpoint rooting method. Biol. J. Linn. Soc. 92, 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D.T. , Chernomor, O. , Von Haeseler, A. , Minh, B.Q. and Vinh, L.S. , 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B. , Zeng, L.‐P. , Yang, X.‐L. , Ge, X.‐Y. , Zhang, W. , Li, B. , Xie, J.‐Z. , Shen, X.‐R. , Zhang, Y.‐Z. , Wang, N. et al., 2017. Discovery of a rich gene pool of bat SARS‐related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W.‐S. , Rhodes, T. , Dang, Q. and Pathak, V. , 2003. Retroviral recombination: review of genetic analyses. Front Biosci. 8, 143–155. [DOI] [PubMed] [Google Scholar]
- Huynh, J. , Li, S. , Yount, B. , Smith, A. , Sturges, L. , Olsen, J.C. , Nagel, J. , Johnson, J.B. , Agnihothram, S. , Gates, J.E. , Frieman, M.B. , Baric, R.S. and Donaldson, E.F. , 2012. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 86, 12816–12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irigoyen, N. , Firth, A.E. , Jones, J.D. , Chung, B.‐Y.‐W. , Siddell, S.G. and Brierley, I. , 2016. High‐resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 12, e1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janies, D. , Habib, F. , Alexandrov, B. , Hill, A. and Pol, D. , 2008. Evolution of genomes, host shifts and the geographic spread of SARSCoV and related coronaviruses. Cladistics 24, 111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus, J.G. , Sacchi, C. , Candido, D.S. , Claro, I.M. , Sales, F.C.S. , Manuli, E.R. , Silva, D.B.B. , Paiva, T.M. , Pinho, M.A.B. , Santos, K.C.O. et al., 2020. Importation and early local transmission of COVID‐19 in Brazil, 2020. Rev. Inst. Med. Trop. São Paulo 62, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, W. , Wang, W. , Zhao, X. , Zai, J. and Li, X. , 2020. Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J. Med. Virol. 92, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainer, D. and Lanfear, R. , 2015. The effects of partitioning on phylogenetic inference. Mol. Biol. Evol. 32, 1611–1627. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B.Q. , Wong, T.K.F. , von Haeseler, A. and Jermiin, L.S. , 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K.‐I. and Miyata, T. , 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. and Standley, D.M. , 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinene, T. , Wainaina, J. , Maina, S. and Boykin, L. , 2016. Rooting Trees, Methods for, Encyclopedia of Evolutionary Biology. p. 489. 10.1016/B978-0-12-800049-6.00215-8. [DOI]
- King, A. , 2020. The hunt for the next potential coronavirus animal host. Available at: https://www.nationalgeographic.com/animals/2020/03/coronavirusanimal‐reservoir‐research/. Accessed 25 January 2021.
- King, A.M. , Lefkowitz, E. , Adams, M.J. and Carstens, E.B. , 2011. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, vol. 9. Elsevier, Amsterdam. [Google Scholar]
- Kluge, A.G. , 1989. A Concern for Evidence and a Phylogenetic Hypothesis of Relationships among Epicrates (Boidae, Serpentes). Syst. Biol, 38 (1), 7–25. 10.1093/sysbio/38.1.7 [DOI] [Google Scholar]
- Kluge, A.G. , 2004. On total evidence: for the record. Cladistics 20, 205–207. [DOI] [PubMed] [Google Scholar]
- Kluge, A.G. and Grant, T. , 2006. From conviction to anti‐superfluity: old and new justifications of parsimony in phylogenetic inference. Cladistics 22, 276–288. [Google Scholar]
- Ksiazek, T.G. , Erdman, D. , Goldsmith, C.S. , Zaki, S.R. , Peret, T. , Emery, S. , Tong, S. , Urbani, C. , Comer, J.A. , Lim, W. et al., 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- Lai, M.M. and Cavanagh, D. , 1997. The molecular biology of coronaviruses, advances in virus research. Elsevier, Vol. 48, pp. 1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, T.‐T.‐Y. , Jia, N. , Zhang, Y.‐W. , Shum, M.‐H.‐H. , Jiang, J.‐F. , Zhu, H.‐C. , Tong, Y.‐G. , Shi, Y.‐X. , Ni, X.‐B. , Liao, Y.‐S. et al., 2020. Identifying SARS‐CoV‐2‐related coronaviruses in Malayan pangolins. Nature 583, 282–285. [DOI] [PubMed] [Google Scholar]
- Lan, J. , Ge, J. , Yu, J. , Shan, S. , Zhou, H. , Fan, S. , Zhang, Q. , Shi, X. , Wang, Q. , Zhang, L. et al., 2020. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature 581, 215–220. [DOI] [PubMed] [Google Scholar]
- Lanfear, R. , Calcott, B. , Ho, S.Y.W. & Guindon, S. , 2012. Partition‐Finder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. [DOI] [PubMed] [Google Scholar]
- Lau, S.K. , Woo, P.C. , Li, K.S. , Huang, Y. , Tsoi, H.W. , Wong, B.H. , Wong, S.S. , Leung, S.Y. , Chan, K.H. and Yuen, K.Y. , 2005. Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 102, 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy, E.M. , Kumulungui, B. , Pourrut, X. , Rouquet, P. , Hassanin, A. , Yaba, P. , Délicat, A. , Paweska, J.T. , Gonzalez, J.‐P. and Swanepoel, R. , 2005. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. [DOI] [PubMed] [Google Scholar]
- Li, B. , Ge, J. and Li, Y. , 2007. Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology 365, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , 2016. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Li, W. , Farzan, M. and Harrison, S.C. , 2005. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science 309, 1864–1868. [DOI] [PubMed] [Google Scholar]
- Li, X. , Giorgi, E.E. , Marichannegowda, M.H. , Foley, B. , Xiao, C. , Kong, X.‐P. , Chen, Y. , Gnanakaran, S. , Korber, B. and Gao, F. , 2020. Emergence of SARS‐CoV‐2 through recombination and strong purifying selection. Science Advances 6, eabb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljander, A. , Meyer, B. , Jores, J. , Müller, M. , Lattwein, E. , Njeru, I. , Bett, B. , Drosten, C. and Corman, V.M. , 2016. MERS‐CoV antibodies in humans, Africa, 2013–2014. Emerg. Infect. Dis. 22, 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Xia, H. , Wahlberg, N. , Belák, S. and Baule, C. , 2009. Phylogeny, classification and evolutionary insights into pestiviruses. Virology 385, 351–357. [DOI] [PubMed] [Google Scholar]
- Liu, P. , Jiang, J.Z. , Wan, X.F. , Hua, Y. , Li, L. , Zhou, J. , Wang, X. , Hou, F. , Chen, J. , Zou, J. et al., 2020a. Are pangolins the intermediate host of the 2019 novel coronavirus SARS‐CoV‐2? PLoS Pathog. 16, e1008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Xiao, X. , Wei, X. , Li, J. , Yang, J. , Tan, H. , Zhu, J. , Zhang, Q. , Wu, J. and Liu, L. , 2020b. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐CoV‐2. J. Med. Virol. 92, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole, K.S. , Bollinger, R.C. , Paranjape, R.S. , Gadkari, D. , Kulkarni, S.S. , Novak, N.G. , Ingersoll, R. , Sheppard, H.W. and Ray, S.C. , 1999. Full‐length human immunodeficiency virus type 1 genomes from subtype c‐infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R. , Zhao, X. , Li, J. , Niu, P. , Yang, B. , Wu, H. , Wang, W. , Song, H. , Huang, B. et al., 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg, J.G. , 1972. Wagner networks and ancestors. Syst. Biol. 21, 398–413. [Google Scholar]
- Luytjes, W. , 1995. Coronavirus gene expression. In: Siddell, S. G. (Ed.), The Coronaviridae. Springer, Boston, MA, pp. 33–54. [Google Scholar]
- Lynch, M. , 2010. Evolution of the mutation rate. Trends Genet. 26, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, D.J. , 2015. YBYRÁ facilitates comparison of large phylogenetic trees. BMC Bioinformat. 16, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison, W.P. , Donoghue, M.J. & Maddison, D.R. , 1984. Outgroup analysis and parsimony. Syst. Biol. 33, 83–103. [Google Scholar]
- Mandelik, R. , Sarvas, M. , Jackova, A. , Salamunova, S. , Novotny, J. and Vilcek, S. , 2018. First outbreak with chimeric swine enteric coronavirus (SeCoV) on pig farms in Slovakia–lessons to learn. Acta Vet. Hung. 66, 488–492. [DOI] [PubMed] [Google Scholar]
- Martin, D.P. , Murrell, B. , Golden, M. , Khoosal, A. and Muhire, B. , 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery, V.D. , Yount, B.L. , Debbink, K. , Agnihothram, S. , Gralinski, L.E. , Plante, J.A. , Graham, R.L. , Scobey, T. , Ge, X.‐Y. , Donaldson, E.F. et al., 2015. A SARS‐like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin, V.N. , Dorman, K.S. , Fang, F. and Suchard, M.A. , 2005. Dual multiple change‐point model leads to more accurate recombination detection. Bioinformatics 21, 3034–3042. [DOI] [PubMed] [Google Scholar]
- Mou, H. , Raj, V.S. , Van Kuppeveld, F.J. , Rottier, P.J. , Haagmans, B.L. and Bosch, B.J. , 2013. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231‐residue region in the spike protein that efficiently elicits neutralizing antibodies. J. Virol. 87, 9379–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moureau, G. , Cook, S. , Lemey, P. , Nougairede, A. , Forrester, N.L. , Khasnatinov, M. , Charrel, R.N. , Firth, A.E. , Gould, E.A. and De Lamballerie, X. , 2015. New insights into Flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS One 10, e0117849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , Von Haeseler, A. and Minh, B.Q. , 2015. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normile, D. , 2004. Viral DNA match spurs China’s civet roundup. Science 303, 292. [DOI] [PubMed] [Google Scholar]
- Nylander, J.A. , Ronquist, F. , Huelsenbeck, J.P. and Nieves‐Aldrey, J. , 2004. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53, 47–67. [DOI] [PubMed] [Google Scholar]
- Oreshkova, N. , Molenaar, R.J. , Vreman, S. , Harders, F. , Munnink, B.B.O. , Hakze‐van der Honing, R.W. , Gerhards, N. , Tolsma, P. , Bouwstra, R. , Sikkema, R.S. et al., 2020. SARS‐CoV‐2 infection in farmed minks, the Netherlands, April and May 2020. Eurosurveillance 25, 2001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink B. B., Sikkema R. S., Nieuwenhuijse D. F., Molenaar R. J., Munger E., Molenkamp R., van der Spek A., Tolsma P., Rietveld A., Brouwer M. et al., 2021. Transmission of SARS‐CoV‐2 on mink farms between humans and mink and back to humans. Science, 371 (6525), 172–177. 10.1126/science.abe5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck, K.M. and Lauring, A.S. , 2018. Complexities of viral mutation rates. J. Virol. 92, e01031‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante, J.A. , Liu, Y. , Liu, J. , Xia, H. , Johnson, B.A. , Lokugamage, K.G. , Zhang, X. , Muruato, A.E. , Zou, J. , Fontes‐Garfias, C.R. et al., 2021. Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature 592, 116–121. 10.1038/s41586-020-2895-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowright, R.K. , Eby, P. , Hudson, P.J. , Smith, I.L. , Westcott, D. , Bryden, W.L. , Middleton, D. , Reid, P.A. , McFarlane, R.A. , Martin, G. et al., 2015. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B 282, 20142124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken, C.B. , Messadi, L. , Feyisa, A. , Ularamu, H. , Godeke, G.‐J. , Danmarwa, A. , Dawo, F. , Jemli, M. , Melaku, S. , Shamaki, D. et al., 2014. Geographic distribution of MERS coronavirus among dromedary camels, Africa. Emerg. Infect. Dis. 20, 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, I. & Bleasby, A. , 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Rindal, E. and Brower, A.V.Z. , 2011. Do model‐based phylogenetic analyses perform better than parsimony? A test with empirical data. Cladistics 27, 331–334. [DOI] [PubMed] [Google Scholar]
- Robertson, D. and Feyertag, F. , 2020. Links to recombinant sequence analysis/detection programs. Available at: http://bioinf.man.ac.uk/robertson/recombination/programs.shtml. Accessed 1 June 2020.
- Rogin, J. , 2020. State Department cables warned of safety issues at Wuhan lab studying bat coronaviruses. Available at: https://www.washingtonpost.com/opinions/2020/04/14/statedepartment‐cables‐warned‐safety‐issues‐wuhan‐lab‐studying‐batcoronaviruses/. Accessed 1 May 2020.
- Roos, R. , 2004. WHO sees more evidence of civet role in SARS. Available at: https://www.cidrap.umn.edu/news‐perspective/2004/01/whosees‐more‐evidence‐civet‐role‐sars. Accessed 1 May 2020.
- Sagulenko, P. , Puller, V. and Neher, R.A. , 2018. TreeTime: maximumlikelihood phylodynamic analysis. Virus Evol. 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán, R. and Domingo‐Calap, P. , 2016. Mechanisms of viral mutation. Cell. Mol. Life Sci. 73, 4433–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán, R. , Nebot, M.R. , Chirico, N. , Mansky, L.M. and Belshaw, R. , 2010. Viral mutation rates. J. Virol. 84, 9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Schneider, A.B. , Jacob Machado, D. , Guirales, S. & Janies, D.A. , 2020. FLAVi: an enhanced annotator for viral genomes of Flaviviridae. Viruses 12, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Wan, Y. , Luo, C. , Ye, G. , Geng, Q. , Auerbach, A. and Li, F. , 2020a. Cell entry mechanisms of SARS‐CoV‐2. Proc. Natl. Acad. Sci. USA 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Ye, G. , Shi, K. , Wan, Y. , Luo, C. , Aihara, H. , Geng, Q. , Auerbach, A. and Li, F. , 2020b. Structural basis of receptor recognition by SARS‐CoV‐2. Nature 581, 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan, T. , Rockx, B. , Donaldson, E. , Sims, A. , Pickles, R. , Corti, D. and Baric, R. , 2008. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J. Virol. 82, 2274–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall, M.E. and Kluge, A.G. , 1997. Probabilism and phylogenetic inference. Cladistics 13, 313–336. [DOI] [PubMed] [Google Scholar]
- Siepel, A.C. , Halpern, A.L. , Macken, C. and Korber, B.T.M. , 1995. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retroviruses 11, 1413–1416. [DOI] [PubMed] [Google Scholar]
- Siepel, A.C. and Korber, B.T. , 1995. Scanning the database for recombinant HIV‐1 genomes. Los Alamos National Laboratory, Los Alamos, N.M., chapter 3, pp. III‐35–III‐60.
- Su, S. , Wong, G. , Shi, W. , Liu, J. , Lai, A.C. , Zhou, J. , Liu, W. , Bi, Y. and Gao, G.F. , 2016. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard, M.A. , Weiss, R.E. , Dorman, K.S. and Sinsheimer, J.S. , 2002. Oh brother, where art thou? A Bayes factor test for recombination with uncertain heritage. Syst. Biol. 51, 715–728. [DOI] [PubMed] [Google Scholar]
- Suchard, M.A. , Weiss, R.E. , Dorman, K.S. and Sinsheimer, J.S. , 2003. Inferring spatial phylogenetic variation along nucleotide sequences: a multiple changepoint model. J. Am. Stat. Assoc. 98, 427–437. [Google Scholar]
- Sun, J. , He, W. , Wang, L. , Lai, A. , Ji, X. , Zhai, X. , Li, G. , Suchard, M.A. , Tian, J. , Zhou, J. et al., 2020a. COVID‐19: epidemiology, evolution, and cross‐disciplinary perspectives. Trends Mol. Med. 26, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, P. , Qie, S. , Liu, Z. , Ren, J. , Li, K. and Xi, J. , 2020b. Clinical characteristics of hospitalized patients with SARS‐CoV‐2 infection: a single arm meta‐analysis. J. Med. Virol. 92, 612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, W. , He, L. , Zhang, X. , Pu, J. , Voronin, D. , Jiang, S. , Zhou, Y. and Du, L. , 2020. Characterization of the receptor‐binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 17, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thézé, J. , Lowes, S. , Parker, J. and Pybus, O.G. , 2015. Evolutionary and phylogenetic analysis of the hepaciviruses and pegiviruses. Genome Biol. Evol. 7, 2996–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A.C. , Park, Y.‐J. , Tortorici, M.A. , Wall, A. , McGuire, A.T. and Veesler, D. , 2020. Structure, function, and antigenicity of the SARSCoV‐2 spike glycoprotein. Cell 181, 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, Y. , Shang, J. , Graham, R. , Baric, R.S. and Li, F. , 2020. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade‐long structural studies of SARS coronavirus. J. Virol. 94, e00127‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N. , Li, S.‐Y. , Yang, X.‐L. , Huang, H.‐M. , Zhang, Y.‐J. , Guo, H. , Luo, C.‐M. , Miller, M. , Zhu, G. , Chmura, A.A. et al., 2018. Serological evidence of bat SARS‐related coronavirus infection in humans, China. Virol. Sin. 33, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar, T. and Zou, Y. , 2020. 2019 nCoV/SARS‐CoV‐2: rapid classification of betacoronaviruses and identification of Traditional Chinese Medicine as potential origin of zoonotic coronaviruses. Lett. Appl. Microbiol. 70, 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watsa, M. and Wildlife Disease Surveillance Focus Group , 2020. Rigorous wildlife disease surveillance. Science 369, 145–147. [DOI] [PubMed] [Google Scholar]
- Watts, J. , 2004. China culls wild animals to prevent new SARS threat. Lancet 363, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel, J. , 2020. Origins of SARS‐CoV‐1 and SARS‐CoV‐2 are often poorly explored in leading publications. Cladistics 36, 374–379. [DOI] [PubMed] [Google Scholar]
- Wildlife Conservation Society , 2020. WCS statement and analysis: on the Chinese government’s decision prohibiting some trade and consumption of wild animals. Available at: https://newsroom.wcs.org/News‐Releases/articleType/ArticleView/articleId/13855/WCS‐Statementand‐Analysis‐On‐the‐Chinese‐Governments‐Decision‐Prohibiting‐Some‐Trade‐and‐Consumption‐of‐Wild‐Animals.aspx. Accessed 1 June 2020.
- Woo, P.C. , Huang, Y. , Lau, S.K. and Yuen, K.‐Y. , 2010. Coronavirus genomics and bioinformatics analysis. Viruses 2, 1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P.C. , Lau, S.K. , Lam, C.S. , Lau, C.C. , Tsang, A.K. , Lau, J.H. , Bai, R. , Teng, J.L. , Tsang, C.C. , Wang, M. et al., 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J. Virol. 86, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P.C. , Lau, S.K. , Lam, C.S. , Tsang, A.K. , Hui, S.‐W. , Fan, R.Y. , Martelli, P. and Yuen, K.‐Y. , 2014. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. J. Virol. 88, 1318–1331. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- World Health Organization , 2015a. MERS outbreak in the Republic of Korea, 2015. Available at: https://www.who.int/westernpacific/emergencies/2015‐mersoutbreak. Accessed 2 July 2020.
- World Health Organization , 2015b. Update 95 ‐ SARS: chronology of a serial killer. Available at: https://www.who.int/csr/don/20030704/en/. Accessed 25 January 2021.
- World Health Organization , 2020a. Disease Outbreak News: SARSCoV‐2 Variants. Available at: https://www.who.int/csr/don/31‐december‐2020‐sars‐cov2‐variants/en/. Accessed 27 January 2021.
- World Health Organization , 2020b. MERS in the Republic of Korea. Available at: https://www.who.int/westernpacific/emergencies/mersin‐the‐republic‐of‐korea. Accessed 1 May 2020.
- World Health Organization , 2020c. Middle East respiratory syndrome coronavirus (MERS‐CoV). Available at: https://www.who.int/emergencies/mers‐cov/en/. Accessed 1 May 2020.
- World Health Organization , 2020d. Middle East respiratory syndrome coronavirus (MERS‐CoV) – The Kingdom of Saudi Arabia. Available at: https://www.who.int/csr/don/24‐february‐2020‐mers‐saudiarabia/en/. Accessed 2 July 2020.
- World Health Organization , 2020e. SARS‐CoV‐2 mink‐associated variant strain – Denmark. Available at: https://www.who.int/csr/don/06‐november‐2020‐mink‐associated‐sars‐cov2‐denmark/en/. Accessed 1 January 2021.
- World Health Organization , 2020f. SARS (severe acute respiratory syndrome). Available at: https://www.who.int/ith/diseases/sars/en/, Accessed 1 May 2020.
- World Health Organization , 2020g. WHO Director‐General’s opening remarks at the media briefing on COVID‐19. Available at: https://www.who.int/dg/speeches/detail/who‐director‐general‐sopening‐remarks‐at‐the‐media‐briefing‐on‐covid‐19—11‐march‐2020. Accessed 25 January 2021.
- World Health Organization , 2021. WHO Coronavirus Disease (COVID‐19) Dashboard. Available at: https://covid19.who.int/. Accessed 30 March 2021.
- Worobey, M. and Holmes, E.C. , 1999. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 80, 2535–2543. [DOI] [PubMed] [Google Scholar]
- Xiao, K. , Zhai, J. , Feng, Y. , Zhou, N. , Zhang, X. , Zou, J.‐J. , Li, N. , Guo, Y. , Li, X. , Shen, X. et al., 2020. Isolation of SARS‐CoV‐2‐related coronavirus from Malayan pangolins. Nature 583, 286–289. [DOI] [PubMed] [Google Scholar]
- Yang, X.‐L. , Hu, B. , Wang, B. , Wang, M.‐N. , Zhang, Q. , Zhang, W. , Wu, L.‐J. , Ge, X.‐Y. , Zhang, Y.‐Z. , Daszak, P. et al., 2016. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J. Virol. 90, 3253–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Z.‐W. , Yuan, S. , Yuen, K.‐S. , Fung, S.‐Y. , Chan, C.‐P. and Jin, D.‐Y. , 2020. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 16, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, C.W. , Hon, C.C. , Shi, M. , Lam, T.T.Y. , Chow, K.Y.C. , Zeng, F. and Leung, F.C.C. , 2009. Phylogenetic perspectives on the epidemiology and origins of SARS and SARS‐like coronaviruses. Infect. Genet. Evol. 9, 1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. , Jiang, S.‐C. and Li, Z.‐L. , 2020. Analysis of possible intermediate hosts of the new coronavirus SARS‐CoV‐2. Front. Vet. Sci. 7, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Wu, Q. and Zhang, Z. , 2020. Probable pangolin origin of SARS‐CoV‐2 associated with the COVID‐19 outbreak. Curr. Biol. 30, 1346–1351.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Herbst, W. , Kousoulas, K. and Storz, J. , 1994. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med. Virol. 44, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.‐Z. and Holmes, E.C. , 2020. A genomic perspective on the origin and emergence of SARS‐CoV‐2. Cell 181, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Zhuang, Z. , Cao, P. , Ran, J. , Gao, D. , Lou, Y. , Yang, L. , Cai, Y. , Wang, W. , He, D. et al., 2020. Quantifying the association between domestic travel and the exportation of novel coronavirus (2019‐nCoV) cases from Wuhan, China in 2020: a correlational analysis. J. Travel Med. 27, taaa022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, N. , Zheng, B. , Li, Y. , Poon, L. , Xie, Z. , Chan, K. , Li, P. , Tan, S. , Chang, Q. , Xie, J. et al., 2003. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 362, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Chen, X. , Hu, T. , Li, J. , Song, H. , Liu, Y. , Wang, P. , Liu, D. , Yang, J. , Holmes, E.C. et al., 2020. A novel bat coronavirus closely related to SARS‐CoV‐2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 30, 2196–2203.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X.‐L. , Wang, X.‐G. , Hu, B. , Zhang, L. , Zhang, W. , Si, H.‐R. , Zhu, Y. , Li, B. , Huang, C.‐L. et al., 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla, A. , Hui, D.S. and Perlman, S. , 2015. Middle East respiratory syndrome. Lancet 386, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. List of supplementary digital materials with file descriptions. In all files containing molecular information from GISAID's EpiCoV database, we masked GISAID data; namely, each nucleotide was replaced by missing data (“?” or “N”), in compliance with that database's policies.
Appendix S1 a. Accession numbers of selected terminals.
Appendix S1 b. Data matrix and partition data.
Appendix S1 c. Scripts used for tree search.
Appendix S1 d. Protocol for recombination analyses.
Appendix S1 e. Graphical abstract.
Appendix S1 f. Best heuristic solutions from parsimony analyses.
Appendix S1 g. Scatter plots and histograms showing bootstrap values and clade sizes from parsimony analysis.
Appendix S1 h. Complete consensus tree from parsimony analyses.
Appendix S1 i. Table and tree describing host transformations.
Appendix S1 j. Digital files resulting from treetime analyses.
Appendix S1 k. Description of results from recombination detection analysis.
Appendix S1 l. Maximum‐likelihood trees.
Appendix S1 m. Subsets used for sensitivity analysis.
Appendix S1 n. Digital files resultong from the phylogenetic analyses of the SARS‐CoV‐2 related clade.
Appendix S1 o. Alignment comparisons in the SARS‐CoV‐2‐related clade.
Appendix S1 p. Alignment comparisons of the repeat binding motif of the spike glycoprotein.
Appendix S2. The complete GISAID acknowledgement table.
Appendix S3. Glossary.