

Key Points
Question
Do copy number variation (CNV) signatures inform prognosis for patients with ovarian cancer?
Findings
In this genetic association study of 564 patients with serous ovarian cancer, an internally validated CNV signature from The Cancer Genome Atlas had more discriminatory ability to prognosticate overall survival than age, clinical stage, grade, and race combined, as well as gross CNV burden, total mutational burden, BRCA status, and open-source candidate genome-wide DNA repair deficiency signatures.
Meaning
These findings suggest that a CNV-based risk score is independent to and stronger than current or near-future ovarian cancer genomic biomarkers, as well as available clinical features, to prognosticate survival.
This genetic association study examines the Cancer Genome Atlas database to assess for genome-wide survival associations agnostic to gene function among patients with serous ovarian cancer.
Abstract
Importance
Tailoring therapeutic regimens to individual patients with ovarian cancer is informed by severity of disease using a variety of clinicopathologic indicators. Although DNA repair variations are increasingly used for therapy selection in ovarian cancer, molecular features are not widely used for general assessment of patient prognosis and disease severity.
Objective
To distill a highly dynamic characteristic, signature of copy number variations (CNV), into a risk score that could be easily validated analytically or repurposed for use given existing US Food and Drug Administration (FDA)–approved multigene assays.
Design, Setting, and Participants
This genetic association study used the Cancer Genome Atlas Ovarian Cancer database to assess for genome-wide survival associations agnostic to gene function. Regions enriched for significant associations were compared to associations from scrambled data. CNV associations were condensed into a risk score, which was internally validated using bootstrapping. The participants were patients with serous ovarian cancer (stages I-IV) diagnosed from 1992 to 2013. Statistical analysis was performed from April to July 2020.
Main Outcomes and Measures
Overall survival (OS).
Results
Among 564 patients with serous ovarian cancer, the mean (SD) age was 59.7 (11.5) years; 34 (6%) identified as Black or African American. A total of 13 genome regions, comprising 14 alterations, were identified as significantly risk associated. Composite risk score was independent of total CNV burden, total mutational burden, BRCA status, and open-source genome-wide DNA repair deficiency signatures. Binned terciles yielded high-, standard-, and low-risk groups with respective median OS estimates of 2.9 (95% CI, 2.3-3.2) years, 4.1 (95% CI, 3.7-4.8) years, and 5.7 (95% CI, 4.7-7.4) years, respectively (P < .001). Associated 5-year survival estimates in each tercile were 15% (95% CI, 10%-22%), 36% (95% CI, 29%-46%), and 53% (95% CI, 45%-62%). The risk score had more discriminatory ability to prognosticate OS than age, clinical stage, grade, and race combined, and was strongly additive to significant clinical features (P < .001). Simulated adaptation of FDA-approved assays showed similar performance. Gene ontology analyses of identified regions showed an enrichment for regulatory miRNAs and protein kinase regulators.
Conclusions and Relevance
This study found that a CNV-based risk score is independent to and stronger than current or near-future ovarian cancer genomic biomarkers to prognosticate OS. CNV regions identified were not strongly associated with canonical ovarian cancer biological pathways, identifying candidates for future mechanistic investigations. External validation of the CNV risk score, especially in concert with more extensive clinical features, could be pursued via existing FDA-approved assays.
Introduction
Ovarian cancer remains the most lethal gynecologic malignancy, with an estimated 21 750 new cases and 13 940 deaths predicted in the United States for 2020.1 Despite notable recent advancements in the use of PARP inhibitors, particularly as maintenance therapy2,3,4 (neo)adjuvant combination platinum-based chemotherapy regimens and surgical cytoreduction have remained frontline treatment for newly diagnosed ovarian cancer for the last 40 years. Breast cancer gene 1 (BRCA1) and BRCA2 variations (germline and somatic) have been reported in up to 20% of patients with high grade serous ovarian cancer, with nearly 50% harboring homologous recombination deficiency (HRD), both associated with significant clinical benefit to PARP inhibitor therapy.2,3,4,5,6 Beyond this, clinical approaches to managing ovarian cancer have not broadly benefited from precision medicine. A range of decisions are required to tailor therapeutic and follow-up regimens to individual patients, all of which are directly or indirectly informed by the general prognosis. The ability to prognosticate survival with standard of care in this patient population, especially long-term survival, is useful to guide therapeutic choices as well as patient follow-up and monitoring.
A number of risk scores and nomograms have been developed to help consolidate and prognosticate ovarian cancer patient risk of death over time. Most clinically used nomograms focus strongly on clinicopathological features.7,8,9 Although inexpensive and ubiquitous, clinical features such as performance status are prone to degrees of subjectivity and considerable interoperator discordance.10,11 It has been suggested that risk assessments that incorporate genomic features might add accuracy and robustness.
Prognostic models incorporating variable genomic parameters have been forwarded to better predict long-term survival in ovarian cancer. Some of these outperform models based solely upon clinicopathological features,12,13,14 but most have not been widely adopted for use because of the complex laboratory infrastructure required. Translating a molecular model or signature to clinical use requires robust, scalable, and readily available diagnostics. The development and implementation of any molecular diagnostics involves a tradeoff between scalability, complexity, and cost.
Different cancer types display varying degrees of copy number variations (CNV), for which serous ovarian cancer has a wide spectrum and diversity of alterations.15,16 CNV assessments can be readily performed on tumor samples, even those that are formalin-fixed paraffin-embedded (FFPE)–derived DNA, using low coverage whole genome sequencing (<20×, <2× reported) with low cost and bioinformatic load.17,18 These approaches are robust relative to protein and mRNA, which require greater preanalytic handling and care. Importantly, a growing number of health care insurers are reimbursing commercially available hybrid capture multigene assays, which both incorporate CNV information and are approved by the FDA.
Given these factors, we were motivated to explore the potential for a signature risk score using CNV that was additive to existing clinicopathological features. Critically, such an approach had to be amenable to the use of simple, cost-effective and/or available and reimbursed molecular diagnostic technology.
Methods
A flowchart of the approach to analyses is provided as eFigure 1 in the Supplement. All specimens were obtained from patients with appropriate consent from the relevant institutional review board. As this study was conducted using publicly available and deidentified data, it did not require institutional review board approval or informed patient consent, in accordance with 45 CFR §46.
Data
The TCGA Firehose Ovarian Cancer data set contains both genomic and clinical outcomes data,19 including both genome-wide copy number assessments for more than 20 000 genes and overall survival data for nearly 600 patients with ovarian cancer, some with more than 10 years of follow-up. Race/ethnicity data were self-reported. The TCGA Firehose database was last accessed on July 2, 2020; analyses reflected these data. CNV data used the provided Genomic Identification of Significant Targets in Cancer (GISTIC)20 calls directly without further tuning.
Statistical Analysis
Model Building
Because it cannot be assumed that a degree of gain or loss of copy number of a gene will have a linear relationship with expression, nor that the linear scale would carry an approximate normal distribution from gene to gene, gene gains and losses were assessed separately as individual dichotomous variables. A GISTIC call of +1 or +2 was considered a gain and −1 or −2 was considered a loss. The patterns of gene gains and losses in ovarian cancer suggest large chromosomal aberrations, as opposed to focal amplifications, as the dominant driving force for CNV alterations.21 Conceptually, we prioritized an outcomes-first approach to model development. We use the term reporter in lieu of gene alteration (including gene gain or gene loss) because the approach is agnostic to the biological causality of specific genes on outcomes. It does not discriminate that causative drivers might be neighboring lesions or aggregate phenomenon rather than the reporter identified. For every gene, 2 reporters were initially assessed: one as a gain and one as a loss. The univariable association with OS was assessed with Cox proportional hazards (PH) models per reporter (gain, loss separately).
To understand the rate and tendencies of false discovery in this data set prior to model building, we evaluated OS associations per reporter separately but with the GISTIC CNV gene calls scrambled within each patient (ie, the GISTIC value for gene X became the value for gene Y, what was Y became Z, randomly, for all genes, per patient). The locations of highly statistically significant alterations in the genome were compared between real and scrambled data.
The distribution of P values generated from the univariable OS assessments of reporters from the scrambled data was used to create a cutoff (above upper 99% CI of −log10 of P values). These reporters were subsequently binned by genomic location, and the reporter at the peak univariable association per region was chosen for the final Cox PH model without further forward or backward feature selection. Orthogonally, LASSO regression22 was used as a complementary technique, and the results were compared. The statistical significance threshold was set at P < .01.
For internal (within data set) validation of the chosen reporters, the CNV Cox PH model was subjected to bootstrapping based on 90:10 (train:test) partitions and 10 000 iterations of resampling with replacement. During each iteration, a model was built with the aforementioned reporters using the training data, which was used to predict a risk score for the held-out test data. The mean risk score per patient from these iterations of held-out data was used in all subsequent univariable and multivariable assessments of the risk score. Leave-one-out cross validation (LOOCV) was used as a secondary technique for internal validation and results were compared.
Model Evaluation
The absolute survival estimate per range of risk score was assessed with the Kaplan-Meier method. The discriminatory ability at specific time points was assessed with the area under the curve (AUC) of time-dependent receiver operating characteristic (ROC) curves. The additive and independent value for prognostication of relative hazards of death relative to existing clinicopathological features in the TCGA database were assessed with a multivariable Cox PH model constructed stepwise. Considered for the model were clinicopathological features with entries available for more than 80% of patients in the TCGA data set with P < .05 univariable association with overall survival. Only clinical stage, age, and risk score met these criteria and were included in the model.
Gene Ontology
The incidence of molecular function ontologies associated with gene alterations identified within vs not within regions of enriched survival associations was evaluated. The molecular function associated with gene alterations enriched in survival-associated regions was evaluated. Genes were scored using the curated Kyoto Encyclopedia of Genes and Genomes Pathway Database.
Software
R software version 3.6.1 (R Project for Statistical Computing) was used for statistical analyses and data visualization with the following packages: survival, survminer, survivalROC, glmnet, biomaRt, ggplot2, gridExtra, clusterProfiler, and forestmodel. KNIME software version 4.0.2 (KNIME AG) was used for data wrangling. Statistical analysis was performed from April to July 2020.
Results
Among 564 patients with serous ovarian cancer, 34 (6%) identified as Black or African American. The mean (SD) age was 59.7 (11.5) years.
Global CNV and OS
Prior to evaluation of signatures, we evaluated whether a simple measure, percentage of genes with CNV, was a prognostic feature. Percentage of CNV was not associated with survival (eFigure 2 in the Supplement).
Model Building
We sought to evaluate the relative risk associated with independent CNV changes observed in ovarian cancer. Scrambled data simulations determined the upper 99th confidence limit −log10(P value) (ie, 99.5th percentile) to be 2.35, and thus we limited our investigation to reporters with −log10(P value) > 2.35.
The frequency of alterations (gains or losses) observed was evaluated on a gene by gene basis across the entire genome. We measured an accompanying univariable association with overall survival per reporter: gene gain or loss (Figure 1A and Figure 1B). The reporters that were significantly associated with overall survival (OS) showed an enrichment in islands of adjacent genes. We identified 13 regions of clustered significance. Among these, only 7q22 was bilaterally associated with OS: both gains and losses were significant. In contrast, an analysis of OS associations with scrambled data did not display clustered associations (Figure 1C and Figure 1D).
Figure 1. Gene Variation Associated With Overall Survival.

OS indicates overall survival.
Orthogonal use of LASSO regression to derive the most important reporters chose 13 of the top 14 reporters from the same regions determined by the clustered approach (eTable 1 in the Supplement). Each of the identified 13 regions had many more significant alterations than the scrambled data (Figure 1D). The identified reporters displayed a high degree of independence in their incidence (Figure 2A) as well as additive and independent contributions to the Cox PH model (Figure 2B).
Figure 2. Copy Number Variation Associated With Cox PH Model.
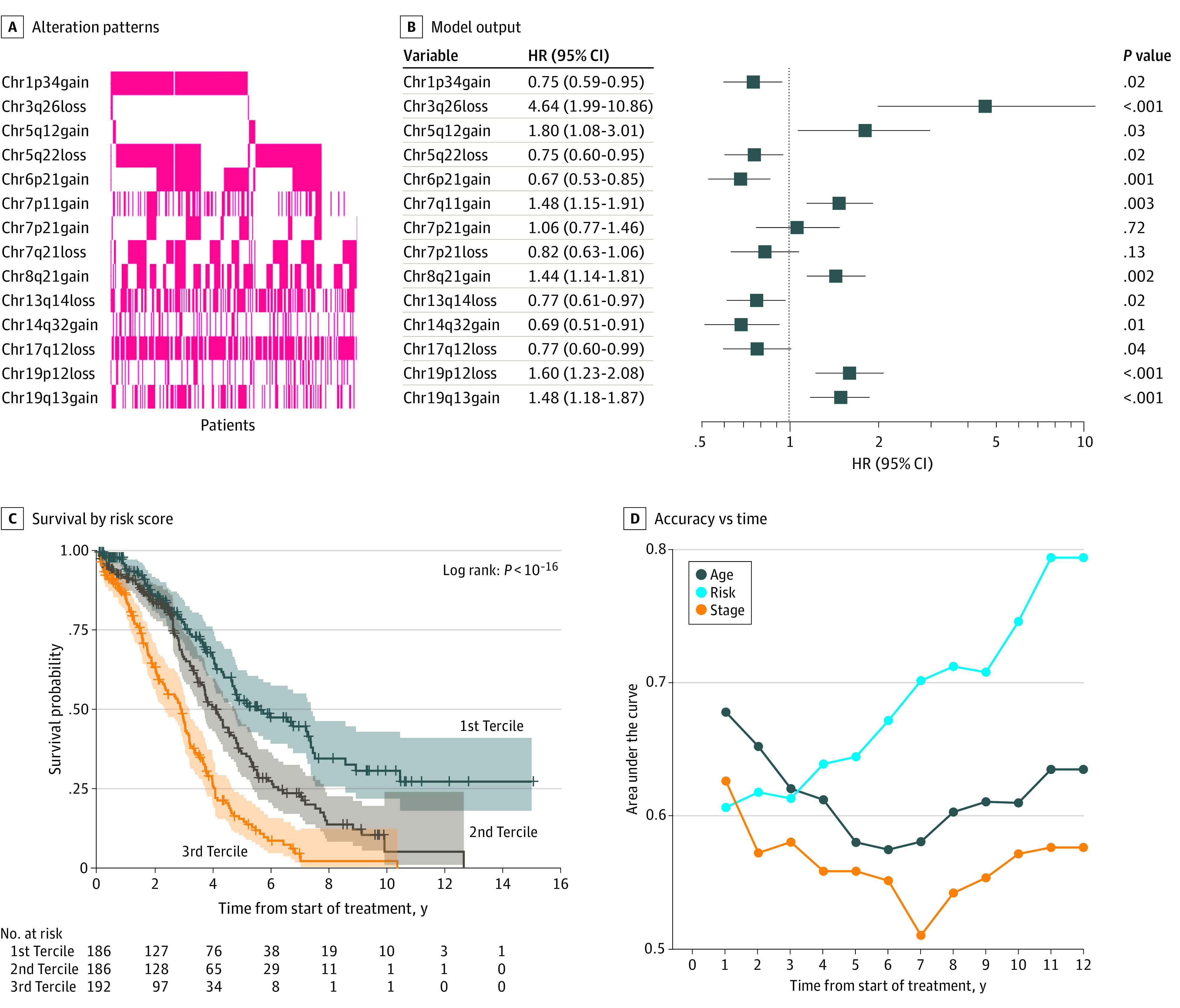
HR indicates hazard ratio.
Univariable Bootstrapped Model Performance
The output from models containing these features (Figure 2B) were next bootstrapped (with replacement) 10 000 times to adjust for optimism bias, and used to generate an internally validated risk score. Use of LOOCV revealed almost identical results (eFigure 4 in the Supplement). Binning the risk score into terciles yielded 3 readily distinguishable risk groups; the highest risk group having a median survival of 2.9 years (95% CI, 2.3-3.2 years) compared to a median survival of the low-risk group of 5.7 years (95% CI, 4.7-7.4 years). Considering only the 5-year time point, Kaplan-Meier estimates of survival were 15% (95% CI, 10%-22%) and 53% (45%-62%), respectively (Figure 2C). When assessed as a continuous variable, the univariable discriminatory power at specific points in time was compared with patient age and clinical stage (Figure 2D). Unlike these clinical features, which remain relatively constant, the discriminatory power increases with respect to time. However, it should be noted that, in part owing to the relative nature of disease, there are few patients available for assessment at later time points as indicated (Figure 2C). At the 10-year time point, the CNV risk score’s AUC was 0.747 (Figure 2D).
Multivariable Bootstrapped Model Performance
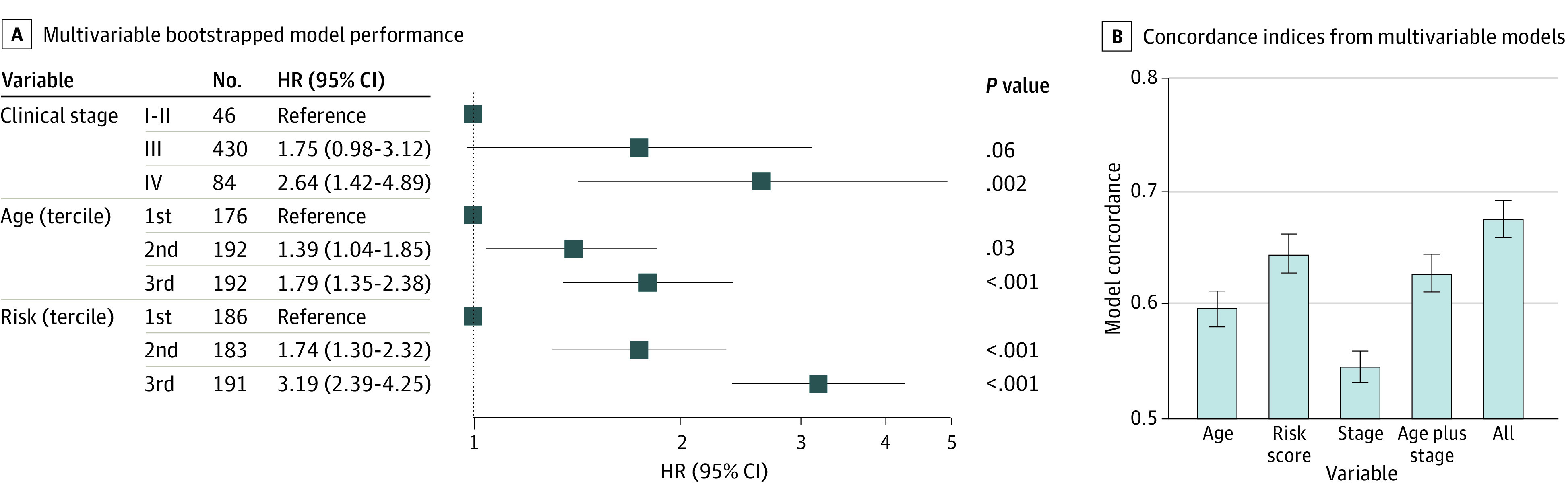
The bootstrapped risk score was evaluated as part of a multivariable Cox PH model in conjunction with age and clinical stage, with additive and independent associations (Figure 3A). Age and the risk score were included in the model as terciles for easier comparison with the clinical stage. Separately, models constructed including age and risk as continuous variables, along with clinical stage, indicate that the risk score alone provided more discriminatory power than both age and clinical stage combined to prognosticate overall survival, and combining the risk score with these features substantially adds to the models (P < .001) (Figure 3B). Sensitivity analyses additionally including grade and race into alternate model builds showed consistent results (eFigure 5 in the Supplement).
Figure 3. Comparison of Hazard Ratio (HR) by Risk Score, Age, and Clinical Stage.
Comparison With Alteration Burden, BRCA Status, and Genomic Scar Signatures
Total genes in the TCGA database with copy number alterations (GISTIC call not equal to 0) was not associated with risk score (Figure 4A) and risk score strength was not diminished in a multivariable model when adjusted for percentage of global CNV alterations (Figure 4B). The risk score was not associated with total mutational burden or BRCA1/2 status (Figure 4C). However, the total variations reported per patient was additive and independent to the risk score to prognosticate OS, even when adjusted for by BRCA1/2 mutational status (Figure 4D).
Figure 4. Cox Proportionate Hazard as a Function of Copy Number Variations (CNV) or BRCA Status.
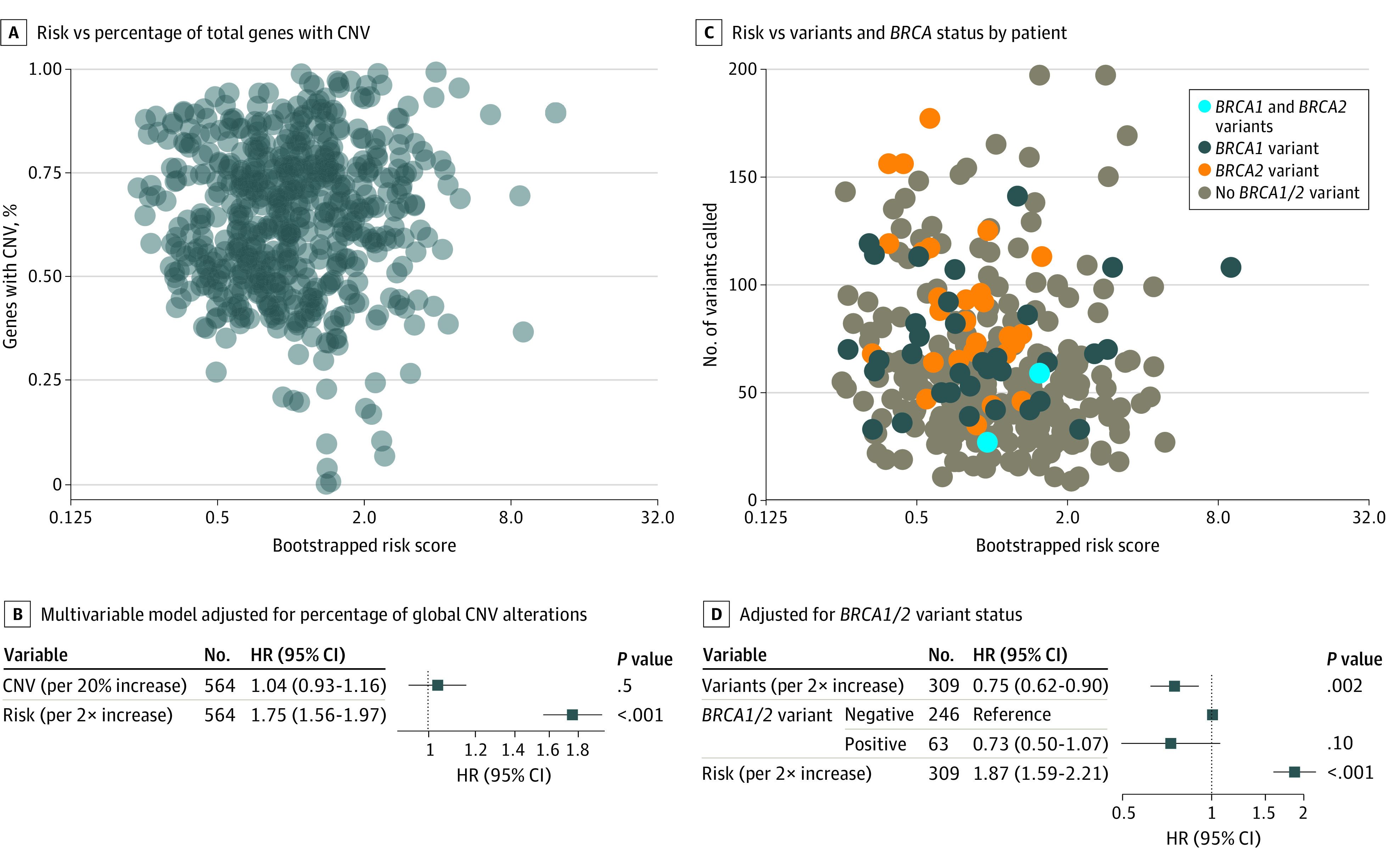
HR indicates hazard ratio.
Patterns of copy number variations as a proxy for genomic scars from DNA repair deficiencies, both proprietary and not, have been investigated as potential predictive biomarkers for PARPi benefit,2,4 as opposed to a general prognostic signature. Open source signatures include: number of discrete chromosomal lesions including telomeric imbalances (NtAI), large scale transitions (LST), and homologous recombination deficiency score loss of heterozygosity (HRD-LOH).21 The risk score was not associated with these genomic scar signatures (eFigure 6 in the Supplement), and had stronger associations with survival than these measures.
Biological Mechanism Associations
Although biological evaluation was not a central focus of this work, we surveyed the known biology of genes (gene ontology) surrounding the identified reporters, as these are prime candidates for future, deeper investigations. In terms of total enrichment within the 13 regions significantly associated with survival, the most significant ontology was mRNA binding involved in posttranscriptional gene silencing (eTable 2 in the Supplement) encompassing 21 miRNAs. Protein kinase regulator activity, an ontology term, was the enriched term most distributed across the regions, present in 6 of 13 regions encompassing the following genes: CCNB1, APC, CDKN1A, PKIA, TCL1B, TCL1A, and PPP1R1B.
Correlation With Commercially Available Multigene Assays
There now exist FDA-approved, commercially available multigene assays that can be ordered by physicians. These assays all include copy number evaluations, but do not evaluate all of the genes in the TCGA database. However, of the identified 13 regions (14 alterations), there exist genes on the assay for 12, and 11, of these regions among the MSK-IMPACT, and FoundationOne CDx assays, respectively (eTable 3 in the Supplement). We reevaluated the resulting change in model performance using the available reporters with each assay. The CNV risk score has a bootstrapped concordance (SE) of 0.644 (0.014). The assays had respective resulting concordance (SE) indices of 0.623 (0.018) and 0.626 (0.018).
Discussion
Prognostication of patient survival in ovarian cancer does not widely use additive genomic features. Using the TCGA database, we sought to develop a risk score that might be complementary to existing nomograms. We chose to focus on copy number variations, because of the dynamic range observed in OVCA, diagnostic considerations, and the availability and ease of potential incorporation to existing commercially available, FDA-approved multigene assays. The nominal drop in discriminative ability using an assay repurpose approach suggests that these tests could potentially be used to prognosticate patient risk.
The ideal comparison of the additive discriminatory power of the CNV risk score would be in conjunction with established clinical features used in current nomograms for ovarian cancer survival, and this would be a logical future extension of the base model presented here. A summary of such models and their contributing clinicopathological features, as well as the availability such features in the TCGA HGSOC firehose database is provided in the Table. Although age, tumor grade, and stage are sufficiently available in the TCGA database, other commonly used features such as preoperative albumin, Ashkenazi ancestry, and maximum diameter of residual tumor (ie, degree of success of surgical resection) were not. Nomograms built from different cohorts do not yield a consistent set of clinical features chosen for final models from cohort to cohort.7,8,9,23 One possible contributing factor might be the (unavoidable) subjective nature of some clinical assessments. However, one of the most consistent strong features observed among these nomograms is the completeness of initial surgery7,8,9,24 (Table), which had insufficient completeness in our data set (9.9%) for comparison. Extending the genetic model to a formal nomogram construction with the CNV risk score would be premature lacking these data. Nonetheless, we felt it could be informative to assess the additive and independent discriminatory value of the CNV risk score in comparison to age, clinical stage, pathological grade, and race.
Table. Prognostic Nomograms for Overall Survival.
| Data collected | TCGA database | Barlin et al, 20117 | Rutten et al, 20149 | Xu et al, 20178 | Hamilton et al, 201824 |
|---|---|---|---|---|---|
| Data source | OVCA Firehose accessed July 2020 (%) | One-site prospective from MSKCC (n = 478) | Retrospective three center (n = 840) | SEER (n = 10 692) | Original collected data (n = 3010) |
| Reported nomogram OS performance | NA | C-index: 0.714 | C-index: 0.71 (95% CI, 0.69- 0.74) | C-index: 0.757 (95% CI, 0.741-0.773) | 10-y AUC: 0.729 |
| Dates of initial treatment | 1992-2013 | 1996-2004 | 1998-2010 | 2004-2013 | Not reported |
| Age | Yes (100%) |
Yes | Yes | Yes | No |
| FIGO tumor grade | Yes (99.3%) |
Yes | Yes | Yes | No |
| Preoperative albumin, g/dL | No | Yes | No | No | No |
| Max diameter residual tumor | No | Yes | Yes | No | Yes |
| Histological subtype | Yes (100%; all serous) |
Yes | Yes | No | No |
| ASA score | No | Yes | No | No | No |
| Breast/ovarian hereditary cancer syndrome | Yes (4.1%) |
Yes | Yes | No | No |
| Race | Yes (94.7%) |
No | No | Yes | No |
| Marital status | No | No | No | Yes | No |
| Tumor location | Yes (100) |
No | No | Yes | No |
| Clinical stage | Yes (99.1) |
No | No | Yes | No |
| LODDS | No | No | No | Yes | No |
| Primary vs interval surgery | No | No | Yes | No | No |
| Performance status | Yes (33.4%) |
No | No | No | No |
| Amount of ascites | No | No | No | No | Yes |
| CA-125 level | No | No | No | No | Yes |
Abbreviations: ASA, American Society of Anesthesiologists; AUC, area under the curve; CA-125, Cancer antigen 125; C-index, concordance index; FIGO, The International Federation of Gynecology and Obstetrics; MSKCC, Memorial Sloan Kettering Cancer Center; LODDS, log of odds of #pos:#neg lymph nodes; OS, overall survival; OVCA, ovarian carcinoma; SEER, Surveillance, Epidemiology, and End Results.
The observed increase in CNV risk score discriminatory ability over time (Figure 2D) is consistent with the notion that with increasing time, it is the biology of the tumor that determines outcome. This phenomenon has also been observed in gene expression prognostic signatures.14 At the 10-year time point, the CNV risk score had an AUC of 0.747. In comparison, Hamilton and colleagues reported a model containing residual tumor burden, CA-125 level, and amount of ascites, with a 10-year ROC AUC of 0.729.24 A suitable cohort combining these features might better predict long-term survivors.
All samples in the TCGA database were collected prior to initiation of systemic therapy, representing a prefirst-line time point. Most patients likely had a combination platinum-taxane chemotherapy as part of their therapeutic regimen, which exploits DNA repair deficiencies. However, with the exception of NBN at 8q21, DNA repair genes were notably absent from regions in the signature.
Several chromosomal alterations shown to be associated with increased risk have been described previously, including regions on chromosome 3, 5 and 7.25,26 The 2 regions on chromosome 19 are noteworthy as they represent previously unrecognized regions associated with ovarian cancer malignancy, although they have been associated with lung, prostate, and breast cancer.27,28,29 Specifically, the miRNA cluster on Chr19q has been implicated as a driver in triple negative breast cancer.29 Genes involved in epithelial to mesenchymal transition (TGFB1 and LTBP4) are present in this region as well. It should be noted that for a feature to be prognostic, it must be altered in some patients and not altered in others. A feature altered in the vast majority, or conversely, almost no patients, would be less likely to have strong prognostic associations, and less likely to arrive in a CNV prognostic signature.
The identified regions could be added to lists of inquiry for drug development that might be currently underappreciated. The survival curves from the patients in the highest at-risk tercile are consistent with modest effect of initial therapy, highlighting the importance of novel drug development and opportunities for improved molecular disease characterization. Conversely, the first tercile may include a cadre of patients that experience prolonged disease remission or are potentially cured.
Limitations and Strengths
Limitations of this study included insufficient clinical features for formal nomogram comparisons, lack of validation with external data sets, serous pathology–only data set, and disproportionate patients in the TCGA database who were self-reported White or European ancestry (82%). Also, the clinical-genomic-outcome associations represent the standard of care from 1992 to 2013; there have been substantial advancements in adjuvant therapy over time, with PARP inhibitors now approved in the front line and recurrent settings as a maintenance and treatment strategy.2,3,4,5,6 Platinum and taxane therapy was the prominent systemic treatment for patients treated in the era collected for the TCGA database and is unlikely to be completely supplanted in the near future.
This study also had several strengths. These included the objectivity and importance of the end point (overall survival), the relative simplicity of the model compared with previous signature work, preanalytic stability of the analyte (CNV) and applicability of the findings, given potential low consumable and bioinformatic load for assay development, or repurposement of existing FDA-approved assays.
Conclusions
This genetic association study found that a CNV-based risk score is independent to and stronger than open-source ovarian cancer genomic biomarkers to prognosticate OS. External validation, especially evaluated in concert with robust clinical features, could provide rapidly accessible additional patient prognostication. Identified genomic regions most tied to OS could inform drug discovery efforts.
eFigure 1. Analysis Flowchart
eFigure 2. Association of Survival with Proportion of Genes with CNV
eFigure 3. CNV Plots and Clustered Regions by Risk Score Tercile
eFigure 4. Bootstrapping Internal Validation Comparison to LOOCV Internal Validation
eFigure 5. Alternate Cox PH Models
eFigure 6. Comparison of Risk Score to Genomic Scar Signatures
eTable 1. Comparison of LASSO-Chosen Features vs the Main Model Features and Performance
eTable 2. FDA-Approved Panel Reporter Approximations
eTable 3. How to Use Risk Score
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi: 10.3322/caac.21442 [DOI] [PubMed] [Google Scholar]
- 2.Ray-Coquard I, Pautier P, Pignata S, et al. ; PAOLA-1 Investigators . Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416-2428. doi: 10.1056/NEJMoa1911361 [DOI] [PubMed] [Google Scholar]
- 3.Coleman RL, Fleming GF, Brady MF, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403-2415. doi: 10.1056/NEJMoa1909707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González-Martín A, Pothuri B, Vergote I, et al. ; PRIMA/ENGOT-OV26/GOG-3012 Investigators . Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391-2402. doi: 10.1056/NEJMoa1910962 [DOI] [PubMed] [Google Scholar]
- 5.Tewari KS, Burger RA, Enserro D, et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J Clin Oncol. 2019;37(26):2317-2328. doi: 10.1200/JCO.19.01009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764-775. doi: 10.1158/1078-0432.CCR-13-2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlin JN, Yu C, Hill EK, et al. Nomogram for predicting 5-year disease-specific mortality after primary surgery for epithelial ovarian cancer. Gynecol Oncol. 2012;125(1):25-30. doi: 10.1016/j.ygyno.2011.12.423 [DOI] [PubMed] [Google Scholar]
- 8.Xu X-L, Cheng H, Tang M-S, et al. A novel nomogram based on LODDS to predict the prognosis of epithelial ovarian cancer. Oncotarget. 2017;8(5):8120-8130. doi: 10.18632/oncotarget.14100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutten MJ, Boldingh JHL, Schuit E, et al. Development and internal validation of a prognostic model for survival after debulking surgery for epithelial ovarian cancer. Gynecol Oncol. 2014;135(1):13-18. doi: 10.1016/j.ygyno.2014.07.099 [DOI] [PubMed] [Google Scholar]
- 10.Ando M, Ando Y, Hasegawa Y, et al. Prognostic value of performance status assessed by patients themselves, nurses, and oncologists in advanced non-small cell lung cancer. Br J Cancer. 2001;85(11):1634-1639. doi: 10.1054/bjoc.2001.2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chow R, Chiu N, Bruera E, et al. Inter-rater reliability in performance status assessment among health care professionals: a systematic review. Ann Palliat Med. 2016;5(2):83-92. doi: 10.21037/apm.2016.03.02 [DOI] [PubMed] [Google Scholar]
- 12.Riester M, Wei W, Waldron L, et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J Natl Cancer Inst. 2014;106(5):dju048. doi: 10.1093/jnci/dju048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Q, Shi X, Xie Y, Huang J, Shia B, Ma S. Combining multidimensional genomic measurements for predicting cancer prognosis: observations from TCGA. Brief Bioinform. 2015;16(2):291-303. doi: 10.1093/bib/bbu003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millstein J, Budden T, Goode EL, et al. ; AOCS Group . Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol. 2020;31(9):1240-1250. doi: 10.1016/j.annonc.2020.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delaney JR, Patel CB, Willis KM, et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat Commun. 2017;8(1):14423. doi: 10.1038/ncomms14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell D, Berchuck A, Birrer M, et al. ; Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609-615. doi: 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kader T, Goode DL, Wong SQ, et al. Copy number analysis by low coverage whole genome sequencing using ultra low-input DNA from formalin-fixed paraffin embedded tumor tissue. Genome Med. 2016;8(1):121. doi: 10.1186/s13073-016-0375-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren T, Suo J, Liu S, et al. Using low-coverage whole genome sequencing technique to analyze the chromosomal copy number alterations in the exfoliative cells of cervical cancer. J Gynecol Oncol. 2018;29(5):e78-e78. doi: 10.3802/jgo.2018.29.e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Lichtenberg T, Hoadley KA, et al. ; Cancer Genome Atlas Research Network . An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400-416.e11. doi: 10.1016/j.cell.2018.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. doi: 10.1186/gb-2011-12-4-r41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquard AM, Eklund AC, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3:9. doi: 10.1186/s40364-015-0033-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1-22. doi: 10.18637/jss.v033.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bookman MA. Can we predict who lives long with ovarian cancer? Cancer. 2019;125(S24)(suppl 24):4578-4581. doi: 10.1002/cncr.32474 [DOI] [PubMed] [Google Scholar]
- 24.Hamilton CA, Miller A, Casablanca Y, et al. Clinicopathologic characteristics associated with long-term survival in advanced epithelial ovarian cancer: an NRG Oncology/Gynecologic Oncology Group ancillary data study. Gynecol Oncol. 2018;148(2):275-280. doi: 10.1016/j.ygyno.2017.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomassen M, Jochumsen KM, Mogensen O, Tan Q, Kruse TA. Gene expression meta-analysis identifies chromosomal regions involved in ovarian cancer survival. Genes Chromosomes Cancer. 2009;48(8):711-724. doi: 10.1002/gcc.20676 [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Yuan Y, Lu KH, Zhang L. Identification of recurrent focal copy number variations and their putative targeted driver genes in ovarian cancer. BMC Bioinformatics. 2016;17(1):222. doi: 10.1186/s12859-016-1085-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Zhang Y, Nilsson CL, et al. Association of chromosome 19 to lung cancer genotypes and phenotypes. Cancer Metastasis Rev. 2015;34(2):217-226. doi: 10.1007/s10555-015-9556-2 [DOI] [PubMed] [Google Scholar]
- 28.Slager SL, Schaid DJ, Cunningham JM, et al. Confirmation of linkage of prostate cancer aggressiveness with chromosome 19q. Am J Hum Genet. 2003;72(3):759-762. doi: 10.1086/368230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jinesh GG, Flores ER, Brohl AS. Chromosome 19 miRNA cluster and CEBPB expression specifically mark and potentially drive triple negative breast cancers. PLoS One. 2018;13(10):e0206008-e0206008. doi: 10.1371/journal.pone.0206008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Analysis Flowchart
eFigure 2. Association of Survival with Proportion of Genes with CNV
eFigure 3. CNV Plots and Clustered Regions by Risk Score Tercile
eFigure 4. Bootstrapping Internal Validation Comparison to LOOCV Internal Validation
eFigure 5. Alternate Cox PH Models
eFigure 6. Comparison of Risk Score to Genomic Scar Signatures
eTable 1. Comparison of LASSO-Chosen Features vs the Main Model Features and Performance
eTable 2. FDA-Approved Panel Reporter Approximations
eTable 3. How to Use Risk Score