

Abstract
The transition of endothelial cells between quiescence and proliferation is essential for regulating the extent of the vasculature that supplies oxygen and nutrients to tissues. A study now shows that the FOXO1 transcription factor regulates endothelial cell proliferation by controlling levels of the metabolite 2-hydroxyglutarate.
The decision whether to cycle and form a new cell is relatively straightforward for single cell organisms such as bacteria and yeast: when nutrients are plentiful, divide; when nutrients are sparse, arrest. For unicellular organisms, the presence or absence of metabolites that can be used as fuels, such as glucose and glutamine, plays a decisive role in cell cycle decisions. In multicellular organisms, by contrast, a more complex system for regulating cell proliferation is required, as it is necessary to ensure that specific cells within an organism can be selectively activated for division, while simultaneously ensuring that cells do not divide excessively if nutrients are plentiful1. Tissue-specific regulation of proliferation in multicellular organisms is largely achieved by deploying signaling pathways that respond to cell type-specific mitogens to trigger cascades that induce proliferation. In the last decade, pioneering studies have shown that proliferation-promoting transcription factors such as MYC and p53 induce not only cell division, but also nutrient utilization by inducing expression of enzymes in metabolic pathways, thus reorganizing metabolism to promote a proliferative or quiescent state2–4. For the endothelial cells that line blood vessels, previous studies have demonstrated the importance of the FOXO1 transcription factor for inducing and maintaining a quiescence5. In a paper now published in Nature Cell Biology, Andrade et al. demonstrate that FOXO1 activation results not only in changes in gene expression, but also an altered metabolome6. In particular, this study shows that the metabolite 2-hydoxyglutarate regulates endothelial proliferation, thereby opening up the possibility of a metabolic regulatory system that functions in parallel to the protein and kinase-based regulatory systems that control a cell’s decision to cycle.
Activity of the FOXO1 pathway in endothelial cells is regulated by its phosphorylation by the PI3K/AKT pathway, an important regulatory pathway for cell proliferation7. FOXO1 inactivation results in excessive endothelial cell proliferation and blood vessel growth, whereas overexpression of FOXO1 results in a loss of blood vessels as endothelial cells reversibly exit the cell cycle and become quiescent8. In their article, Andrade et al. investigate the mechanism whereby FOXO1 affects endothelial cell proliferation. Using a mutated version of FOXO1 in which three AKT phosphorylation sites are replaced with alanines, the authors performed a metabolomics analysis that revealed that endothelial cells with constitutive nuclear FOXO1 contain higher levels of branched chain amino acid metabolites, including the metabolite 2-hydroxyglutarate. 2-hydroxyglutarate levels were also higher when FOXO1 levels were induced by a physiological quiescence and FOXO1-inducing signal, contact inhibition. 2-hydroxyglutarate is a metabolite formed by the oncogenic, mutated form of isocitrate dehydrogenase9. 2-hydroxyglutarate can inhibit the function of enzymes that normally bind alpha-ketoglutarate, which includes demethylases10, 11 and prolyl hydroxylases12. However, oncogenic forms of isocitrate dehydrogenase induce the R-enantiomer13, whereas Andrade et al. found that the S-enantiomer was induced by FOXO1. In investigating the mechanism whereby 2-hydroxyglutarate accumulates in cells with FOXO1 activation, the authors discovered reduced activity of the oxoglutarate dehydrogenase (OGDH) complex. This complex catalyzes conversion of 2-oxoglutarate to succinyl CoA, and when its activity was reduced, there was an accumulation of the substrate, 2-oxoglutarate. This increased pool of 2-oxoglutarate could be converted to 2-hydroxyglutarate.
To better understand the mechanism whereby FOXO1 affects OGDH activity, Andrade et al. performed RNA-seq. The RNA-seq analysis indicated that FOXO1 did not affect OGDH transcript or protein levels. Instead, RNA-seq revealed that FOXO1 induces enzymes that regulate catabolism of branched chain amino acids, while metabolomics analysis revealed that FOXO1 induces the accumulation of multiple intermediates in branched chain amino acid catabolism, which are known inhibitors of OGDH14. Andrade et al showed that treatment with one of these branched chain amino acid breakdown products, 3-methyl-2-oxovalerate (KMV), reduced OGDH activity and caused an increase in S-2HG.
To assess the effects of S-2HG treatment of endothelial cells, the authors monitored the cell cycle and found that treating endothelial cells with S-2HG resulted in arrest in the G0/G1 phase of the cell cycle that was reversed when S-2HG was removed. The quiescent state induced by S-2HG shared characteristics with endothelial quiescence, including downregulation of genes associated with cell cycle progression, decreased metabolic activity and reduced RNA and protein synthesis. S-2HG also made the endothelial cells less mobile in scratch assays. Three-dimensional endothelial spheroid cultures made fewer sprouts and mouse model experiments demonstrated that pups treated with S-2HG grew less developed vascular networks in their eyes. Further demonstrating a role for OGDH in endothelial cell proliferation, Andrade et al. knocked out OGDH from endothelial cells in mice and found that the mice had a sparse blood vessel network, consistent with a greater likelihood that endothelial cells enter quiescence when OGDH is inactivated.
The results of this study shed light on a poorly understood signaling system based on metabolites and their effects. While previous studies have indicated changes in metabolism associated with the transition between proliferation and quiescence, these changes largely involved molecules that affect flux through metabolic pathways that are involved in the acquisition and metabolism of nutrients, and the ensuing generation of ATP and energy15. The induction of S-2-hydroxyglutarate by FOXO1 is more surprising because 2-hydroxyglutarate is not part of a metabolic pathway leading to nutrient utilization, TCA cycle or oxidative phosphorylation. The authors suggest S-2-hydroxyglutarate may serve to modulate the activity of the endothelial cell proliferation regulator hypoxia inducible factor (HIF), but its roles in modifying the activity of enzymes that post-translationally modify histones and DNA may also be involved. The findings thus demonstrate that transcription factors can modulate cell fate through increasing expression of specific target genes, leading to changes in the levels of metabolites, including metabolites that are not directly involved in ATP production. These metabolites can serve as signaling molecules that regulate cell fate decisions, such as the proliferation-quiescence transition, and consequently the expansion or contraction of specific tissues within an organism.
These findings by Andrade et al. raise fascinating questions for future investigation. It will be important to determine the specific mechanism whereby 2-hydroxyglutarate induces quiescence in endothelial cells. The physiological significance of this signaling pathway also remains to be established. Under what conditions is signaling through 2-hydroxyglutarate the predominant determinant of blood vessel expansion or contraction, and in what conditions are other pathways of greater importance. Does 2-hydroxyglutarate contribute to the development of vasculature during normal development, or to the growth of new vessels in response to hypoxia? Is 2-hydroxyglutarate a more important signaling molecule for some types of blood vessels than others, or for blood vessels within some organs than others? The research also raises broader questions. Do other transcription factors affect cell fate by modulating levels of metabolites in addition to RNA and protein? Are there additional metabolites that have the capacity to regulate the transition between proliferation and quiescence. Finally, future studies will be required to determine whether metabolites play a role in proliferation or quiescence in other cell types, and therefore regulate the expansion or contraction of other tissues.
Figure 1.
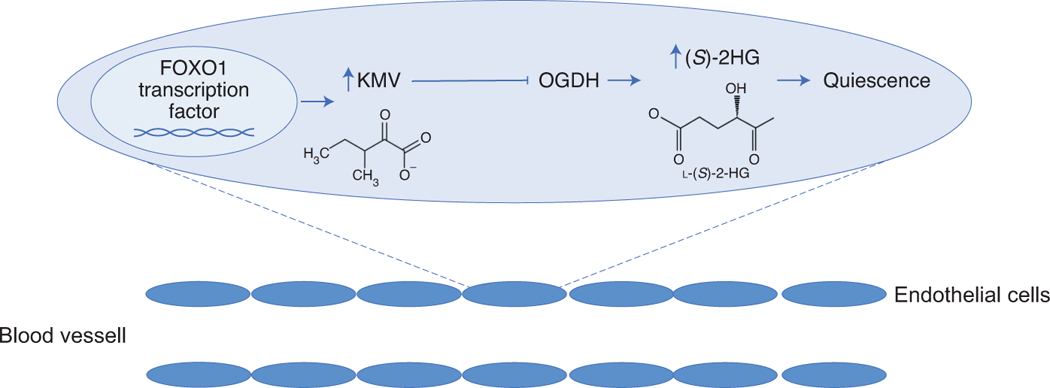
Schematic of proposed regulatory pathway. In Andrade et al., the authors propose a regulatory pathway in the endothelial cells that line blood vessels. The FOXO1 transcription factor increases the expression of metabolic enzymes that generated catabolic intermediates of branched chain amino acids such as 3-methyl-2-oxovalerate (KMV). Elevated levels of these intermediates inhibit the activity of the oxoglutarate dehydrogenase (OGDH) complex. Reduced activity of this complex results in an accumulation of S-2-hydroxyglutarate (S-2HG), which can promote endothelial cell quiescence.
Acknowledgements
The author would like to acknowledge support from NCI R01 CA221296, R01 AR070245, the Melanoma Research Alliance, the Cancer Research Institute, the Broad Stem Cell Center, and the Jonsson Comprehenisve Cancer Center.
References
- 1.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wise DR et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105, 18782–18787 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao P et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bensaad K et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Potente M et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 115, 2382–2392 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrade J. et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nature Cell Biology. doi: 10.1038/s41556-021-00637-6. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzivion G, Dobson M & Ramakrishnan G FoxO transcription factors; Regulation by AKT and 14–3-3 proteins. Biochim Biophys Acta 1813, 1938–1945 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Wilhelm K et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross S et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207, 339–344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarhonskaya H et al. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun 5, 3423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaturvedi A et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia 30, 1708–1715 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel MS Inhibition by the branched-chain 2-oxo acids of the 2-oxoglutarate dehydrogenase complex in developing rat and human brain. Biochem J 144, 91–97 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valcourt JR et al. Staying alive: metabolic adaptations to quiescence. Cell Cycle 11, 1680–1696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]