

Abstract
Background:
Autosomal recessive ichthyosis with hypotrichosis (ARIH; MIM 602400) syndrome is characterized by diffused congenital ichthyosis and generalized non-scarring hypotrichosis. The underlying genetic cause of ARIH syndrome has been associated with sequence variants of the gene ST14, encoding type II transmembrane serine protease matriptase, which maps to chromosome 11q24.3.
Objectives:
The current report aimed to investigate the clinical features and genetic cause of ARIH syndrome in a large consanguineous family of Pakistani origin.
Materials & methods:
The technique of homozygosity mapping with highly polymorphic microsatellite markers was employed to establish linkage within the family. Sanger sequencing of exons and intron-exonboundaries of ST14 was performed to identify the potential pathogenic sequence variants, followed by structural analysis of the mutated protein.
Results:
Linkage was established to chromosome 11q24.3, comprising the gene ST14. Sequence analysis led to the identification of a novel homozygous missense variant (c.1315G>A, p.Gly439Ser) in the ST14 gene that co-segregated with the disease phenotype in all affected members. Homology modelling and molecular docking analysis of ST14 with wild-type TMEFF1 protein was performed which revealed that glycine at position 439 is crucial for maintaining normal structural confirmation and interaction with the EGF domain of TMEFF1 protein.
Conclusion:
Taken together, the data strongly advocate this ST14 variant as the underlying genetic cause of ARIH syndrome in this first reported affected family from Pakistan. Moreover, the present study adds to the spectrum of mutations in the ST14 gene, implicating them in the pathogenesis of ARIH syndrome.
Keywords: autosomal recessive ichthyosis with hypotrichosis syndrome, ST14 gene, novel missense variant
The term autosomal recessive congenital ichthyosis (ARCI) is a large heterogeneous group of genodermatoses with abnormal skin manifestations as a result of defective keratinocyte differentiation and desquamation in the upper layer of the epidermis. This cornification disorder may be non-syndromic, confined to skin only or syndromic, and associated with extra-cutaneous findings [1]. Only limited data are available on the epidemiology of ARCI. According to the Foundation for Ichthyosis and Related Skin Types, ARCI affects approximately 1:200,000 individuals in the USA [2].
To date, mutations in nine genes have been shown to be involved in the aetiology of non-syndromic ARCI [3]. These include ABCA12 (MIM 601277), TGM1 (MIM 242300), ALOXE3 (MIM 607206), ALOX12B (MIM 603741), PNPLA1 (MIM 612121), CYP4F22 (MIM 604777), LIPN (MIM 613924), NIPAL4 (MIM 612281) [4, 5], and CERS3 (MIM 615276) [6]. Congenital recessive syndromic forms of ichthyosis, in association with hair abnormalities, include: Netherton syndrome (NTS, MIM 256500), caused by mutations in SPINK5 (MIM 605010, chromosome 5q32) [7]; congenital ichthyosis, follicular atrophoderma, hypotrichosis, and hypohidrosis syndrome (IFAH, MIM 602400), characterized by generalized and diffuse body scaling, follicular atrophoderma (a rare disorder consisting of enlarged funnel-shaped depressions of the pilosebaceous orifices), generalized, diffuse and non-scarring hypotrichosis, and hypohidrosis (reduced sweating), which is limited to various parts of the body [8]; autosomal recessive ichthyosis with hypotrichosis (ARIH, MIM 602400) syndrome, characterized by congenital generalized and diffused scaling (ichthyosis) on the body associated with generalized and diffused hypotrichosis, caused by mutation in the ST14 gene (MIM 606797, chromosome 11q24.3), encoding serine protease matriptase [9]; trichothiodystrophy with congenitalichthyosis, characterized by ichthyosis, brittle hair, intellectual impairment, decreased fertility, and short stature (IBIDS MIM 601675) due to mutations in ERCC3/XPB (MIM 133510) [10] and ERCC2/XPD (MIM 126340) [11]; ichthyosis, split hairs and amino aciduria syndrome (MIM 242550) which also includes intellectual disability [12]; and ichthyosis-sclerosing cholangitis (IHSC, MIM 607626) due to variants in CLDN1 (MIM 603718, chromosome 3q28) [13].
In this study, we describe a large consanguineous Pakistani family displaying the hallmarks of ARIH syndrome, characterized by congenital ichthyosis associated with abnormal hair. Genotyping followed by DNA sequence analysis led to the identification of a novel ST14 variant (c.1315G>A, p.Gly439Ser) co-segregating with the disease phenotype.
Materials and methods
Family recruitment and ethical approval
Approval for the study was obtained from the Institutional Review Board (IRB) of Quaid-i-Azam University, Islamabad, Pakistan. Written informed consent for the research work was provided by all participants and legal guardians. The large consanguineous kindred (figure 1A) was from a remote area of Sindh province, Pakistan. The family’s pedigree indicated an autosomal recessive inheritance of syndromic ARCI. Affected individuals were referred to dermatologists for clinical examination at local government hospitals.
Figure 1.
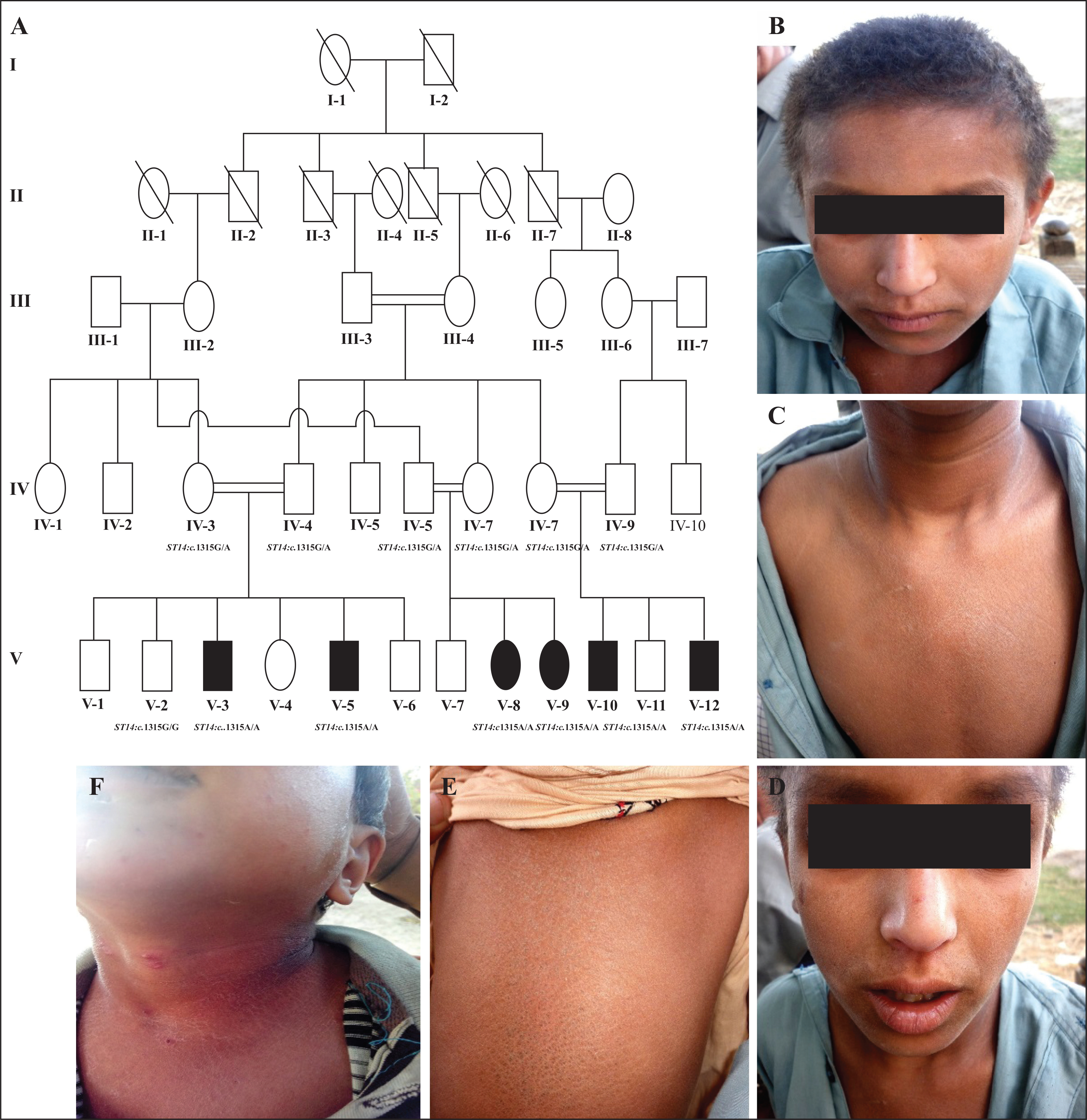
A) Pedigree of the five-generation consanguineous Pakistani family, displaying the features of autosomal recessive ichthyosis with hypotrichosis (ARIH) syndrome. Consanguineous unions are represented by double lines. Males are represented by squares and females by circles. Filled symbols indicate affected family members and unaffected individuals are represented by clear symbols. A diagonal line indicates a deceased individual. B, C, D, E, F) The clinical appearance of affected individuals with the features of ARIH syndrome. B) Hypotrichosis; scalp hairs appear light-brown, woolly, curly, and lustreless with receding frontal hair line and sparse eyebrows and eyelashes. C) Chest area with diffuse and dry scales. D) Facial area showing sparse eyebrows and eyelashes, dry facial skin, with mild and diffuse scales. (E) Lower back area, severely affected with dark brown scales. F) Neck area, severely affected by dry and diffuse scales.
DNA extraction and genotyping of microsatellite markers
We extracted genomic DNA from peripheral leucocytes from six affected (V-3, V-5, V-8, V-9, V-10, and V-12) and seven unaffected (IV-3, IV-4, IV-6, IV-7, IV-8, IV-9, and V-2) (figure 1A) family members using the GenElute™ blood genomic DNA kit (Sigma-Aldrich, St. Louis, MO, USA). DNA quantification was performed using Nanodrop-1000 spectrophotometer (Thermal scientific, Wilmington, MA, USA) to calculate the optimal density at 260 nm and dilute to 40–50 ng/μL for amplification by polymerase chain reaction (PCR).
Based on clinical features observed in the affected family members as well as the autosomal recessive inheritance pattern, linkage was tested by genotyping highly polymorphic microsatellite markers flanking the ST14 gene at the ARCI11 locus on chromosome 11q24.3 (D11S975, D11S4110, D11S1351, D11S4091, D11S4123, D11S4150, D11S4131, D11S874, D11S1304, and D11S969), the SPINK5 gene on chromosome 5q32 (D5S436, D5S2099, D5S2033, D5S638, D5S2847, D5S2490, D5S434, D5S1469, D5S2013, and D5S2014), and the CLDN1 gene on chromosome 3q28 (D3S3686, D3S3628, D3S3596, D3S1580, D3S3530, D3S1294, D3S1314, D3S2747, D3S3054, and D3S2418). A standard PCR procedure was followed for the amplification of microsatellite markers using genomic DNA in a total reaction volume of 25 μL, as previously described [14].
The PCR products were resolved on 8% non-denaturing polyacrylamide gels stained with ethidium bromide and genotypes were assigned by visual inspection.
Sequencing the ST14 gene
After linkage was established to the ST14 gene at ARCI11 locus on chromosome 11q24.3, the ST14 gene (NM_021978.3) was sequenced for all the available affected and normal individuals of the family. For sequencing of the ST14 gene, primers were designed for 19 exons and splice junctions using Primer3 software [15]. After DNA purification with commercially available kits (Axygen Inc., CA, USA), sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit, together with an ABI Prism 310 Genetic Analyzer (Applera, Foster City, CA, USA). Sequence variants were identified via Bioedit sequence alignment editor version 6.0.7.
Protein modelling and docking
Matriptase/ST14 is co-expressed with TMEFF1 in several tissues, suggesting TMEFF1 is a substrate of ST14. In order to characterize the possible functional effect of a novel variant of ST14, homology modelling and docking of the complement sub-component C1r/s (CUB) domain of ST14 with the EGF domain of TMEFF1 was performed.
The three-dimensional structure of the CUB domain of human ST14 and EGF domain of TMEFF1 protein was constructed using the Molecular Operating Environment (MOE). The crystallographic structure of the catalytic domain of human plasma kallikrein (PDB code 2ANY; Resolution 1.4 Å) was selected as a template to model the native and mutant CUB functional domain [16]. PDB code 1XDT was used as template to model the EGF domain of TMEFF1 protein [17]. Qualities of models were validated using Ramachandran Plot and ProSA server [18]; ProSA returns z-score that indicates overall model quality based on the Cα positions in 3D space. The protein structure was visually inspected using PYMOL viewer (http://www.pymol.org).
The interaction between wild-type or mutant ST14 and TMEFF1 protein was investigated using docking studies. For protein-protein docking, we used unrestrained pairwise rigid body docking for ST14 and TMEFF1. Energy minimization was carried out using AMBER99 force field implemented in MOE software. ZDOCK server was utilized for protein-protein docking to predict and evaluate the interactions in the ST14-TMEFF1 complex [19].
Results
Clinical profile
Affected members of the family were born full-term without any complications during pregnancy. At birth, no ectropion, eclabium or collodion membrane was observed, but mild diffused hypotrichosis was present and gradually progressed with age. Body hair appeared sparse, woolly, curly, and lustreless with very fine texture and light brown colour (figure 1B). Eyebrows and eyelashes were also sparse (figure 1D). Plucked hair did not reveal any gross abnormality. Slow-growing scalp hair with receding frontal hairline was observed. Skin showed generalized scaling that appeared very dry, diffuse, and affected the scalp, face, neck, chest, and lower back (figure 1B–F). Scales were more prominent and severe at the lower back with dark brown colour (figure 1E). There were no erythema, palmoplantar hyperkeratosis, mucosal changes, abnormal sweating, hyperthermia, blepharitis, intellectual disability, or tooth, nail, visual or hearing abnormalities. Except for the additional feature of woolly hair, clinical features were consistent with previous reports of ST14-related ARIH. Heterozygous carriers were unaffected and clinically indistinguishable from the wild-type individuals in the family.
Genotyping and mutation screening
A minimum of eight to ten microsatellite markers were genotyped. Analysis of microsatellite markers (D11S975, D11S4110,D11S1351,D11S4091,D11S4123,D11S4150, D11S4131, D11S874, D11S1304, and D11S969) established linkage to the ST14 gene at ARCI11 on chromosome 11q24.3. There was no evidence of linkage to either the SPINK5 or CLDN1 region.
Sanger sequencing of the ST14 gene was performed using DNA samples from all available family members which revealed a novel transition mutation, c.1315G>A (p.Gly439Ser), and this segregated in the family with the disease phenotype (figure 2A–C). Affected individuals were homozygous for the altered allele (AA), the parents were heterozygous carriers (GA), and unaffected children were either heterozygous (GA) or homozygous for the wild-type allele (GG) (figure 2A–C). The disease-causing mutation (c.1315G>A) resides in exon 11 of the ST14 gene (NM_021978.3) and causes glycine to serine substitution at position 439 (p.Gly439Ser) within the amino acid sequence of the translated ST14 protein. This mutation occurs at a highly conserved residue (figure 3), has a CADD score of 29.3, and is predicted to be damaging according to PolyPhen2 (http://genetics.bwh.harvard.edu/pph/ ), SIFT/PROVEAN (http://sift.jcvi.org), MutationAssessor and Mutation-Taster (http://www.mutationtaster.org/). Additionally, the c.1315G> A (p.Gly439Ser) mutation is absent in the Exome Aggregation Consortium (ExAC) database, in 177 in-house exome sequences from unrelated Pakistani individuals, and in sequences from a total of 200 healthy Pakistani controls without ichthyosis and hypotrichosis.
Figure 2.
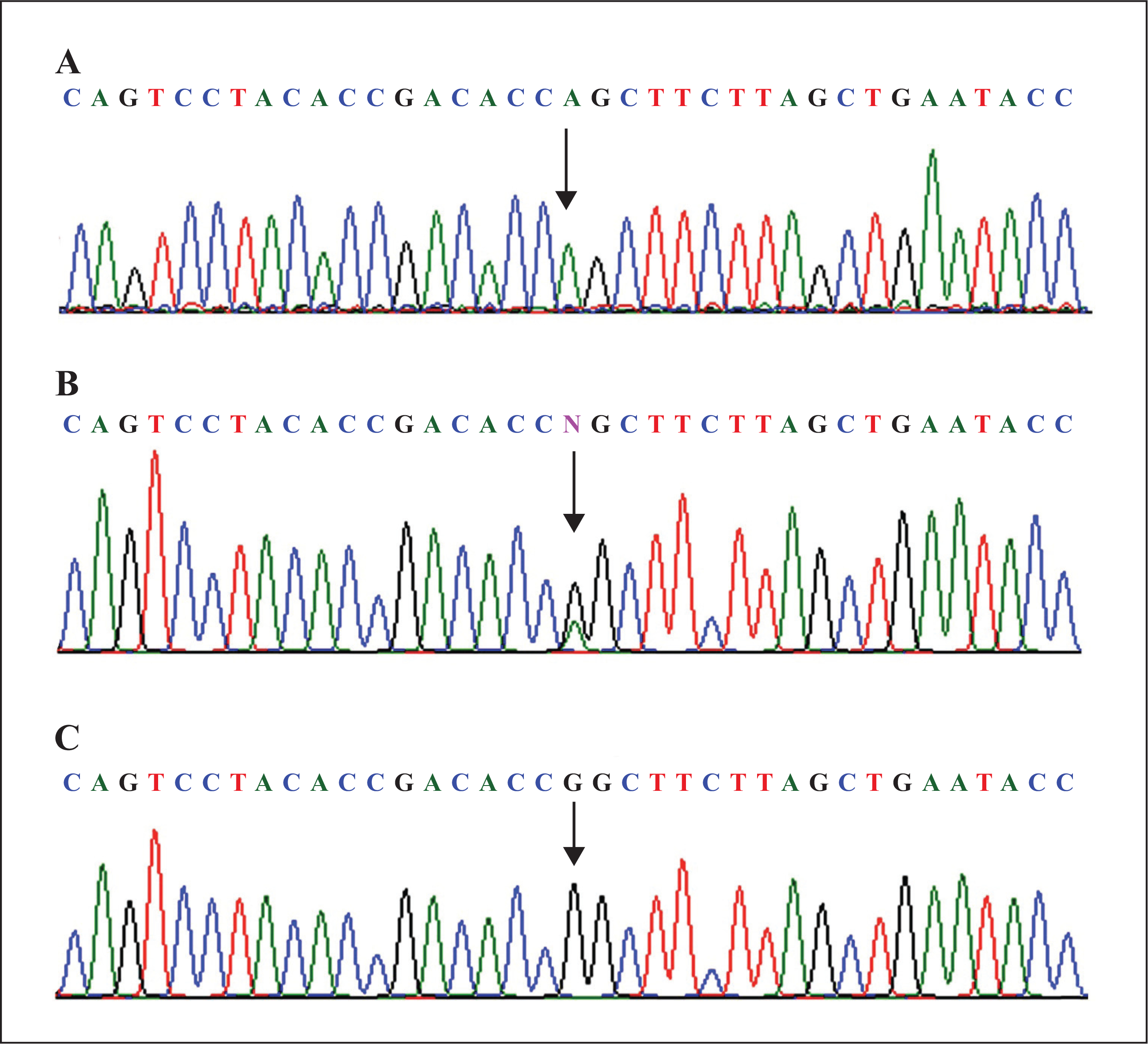
Sequence analysis of the ST14 gene revealing the mutation c.1315G>A (p.Gly439Ser). Upper panel (A) represents the nucleotide sequence of an affected individual, middle panel (B) a heterozygous carrier, and lower panel (C) a homozygous normal individual. The arrow indicates the missense mutation.
Figure 3.
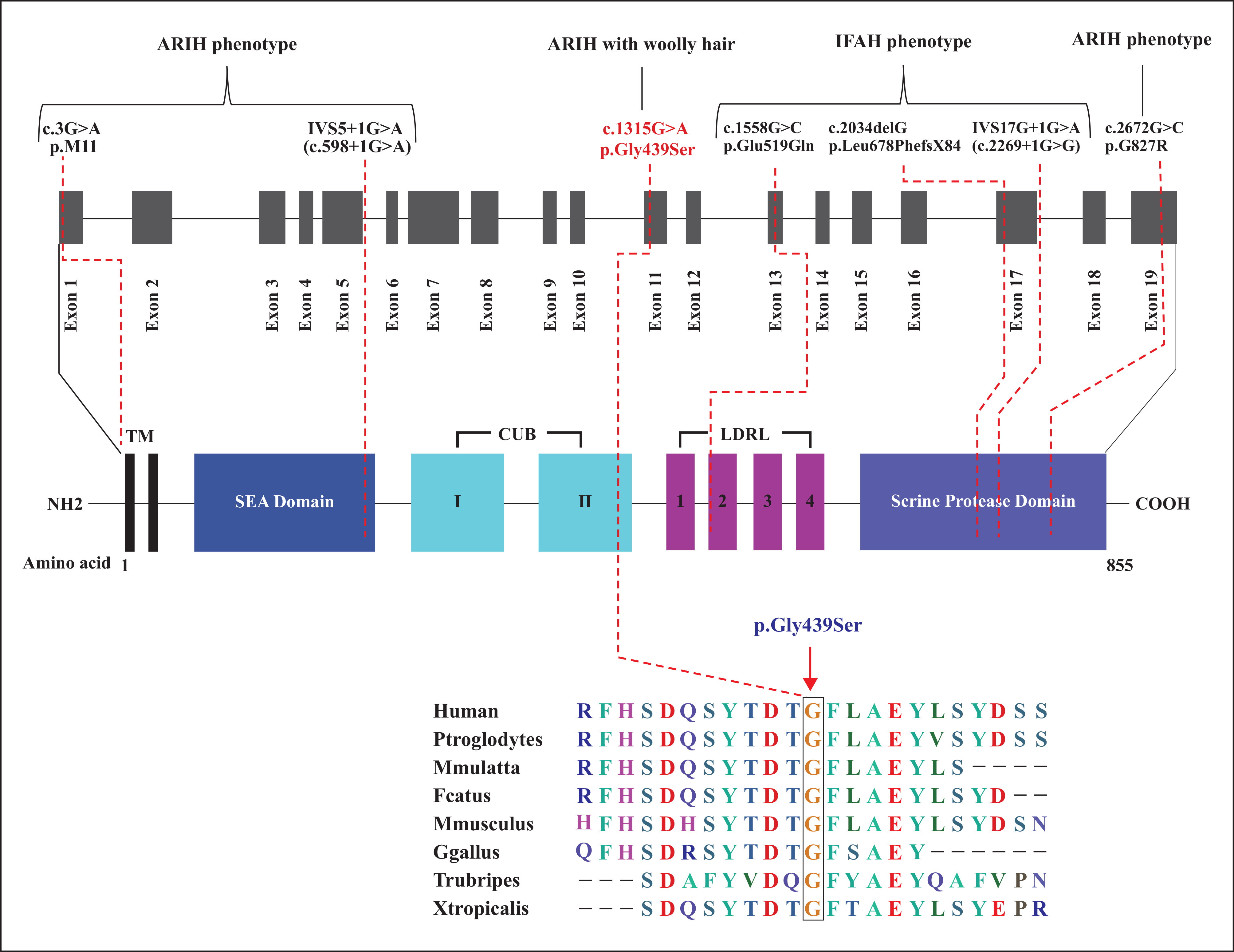
Upper panel: schematic representation of the coding region of the ST14 gene alongside known protein structural and functional domains (intronic regions are not drawn to scale). The location of all mutations known to date is also indicated. The novel missense mutation c.1315G>A (p.Gly439Ser), identified in the present report, is indicated in red; this affects the second CUB domain of the mature ST14 protein. Lower panel: Sequence comparison of the ST14 gene across different species, highlighting homology and the conserved glycine residue at position 439.
Homology modelling and docking analysis
ST14 has a transmembrane N-terminal domain, a conserved extracellular C-terminal serine protease domain, a SEA domain, two tandem repeats of the CUB domains, and four tandem repeats of the low-density lipoprotein receptor class A (LDLRA) domain (figure 3). The ST14 CUB domain was modelled according to the crystallographic structure of the catalytic domain of human plasma kallikrein (PDB: 2ANY), while the known structure of TMEFF1 EGF domain (PDB: 1XDT) was used for docking studies. The ST14 Gly439 residue lies within the beta-sheet region of the C-terminal of the second CUB domain (figure 4B) and is close to a putative proteolytic activation site, Arg442, within an Arg-Val-Val-Gly-Gly motif. The p.Gly439Ser mutation is predicted to result in an extra disulphide bond with Ser353 (figure 4C), which is adjacent to Asn352, a residue identified to participate in TMEFF1 docking. The p.Gly439Ser mutation is therefore predicted to cause not only local perturbation of loop conformation (figure 4A–C), but also atypical interaction with TMEFF1 (figure 5A–D).
Figure 4.
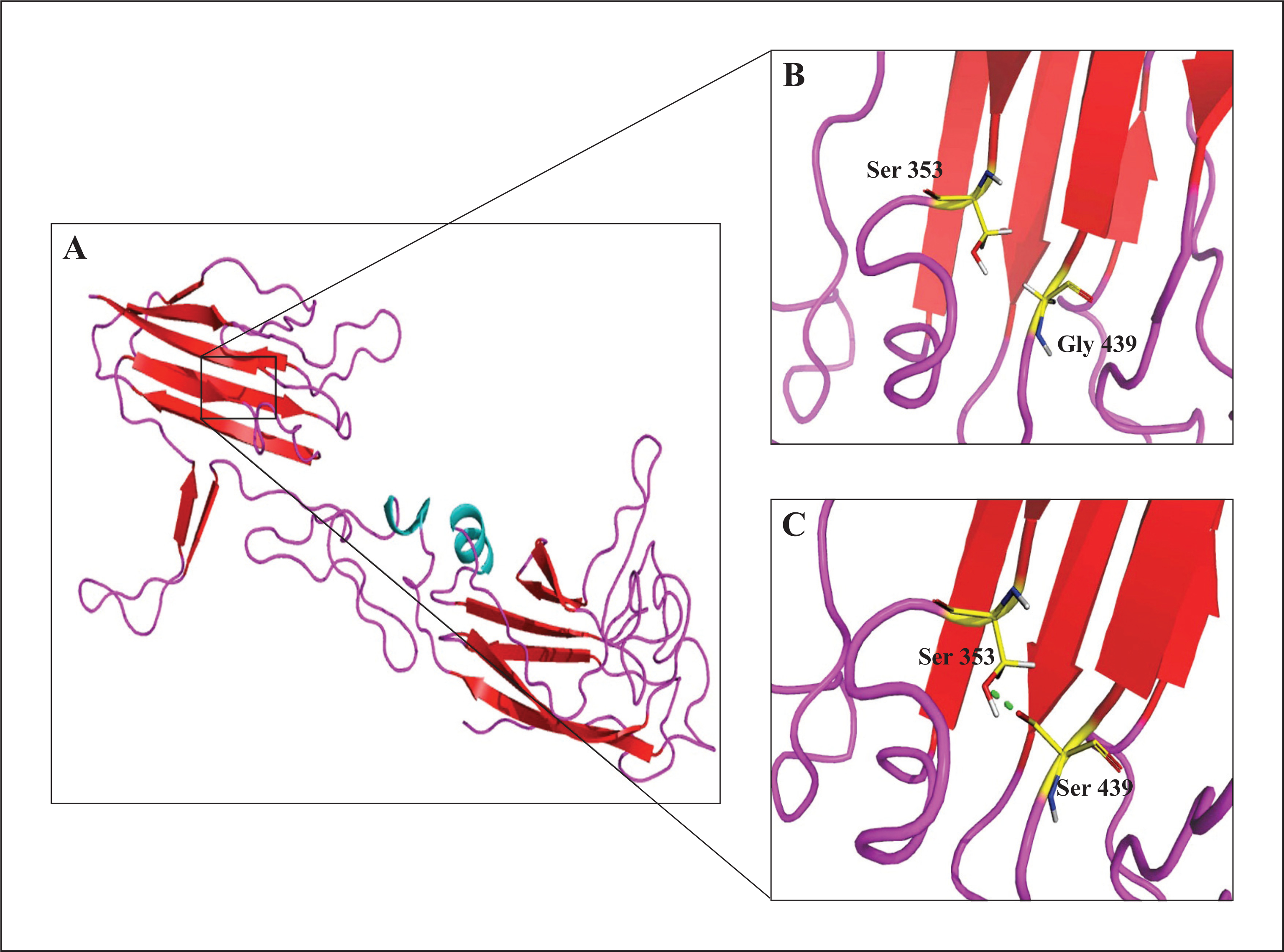
(A) Representation of predicted structure of the CUB domain of human ST14 protein, with beta sheet in red, alpha helices in cyan, and loop in pink. Close-up view showing (B) the wild-type domain with Gly439 and (C) the mutant domain with Ser439; in the latter, a hydrogen bond is depicted (green dotted line) between Ser439 and Ser353.
Figure 5.
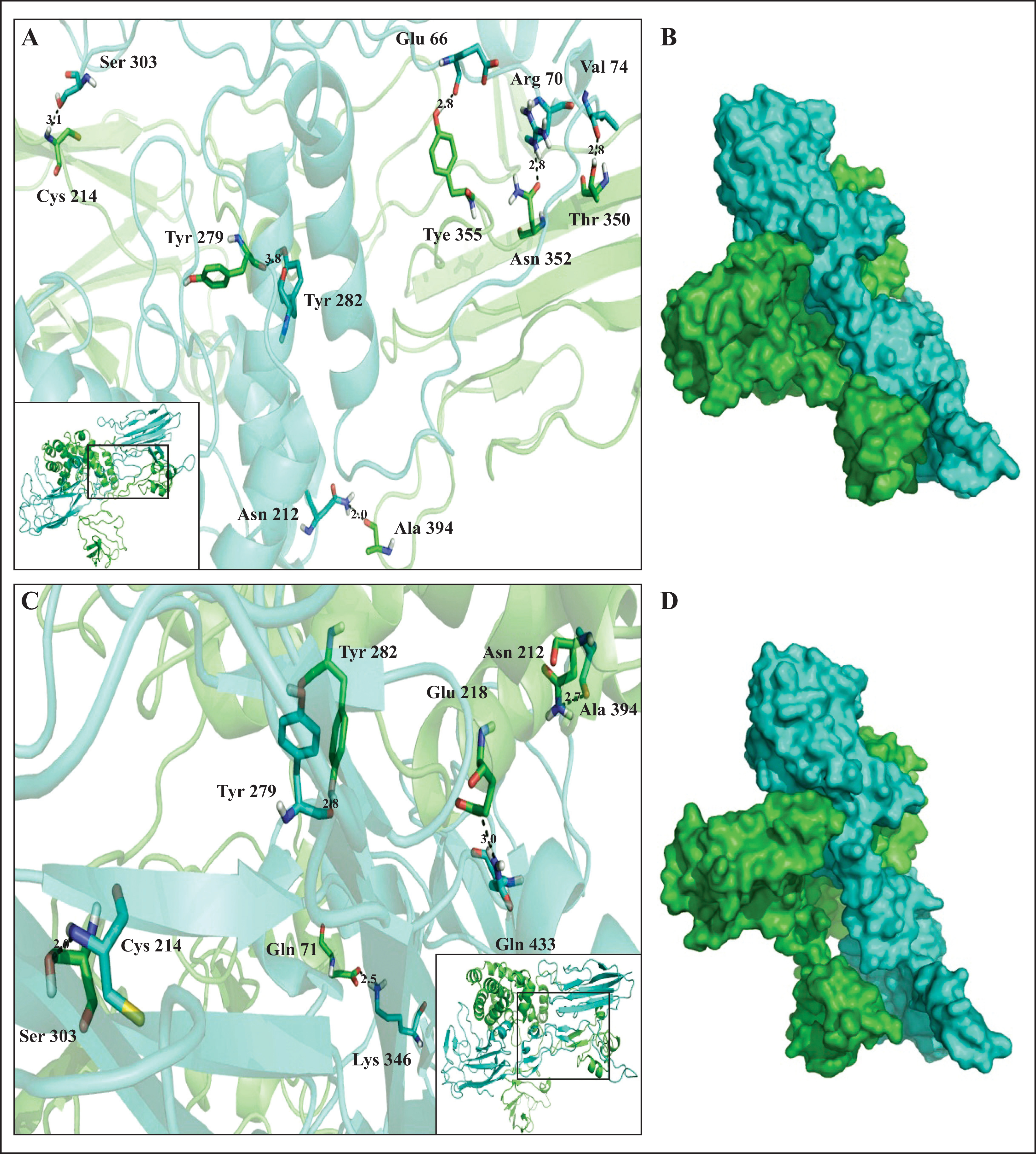
Visualization of optimal docking for the ST14 (CUB domain)-TMEFF1 complex. A) Detailed structure and (B) surface view of wild-type ST14 interacting with TMEFF1; C) detailed structure and (D) surface view of mutated ST14 interacting with TMEFF1 (ST14 is depicted in cyan and TMEFF1 in green). Interacting residues are shown in stick representation and hydrogen bonds are highlighted by a black dotted line.
Discussion
In the present study, we describe a large consanguineous Pakistani kindred sharing common features of ARIH syndrome caused by a novel missense mutation (c.1315G>A [p.Gly439Ser]) in the ST14 gene that encodes the serine protease matriptase. This is the first report of a novel disease-causing sequence variant of the ST14 gene in the Pakistani population and its association with autosomal recessive congenital ichthyosis with hypotrichosis. Previous studies have reported the ST14 gene to be involved in the aetiology of ARIH and IFAH syndromes in Arab [20], Turkish [20, 21], Rumanian [22], Israeli Arab [9], and Bedouin [23] families.
The clinical features observed in the family reported here show a resemblance to the previously reported phenotypes of ARIH patients with variants in the ST14 gene, as described previously [9, 21] (table 1). Clinically, affected members had characteristic features of generalized and diffuse scaling on the body with dark scales on the lower back, as well as diffuse and generalized hypotrichosis with sparse eyebrows and eyelashes. Hair phenotype appeared sparse, light brown, woolly and curly, and lustreless. Features, such as photophobia or corneal opacities, follicular atrophoderma, palmoplantar keratoderma, or hyperlinearity, were not observed. Healthy teeth and nails were observed in the affected members. Patients displayed normal features of mucosal surfaces and sweating.
Table 1.
Clinical features of affected individuals with autosomal recessive ichthyosis and hypotrichosis.
| Clinical Characteristics | Clinical features in the present study | First family reported by Basel-Vanagaite et al. [9] | Clinical findings of a child described by Avrahami et al. [21] |
|---|---|---|---|
| Skin abnormalities: vernix-like scaling, ichthyosis | + | + | + |
| Scalp hair: light brown, curly, coarse; improving with age | + | + | + |
| Woolly scalp hair | + | − | − |
| Eyebrows and eyelashes: Sparse, curly, light-coloured | + | + | + |
| Eyes: photophobia, corneal opacity | − | + | − |
| Blepharitis | − | − | + |
| Teeth: notching | − | + | − |
| Body hair: sparse | + | + | + |
+: presence of feature; −: absence of feature.
Two different conditions (ARIH and IFAH) have been described to be caused by mutation in the ST14 gene. ARIH syndrome was identified by Basel-Vanagaite et al. [9] and a new type of genodermatosis with congenital ichthyosis, follicular atrophoderma, hypotrichosis, and hypohidrosis (IFAH) was described by Lestringant et al [8]. To date, only six pathogenic sequence mutations of the ST14 gene have been identified as the cause of ARIH and IFAH (figure 3). These include p.Gly827Arg [9], p.MetlILeu [21], and IVS5+1G>A [23] which cause ARIH syndrome and the remaining three, IVS17+1G>A, p.Leu678PhefsX84 [20], and p.Glu519Gln [22], which cause the IFAH phenotype. Here, we report on another novel missense mutation (p.Gly439Ser) in the ST14 gene that causes ARIH syndrome with an additional phenotype of woolly scalp hair which was absent in the previous reports of ARIH syndrome.
Like other type II transmembrane serine proteases of the S1 trypsin-like protein family [24], ST14/matriptase is synthesized as a zymogen precursor that requires proteolytic cleavage and shedding from the cell surface for activation [25]. It is involved in matrix degradation with trypsin-like specificity [26]. Matriptase is widely expressed in various tissues, including the epithelium and thymic stroma, and plays a crucial role in the normal development of the epidermis, stratum corneum, hair follicles, and the immune system. Matriptase-deficient mice display dry, red, shiny, and wrinkled skin with dysmorphic corneocytes within the stratum corneum. In these mice, deficiency of vesicular bodies in the transitional layers results in inward and outward epidermal barrier dysfunction, excessive and fatal dehydration, and postnatal lethality. Additionally, matriptase deficiency contributes to abnormal hair follicle development as well as apoptosis and marked depletion of thymocytes [27].
The novel missense mutation (c.1315G>A; p.Gly439Ser) identified in the present study gives rise to a protein product which is anticipated to lack a highly conserved amino acid glycine at position 439 of the 855-amino acid protein. The change from glycine to serine lies in the C-terminal of the second CUB domain (figure 3) of the MT-SP1 protein which is thought to be involved in protein-protein interactions or protein-ligand interactions. The amino acid substitution (p.Gly439Ser) results in the formation of an extra hydrogen bond, as predicted by the model, and may cause local perturbation in structural conformation. Moreover, the glycine at position 439 is highly conserved and is crucial due to its small and neutral side chain which allows for sharp turns or location within loops of the protein structure, providing high flexibility to these regions. Thus, the substitution of the single hydrogen of glycine with the bulky, polar serine side group is predicted to compromise normal structural conformation and results in loss of ST14 function or protein-protein interactions that are necessary for ST14 activation and cleavage.
The CUB domain-interacting protein, TMEFF1, was docked with normal and mutated ST14 which suggests an atypical interaction between normal or mutant ST14 protein and TMEFF1 (figure 5A–D). Although evidence indicates that TMEFF1 and ST14 interact directly and are co-expressed (e.g. in human breast cancer cells [28]), the precise mechanisms and pathway(s) involved which lead to the development of ARIH syndrome are yet to be explored.
In conclusion, we have identified a novel homozygous missense variant (p.Gly439Ser) of the ST14 gene that causes ARIH syndrome in a large consanguineous Pakistani kindred. The position and the predicted effect of the p.Gly439Ser substitution in the second CUB domain of ST14 suggest that the mutation compromises normal structural conformation of the second CUB domain, which is thought to be involved in protein-protein or protein-ligand interactions. The effect of this mutation is further strengthened by the atypical interaction shown by wild-type CUB domain-interacting protein TMEFF1 with normal or mutated ST14 protein.
Taken together, our findings further strengthen the role of ST14 mutation as a cause of ARIH syndrome, and provides further evidence for the lack of genotype-phenotype correlation and clinical variability in patients with ST14 mutation and ARIH syndrome.
Acknowledgments:
We are grateful to all the members of the family who participated in this study. The work presented was funded by the Higher Education Commission (HEC), Islamabad, Pakistan.
Footnotes
Financial support: none.
Conflict of interest: none.
References
- 1.Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010; 63: 607–41. [DOI] [PubMed] [Google Scholar]
- 2.Rodríguez-Pazos L, Ginarte M, Vega A, Toribioa J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr 2013; 104: 270–84. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad F, Ansar M, Mehmood S, et al. A novel missense variant in the PNPLÁ1 gene underlies congenital ichthyosis in three consanguineous families. J Eur Acad Dermatol Venereol 2016; 12: e210–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer J Autosomal recessive congenital ichthyosis. J Invest Dermatol 2009; 129:1319–21. [DOI] [PubMed] [Google Scholar]
- 5.Grall A, Guaguère E, Planchais S, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet 2012;44: 140–7. [DOI] [PubMed] [Google Scholar]
- 6.Radner FP, Marrakchi S, Kirchmeier P, et al. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet 2013;9:e1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000; 25: 141–2. [DOI] [PubMed] [Google Scholar]
- 8.Lestringant GG, Kuster W, Frossard PM, Happle R. Congenital ichthyosis, follicular atrophoderma, hypotrichosis, and hypohidrosis: a new genodermatosis? Am J Med Genet 1998; 75: 186–9. [DOI] [PubMed] [Google Scholar]
- 9.Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am J Hum Genet 2007; 80:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weeda G, Eveno E, Donker I, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 1997; 60: 320–9. [PMC free article] [PubMed] [Google Scholar]
- 11.Broughton BC, Berneburg M, Fawcett H, et al. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Molec Genet 2001; 10: 2539–47. [DOI] [PubMed] [Google Scholar]
- 12.Yesudian P, Srinivas K. Ichthyosis with unusual hair shaft abnormalities in siblings. Brit J Dermatol 1977; 96: 199–203. [DOI] [PubMed] [Google Scholar]
- 13.Feldmeyer L, Huber M, Fellmann F, et al. Confirmation of the origin of NISCH syndrome. Hum Mutat 2006;27:408–10. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad F, Ali RH, Muhammad D, et al. Novel homozygous sequence variants in the CDH3 gene encoding P-cadherin underlying hypotrichosis with juvenile macular dystrophy in consanguineous families. Eur J Dermatol 2016; 26:610–2. [DOI] [PubMed] [Google Scholar]
- 15.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–86. [DOI] [PubMed] [Google Scholar]
- 16.Tang J, Yu CL, Williams SR, et al. Expression, crystallization, and three-dimensional structure of the catalytic domain of human plasma kallikrein. J Biol Chem 2005; 280:41077–89. [DOI] [PubMed] [Google Scholar]
- 17.Louie GV, Yang W, Bowman ME, Choe S. Crystal structure of the complex of diphtheria toxin with an extracellular fragment of its receptor. Mol Cell 1997; 1: 67–78. [DOI] [PubMed] [Google Scholar]
- 18.Lovell SC, Davis IW, Arendall WB, WB 3rd, et al. Structure validation by C alpha geometry: phi, psi and C beta deviation. Proteins 2002;50:437–50. [DOI] [PubMed] [Google Scholar]
- 19.Pierce BG, Wiehe K, Hwang H, et al. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014; 30: 1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alef T, Torres S, Hausser I, et al. Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. J Invest Dermatol 2009; 129:862–9. [DOI] [PubMed] [Google Scholar]
- 21.Avrahami L, Maas S, Pasmanik-Chor M, et al. Autosomal recessive ichthyosis with hypotrichosis syndrome: further delineation of the phenotype. Clin Genet 2008;74:47–53. [DOI] [PubMed] [Google Scholar]
- 22.Neri I, Virdi A, Tortora G, et al. Novel p.Glu519Gln missense mutation in ST14 in a patient with ichthyosis, follicular atrophoderma and hypotrichosis and review of the literature. J Dermatol Sci 2016;81:63–6. [DOI] [PubMed] [Google Scholar]
- 23.Takeichi T, Nanda A, Aristodemou S, et al. Whole-exome sequencing diagnosis of two autosomal recessive disorders in one family. BrJ Dermatol 2015; 172: 1407–11. [DOI] [PubMed] [Google Scholar]
- 24.Hooper JD,Clements JA,Quigley JP, Antalis TM. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem 2001; 276: 857–60. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem 2001; 275: 26333–42. [DOI] [PubMed] [Google Scholar]
- 26.Lin CY, Anders J, Johnson M, Sang QA, Dickson RB. Molecular cloning of cDNA for matriptase, a matrix-degrading serine protease with trypsin-like activity. J Biol Chem 1999; 274: 18231–6. [DOI] [PubMed] [Google Scholar]
- 27.List K, Haudenschild CC, Szabo R, et al. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymichomeostasis. Oncogene 2002; 21: 3765–79. [DOI] [PubMed] [Google Scholar]
- 28.Ge W, Hu H, Ding K, et al. Protein interaction analysis of St14 domains and their point and deletion mutants. J Biol Chem 2006;281:7406–12. [DOI] [PubMed] [Google Scholar]