

Abstract
Traces of Neandertal and Denisovan DNA persist in the modern human gene pool, but have been systematically purged by natural selection from genes and other functionally important regions. This implies that many archaic alleles harmed the fitness of hybrid individuals, but the nature of this harm is poorly understood. Here, we show that enhancers contain less Neandertal and Denisovan variation than expected given the background selection they experience, suggesting that selection acted to purge these regions of archaic alleles that disrupted their gene regulatory functions. We infer that selection acted mainly on young archaic variation that arose in Neandertals or Denisovans shortly before their contact with humans; enhancers are not depleted of older variants found in both archaic species. Some types of enhancer appear to have tolerated introgression better than others; compared with tissue-specific enhancers, pleiotropic enhancers show stronger depletion of archaic single-nucleotide polymorphisms. To some extent, evolutionary constraint is predictive of introgression depletion, but certain tissues’ enhancers are more depleted of Neandertal and Denisovan alleles than expected given their comparative tolerance to new mutations. Foetal brain and muscle are the tissues whose enhancers show the strongest depletion of archaic alleles, but only brain enhancers show evidence of unusually stringent purifying selection. We conclude that epistatic incompatibilities between human and archaic alleles are needed to explain the degree of archaic variant depletion from foetal muscle enhancers, perhaps due to divergent selection for higher muscle mass in archaic hominins compared with humans.
Although hybrids between humans and archaic hominins were once viable, fertile and numerous1–4, Neandertal and Denisovan alleles have been systematically depleted from the most functionally important regions of the human genome5–7. This pattern implies that archaic introgression often had deleterious consequences for human populations, but it is challenging to fine-map the locations of detrimental archaic alleles and determine the nature of their fitness effects. Petr et al. recently found that promoters were actually more depleted of introgression than the coding sequences that lie immediately downstream8, lending weight to the longstanding hypothesis that gene regulatory mutations underlie much of the functional divergence between closely related lineages of hominins9–11. Two other recent studies found that introgressed alleles are associated with gene expression variation more often than is expected by chance12,13, implying that even the archaic regulatory variation that remains in the human gene pool is not necessarily benign.
Previous work comparing rates of amino acid change with rates of substitution at synonymous sites showed that selection was probably relaxed within the Neandertal exome, leading to the accumulation of deleterious mutations14. However, it is less straightforward to perform similar analyses on non-coding variation because of gaps in our understanding of the grammar relating sequence to regulatory function15,16. Allele frequency spectra and patterns of sequence divergence can sometimes provide information about the mode and intensity of selection acting on non-coding regions17–21, but introgressed variants have an unusual distribution of allele ages and frequencies that can confound the efficacy of standard methods that assume simple population histories22. Reporter assays can directly measure the impact of archaic variants on gene expression in vitro23–25, but they cannot translate gene expression perturbations into the subtle effects on survival and reproduction that probably determined which archaic variants were purged from human populations.
Petr et al. used a direct f4 ratio test to conclude that promoters and other conserved non-coding elements harboured less Neandertal DNA than the genome as a whole, but found no similar depletion in enhancers8. However, a subsequent study by Silvert et al.26 came to somewhat different conclusions using different methodology, which involved quantifying the distribution of alleles flagged as probably Neandertal in origin based on their presence in the Altai Neandertal reference and their absence from an African reference panel. Most such alleles are presently rare (<2% frequency in modern Eurasians) and Silvert et al.26 found these rare archaic alleles to be significantly depleted from enhancers. However, archaic variants present at population frequencies of 5% or more were found to occur in enhancers more often than would be expected by chance. Enhancers containing these common archaic alleles were found to be preferentially active in T cells and mesenchymal cells, perhaps due to positive selection for alleles that alter gene expression in the immune system26.
The results of these two previous papers are consistent with a model in which most introgressed enhancer sequences have been segregating neutrally within the human gene pool, but in which archaic haplotypes containing private Neandertal enhancer variation were more often selected against than introgressed haplotypes containing private Neandertal variants outside regulatory regions. To interrogate this model more directly, we leveraged a set of archaic variant calls that were previously generated using a conditional random field (CRF) approach5,7. The CRF introgression calls are organized hierarchically in a way that correlates with age: some of these alleles are confidently inferred to be either Neandertal or Denisovan in origin, while others might have originated in either archaic species and probably segregated for a longer period of time before human secondary contact. We quantified the abundance of young versus old archaic alleles in enhancers as a function of tissue activity, controlling for the amount of background selection enhancers experience, to estimate whether selection acted to remove certain classes of archaic variants from regulatory regions.
Results
Enhancers appear depleted of Neandertal alleles compared with control regions affected by similar levels of background selection.
We intersected the ENCODE RoadMap enhancer calls with the high-confidence Neandertal and Denisovan CRF introgression calls generated from the Simons Genome Diversity Project (SGDP) data7,27. Of the two available call sets, we used variant set number 2, which identifies Eurasian haplotypes as Neandertal if they appear closer to the Altai Neandertal reference than to either an African reference panel or the Altai Denisovan reference. (Similarly, haplotypes are identified as Denisovan if they appear closer to the Altai Denisovan reference than to an outgroup consisting of Africans plus the Altai Neandertal reference).
Since Neandertal variants in enhancers might have been purged due to selection against nearby linked coding variants, we devised a method to measure archaic allele depletion while controlling for the strength of background selection, as quantified by McVicker and Green’s B statistic5,7,28. We randomly paired each Neandertal variant with two control variants matched for both B statistic decile and allele frequency (Fig. 1a), then computed the proportion of archaic versus control alleles occurring within enhancers.
Fig. 1 |. Introgressed variants are depleted from enhancers and exons relative to matched control variants.
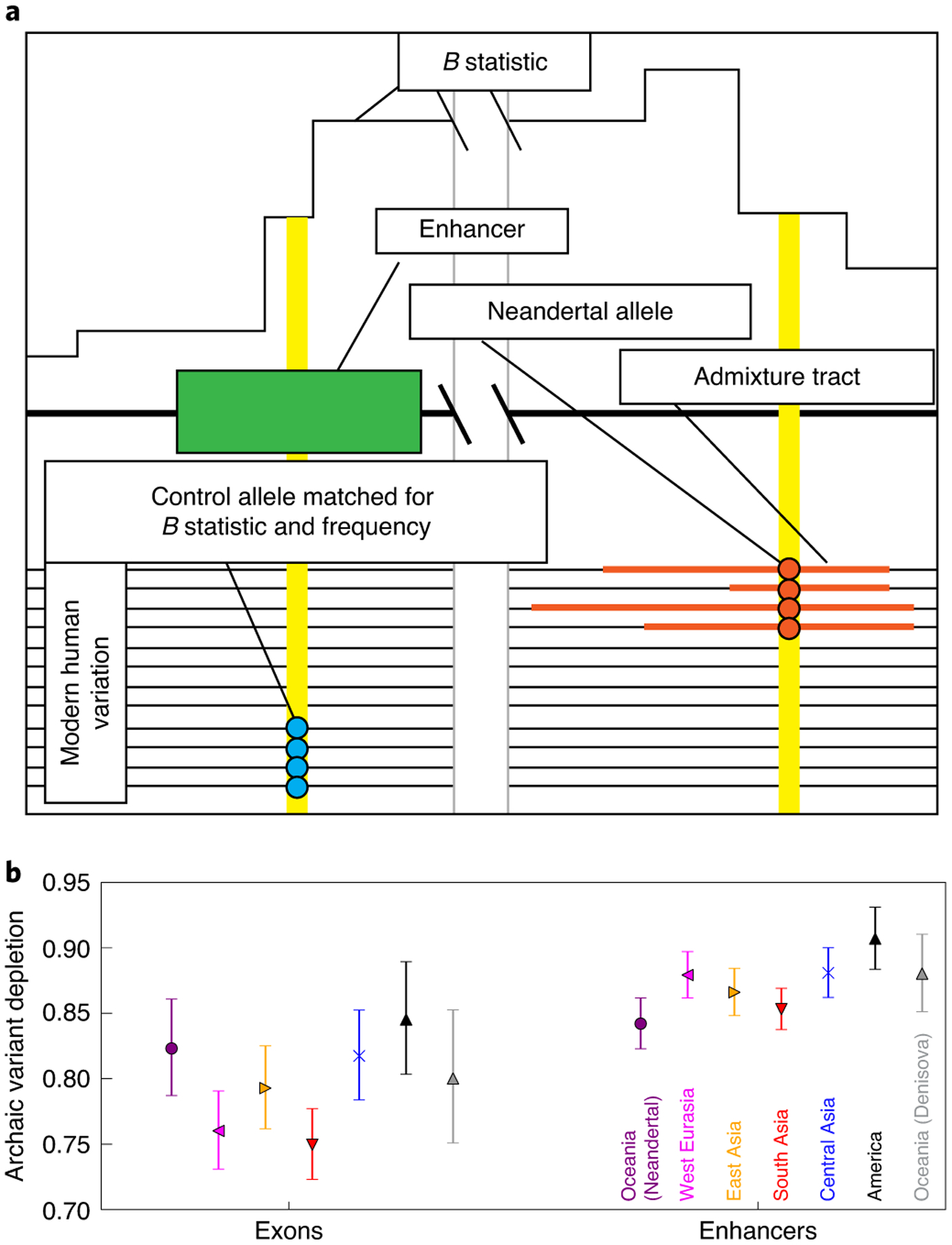
a, Schematic illustrating the process of matching archaic variants to control variants with identical allele frequencies and B statistic values. b, Introgressed-to-control variant odds ratios showing depletion of Neandertal variants from both exons and enhancers in every population sequenced by the SGDP. In the case of Oceanians, a similar pattern holds for Denisovan variant calls. error bars span 95% binomial confidence intervals.
In every population, we found that control variants occur within enhancers significantly more often than introgressed variants do (Fig. 1b), with depletion odds ratios ranging from 0.84–0.91 and 95% binomial confidence intervals excluding an odds ratio of 1. As expected, this method also detects negative selection against introgression in exons. Enard and Petrov29 recently used a related approach to quantify Neandertal introgression in proteins that interact with viruses. To ensure that the linkage structure of the archaic SNPs was not contributing to this result, we sampled an alternative set of controls with a similar linkage block structure. Upon substituting these controls for our originally sampled controls, we observed a nearly identical landscape of introgression depletion (Extended Data Fig. 1).
Highly pleiotropic enhancers harbour fewer archaic variants than tissue-specific enhancers.
The enhancers annotated by RoadMap exhibit wide variation in tissue specificity. Some are active in only one or two tissues, while others show activity in 20 tissues or more30. When we stratified enhancers by pleiotropy number (that is, the number of tissues in which the enhancer is active), we found pleiotropy to be correlated with the magnitude of archaic variant depletion (Fig. 2).
Fig. 2 |. Archaic variant depletion is correlated with the number of cell types in which an enhancer is active.
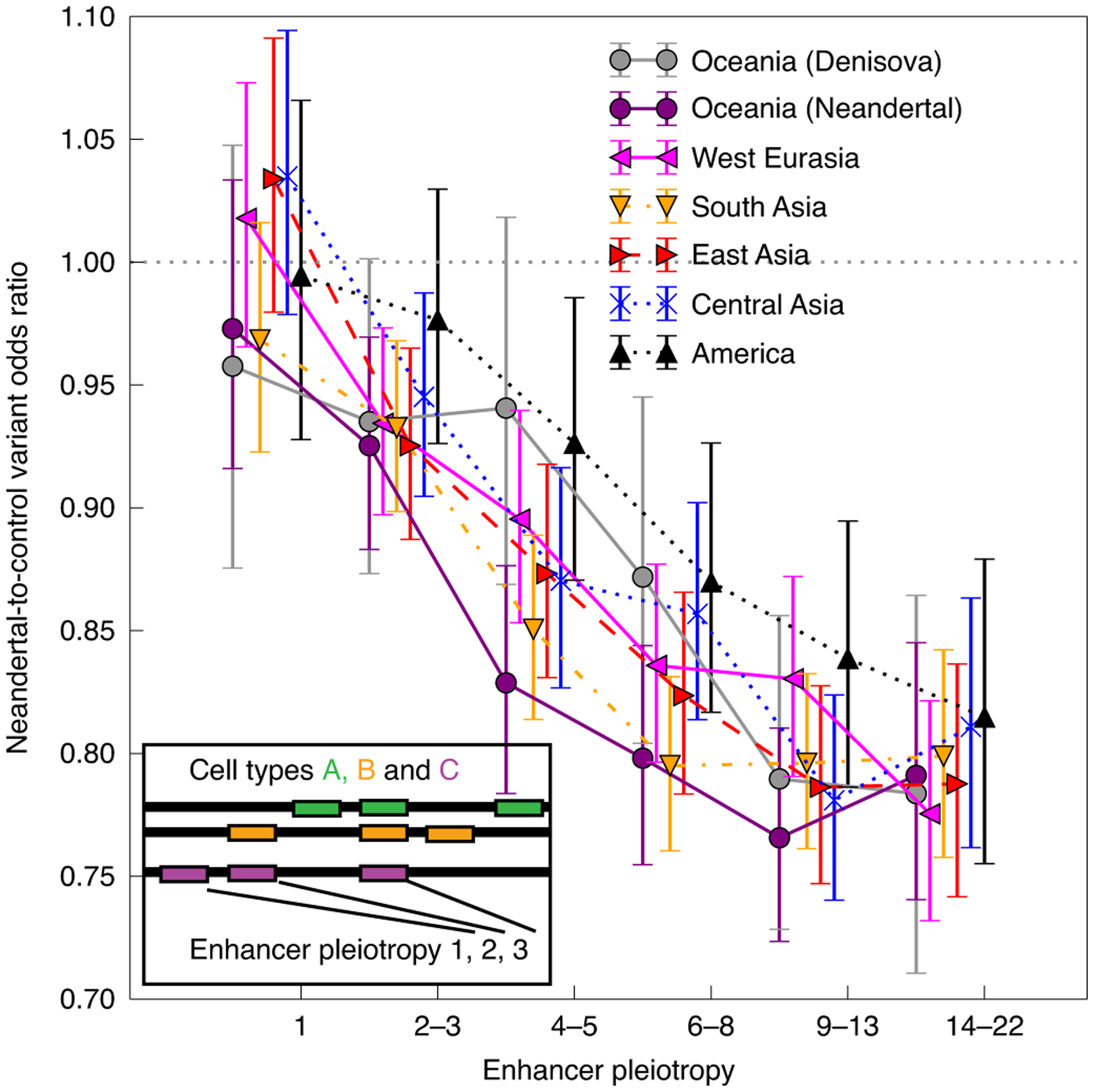
The pleiotropy number of an enhancer is defined as the number of tissues in which the enhancer is active (as illustrated by the inset panel). enhancers are grouped into bins of increasing pleiotropy number such that each bin contains a roughly equal number of enhancers; within each bin, we computed the odds ratio of archaic SNPs relative to matched control SNPs in each of the SGDP continental groups. error bars span 95% binomial confidence intervals. The dotted line on the y axis marks an archaic-to-control variant odds ratio of 1.0, meaning that every confidence interval lying completely below this line corresponds to a set of enhancers that are depleted of archaic variation at the P < 0.05 significance level. enhancers active in only a single cell type do not appear depleted of archaic SNPs, whereas enhancers that are active in multiple cell types contain up to 20% fewer archaic variant calls than expected.
If high-pleiotropy enhancers exhibited high sequence similarity between humans, Neandertals and Denisovans, this could make it difficult to detect archaic introgression in these regulatory regions and could create the false appearance of selection against introgression. However, we found that the human and archaic reference sequences were actually more divergent in high-pleiotropy enhancers than in other regions (Extended Data Fig. 2a), making selection against introgression more likely to be responsible for the observed depletion gradient. Enhancer activity is known to increase the mutation rate by inhibiting DNA repair31,32, which may explain why highly active enhancers have been diverging between hominid species at an accelerated rate.
We found substantial variation between tissues in the magnitude of archaic SNP depletion (Fig. 3a and Extended Data Fig. 3), as well as correlation across tissues between depletion of Neandertal variants and depletion of Denisovan variants (r2 = 0.537; P < 4 × 10−5). Enhancers active in foetal muscle, foetal brain and neurosphere cells are the most strongly depleted of introgressed variation, while enhancers active in foetal blood cells and T cells, as well as mesenchymal cells, appear the least depleted. We observed no correlation across tissues between the degree of archaic variant depletion and the genetic divergence between archaic and human reference sequences (Extended Data Fig. 2b,c). Mesenchymal cells, T cells and other blood cells are among the cell types in which some adaptively introgressed regulatory variants are thought to be active26,33–35, but our results suggest that selection overall decreased archaic SNP load even within the regulatory networks of these cells. The excess archaic SNP depletion in brain and foetal muscle is a pattern that holds robustly across populations (Extended Data Fig. 4). Although exons are slightly more depleted of archaic SNPs than enhancers as a whole (see the non-overlapping 95% confidence intervals in Fig. 1b), they are actually less depleted of archaic variation than brain or foetal muscle enhancers.
Fig. 3 |. Neandertal and Denisovan variant depletion varies between enhancers active in different tissues.
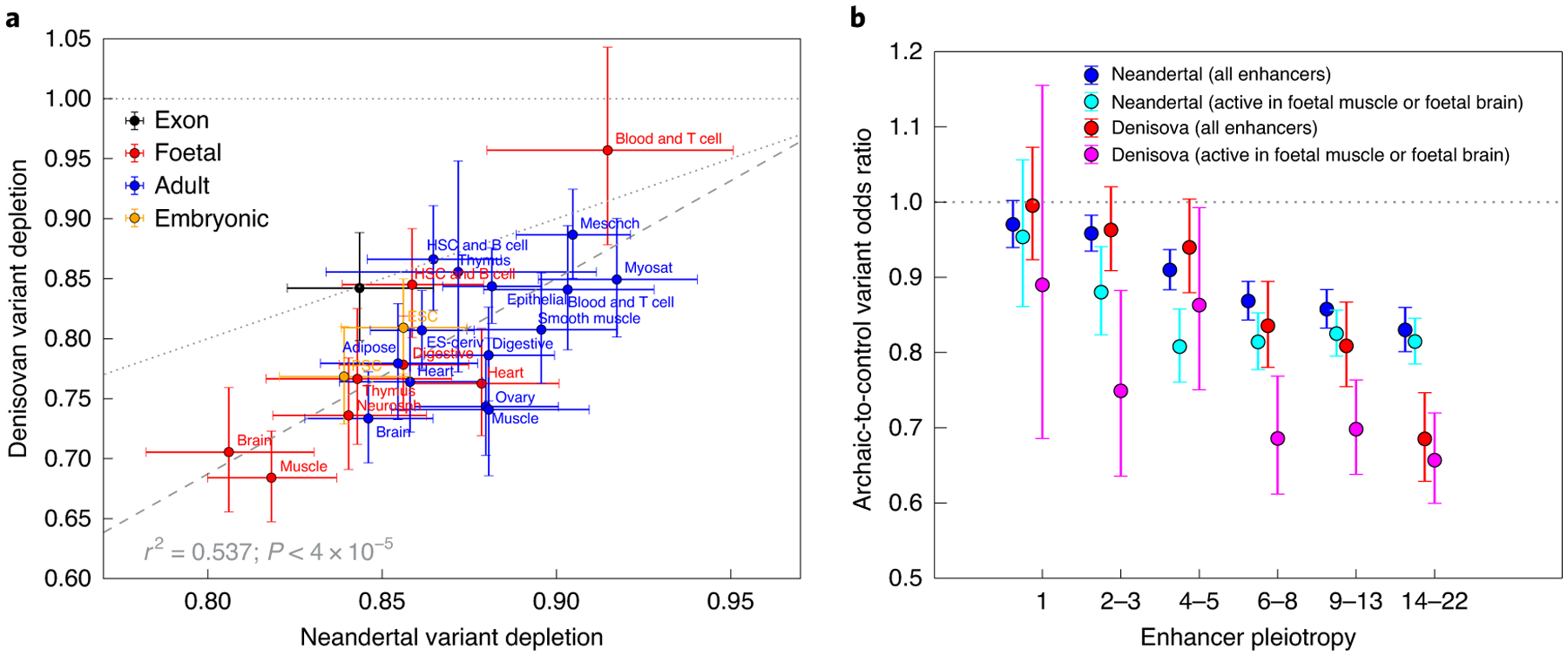
a, The set of enhancers active in each Roadmap cell line is significantly depleted of both Neandertal and Denisovan variation, with the exception of blood and T cells, whose Denisovan depletion confidence interval overlaps an odds ratio of 1.0 (horizontal dotted line). Data points that lie below the diagonal dotted line correspond to tissues whose enhancers are more depleted of Denisovan SNPs compared with Neandertal SNPs. The slope of the dashed regression line is significantly positive (P < 4 × 10−5), implying that Neandertal variant depletion and Denisovan variant depletion are correlated across tissues (r2 = 0.537). b, even after restricting to enhancers active in foetal muscle or foetal brain (the two tissue types most depleted of introgressed variation), pleiotropy remains negatively correlated with archaic SNP depletion. The difference between these two tissues and other tissues is driven mainly by enhancers of intermediate pleiotropy. All error bars span 95% binomial confidence intervals. eSC, embryonic stem cell; HSC, haematopoietic stem cell; mesench, mesenchymal; myosat, myosatellite cell; iPSC, induced pluripotent stem cell; eS-deriv, cell line derived from embryonic stem cells; neurosph, neurosphere cell.
Although foetal muscle and foetal brain enhancers are more depleted of archaic SNPs than enhancers active in other tissues, selection acting in these two tissues alone is not sufficient to explain the apparent depletion of archaic SNPs from other tissues’ enhancers. When we computed the magnitude of archaic SNP depletion as a function of pleiotropy in the subset of enhancers that are active in foetal brain, foetal muscle or both, we still found pleiotropy to be predictive of introgression depletion (Fig. 3b).
For enhancers active in six or more tissues, depletion of Denisovan variants is notably stronger than depletion of Neandertal variants. One possible culprit is a discrepancy in the power of the CRF to ascertain Neandertal versus Denisovan introgression. The Denisovans who interbred with modern humans were quite genetically differentiated from the Altai Denisovan reference individual, while the Altai Neandertal reference is more modestly divergent from Neandertal introgressed tracts36, and this probably created differences between species in the sensitivity and specificity of archaic SNP detection.
Old variation shared by Neandertals and Denisovans was probably less deleterious to humans than variation that arose in these species more recently.
To further test the hypothesis that selection acted to purge young, rare archaic variation from enhancers, we leveraged the difference between two introgression call sets that Sankararaman et al.7 generated from the SGDP data. As mentioned earlier, we conducted all previous analyses using call set 2, which was constructed to minimize the misidentification of Neandertal alleles as Denisovan and vice versa. To generate Neandertal call set 2, Sankararaman et al.7 used an outgroup panel that contained the Altai Denisovan as well as several Yoruban genomes. Similarly, Denisovan call set 2 was generated using a panel that included Yorubans plus the Altai Neandertal. In contrast, call set 1 was generated using an outgroup panel composed entirely of Africans, and this procedure identifies more archaic SNPs overall.
Compared with set 2, we hypothesized that the more inclusive set 1 calls should contain more old variation that arose in the common ancestral population of Neandertals and Denisovans (Fig. 4a). We posited that this older variation might be better tolerated in humans because it rose to high frequency in an ancestral population that was not as divergent from humans as later Neandertal and Denisovan populations. Neandertals and Denisovans also suffered from increasingly severe inbreeding depression as time went on, further increasing the probability that younger variants could have deleterious effects2,14.
Fig. 4 |. Different landscapes of young and old introgressed archaic variation.
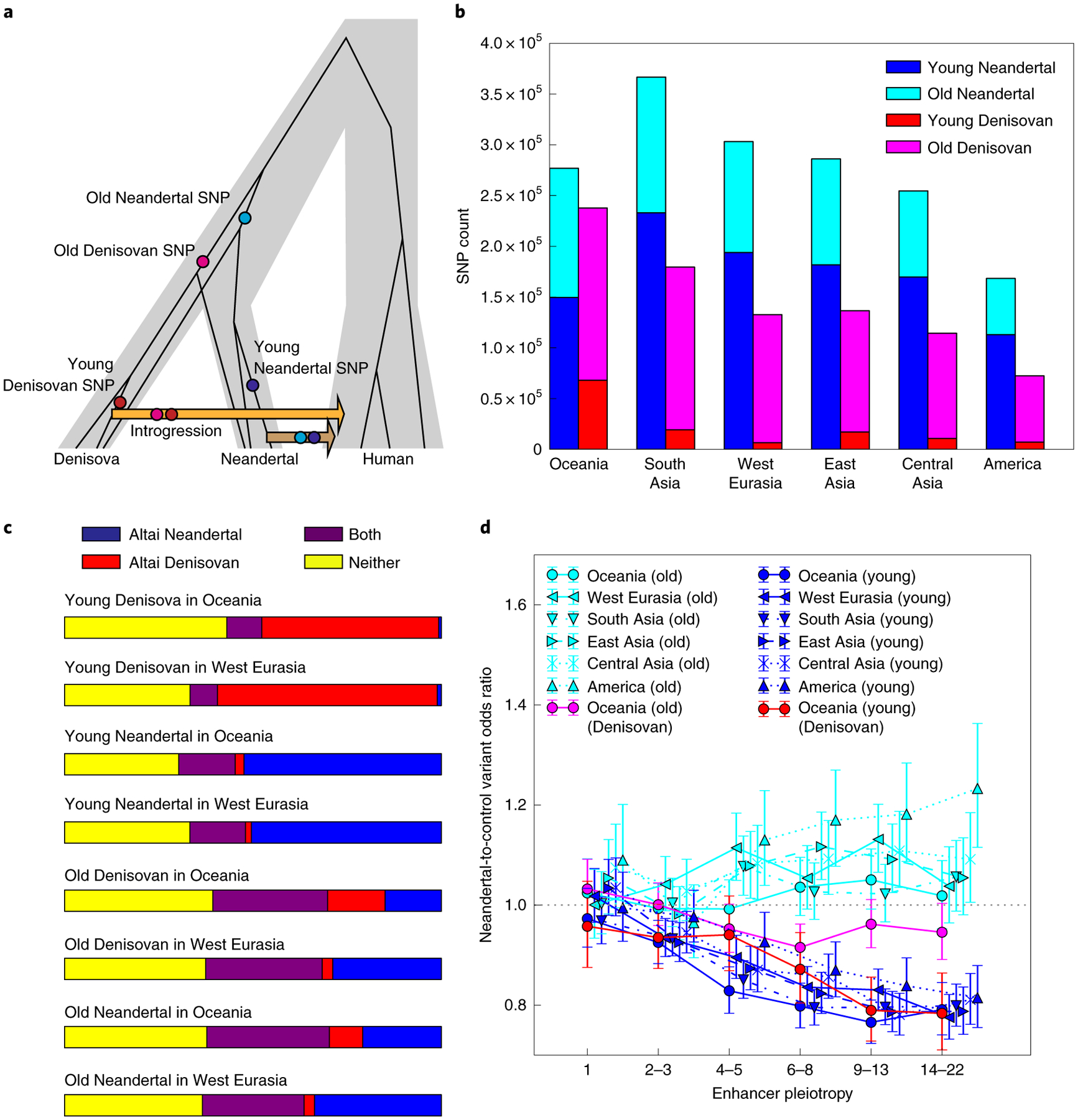
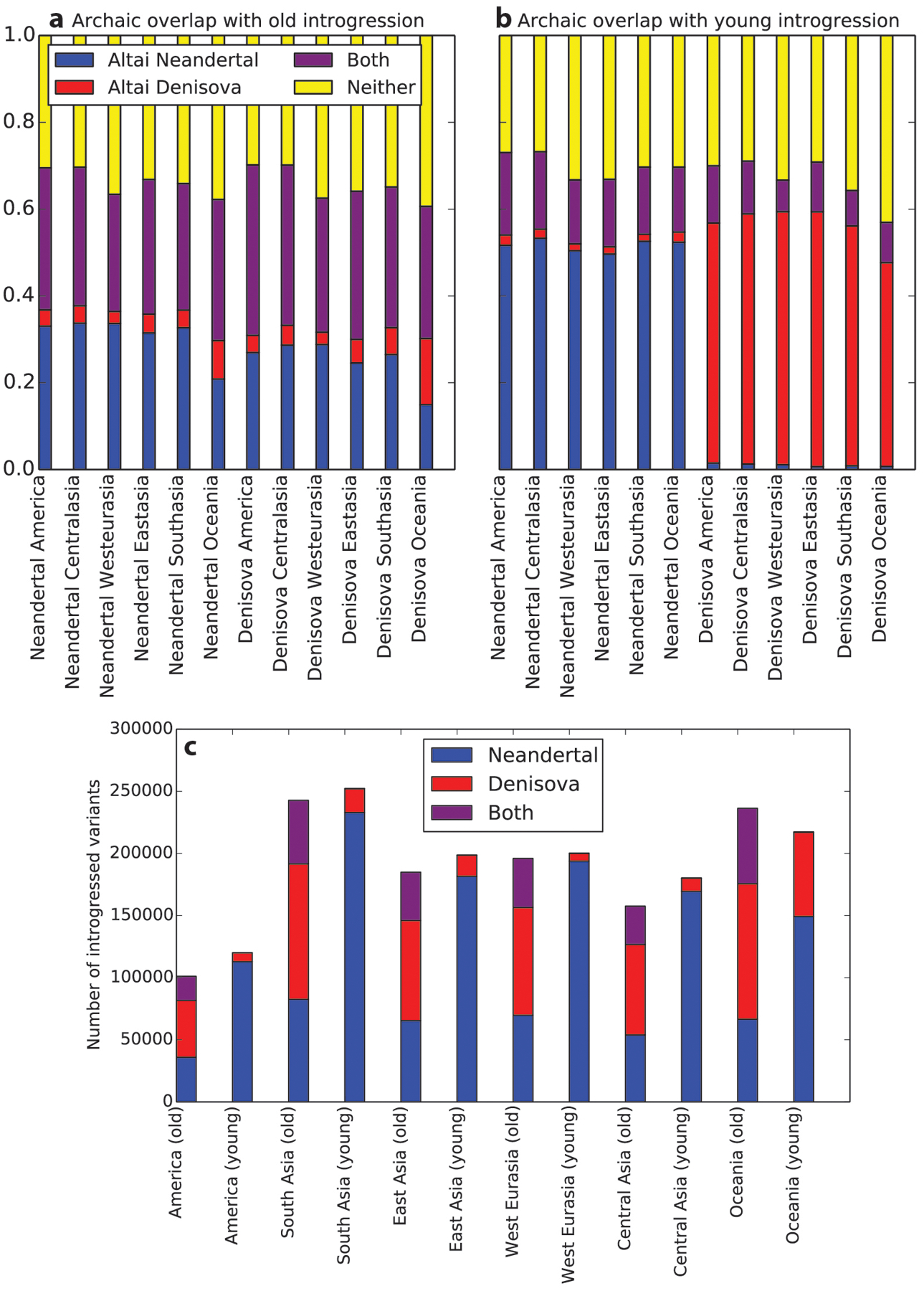
a, We classified introgression calls as old or young based on their presence in call sets 1 and 2 generated by Sankararaman et al.7. By design, the old alleles (present in call set 1 but not call set 2) are more often shared between Neandertals and Denisovans, and we hypothesize that many of these alleles arose before the Neandertal/Denisovan divergence, as pictured, or else crossed between the two species via Neandertal/Denisovan gene flow. In contrast, we hypothesize that the young alleles most often arose after Neandertals and Denisovans had begun to diverge. b, Numerical counts of old and young introgressed variants in the SGDP human genomes. Young Denisovan variants are probably rare because the Altai Denisovan was not closely related to the Denisovan population that primarily interbred with humans36. c, We computed the fraction of CRF introgression calls that occur as derived alleles in the Altai Neandertal genome and/or the Altai Denisovan genome. As expected, old variants are two- to fourfold more likely than young variants to occur in both archaic reference genomes (see extended Data Fig. 5a,b for more data on allele sharing between introgression calls and the reference archaic genomes). d, In contrast with the young archaic variation considered elsewhere in this paper, old archaic variation is not measurably depleted from enhancers—even enhancers active in numerous tissues. All error bars span 95% binomial confidence intervals; confidence intervals that do not intersect the dotted line (shown at an odds ratio of 1.0) indicate significant depletion of archaic variants relative to matched controls.
To test our hypothesis that set 2 might be enriched for deleterious variation, we compiled sets of old Neandertal and Denisovan variation comprising their respective set 1 introgression calls and excluding all set 2 introgression calls. In every population, young Neandertal variants outnumber old Neandertal variants, but conversely, old Denisovan variants outnumber young Denisovan variants (Fig. 4b and Extended Data Fig. 5c). The CRF may have been better powered to detect young Neandertal variants compared with young Denisovan variants due to the aforementioned closer relationship of the reference Neandertal to archaic individuals who interbred with humans36. As expected, old variants are more likely than young variants to be present in both archaic reference genomes rather than just one, although more than 30% of calls in each category are absent from both archaic references and are presumably identified as archaic due to patterns of linkage disequilibrium (Fig. 4c, Extended Data Fig. 5a,b).
In contrast with the young set 2 introgression calls, old calls are not measurably depleted from enhancers compared with control variants matched for allele frequency and B statistic (Fig. 4d). Old introgressed variants also have higher mean allele frequencies, which could indicate that they have experienced less negative selection following introgression (Extended Data Fig. 6). These patterns suggest that the introgression landscape was shaped mainly by selection against Neandertal and Denisovan variants that arose relatively close to the time that gene flow occurred, not variation that arose soon after their isolation from humans. Many populations actually show a slight enrichment of old archaic variants in enhancers compared with controls, as shown in Fig. 4d (95% confidence intervals that exclude an odds ratio of 1). These sets of old, shared variants are possibly enriched for beneficial alleles that swept to high frequency in the common ancestor of Neandertals and Denisovans. They should at least be depleted of deleterious variation compared with our control alleles that probably arose more recently in humans. In several cases, the odds ratio enrichment of old archaic variation in enhancers actually trends upward with increasing pleiotropy, possibly because the highest-pleiotropy enhancers show the most divergence between human and archaic reference genomes (Extended Data Fig. 2a). If archaic SNPs in enhancers were generally neutral or selectively favoured, the positive correlation between pleiotropy and human/archaic divergence would lead us to predict the odds ratio trend that is observed for old archaic variants, not the opposite correlation with pleiotropy that is observed for young archaic variants.
Neandertals and Denisovans are thought to have begun diverging about 640,000 years ago37. Since this is long enough to efficiently purge deleterious variation, any surviving archaic variation that predates this split is likely to have nearly neutral or beneficial fitness effects, assuming no negative epistasis with human variation. We can see this from a simple population genetic calculation: assuming that the Neandertal/Denisovan effective population size was about 4,000 (ref.2) and their generation time is 30 years, 4Ne for these species would be 480,000. This implies that more than half of the variation that segregated neutrally in the ancestral Neandertal/Denisovan population would have been fixed or lost by the time the two species interbred with humans, leaving ample time for deleterious ancestral variation to be purged. 480,000 years also predates the estimate of the start of the bottlenecks that affected Neandertals and Denisovans2, so variation that predates this period may have been efficiently purged of deleterious alleles that would have segregated neutrally if they had arisen after the start of the bottleneck period. Some old variants might be younger than the Neandertal/Denisovan split if they crossed between the boundaries of these species by introgression; Neandertals and Denisovans are known to have interbred with each other while still maintaining distinct gene pools. This population history suggests that gene flow between Neandertals and Denisovans may be enriched for variants that are benign on a variety of genetic backgrounds38, making them more likely to be benign on a human background as well.
Introgressed variants and recent mutations have been differently selected against as a function of enhancer activity.
Next, we investigated whether the enhancers most depleted of young archaic variants are simply the enhancers most intolerant to new mutations, leveraging the fact that natural selection allows neutral and beneficial mutations to reach high frequencies more often than deleterious mutations do39,40 (Fig. 5a). Working with the site frequency spectrum (SFS) of African enhancer variation from the 1000 Genomes project, we computed the proportion of variants segregating in enhancers that are singletons and compared this with the proportion of singletons in the immediately upstream enhancer-sized regions (Fig. 5b). Neandertals contributed much less genetic material to sub-Saharan Africans compared with non-Africans1,4, meaning that Neandertal alleles should have little direct effect on the African SFS.
Fig. 5 |. Rare variant enrichment reveals that enhancer sequences are weakly selectively constrained.
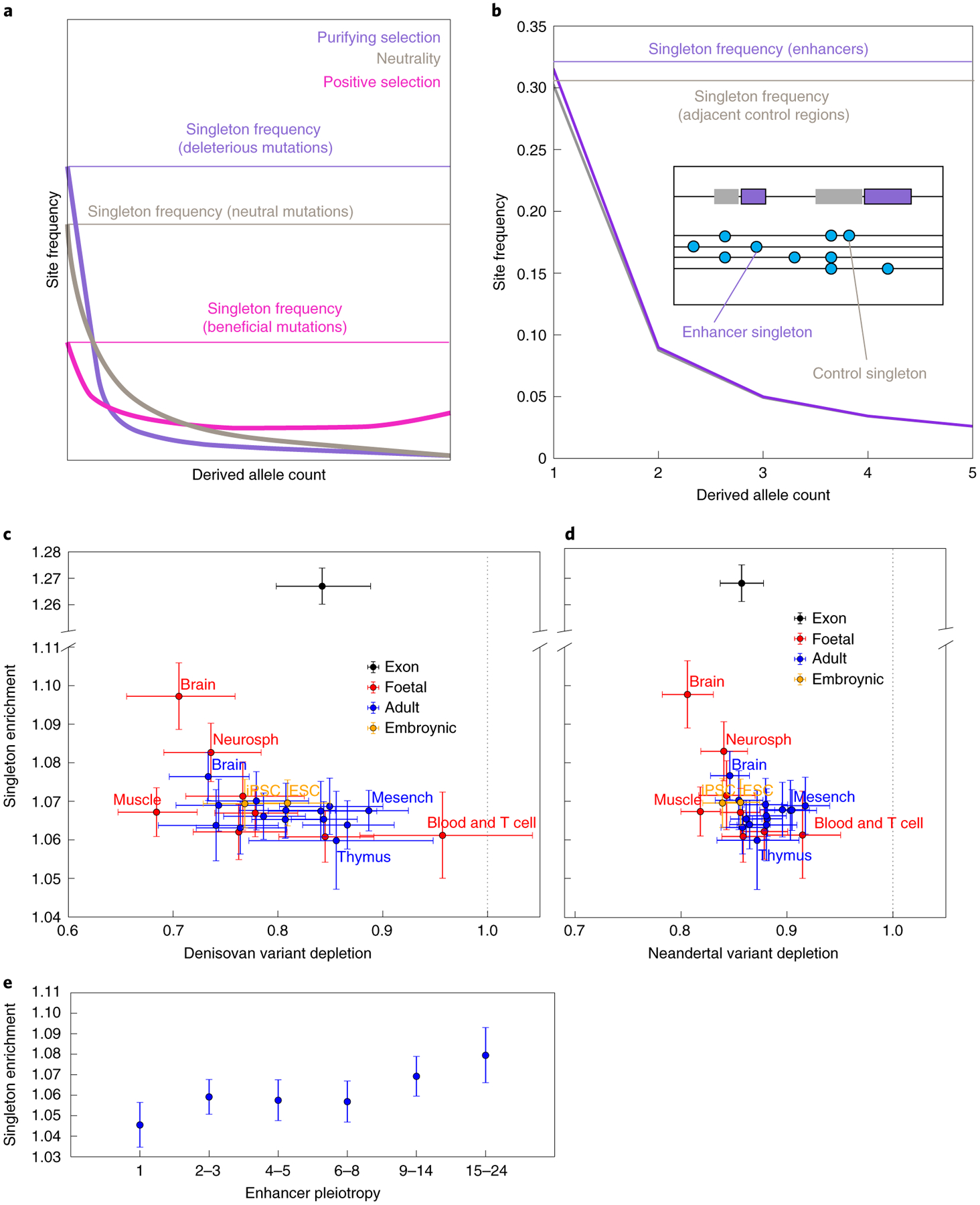
a, Theory predicts that the SFS becomes skewed towards rare variants by the action of purifying selection. b, In African data from the 1000 Genomes project, the enhancer SFS has a higher proportion of singletons compared with control regions adjacent to enhancers. c, every tissue type’s enhancer complement is enriched for singletons compared with adjacent control regions. This comparison of singleton enrichment odds ratios versus Denisovan depletion odds ratios shows that foetal brain, neurosphere cells and adult brain are outliers under stronger constraint. The yaxis has been split to accommodate the magnitude of singleton enrichment in exons and the vertical dotted line demarcates a Denisovan-to-control variant ratio of 1.0. error bars span two binomial test standard errors. d, Comparison of the singleton enrichment landscape with the Neandertal depletion landscape. e, enhancer pleiotropy is negatively correlated with singleton enrichment, although even enhancers of pleiotropy 1 have a singleton enrichment odds ratio significantly greater than 1. All error bars span 95% binomial confidence intervals.
One caveat is that this strategy will not detect the effects of strongly deleterious mutations that do not segregate long enough to affect the frequency spectrum’s shape. However, strongly deleterious mutations are not expected to contribute to mutation load differences between populations, making it appropriate to focus on identifying regions whose variation is affected by selection against weakly deleterious mutations.
By comparing enhancers with immediately adjacent regions, we control for the potentially confounding effects of recombination rate, background selection and sequencing read depth. Although enhancers probably have elevated mutation rates because transcription factor binding impairs DNA repair31, the proportion of variants that are singletons is independent of mutation rate as long as the mutation rate has remained constant over time41. Enhancers admittedly have higher GC content than adjacent control regions, but the enrichment of singletons in the enhancer SFS holds separately for SNPs with AT ancestral alleles and SNPs with GC ancestral alleles (Extended Data Fig. 7a). This suggests that the SFS is not enriched for singletons because of a force such as biased gene conversion, which only depresses the frequencies of mutations from GC to AT and instead increases the frequencies of mutations from AT to GC42. There is also no apparent correlation across tissues between GC content and the enrichment of rare variants in enhancers (Extended Data Fig. 7b). We conclude that purifying selection is probably driving the difference between the SFSs of enhancers and control regions, not base composition or biased gene conversion.
Although enhancers broadly show evidence of purifying selection against both archaic variation and new mutations, the strength of selection against these two types of perturbation is poorly correlated among tissues (Fig. 5c,d). Although singleton enrichment appears to be nominally correlated with Neandertal depletion (r2 = 0.31; P < 0.004) and Denisovan depletion (r2 = 0.27; P < 0.009), this correlation disappears when brain tissues are excluded (Neandertal P < 0.42; Denisovan P < 0.10; see Extended Data Fig. 8). Foetal brain, neurosphere cells and (to a lesser extent) adult brain are the tissues whose active enhancers show the most singleton enrichment, suggesting that mutations perturbing brain development have a high probability of deleterious consequences. In contrast, foetal muscle enhancers show no evidence of unusual selective constraint despite their strong depletion of both Neandertal and Denisovan ancestry. We obtain categorically similar results when we estimate selective constraint using phastCons scores rather than singleton enrichment (Extended Data Fig. 9).
Enhancer pleiotropy is positively correlated with singleton enrichment as well as the depletion of archaic alleles (Fig. 5e). This observation may be related to experimental evidence that the most highly pleiotropic enhancers tend to have the most consistently conserved functioning across species43. One difference, however, is that enhancers active in only a single tissue (pleiotropy number 1) still show significant evidence of selection against new mutations despite their lack of any evidence for selection against archaic introgression (odds ratio 95% confidence interval excludes 1).
Discussion
Most methods for identifying introgressed archaic haplotypes rely on putatively unadmixed outgroup data. Chen et al.4 recently showed that the use of an African outgroup can confound measurements of introgression fraction differences between populations, causing less introgression to be detected in Europeans compared with Asians because Europeans exchanged more recent migrants with Africa4. Our analysis of young versus old CRF-based calls shows that the choice of outgroup can also affect the distribution of archaic allele calls across functional versus putatively neutrally evolving genomic regions. This implies that outgroup panel use can interfere with efforts to estimate unbiased Neandertal and Denisovan admixture fractions, but does not imply that unbiased admixture fractions are necessarily the most powerful statistic for detecting the footprints of selection against archaic alleles. The subset of archaic haplotypes that are most divergent from outgroup panels are by definition enriched for mutations that may have detectable fitness effects, whereas archaic haplotypes that are less divergent and more difficult to detect computationally are more likely to segregate neutrally in human populations. In reaching such conclusions, proper care must be taken to control for rates of human/archaic reference divergence, which can vary across the genome. In enhancers, however, we found archaic/human divergence to be elevated, which probably enhanced the power of the CRF to discover introgression overlapping these regions. This suggests that selection is needed to explain the observed depletion of young archaic variants from enhancers.
Two sources of dysfunction are thought to drive selection against archaic introgression: excess deleterious mutation load in inbred Neandertal and Denisovan populations44,45; and accumulation of epistatic incompatibilities due to divergent selective landscapes5,7,46. Both forces have the potential to affect enhancers, and our results confer some ability to distinguish between the two. In particular, the weakness of the correlation between archaic allele depletion and singleton enrichment furnishes useful insights into the fitness effect differences between de novo human mutations and young introgressed archaic alleles. This difference appears starkest when comparing enhancers with exons, which are known to evolve more slowly than enhancers over phylogenetic timescales47–49, implying that selection acts more strongly against new coding mutations compared with new regulatory mutations. However, despite their different levels of selective constraint against new mutations, exons and enhancers show evidence for selection against archaic alleles (Fig. 5c,d), suggesting that regulatory effects may have played a significant role in shaping the landscape of Neandertal and Denisovan introgression.
When a set of regulatory elements is more depleted of introgression than expected given their level of selective constraint, this suggests that the Neandertal and Denisovan selective landscape may have diverged from the human one in these regions. Foetal muscle enhancers appear to fit this profile, with unremarkable singleton enrichment and phastCons scores but strong depletion of young archaic variants. Archaeological evidence indicates that Neandertals had higher muscle mass, strength and anatomical robustness compared with humans50,51, supporting the idea that the two species had different foetal muscle growth optima. We have no direct knowledge of Denisovan muscle anatomy, but the depletion of Denisovan DNA from muscle enhancers may suggest that they shared Neandertals’ robust phenotype, assuming that phenotype is mediated by gene regulation in foetal muscle.
In contrast with muscle, mutation load is a more attractive candidate cause for the depletion of archaic alleles from brain enhancers. Our conclusion that brain enhancers experience high deleterious mutation rates is bolstered by previous knowledge of many de novo mutations in these regions that cause severe developmental disorders52–54.
Both genetic load and hybrid incompatibilities might drive the correlation we have found between enhancer pleiotropy and archaic allele depletion. Steinrücken et al.55 noted that epistatic incompatibilities are most likely to arise in genes with many interaction partners; when a gene is active in multiple tissues, it must function as part of a different expression network in each tissue, which could create additional constraints on enhancers that must coordinate expression correctly in several different contexts. Our results thus imply that introgression is most depleted from enhancers that must function within a variety of cell-specific regulatory networks. We also know that genes expressed in many tissues evolve more slowly than genes expressed in few tissues because they have greater potential for functional tradeoffs11,56, and a mutation that disrupts the balance of a functional tradeoff is likely to have a deleterious effect. This idea is corroborated by our finding that pleiotropic enhancers are more constrained. One caveat is that highly pleiotropic enhancers may be the easiest to experimentally identify. If the RoadMap call sets of tissue-specific enhancers contain a higher proportion of false positives, this might inflate our estimate of the correlation between pleiotropy and selective constraint.
Both genetic load and epistatic incompatibilities are expected to snowball over time, making young archaic variation more likely to be deleterious in hybrids compared with older, high-frequency archaic variation. Part of this effect might be due to positive selection on beneficial introgressed alleles that have risen to high frequency in multiple populations. As more methods for inferring admixture tracts are developed, our results underscore the importance of investigating how they might be biased towards young or old archaic variation and using this information to update our understanding of how selection shapes introgression landscapes. Regulatory mutations appear to have created incompatibilities between many species that are already in the advanced stages of reproductive isolation57–60, and our results suggest that they also harmed the fitness of human/Neandertal hybrids during the relatively early stages of speciation between these hominids. As more introgression maps and functional genomic data are generated for hybridizing populations of non-model organisms, it should be possible to measure the prevalence of weak regulatory incompatibility in more systems that exist in the early stages of reproductive isolation and to test how many of the patterns observed in this study occur repeatedly outside the hominoid speciation continuum.
Methods
Extraction of Neandertal and Denisovan variant sets.
Neandertal and Denisovan variant call sets were downloaded from https://sriramlab.cass.idre.ucla.edu/public/sankararaman.curbio.2016/summaries.tgz. These files classify a haplotype as archaic if it is classified as archaic with ≥50% probability. Using these summaries, we classify a variant as archaic if 100% of the haplotypes on which it appears are classified as such. Unless otherwise stated, all Neandertal and Denisovan variants are obtained from the respective summary call set 2, which we refer to in the text as the young call sets. To construct the old Neandertal call set analysed in Fig. 4, we included all variants from Neandertal set 1 except any variants that also appeared in Neandertal set 1 or Denisovan set 1. Similarly, the old Denisovan call set included all variants present in Denisovan set 1 except those variants also present in Neandertal set 2 or Denisovan set 2. Chromosome X was excluded given its unique systematic depletion of Neandertal and Denisovan variants. Across SGDP populations, the number of SNPs identified as Neandertal in origin ranges from 109,253 (in West Eurasia) to 233,013 (in South Asia). The number of introgressed Denisovan SNPs ranges from 6,437 (in West Eurasia) to 68,061 (in Oceania).
Classifying enhancers by tissue type and pleiotropy number.
Cell lines were classified into tissue types using the tissue assignment labels from the July 2013 RoadMap data compendium, available at https://personal.broadinstitute.org/meuleman/reg2map/HoneyBadger2_release/DNase/p10/enh/state_calls.RData. Whenever a tissue type contained both foetal and adult cell lines, we further subdivided that tissue type into adult and foetal. We then computed a pleiotropy number for each enhancer by counting the number of distinct tissue type labels in the cell lines where that enhancer is annotated as active. Three separate states are used to denote enhancer activity in the honey badger model (states 6, 7 and 12 denote genic enhancers, enhancers and bivalent enhancers, respectively) and we considered each of these states as equivalent evidence of enhancer activity. Foetal and adult tissue types are counted as distinct tissues for the purpose of this computation.
Testing for depletion of archaic variation relative to matched control variation.
To estimate the strength of background selection experienced by human genomic loci, B statistic values ranging on a scale from 1–1,000 were downloaded at http://www.phrap.org/software_dir/mcvicker_dir/bkgd.tar.gz. We quantized these values by rounding them down to the nearest multiple of 50 B statistic units, then lifted them over from hg18 coordinates to hg19 coordinates. Each SNP in the SGDP data was assigned the B statistic value of the closest site annotated by McVicker et al.28.
Our tests for depletion of archaic variation are computed relative to non-archaic control SNPs that have the same joint distribution of allele frequency and B statistic as the SNPs annotated as archaic in origin (see the section ‘Detailed sampling procedure for matched control SNPs’ for more information on how these matched control sets are obtained).
Assume that is a set of A archaic SNPs and is a set of 2 × A-matched controls (we chose to sample 2A controls rather than A controls to reduce the stochasticity of the control set and decrease the size of the confidence intervals on all computed odds ratios). To test whether archaic variation of this type is enriched or depleted in a set of genomic regions, we start by counting the number AG of archaic SNPs contained in and the number CG of control SNPs contained in . We say that this type of archaic variation is depleted from if the odds ratio (AG/(A − AG))/(CG/(2A − CG)) is less than 1.
To assess the significance of any enrichment or depletion we measure, we ask whether the corresponding log odds ratio, log [AG] + log [2A – CG] − log [A – AG] − log [CG]I, is more than two standard errors away from zero. The standard error of this log odds ratio is In each forest plot presented in the manuscript, this formula was used to draw error bars that span two standard errors in each direction.
Detailed sampling procedure for matched control SNPs.
For each archaic SNP set (Neandertal 1, Neandertal 2, Denisovan 1 and Denisovan 2) and each population p, we counted the number Ap(b, c) of alleles with B statistic value b and derived allele count c in population p, counting the allele as archaic if all derived alleles were annotated as present on archaic haplotypes in the relevant call set of population p. We then counted the number Np(b, c) of non-archaic alleles with B statistic b and derived allele count c. In order for a SNP to count as non-archaic, none of its derived alleles could be present on a haplotype from population p that was called as archaic in either call set 1 or call set 2. A set of 2 × Ap(b, c) control SNPs was then sampled uniformly at random without replacement from the Np(b, c) control candidate SNPs. In the rare event that Np(b, c) < 2 × Ap(b, c), the control set was defined to be the entire set Np(b, c) and an extra 2 × Ap(b, c) − Np(b, c) SNPs from the control set were chosen uniformly at random to be counted twice in all analyses.
Several analyses in the paper were performed on a merged set of archaic variation compiled across populations (see Extended Data Fig. 10 for a schematic illustrating how controls were sampled for this variant set). To form the archaic SNP set , we merged together the archaic SNP sets across all populations p. For each site where the derived allele was present in two or more populations, it was randomly assigned one population of origin. This population assignment process yielded new archaic allele counts A′(b, c) Ithat might be less than the counts Ap(b, c) due to the deletion of duplicate SNPs. For each population p, we sampled 2 × A′(b, c) control SNPs from population p, as before, and merged all of these control sets together to obtain a merged control set . In the unlikely event that a single control allele was sampled in two or more populations, this control SNP would simply be counted two or more times during downstream analyses.
To obtain sets of old archaic SNPs and controls, we must be careful about how we subtract call set 2 from call set 1. We want to sample control SNPs such that no control SNP is part of call set 2 for any archaic species in any population. To achieve this, the set of distal Denisovan SNPs is defined as the set of all SNPs that are present in Denisovan call set 1 but absent from both the Denisovan call set 2 and the Neandertal call set 2 . To generate the corresponding control set , we first look within each population to generate the superset of matched control SNPs . is defined as the set of all SNPs present in population p in Denisovan call set 1 but absent from the population-merged sets of non-archaic variants from Neandertal set 2 plus Denisovan set 2 . Once we have the population-specific candidate control sets , we randomly assign each archaic SNP from to one of the populations where the derived allele is called as archaic, obtaining population-specific call sets that each contain SNPs. As described earlier, we sample 2 × A′(D1–2)(b, c) control SNPs uniformly at random from each set and merge these control sets together to obtain a merged set of distal controls .
Sampling an alternative set of controls to approximate the clustering and linkage disequilibrium structure of introgressed variation.
The archaic variants present in the human population do not have independent demographic and selective histories, but are in many cases organized into linked archaic haplotypes. To measure whether this linkage disequilibrium structure might affect the apparent depletion of archaic alleles from enhancers, we sampled an alternate set of control SNPs whose linkage disequilibrium structure is more similar to the linkage disequilibrium structure of the introgressed SNPs. Linkage disequilibrium has the effect of organizing introgressed SNPs into clusters of close-together variants that have similar allele frequencies, and such clustering could increase the probability that a short enhancer sequence might fall into a gap between introgressed SNPs.
To enable sampling of control SNPs in a way that matches the clustering of archaic SNPs, we first organized the introgressed SNPs into blocks, considering two SNPs to be part of the same block if they were less than 20 kilobases apart and had minor allele counts that differed by at most 1. After organizing the archaic SNPs into these blocks, we counted blocks of control SNPs from the same population variant cell format file (VCF) that had approximately the same allele frequency and B statistic value. To find enough matched control blocks, we relaxed the assumption that archaic and control SNPs should have exactly the same allele frequency and B statistic. Instead, we binned minor allele count into log2 spaced bins (minor allele count 1, 2, 3–4, 5–8, 9–16 and 17+) and required each control SNP cluster to match the minor allele count bins of the matched archaic cluster. Specifically, given a block of k archaic SNPs that we inferred to be a haplotype block, we assigned the minor allele count bin of that block to be the most common bin occupied by the k SNPs. We assigned the B statistic of the block to be the median B statistic of the k SNPs. We then counted the number of blocks of k consecutive non-introgressed SNPs that had the same minor allele count (plus or minus 1) and the same median B statistic, and whose genomic span in base pairs was within a factor of two of the span in base pairs of the archaic SNP set. We selected one of these blocks uniformly at random to be the control SNP block matched to the archaic SNP block.
Quantifying singleton enrichment in the 1000 Genomes SFS.
Let be a set of enhancers or other genomic regions. To test whether is under stronger purifying selection than its immediate genomic neighbourhood, we compared its SFS with the SFS of a region set , defined as follows: can always be defined as a collection of genomic intervals , where each is a pair of genomic coordinates delineating a piece of DNA contained entirely within the set . We define to be the collection of genomic intervals, , that is the set of intervals immediately adjacent on the left to the intervals that make up . (We are slightly abusing notation here by failing to note that different chromosomes have different coordinate systems).
We computed folded SFSs for and using the African individuals in the 1000 Genomes Phase 3 VCF, excluding SNPs that did not pass the VCF’s default quality filter. Let SG and SG′ be the numbers of singletons that fall into the regions and , respectively, and let NG and NG′ be the numbers of non-singleton variants that fall into these regions. We say that is enriched for singletons if the odds ratio (SG/NG)/(SG′/NG′) is greater than 1. To assess the significance of any enrichment or depletion, we use the fact that the standard error of this binomial test is . All singleton enrichment plots in this manuscript contain error bars that span two standard errors above and below the estimated odds ratio.
Comparing hominin reference divergence among enhancers, exons and control regions.
If human and archaic genomes were less diverged from each other in high-pleiotropy enhancers than within other regions of the genome, this could in theory explain why introgressed archaic SNPs are depleted from high-pleiotropy enhancers. To test whether this divergence pattern holds, we let πNean(p) be the pairwise divergence between a Neandertal haplotype (averaged between the two haplotypes of the Altai Neandertal reference) and the human reference genome measured across the set of enhancers of pleiotropy p, and let π′(p) be the pairwise divergence between Neandertal and human within the adjacent matched control regions described in the section ‘Quantifying singleton enrichment in the 1000 Genomes SFS’. We similarly measured the average divergence of the human reference genome from an Altai Denisovan reference haplotype and from the set of African genomes sequenced as part of the 1000 Genomes project. The results, πNean(p)/π′(p), πDeni(p)/π′(p) and πAFR(p)/π′(p), are plotted in Extended Data Fig. 2, together with analogous ratios comparing hominid reference divergence within exons and their adjacent control regions.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Replication of archaic SNP depletion after sampling control SNPs in clusters.
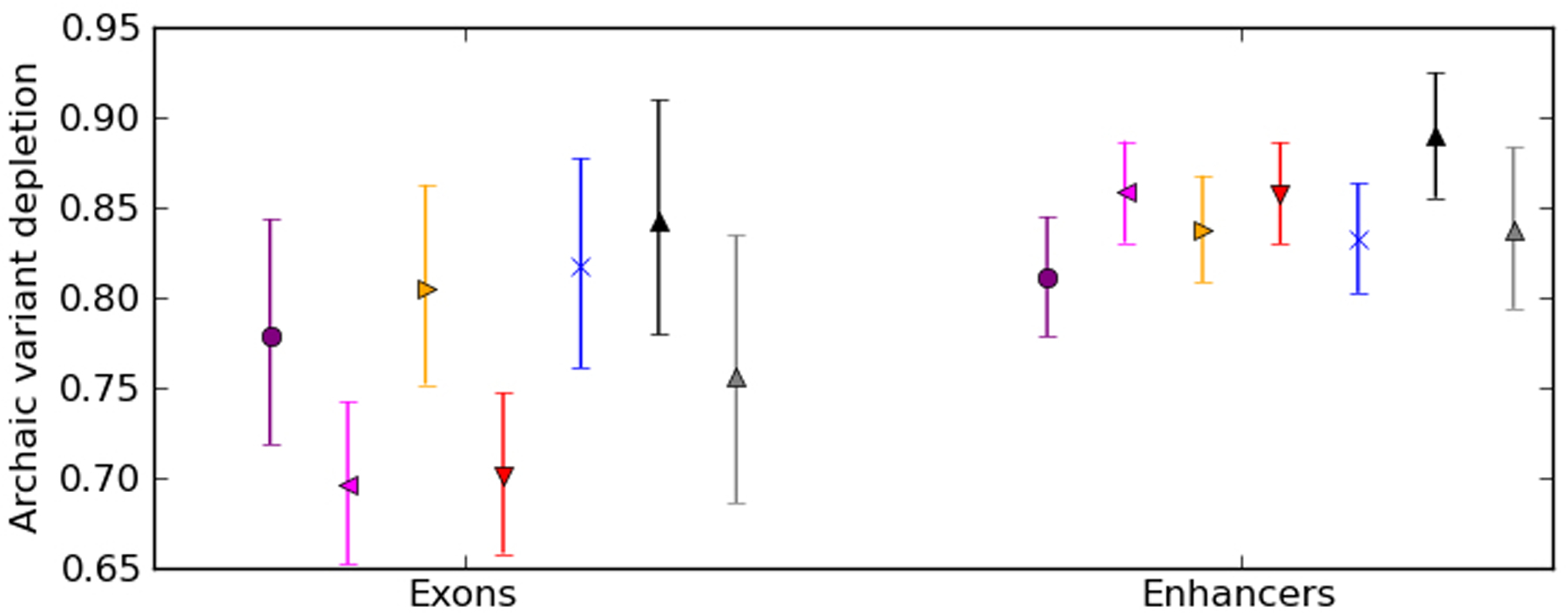
This plot replicates the analysis from Fig. 1b using archaic SNPs and controls sampled to match the clustering induced by LD structure. The depletion of archaic SNPs from exon and enhancers is nearly identical to the depletion measured using controls not sampled to match the spatial clustering of introgressed SNPs.
Extended Data Fig. 2 |. Human versus archaic reference sequence divergence as a function of enhancer pleiotropy and tissue activity.
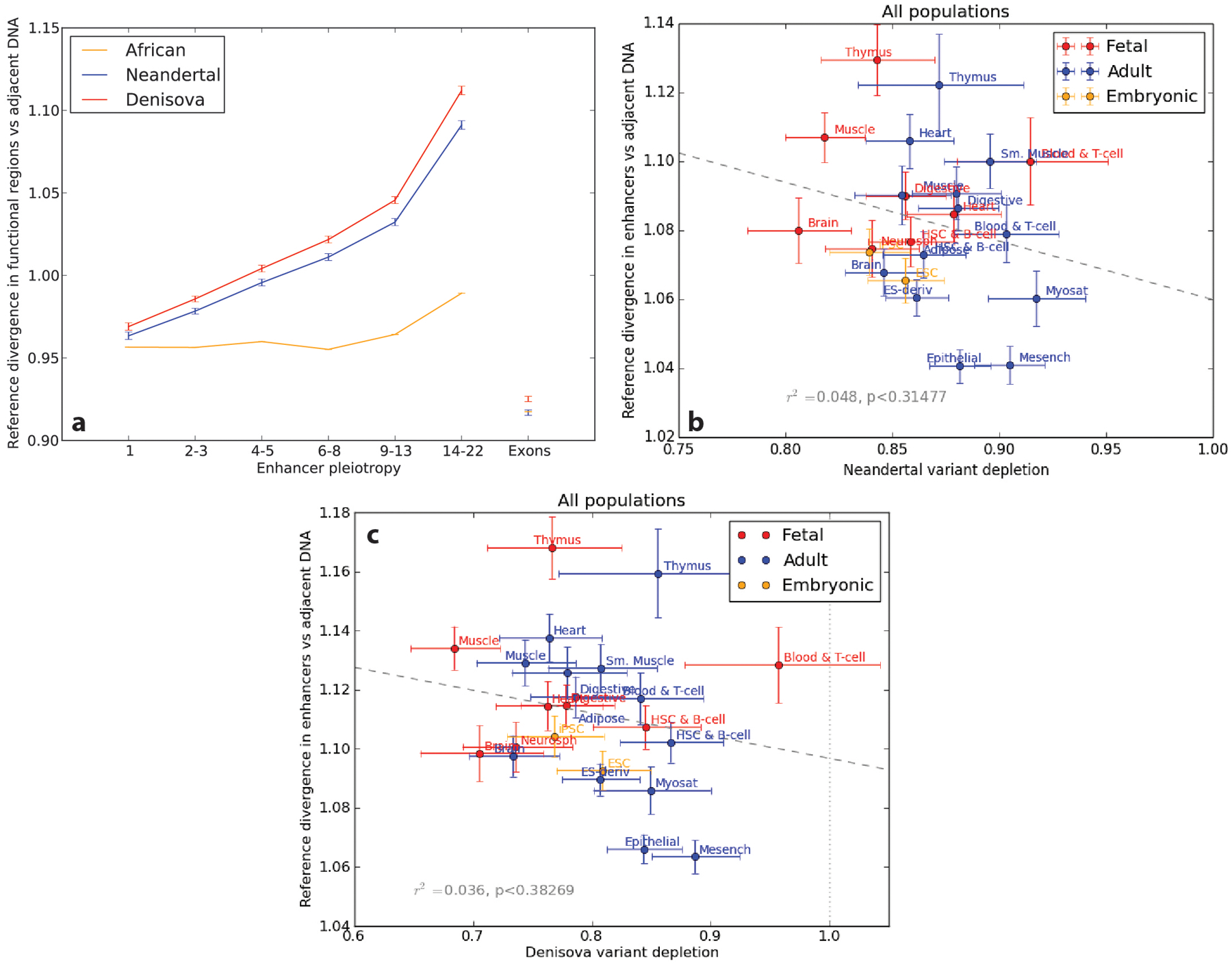
a, Within enhancers and exons, we measured divergence of the human reference from the Altai Neandertal, the Altai Denisovan, and the YRI African genomes, then normalized each by the divergence of the same genomes within adjacent control regions. exon divergence ratios <1 indicate that purifying selection has slowed down their sequence evolution compared to less constrained adjacent regions. In contrast, divergence is accelerated in enhancers relative to control regions. This acceleration is positively correlated with pleiotropy and is stronger for archaic vs. human comparisons than for the African vs. human reference genome comparison. In the absence of selection against archaic enhancer variation, this divergence pattern should cause archaic SNPs to be enriched within high-pleiotropy enhancers, not depleted as we in fact observe. b, We see no correlation across tissue-specific enhancer sets between Neandertal divergence from the human reference and archaic allele depletion from enhancers. This suggests that differences between tissues in the depletion of introgressed archaic variants are not driven by differences in divergence between reference genomes. c, We see no correlation across tissue-specific enhancer sets between Denisovan divergence from the human reference and archaic allele depletion from enhancers. All error bars in panels a–c are 95% confidence intervals derived from the binomial approximation to the Bernoulli distribution.
Extended Data Fig. 3 |. Neandertal and Denisovan variant depletion as a function of enhancer tissue activity.
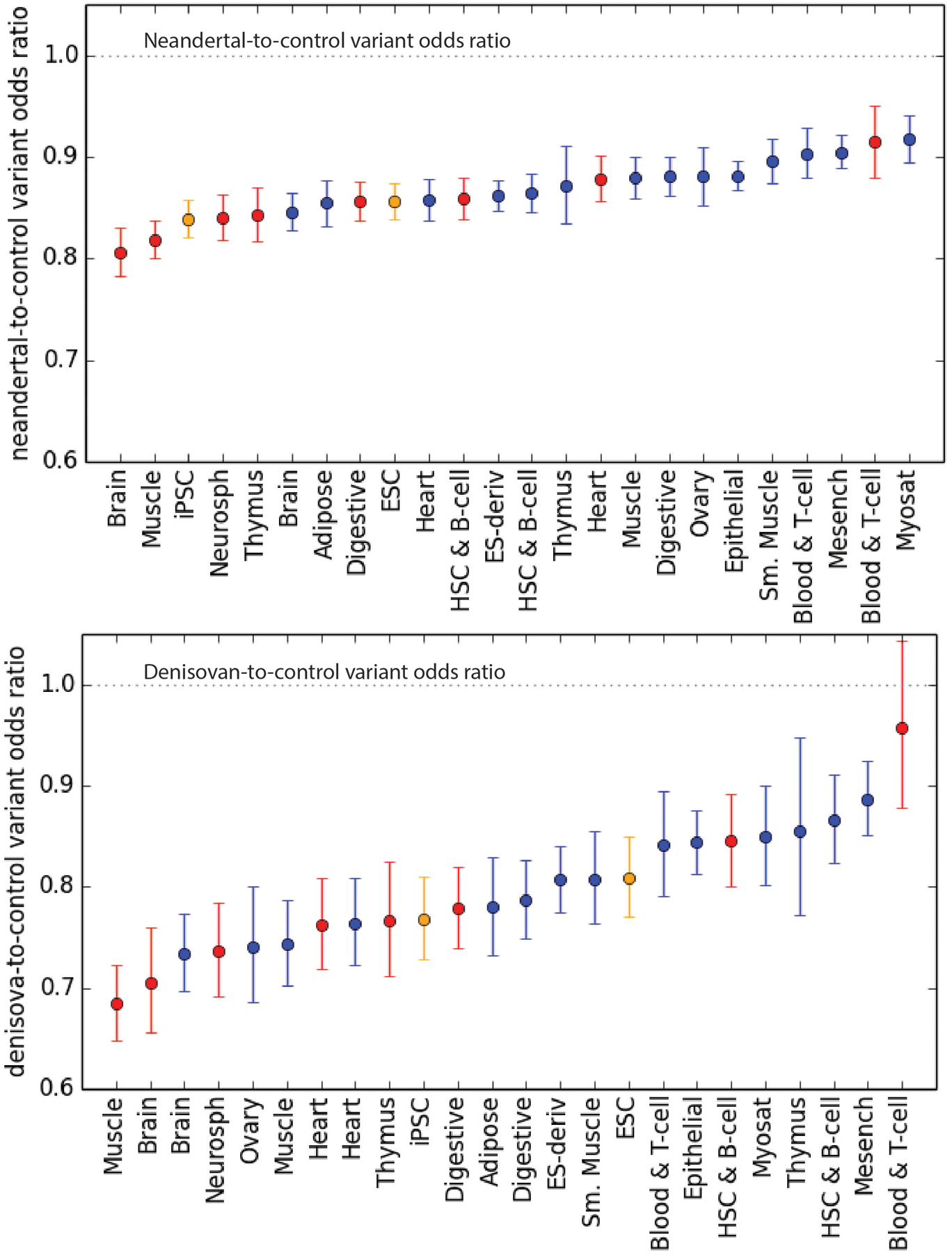
These plots show the data from Fig. 3a with Neandertal and Denisovan odds ratios on separate plots for clarity.
Extended Data Fig. 4 |. Joint distributions of Neandertal and Denisovan SNP depletion within each SgDP population.
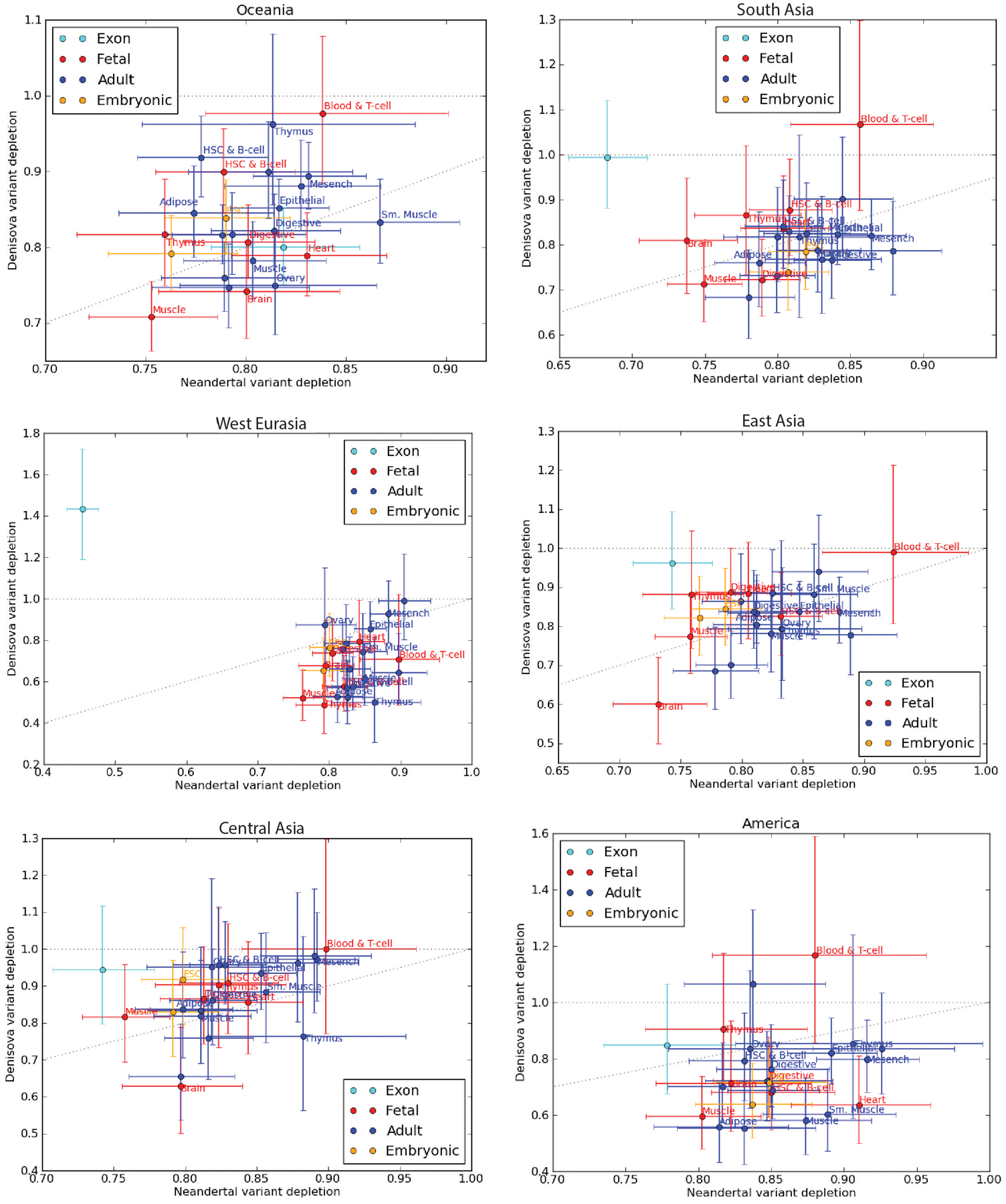
Although there are differences between populations, particularly since Denisovan introgression is sparse and noisy, all show that brain and fetal muscle enhancers are the most depleted of introgression. In most populations the ‘Blood & T-cell’ tissue is least depleted of introgression.
Extended Data Fig. 5 |. Counts of young and old archaic alleles present in modern populations and shared by archaic reference genomes.
a, Recall that ‘young’ introgression calls are SNPs that appear in Call Set 2 generated by Sankararaman, et al. while ‘old’ calls appear in Call Set 1 for at least one archaic species but not in either set of young calls. In every modern human population, we find that 20–30% of old introgressed SNPs are shared with both the Altai Neandertal and Altai Denisovan, suggesting they likely predate the divergence of Neandertals and Denisovans or are at least old enough to have passed between the two species by gene flow. b, In contrast, only 10–20% of young introgressed SNPs are present in both archaic reference genomes. Over 45% of young Neandertal alleles are shared with the Altai Neandertal but not the Altai Denisovan; conversely, over 45% of young Denisovan alleles are shared with only the Denisovan reference. Compared to the sets of young Neandertal and Denisovan alleles, old Neandertal and Denisovan alleles look more similar to each other in their archaic reference sharing profiles: each contains 10–25% Neandertal-specific alleles and 2–10% Denisovan-specific alleles. These patterns support our hypothesis that the old calls are indeed older than the young calls. c, This panel shows the numbers of introgressed SNPs classified as young versus old within each population. each SNP set is further subdivided into SNPs that appear in the Neandertal call set only, the Denisovan call set only, or the intersection of both call sets.
Extended Data Fig. 6 |. Site frequency spectra of introgressed SNPs classified as young versus old in each SgDP population.
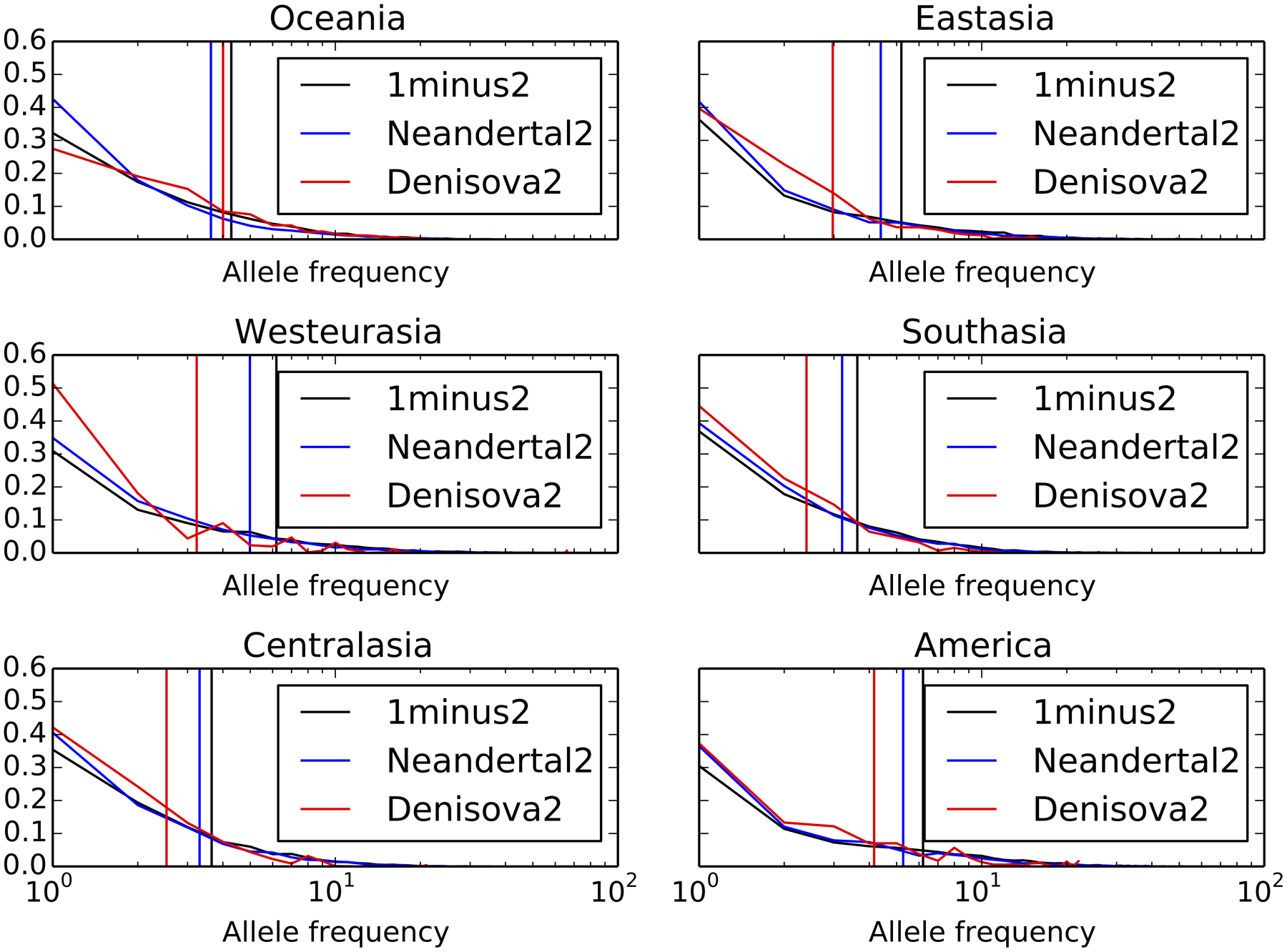
For each call set, the corresponding vertical line demarcates the mean allele frequency of that category. In each population, the old ‘1 minus 2’ call set has the highest mean allele frequency, adding support to our hypothesis that these variants are older and/or less deleterious than either population-specific Call Set 2.
Extended Data Fig. 7 |. GC content cannot explain differences in singleton enrichment between tissues.
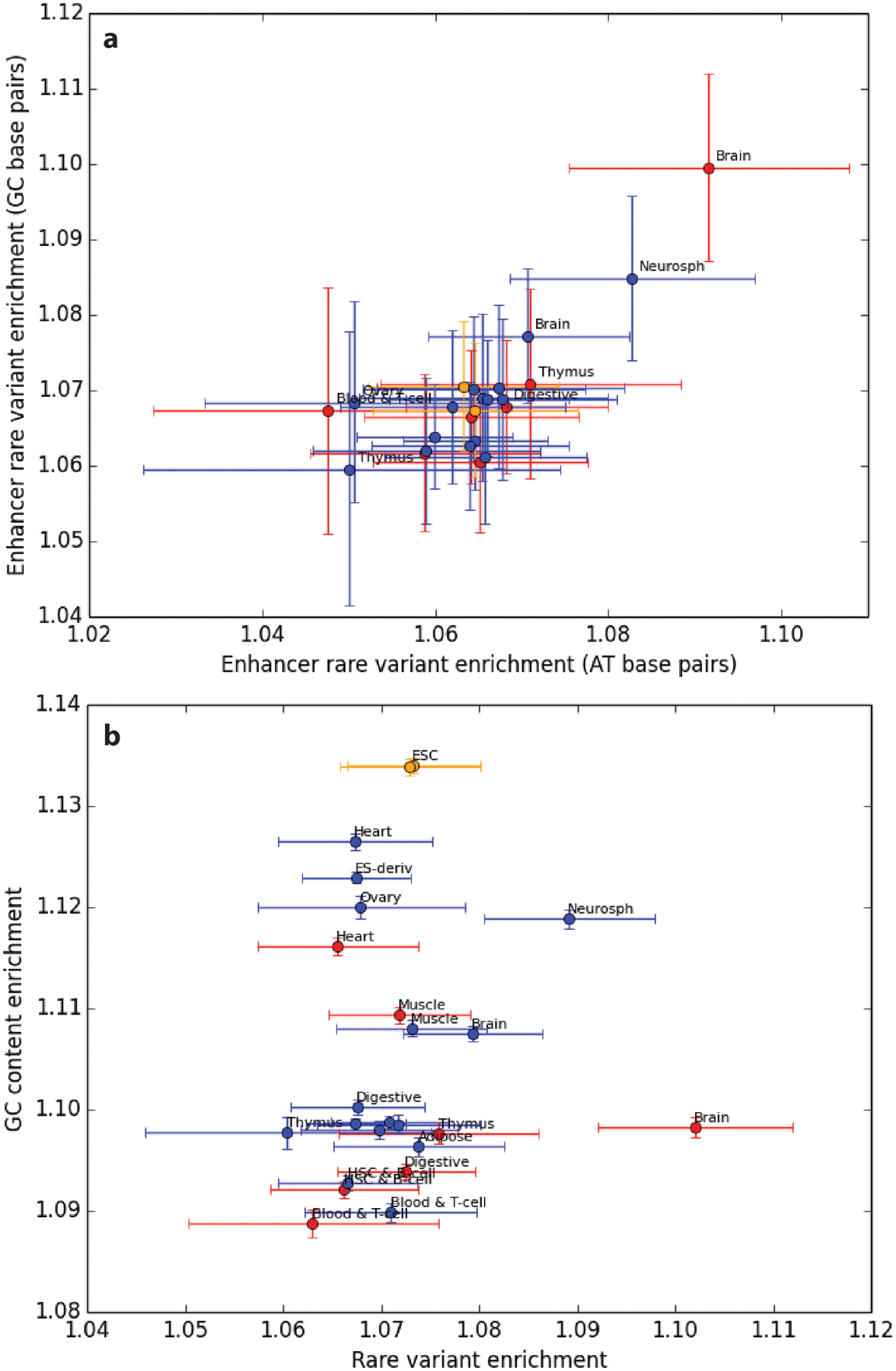
a, We partitioned the site frequency spectra of enhancers into SNPs that have GC ancestral alleles and SNPs that have AT ancestral alleles. Using each of these disjoint variants sets, we then computed singleton enrichment in enhancers versus adjacent control regions. GC-biased gene conversion is expected to have opposite effects on the two frequency spectra, increasing the proportion of GC-ancestral singletons and decreasing the proportion of AT-ancestral singletons. Despite this confounder, the finding that brain enhancers are enriched for singletons holds up when we restrict to either GC-ancestral SNPs or ATancestral SNPs. b, Across tissues, enhancers are enriched for GC base pairs compared to adjacent genomic regions. However, there is no correlation between GC content enrichment and the singleton enrichment that we attribute to purifying selection.
Extended Data Fig. 8 |. Joint distribution across tissues of enhancer singleton enrichment and introgression depletion.
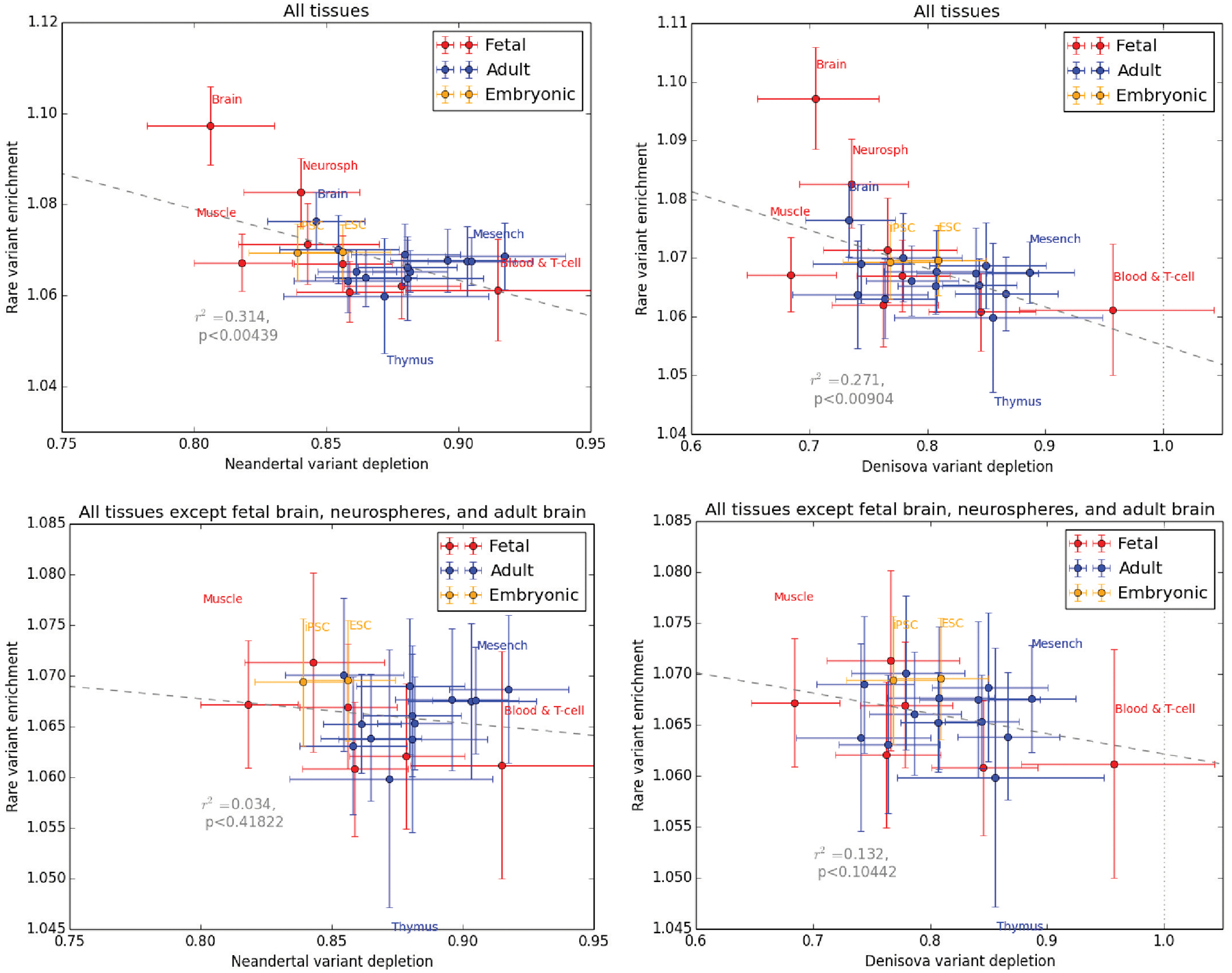
Although singleton enrichment is correlated with depletion of young Neandertal alleles and young Denisovan alleles, the significance of this correlation disappears when all brain related tissues are excluded from the regression.
Extended Data Fig. 9 |. Mean enhancer phastcons scores partitioned by tissue activity.
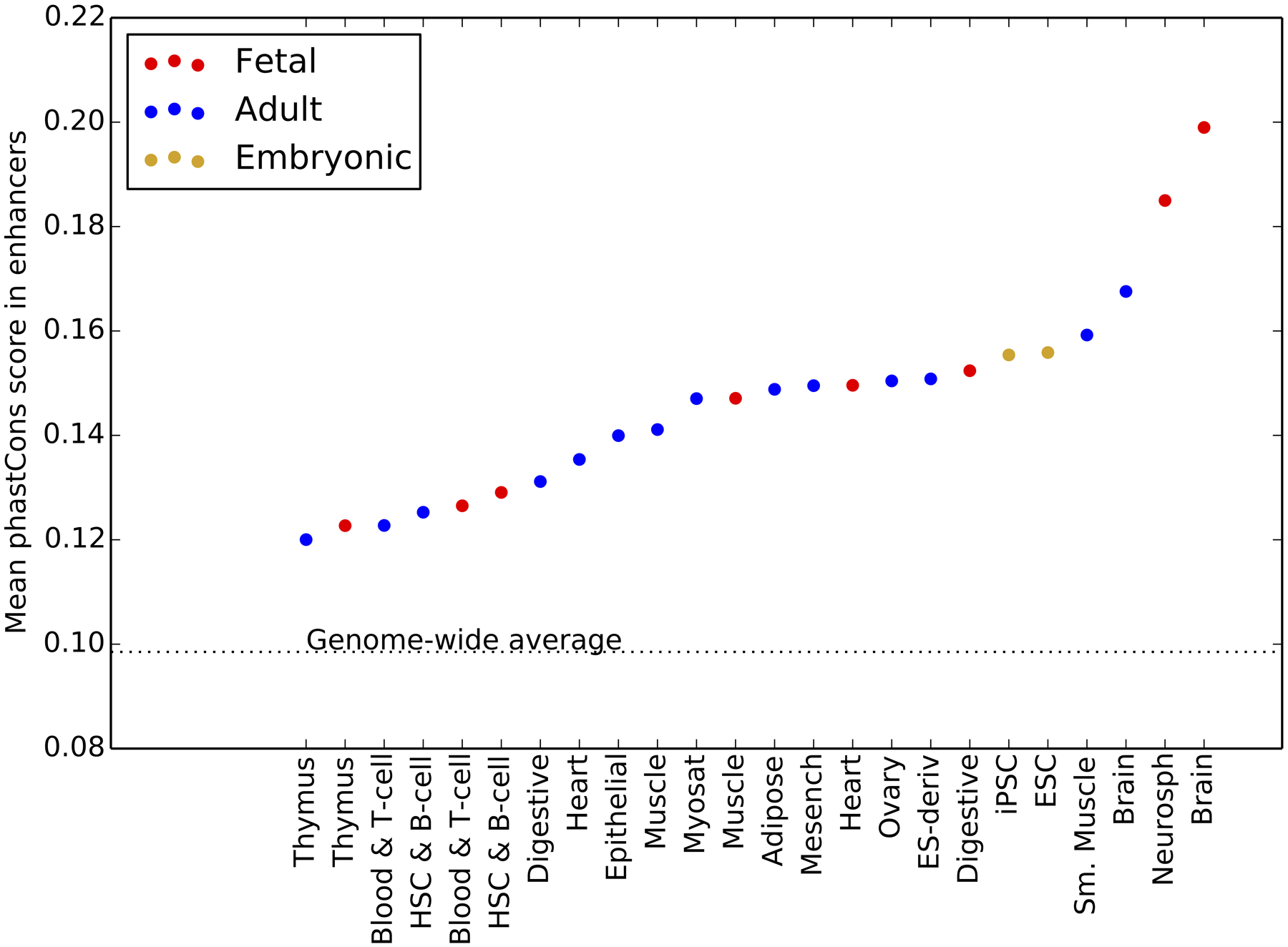
Across all tissues, enhancers have a mean phastCons score that is slightly elevated above the genomic mean, indicating that these regions are slightly conserved over phylogenetic timescales. Fetal brain and neurosphere enhancers have a higher mean phastCons score than enhancers active in any other tissues. This result mirrors our findings on the landscape of recent purifying selection as measured by site frequency skew: fetal brain enhancers are more conserved than other regulatory elements, but fetal muscle enhancers are not.
Extended Data Fig. 10 |. Random sampling of control SNPs to match introgressed SNPs that have been pooled across populations.
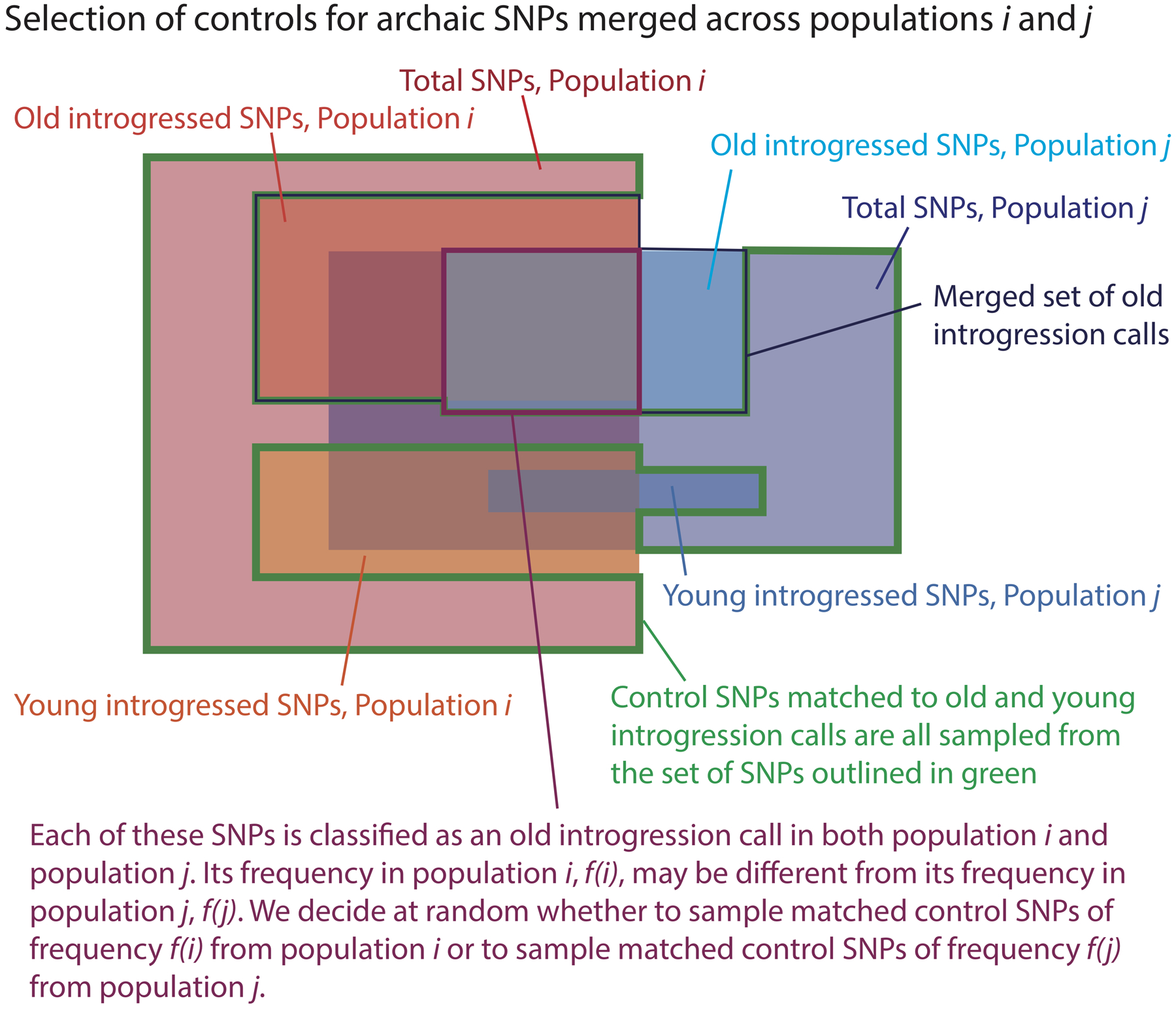
This figure illustrates how we construct the set of SNPs that are eligible to be chosen as control SNPs that match introgressed SNPs for allele frequency and B statistic after these introgressed SNPs have been pooled across all SGDP populations.
Acknowledgements
We are grateful to J. Pritchard, S. Sankararaman, J. Schraiber and members of the Harris Laboratory for helpful discussions. We thank R. Nielsen and B. Vernot for manuscript comments. We acknowledge financial support from the following grants awarded to K.H.: NIH grant 1R35GM133428-01; a Burroughs Wellcome Fund Career Award at the Scientific Interface; a Searle scholarship; a Sloan Research fellowship; and a Pew Biomedical scholarship.
Footnotes
Data availability
All datasets analysed here are publicly available at the following websites: CRF introgression calls (https://sriramlab.cass.idre.ucla.edu/public/sankararaman.curbio.2016/summaries.tgz); SGDP (https://www.simonsfoundation.org/simons-genome-diversity-project/); RoadMap (https://personal.broadinstitute.org/meuleman/reg2map/HoneyBadger2_release/); and 1000 Genomes Phase 3 (http://www.1000genomes.org/category/phase-3/).
Code availability
Summary data files and custom python scripts for reproducing the paper’s main figures are available at https://github.com/kelleyharris/hominin-enhancers/.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41559-020-01284-0.
Supplementary information is available for this paper at https://doi.org/10.1038/s41559-020-01284-0.
References
- 1.Green RE et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prüfer K et al. The complete genome sequence of a Neanderthal from the Altai mountains. Nature 505, 43–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vernot B et al. Excavating Neanderthal and Denisovan DNA from the genomes of Melanesian individuals. Science 352, 235–239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Wolf A, Fu W, Li L & Akey J Identifying and interpreting apparent Neandertal ancestry in African individuals. Cell 180, 677–687 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Sankararaman S et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vernot B & Akey JM Resurrecting surviving Neandertal lineages from modern human genomes. Science 343, 1017–1021 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Sankararaman S, Mallick S, Patterson N & Reich D The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Biol 26, 1241–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petr M, Pääbo S, Kelso J & Vernot B Limits of long-term selection against Neandertal introgression. Proc. Natl Acad. Sci. USA 116, 1639–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King M & Wilson A Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975). [DOI] [PubMed] [Google Scholar]
- 10.Enard W et al. Intra- and interspecific variation in primate gene expression patterns. Science 296, 340–343 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Wray G The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet 8, 206–216 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Dannemann M, Prüfer K & Kelso J Functional implications of Neanderthal introgression in modern humans. Genome Biol 18, 61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCoy R, Wakefield J & Akey J Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell 168, 916–927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellano S et al. Patterns of coding variation in the complete exams of three Neanderthals. Proc. Natl Acad. Sci. USA 111, 6666–6671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn M Detecting natural selection on cis-regulatory DNA. Genetica 129, 7–18 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Long H, Prescott S & Wysocka J Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong W & Nielsen R Detecting selection in noncoding regions of nucleotide sequences. Genetics 167, 949–958 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torgerson D et al. Evolutionary processes acting on candidate cis-regulatory regions in humans inferred from patterns of polymorphism and divergence. PLoS Genet 5, e1000592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward L & Kellis M Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science 337, 1675–1678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith J, McManus K & Fraser H A novel test for selection on cis-regulatory elements reveals positive and negative selection acting on mammalian transcriptional enhancers. Mol. Biol. Evol 30, 2509–2518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arbiza L et al. Genome-wide inference of natural selection on human transcription factor binding sites. Nat. Genet 45, 723–729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huerta-Sánchez E et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prabhakar S et al. Human-specific gain of function in a developmental enhancer. Science 321, 1346–1350 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capra J, Erwin G, McKinsey G, Rubenstein J & Pollard K Many human accelerated regions are developmental enhancers. Phil. Trans. R. Soc. B 368, 20130023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinker D et al. Neanderthal introgression reintroduced functional ancestral alleles lost in Eurasian populations. Nat. Ecol. Evol 10.1038/s41559-020-1261-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silvert M, Quintana-Murci L & Rotival M Impact and evolutionary determinants of Neanderthal introgression on transcriptional and post-transcriptional regulation. Am. J. Hum. Genet 104, 1241–1250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McVicker G, Gordon D, Davis C & Green P Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet 5, e1000471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enard D & Petrov D Evidence that RNA viruses drove adaptive introgression between Neanderthals and modern humans. Cell 175, 360–371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A & Lopez-Bigas N Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature 532, 264–267 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Kaiser V, Taylor M & Semple C Mutational biases drive elevated rates of substitution at regulatory sites across cancer types. PLoS Genet 12, e1006207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quach H et al. Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell 167, 643–656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nédélec Y et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Dannemann M, Andrés A & Kelso J Introgression of Neanderthal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet 98, 22–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Browning S, Browning B, Zhou Y, Tucci S & Akey J Analysis of human sequence data reveals two pulses of archaic Denisovan admixture. Cell 173, 53–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reich D et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slon V et al. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature 561, 113–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawyer S & Hartl D Population genetics of polymorphism and divergence. Genetics 132, 1161–1176 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyko A et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4, e1000083 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffiths R The frequency spectrum of a mutation, and its age, in a general diffusion model. Theor. Popul. Biol 64, 241–251 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Meunier J & Duret L Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol 21, 984–990 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Fish A, Chen L & Capra J Gene regulatory enhancers with evolutionarily conserved activity are more pleiotropic than those with species-specific activity. Genome Biol. Evol 9, 2615–2625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris K & Nielsen R The genetic cost of Neanderthal introgression. Genetics 203, 881–891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juric I, Aeschbacher S & Coop G The strength of selection against Neanderthal introgression. PLoS Genet 12, e1006340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schumer M et al. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360, 656–660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone J & Wray G Rapid evolution of cis-regulatory sequences via local point mutations. Mol. Biol. Evol 18, 1764–1770 (2001). [DOI] [PubMed] [Google Scholar]
- 48.MacArthur S & Brookfield J Expected rates and modes of evolution of enhancer sequences. Mol. Biol. Evol 21, 1064–1073 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Nord A et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruff C, Trinkaus E & Holliday T Body mass and encephalization in Pleistocene Homo. Nature 387, 173–176 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Churchill S in Neanderthals Revisited: New Approaches and Perspectives (eds Hublin J et al.) 113–133 (Springer, 2006). [Google Scholar]
- 52.Gulsuner S et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner T et al. Genomic patterns of de novo mutation in simplex autism. Cell 171, 710–722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Short P et al. De novo mutations in regulatory elements in neurodevelopment disorders. Nature 555, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinrücken M, Spence J, Kamm J, Wieczorek E & Song Y Model-based detection and analysis of introgressed Neanderthal ancestry in modern humans. Mol. Ecol 27, 3873–3888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khaitovich P et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309, 1850–1854 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Coolon J, McManus C, Stevenson K, Graveley B & Wittkopp P Tempo and model of regulatory evolution in Drosophila. Genome Res 24, 797–808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner L, White M, Tautz D & Payseur B Genomic networks of hybrid sterility. PLoS Genet 10, e1004162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mack K, Campbell P & Nachman M Gene regulation and speciation in house mice. Genome Res 26, 451–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewis J, van der Burg K, Mazo-Vargas A & Reed R ChIP-Seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in Lepidoptera. Cell Rep 16, 2855–2863 (2016). [DOI] [PubMed] [Google Scholar]