

Abstract
Cardiovascular diseases (CVD) are the leading cause of death worldwide, and age is by far the greatest risk factor for developing CVD. Vascular dysfunction, including endothelial dysfunction and arterial stiffening, is responsible for much of the increase in CVD risk with aging. A key mechanism involved in vascular dysfunction with aging is oxidative stress, which reduces the bioavailability of nitric oxide (NO) and induces adverse changes to the extracellular matrix of the arterial wall (e.g., elastin fragmentation/degradation, collagen deposition) and an increase in advanced glycation end products, which form crosslinks in arterial wall structural proteins. Although vascular dysfunction and CVD are most prevalent in older adults, several conditions can “accelerate” these events at any age. One such factor is chemotherapy with anthracyclines, such as doxorubicin (DOXO), to combat common forms of cancer. Children, adolescents and young adults treated with these chemotherapeutic agents demonstrate impaired vascular function and an increased risk of future CVD development compared with healthy age-matched controls. Anthracycline treatment also worsens vascular dysfunction in mid-life (50-64 years of age) and older (65 and older) adults such that endothelial dysfunction and arterial stiffness are greater compared to age-matched controls. Collectively, these observations indicate that use of anthracycline chemotherapeutic agents induce a vascular aging-like phenotype and that the latter contributes to premature CVD in cancer survivors exposed to these agents. Here, we review the existing literature supporting these ideas, discuss potential mechanisms as well as interventions that may protect arteries from these adverse effects, identify research gaps and make recommendations for future research.
Keywords: endothelial dysfunction, arterial stiffness, oxidative stress
Graphical Abstract
1. INTRODUCTION
1.1. Cardiovascular disease risk following anthracycline chemotherapy.
Cardiovascular diseases (CVD) and cancer are the two leading causes of death in developed and developing nations(1). In the year 2021, the National Cancer Institute estimates there will be approximately 2 million new cancer cases and approximately 600,000 cancer-related deaths in the United States(2). Of the new cancer cases, approximately 650,000 patients are projected to undergo chemotherapy(3). Although chemotherapy is a highly effective treatment for selective cancers, it comes with severe CV side effects(4). As a result, CVD are the leading cause of later morbidity and early mortality among chemotherapy-treated cancer survivors(5). Worsening cardiovascular risk factors also increase the risk of developing cancer in the future(6), which may further influence increased morbidity and mortality in this patient population. In particular, anthracyclines, which are first-line chemotherapeutic agents for several common cancers (e.g., breast, prostate, lymphomas and leukemias), have toxic effects on the CV system(7, 8). Anthracycline chemotherapeutic agents include daunorubicin, doxorubicin (DOXO), epirubicin, idarubicin, mitoxantrone and valrubicin(9).
1.2. DOXO – the most commonly administered anthracycline chemotherapeutic agent.
The mostly commonly administered anthracycline chemotherapeutic agent is DOXO or alternatively referred to as Adriamycin. This compound, among other anthracyclines, is thought to be the major culprit for inducing toxic effects on the CV system(9). As such, a majority of the preclinical work in this field has used DOXO to understand the potential mechanisms by which anthracycline chemotherapeutic agents cause cardiovascular dysfunction, which is discussed later in this review.
A primary concern for patients exposed to chemotherapy with anthracyclines who survive their cancer is so-called “cardiotoxicity” manifesting clinically as heart failure. The overall incidence of heart failure in cancer patients treated with DOXO is exponentially increased relative to age-matched healthy controls(10), with the risk being proportional to the cumulative dose of DOXO received throughout chemotherapy. Such effects have led the National Cancer Institute to stated that “the cancer patient of today will likely be the cardiac patient of tomorrow.”
Given this link to heart failure, much of the work to date in the emerging field of “cardio-oncology” has understandably focused on anthracycline-induced cardiomyopathy. However, anthracyclines are administered as a systemic therapy and the vasculature is the first tissue exposed to the potentially toxic effect of these drugs(11). Vascular dysfunction is a key antecedent for the development of future CVD(12), so it is entirely possible that DOXO-induced damage to arteries plays an important role in the increased risk of CVD observed in patients treated with these agents (Figure 1). Thus, understanding the impact of anthracyclines on vascular function and the underlying mechanisms of action, is an important biomedical research gap, as this research could lead to the development of novel targeted therapeutics which could significantly reduce the risk of CVD in patients receiving anthracycline chemotherapy. In this review, we synthesize what is known about the influence of anthracycline chemotherapy on vascular function and health, the responsible pathophysiological mechanisms, potential therapeutic targets for maintaining vascular health with anthracycline treatment, and existing research gaps and future directions.
Figure 1. Vascular dysfunction may be an antecedent to the development of cardiovascular diseases (CVD) with anthracycline chemotherapeutic agents.

Treatment with anthracycline chemotherapeutic agents increases the risk of developing CVD. Vascular dysfunction is a well-established risk factor for the development of CVD and the vasculature is the first point of contact for anthracyclines. As such, vascular dysfunction may precede CVD with anthracyclines.
2. Overview of vascular function
2.1. Large elastic artery function and dysfunction.
Arterial dysfunction can be defined in multiple ways, but two manifestations of this dysfunction, which are highly associated with clinical CVD risk will be emphasized here. The first is the stiffening of the large elastic arteries, i.e., the aorta and carotid arteries(13, 14). As the nomenclature suggests, these arteries are designed to expand as they accept the left ventricular stroke volume with each contraction of the heart, and then recoil to create the necessary kinetic energy to drive the blood distally to our tissues and cells(15). Moreover, the elastic recoil of the aorta aids in maintaining perfusion of the heart during diastole(15). The stiffening of these arteries leads to numerous pathophysiological effects that collectively increase risk of CVD, including increases in arterial systolic and pulse pressures, left ventricular hypertrophy (caused by repeatedly ejecting blood out into stiff arteries), and tissue damage as a result of microvascular damage due to increases in pulsatile flow(12), especially in high-flow vital organs such as the brain(16, 17) and kidneys(18).
Structural changes to arteries, functional influences (i.e., factors influencing vascular smooth muscle tone) and the intrinsic stiffness of vascular smooth muscle cells may all contribute to large elastic artery stiffening in any particular pathological setting(15). The primary structural changes mediating arterial stiffening occur in the extracellular matrix and include degradation/fragmentation of elastin, an increase in the deposition of collagen (fibrosis) and formation of advanced glycated end products (AGEs), which cross-link collagen fibers, further increasing their stiffness. Increased vascular smooth muscle tone is a consequence of changes in the molecules produced by endothelial cells (e.g., reductions in nitric oxide [NO] and increases in endothelin-1), increased sympathetic nervous system activity and release of norepinephrine, and renin angiotensin aldosterone system activity(12, 19, 20). These factors also influence the intrinsic stiffness of the vascular smooth muscle cells, which adds to the stiffness of the arterial wall(21).
2.2. Endothelial function and dysfunction.
A second clinically important expression of arterial dysfunction is impaired endothelium-dependent dilation (EDD)(22, 23). The vascular endothelium is a single cell layer at the interface between the flow of blood and the lumen of the artery and the walls of the artery(24). Once believed to be primarily a physical barrier charged with filtering solutes moving between the blood and arterial wall, the vascular endothelium is currently understood to synthesize and release a wide array of biologically active molecules that act in autocrine and/or paracrine fashion to influence the function and health (resistance to disease) of arteries and their surrounding tissues. The most important of these endothelial derived molecules is NO, which exerts a pro-vasodilatory and anti-coagulative, -proliferative, and -inflammatory protective effect on arteries(24). Experimentally, NO-mediated EDD can be evoked either a mechanical (i.e., increase blood flow) or a chemical (e.g., acetylcholine [ACh]) stimulus, both which activate the enzyme NO synthase (eNOS). eNOS catalyzes the generation of NO from L-arginine and oxygen, with NO subsequently diffusing to vascular smooth muscle cells where it induces vascular smooth muscle relaxation and vasodilation(24). Endothelial dysfunction is characterized by a decline in EDD, largely as a consequence of reductions in NO, although changes in concentrations of vasoactive factors such as prostaglandins, endothelin-1, norepinephrine and angiotensin II also may contribute(24).
3. Measuring vascular function
3.1. Large elastic artery stiffness.
In vivo, arterial stiffness can be assessed in preclinical settings and humans using pulse wave velocity (PWV), which is a measure of the (regional) speed of the pulse wave generated by the heart when blood is ejected into the arterial system(15). Aortic PWV, measured as the PWV between the aortic arch and abdominal aorta, is the predominant measure used in rodents and carotid-femoral PWV is the reference standard measure of aortic stiffness in humans(15). The local distensibility of the carotid artery can also be determined in humans by measuring carotid artery compliance (the change in artery diameter for a given change in arterial pressure) and expressing inversely as carotid β stiffness(25). The intrinsic mechanical wall stiffness of the large elastic arteries, most commonly the aorta, can be determined ex vivo in preclinical models via stress-strain curves(26). These methods are illustrated in Figure 2.
Figure 2. Methods for assessing large elastic artery stiffness.

Aortic pulse wave velocity in mice measured between the aortic arch and the abdominal aorta (left) and in humans between the carotid and femoral arteries (middle). Intrinsic mechanical wall stiffness of isolated aorta rings can be accessed via stress-strain testing (right).
3.2. Endothelial function.
NO-mediated EDD is measured in pre-clinical models by assessing changes in artery diameter in response to flow in vivo or changes in diameter of isolated artery segments ex vivo in response to mechanical or pharmacological stimuli, as mentioned above(27-30). In humans, the gold-standard non-invasive assessment of NO-mediated EDD is brachial artery flow-mediated dilation (FMD), in which the change in brachial artery diameter in response to blood flow (shear rate)-induced increases in NO production is determined(31). Brachial artery FMD primarily assesses macrovascular (conduit artery) function(31). Microvascular (resistance vessels) function can be determined by measuring changes in blood flow in response to intra-arterial infusions of ACh(32). Endothelial dysfunction is the major antecedent of atherosclerosis and both reduced brachial artery FMD and resistance artery blood flow responses to ACh are independent predictors of CV events and CVD in large, community-based cohorts free from clinical disease(33). These methods are illustrated in Figure 3.
Figure 3. Methods for assessing vascular endothelial function.

Endothelial function in mice can be assessed using ex vivo carotid artery endothelium-dependent dilation in response to acetylcholine (left), whereas endothelial function is most commonly assessed in humans using brachial artery flow-mediated dilation (right).
3.3. Influence of advancing age.
It is well established that aortic PWV and EDD increase and decrease, respectively, as a result of advancing age(34). Indeed, old mice(28, 35, 36) and healthy older adults(37) have ~50% greater aortic PWV and ~50% lower EDD, relative to young controls. As such, any form of pathophysiological stress (e.g., environmental pollution, hypertension and other CVD risk factors, psycho-social stress) that causes aortic stiffening or reduced EDD in early life can be viewed as a model of accelerated vascular aging. Below, we provide a description of the published human/clinical studies, which establish anthracycline chemotherapy as a model of accelerated vascular aging. Preclinical data on the topic will be described in subsequent sections of this review.
4. Clinical changes in vascular function with anthracycline chemotherapy
4.1. Large elastic artery stiffening.
Children and adolescent/young adult patients receiving anthracycline chemotherapy.
Most of the adverse long-term effects of anthracycline treatment on arterial health have been reported in childhood cancer survivors. Children who were treated with anthracycline chemotherapy demonstrate greater arterial stiffness when compared with healthy age-matched controls (individuals who did not have cancer and were not treated with anthracyclines) upon follow-up ranging from 1 to 20 years(38, 39). Herceg-Cavrak et al.(40) showed that children and adolescents (6-20 years of age) treated with anthracycline chemotherapy had an ~15% greater aortic PWV compared with healthy age- and sex-matched controls 2 years following the completion of treatment. Conversely, Krystal et al.(41) found no differences in carotid-femoral PWV between age- and sex-matched controls and adolescent childhood cancer survivors with an average follow-up time of 7 years following the completion of treatment. However, in a subgroup analysis, patients >18 years of age had 10% greater carotid-femoral PWV compared with age-matched controls, which suggests that aortic stiffening may manifest 5-10 years following the completion of treatment in some adolescent/young adult cancer survivors (41). With regard to more localized large elastic artery stiffness, Jenei et al.(42) showed a 3-fold greater carotid artery β stiffness within a 10-year follow-up period in adolescent childhood cancer survivors treated with anthracyclines when compared with age- and sex-matched controls. Consistent with this observation, Arnold et al.(43) demonstrated that participants (30-45 years of age) from the CVSS (Cardiac and Vascular Late Sequalae in Long-Term Survivors of Childhood Cancer) study that were treated with anthracyclines prior to 15 years of age had ~20% higher β stiffness compared to age-matched healthy controls. Overall, these findings support conclusion that anthracycline treatment induces stiffening of the large elastic arteries in childhood and adolescent/young adult cancer survivors.
Middle-aged adults receiving anthracycline chemotherapy.
Middle-aged patients also appear to undergo arterial stiffening in response to treatment with anthracyclines. For example, Frye et al.(44) recently demonstrated ~20% greater carotid artery β stiffness in middle-aged (mean age: 56 years) patients receiving anthracycline chemotherapy relative to age-matched controls. In another study, Chaosuwannakit et al.(45) showed that 4 months following completion of anthracycline treatment in middle-aged patients, there was a 3-fold increase in carotid-femoral PWV from baseline relative to controls. Likewise, Drafts et al.(46) reported a rapid increase in arterial stiffness in the first month following the completion of anthracycline treatment in middle-aged patients, with an ~50% increase 6 months following treatment. In contrast, Mizia-Stec et al.(47) found no differences in carotid-femoral PWV in middle-aged patients from baseline to 6 months following the completion of the treatment. However, ~50% of the individuals in that study were on CV-related medications (e.g., beta blockers, angiotensin converting enzyme inhibitors, calcium channel blockers) or were supplemented with tamoxifen (estrogen receptor agonist), all of which have shown to promote favorable vascular effects that could have counteracted the detrimental impact of anthracycline chemotherapy on arterial stiffness.
Summary.
In summary, cancer therapy with anthracyclines significantly increases arterial stiffness in children, adolescents, young adults and middle-aged adults. The differences in arterial stiffness between cancer survivors and age- and sex-matched non-patient controls are similar to those reported between older (≥ 65 years of age) compared with young adults without a history of cancer(25, 37), highlighting anthracycline chemotherapy as a model of accelerated vascular aging. A recent systematic review and meta-analysis on the topic of anthracycline chemotherapy and arterial stiffness suggests that regular assessment of large elastic artery stiffness should be used as part of a targeted vascular health monitoring strategy for the identification of early CV risk both during anthracycline chemotherapy and in longer-term survivors(38). Importantly, arterial stiffness is an independent risk factor for the development of cognitive impairment(16, 17) and kidney dysfunction(18), both of which are common side effects of anthracycline chemotherapy. Thus, monitoring arterial stiffness also might be useful in preventing the development of other comorbidities associated with anthracycline chemotherapy.
4.2. Vascular endothelial dysfunction.
Children, adolescent and adult patients receiving anthracycline chemotherapy.
Endothelial dysfunction appears to be another undesirable side effect of anthracycline chemotherapy in younger individuals. Children (mean age: 10.3 years) who received anthracycline chemotherapy for acute lymphoblastic leukemia were reported to have an ~4-fold lower brachial artery FMD relative to age- and sex-matched controls when assessed 2 months to 7 years following the final anthracycline treatment(48). The impairments in FMD were independent of the time elapsed since the final anthracycline treatment(48). Similarly, Chow et al.(49) demonstrated that brachial FMD was ~2-fold lower in children (mean age: 14.5 years) 2 months to 5 years following the completion of anthracycline chemotherapy for the treatment of T-cell acute lymphoblastic leukemia, relative to age-, sex- and blood pressure-matched controls. In another investigation, FMD was ~2-fold lower in older children (mean age: 14.9 years) who had been treated with anthracyclines for a variety of childhood cancers (mean treatment age: 4.8 years), relative to age- and sex-matched controls(42). In agreement with these observations, brachial FMD was ~40% lower in young/early middle-aged adults (mean age: 30.2 years) who received anthracycline chemotherapy for the treatment of childhood acute lymphoblastic leukemia (average treatment age: 4.7 years), relative to age-, sex- and body weight-matched controls(50).
Recently, Hader et al.(51) assessed the influence of DOXO on endothelial function in isolated microvessels (small arteries – arterioles) from pediatric patients (mean age: 6.0 years), collected at the time of surgery to repair congenital heart defects. It was determined that microvessels treated with DOXO had lower endothelial function (EDD) relative to vessels treated with a standard culture medium control, assessed in response to both flow and ACh. Similar results were observed in microvessels obtained from adult patients.
Summary.
Taken together, cancer patients who have undergone anthracycline chemotherapy have lower endothelial function relative to age- and sex-matched controls. As noted with arterial stiffening, the magnitude of endothelial dysfunction in anthracycline-treated cancer survivors is similar if not greater than what is observed in older adults relative to young, further supporting the idea of anthracycline chemotherapy as a model of accelerated vascular aging.
4.3. Current gaps in clinical research.
Future work is needed to understand: 1) the influence of childhood anthracycline chemotherapy on large elastic artery stiffness and EDD in mid to late life, assessed longitudinally following treatment; 2) the effects of mid-life anthracycline chemotherapy on age-related changes in large elastic artery stiffness and EDD; 3) the interaction between baseline (before the onset of treatment) aortic stiffness and EDD (and other indices of CV health) and anthracycline chemotherapy-mediated arterial stiffening and impaired EDD; and 4) the influence of sex on anthracycline-mediated arterial stiffening and endothelial function.
Although it is clear that anthracycline chemotherapy can induce an aging-like phenotype in the vasculature, the underlying mechanisms for why this occurs are incompletely understood. In the subsequent sections of this review, we will describe: 1) the underlying mechanisms of vascular aging; 2) how vascular aging mechanisms relate to anthracycline chemotherapy; 3) mechanistic insight gained from preclinical studies; and 4) how we might be able to target common hallmarks of aging to improve vascular health with anthracycline chemotherapy.
5. Commonalities between anthracycline chemotherapy and the mechanisms underlying vascular aging: potential therapeutic targets
5.1. Oxidative stress and inflammation.
Two macro-mechanistic processes underlying vascular aging(24, 52, 53), which are also present in the setting of anthracycline chemotherapy, are excessive reactive oxygen species (ROS)-associated oxidative stress(27) and chronic, low grade inflammation(54) (Figure 4). As frequently observed with chronological aging, excessive production of ROS (primarily superoxide) in combination with unchanged or decreased abundance/activity of antioxidant enzymes (e.g., superoxide dismutase, SOD) results in the development of oxidative stress in arteries with anthracycline chemotherapy(27). Excess superoxide rapidly reacts with NO to form the secondary reactive species peroxynitrite (ONOO−), decreasing the bioavailability of NO(55), ultimately causing endothelial dysfunction(24). Peroxynitrite also reacts with and oxidizes tetrahydrobiopterin (BH4), an essential co-factor for NO production by eNOS(56). Excess ROS also can activate pro-inflammatory networks such as those regulated by the transcription factor nuclear factor kappa B (NFκB), which upregulates the production of pro-inflammatory cytokines that can impair vascular function and activate other ROS producing systems and enzymes, creating a vicious feed-forward cycle of inflammation and oxidative stress(57, 58).
Figure 4. Mechanisms of anthracycline chemotherapy-mediated vascular dysfunction and related clinical disorders.

Anthracycline chemotherapy is associated with vascular inflammation (pro-inflammatory cytokine signaling) and excessive mitochondrial reactive oxygen species (ROS)-derived oxidative stress. In the vasculature, oxidative stress and inflammation are largely induced by higher nuclear factor kappa-B (NFκB), transforming growth factor beta (TGF-β), matrix metalloproteinases (MMPs), ROS, peroxynitrite and nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase). Together, these processes induce vascular dysfunction, featuring: (top) vascular endothelial dysfunction characterized by reduced nitric oxide (NO) bioavailability and endothelium-dependent dilation; and (bottom) large elastic artery stiffening mediated by degradation of elastin fibers (blue) and greater cross-linking of collagen (brown) by advanced glycation end products (dashed connecting lines). These and other changes in arteries, in turn, increase risk of developing cardiovascular diseases, chronic kidney disease, and Alzheimer’s disease and related dementias.
This overall state of oxidative stress and inflammation also contributes to arterial stiffening by inducing remodeling of the extracellular matrix, which alters structural properties of the arterial wall(15). For example, production of collagen by fibroblasts is stimulated by superoxide-related oxidative stress(59). Matrix metalloproteinases (MMPs) are also are upregulated and elastin content is lower in aorta of SOD-deficient mice, consistent with the concept that elastin degradation is induced by oxidative stress(60). Vascular oxidative stress also promotes transforming growth factor β (TGF-β) signaling and this, in turn, stimulates inflammation, which further reinforces arterial stiffness via activation of the pro-oxidant enzyme, nicotinamide adenine dinucleotide phosphate oxidase(20). AGEs interact with the receptor for AGEs to activate NFκB-regulated pro-inflammatory pathways and oxidative stress, which ultimately perpetuates arterial stiffening and further production of AGEs(20).
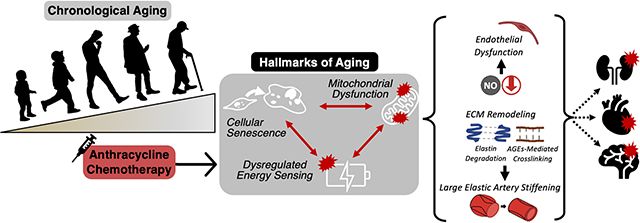
The remaining sections of this article will focus on three hallmarks of aging(61) that could be targeted to prevent excess oxidative stress and inflammation following anthracycline chemotherapy: 1) mitochondrial dysfunction; 2) cellular senescence; and 3) reduced energy sensing (e.g., sirtuins, AMP-activated protein kinase [AMPK] and the mammalian target of rapamycin [mTOR]), as potential drivers of anthracycline-induced vascular dysfunction (Figure 5). We then will review current evidence for prevention/treatment of anthracycline-induced vascular dysfunction via lifestyle and pharmacological strategies that improve vascular health through these pathways. We also discuss current research gaps and future directions for the field.
Figure 5. The interaction of aging hallmarks (mitochondrial dysfunction, cellular senescence and dysregulated energy sensing) and how they may influence vascular function.

Anthracycline chemotherapy is associated with mitochondrial dysfunction, cellular senescence and dysregulated energy sensing, all of which are key hallmarks of aging. Independently and/or interactively, these hallmarks can influence cellular mechanisms associated with vascular dysfunction including stimulation of the mammalian target of rapamycin (mTOR), reactive oxygen species (ROS)-mediated oxidative stress and the senescence associated secretory phenotype (SASP). These hallmark processes also inhibit AMP-activated protein kinase (AMPK), sirtuin-1 (SIRT-1), endothelial nitric oxide synthase (eNOS), nicotinamide adenine dinucleotide (NAD+), and nitric oxide (NO) bioavailability.
6. Impaired vascular mitochondrial function
Mitochondria are cytoplasmic organelles that are present in the majority of cell types in the human body, including vascular endothelial and smooth muscle cells. Mitochondria are often referred to as the “powerhouse” of the cell for their role in ATP production. The latter occurs via oxidative phosphorylation, which is a series of electron transfers through the respiratory chain in the mitochondrial inner membrane. However, mitochondria are also vital for a number of other cellular processes, including regulation of metabolism, calcium homeostasis, immune function, and control of cell death pathways. Although mitochondrial density in vascular tissues is considerably lower than other tissues such as skeletal muscle, liver and heart(62, 63), increasing evidence indicates that these organelles are critical for maintenance of cellular and tissue homeostasis in the vasculature. This topic has been reviewed in detail elsewhere(64-68), but below we briefly summarize some of the key roles of mitochondria in the vasculature and how it is influenced by anthracycline chemotherapy.
An important distinction is to consider the vascular cell type in question, as the density and subcelluar distribution of mitochondria vary between endothelial and vascular smooth muscle cells, and among the same cell types in different vascular beds(64, 67). In general, unlike in highly metabolically active tissues with greater ATP demand, the principal role of mitochondria in the vasculature appears to be cellular signaling rather than energy provision(64).
Cellular energy demand is relatively low in vascular endothelial cells, and ATP demand is met primarily via glycolysis. However, endothelial mitochondria are critical in the regulation of calcium homeostasis, apoptosis/necrosis, cellular response to stress, and immune and inflammatory pathways. An essential feature of these roles is the regulated production of signaling molecules (reactive oxygen, nitrogen, and other species), mitochondrial DNA, mitochondria derived peptides and damage-associated molecular pattern molecules (DAMPs)(69). Importantly, there is crosstalk between mitochondrial and nuclear signaling pathways, such that mitochondria-derived signaling is influenced by and can influence nuclear events including gene expression.
Mitochondria have a similarly important role in cellular signaling in vascular smooth muscle cells. Mitochondria are involved in signaling pathways for regulation of vascular smooth muscle cell growth and proliferation (e.g., TGF-β activity)(70), as well as maintenance of the dynamic balance among synthesis and breakdown of extracellular structural proteins, including collagen and elastin, e.g., via modulation of MMP enzyme expression and activity(71). There is also emerging evidence demonstrating interplay between mitochondrial ROS signaling and inflammatory pathways known to be important for regulating vascular smooth muscle cell function, including those involved with NFκB and the NLRP3-inflammasome(72-75), further highlighting the crucial role of mitochondrial ROS in vascular homeostasis.
6.1. Mitochondria as a target of anthracycline chemotherapy.
The signaling functions of vascular mitochondria are thought to be mediated in large part by the production of ROS at low, physiological levels. However, the dysregulation of this mitochondrial ROS production may lead to pathophysiological sequelae that disrupt other mitochondrial functions, cellular homeostasis, and ultimately vascular function. The anti-oncogenic properties of anthracycline chemotherapeutic agents are primarily driven by their ability to target mitochondria. Anthracyclines accumulate in mitochondria, resulting in mitochondrial DNA damage and impaired electron transport system function(76). Specifically, ROS producing enzymes in the mitochondria can convert anthracyclines to a semiquinone radical through one electron reduction of the quinone moiety(77). This semiquinone can readily react with oxygen to generate the anion superoxide (O2.−). Anthracyclines also reduce the abundance of key mitochondrial antioxidant defense enzymes(27, 78, 79), which is thought to serve a functional role since the anthracycline-induced ROS from the mitochondria serves to clear cancer cells by initiating apoptosis(76). Unfortunately, this excessive state of mitochondrial ROS coupled with reduced mitochondrial antioxidant defenses also occurs in the vasculature, which has severe adverse effects on vascular function(27).
6.2. Functional implications of vascular mitochondrial dysfunction and excessive mitochondrial ROS.
Mitochondrial ROS and arterial stiffening.
Oxidative stress is a critical upstream mechanism driving arterial stiffening with advancing age(80), and there is growing evidence that excessive mitochondrial ROS production is a key source of this oxidative stress (36, 81, 82). Excessive mitochondrial ROS in vascular smooth muscle cells may induce aberrant signaling in growth factor (e.g., TGF-β)(70) and proteolytic enzyme (e.g., MMP)(71) pathways that leads to overproduction of collagen and accelerated elastin degradation. Furthermore, mitochondrial ROS are now recognized as important activators of pro-inflammatory signaling in vascular smooth muscle cells implicated in mediating adverse structural changes in arteries(82, 83). Finally, excessive levels of mitochondrial ROS may also contribute to oxidative stress-driven formation of AGEs and subsequent cross-linking of collagen in the arterial wall(82).
Anthracycline-mediated increase in vascular mitochondrial ROS and arterial stiffening.
Recently, we performed reverse translation experiments in young adult C57BL6/J wild-type mice to determine the mechanism by which DOXO chemotherapy causes arterial stiffening(54). We demonstrated that young adult mice administered DOXO had higher aortic PWV (in vivo aortic stiffness) relative to saline-treated controls, which was mediated by greater intrinsic mechanical wall stiffness of the aortic wall. The latter was, at least in part, due to the combination of elastin degradation and greater formation of AGEs, inflammation and greater mitochondrial ROS production(27, 54). Therefore, we aimed to determine whether mitochondrial-targeted antioxidant supplementation following DOXO administration could prevent DOXO-mediated aortic stiffening. We found that DOXO-treated mice that received 4 weeks of oral supplementation (in drinking water) with the mitochondrial-targeted antioxidant MitoQ (also referred to as Mitoquinone or Mitoquinol Mesylate) had lower aortic intrinsic mechanical wall stiffness relative to DOXO-treated mice that received standard drinking water(84) (Figure 6). These observations suggest that mitochondrial ROS may be a novel therapeutic target for prevention of DOXO-mediated arterial stiffening. However, more work is needed to understand how MitoQ or other mitochondrial-targeted therapies influence aortic PWV in DOXO-treated mice. Regarding clinical translation, MitoQ is commercially available and safe for use in humans(85, 86) and our laboratory recently showed that oral MitoQ supplementation can reduce carotid-femoral PWV in healthy middle-aged/older adults free of overt CVD(81). Therefore, MitoQ supplementation may be an effective therapeutic strategy for reducing arterial stiffness in patients that have received anthracycline chemotherapy.
Figure 6. Anthracycline chemotherapy (doxorubicin [DOXO])-mediated aortic stiffening and mitochondrial ROS production.

A) Aortic pulse wave velocity in young mice increases following DOXO administration to levels observed in old mice, which is associated with B) greater aortic mitochondrial ROS production and C) aortic intrinsic mechanical wall stiffness (elastic modulus). D) Chronic oral supplementation with the mitochondrial targeted antioxidant MitoQ prevented the DOXO-mediated increase in aortic elastic modulus. *P < 0.05 vs. Young Control; ^P < 0.05 vs Old Control. Data from(27, 28, 54).
Mitochondrial ROS and vascular endothelial function.
Excessive production of mitochondrial ROS may contribute to vascular oxidative stress and reduce the bioavailability of NO, either directly via formation of peroxynitrite or indirectly by uncoupling of eNOS and reducing NO production as a result of oxidation of the eNOS co-factor BH4. These events are further propagated by peroxynitrite inhibition of an appropriate upregulation of the mitochondrial antioxidant enzyme manganese (Mn) SOD(60). Reduced NO bioavailability leads to impairments in endothelial function (as described above) but may also contribute to further mitochondrial dysregulation. NO has a key regulatory role in PGC-1α signaling and mitochondrial biogenesis(87). Moreover, NO acts as a tonic inhibitor of complex IV of the mitochondrial electron transport system(88); as such, decreases in NO bioavailability may also augment mitochondrial superoxide production by the electron transport system as this tonic inhibition is removed. Mitochondria-derived hydrogen peroxide is a key signaling molecule in the vasculature, as a compensatory vasodilatory mechanism for reduced NO bioavailability in the microvasculature and coronary arterioles in atherosclerotic heart disease(89). However, as with superoxide, excessive levels of hydrogen peroxide production, either de novo or as a result of superoxide dismutation, can disrupt vascular homeostasis, including activation of NFκB with resultant pro-thrombotic and pro-inflammatory effects(89). Furthermore, reduced NO formation as a result of excess mitochondrial ROS may initiate a pro-inflammatory response in the vasculature, as normal physiological levels of NO can inhibit a variety of pro-inflammatory responses (i.e., adhesion molecule expression, cytokine and chemokine synthesis and leukocyte adhesion and transmigration) known to cause endothelial dysfunction(90).
DOXO-mediated increase in vascular mitochondrial ROS and endothelial dysfunction.
DOXO can directly increase the production of mitochondrial ROS from vascular cells in vitro and reduce the abundance of the mitochondrial isoform of SOD (Manganese [Mn] SOD)(91, 92). However, until recently, there had been no systematic investigations regarding the influence of mitochondrial ROS on vascular endothelial dysfunction induced by DOXO. We showed that DOXO reduces vascular MnSOD and that we can reverse endothelial dysfunction in carotid arteries from mice treated with DOXO via ex vivo treatment of arteries with MitoQ(27). Furthermore, we demonstrated that chronic oral supplementation (4 weeks in drinking water) with MitoQ could preserve NO bioavailability and prevent mitochondrial ROS-related suppression of EDD following DOXO treatment(27) (Figure 7). Collectively, these findings strongly suggest that mitochondrial ROS is a promising therapeutic target for the prevention of DOXO-mediated endothelial dysfunction. Importantly, our laboratory has shown that oral MitoQ supplementation can increase brachial FMD in healthy middle-aged/older adults free of overt CVD(81). Thus, MitoQ supplementation may be a treatment strategy for preserving endothelial function in patients that have received anthracycline chemotherapy.
Figure 7. Anthracycline chemotherapy treatment (doxorubicin [DOXO])-mediated vascular endothelial dysfunction occurs as a result of excess vascular mitochondria-derived reactive oxygen species (ROS) and a reduction in mitochondrial antioxidant defenses.

A) Vascular endothelium-dependent dilation is lower in young DOXO-treated mice compared to young (Young Control) and chronologically aged controls (Old Control). B) Vascular manganese (Mn) SOD (mitochondrial isoform) is lower in young DOXO-treated mice relative to young controls and to levels similar to old controls. C) Peak endothelium-dependent in young DOXO-treated mice is rescued (back to young control levels) when exposed to an acute dose of the mitochondrial-targeted antioxidant, MitoQ. Additionally, chronic (4 weeks) oral supplementation (in the drinking water) with MitoQ prevents DOXO-mediated vascular endothelial dysfunction by suppressing tonic mitochondria-derived ROS (last bar; no further dilation when exposed to acute MitoQ). *P < 0.05 vs. Young Control; ^P < 0.05 vs. Old Control. Data from(27, 28, 157).
7. Cellular senescence
Cellular senescence is a multifaceted stress response that causes an essentially irreversible arrest of the cell cycle(93); however, senescent cells remain metabolically active and secrete a variety of inflammatory cytokines, chemokines, proteases and growth factors, which is collectively referred to as the senescence-associated secretory phenotype (SASP). Therefore, it is plausible that the SASP may be an upstream regulator of tonic oxidative stress and chronic low-grade inflammation -- key mechanistic features of vascular dysfunction, as described above. Cellular senescence is considered one of the hallmarks of aging(61) and is an emerging topic in the field aging-related CVD (reviewed in detail elsewhere(94)), as there is an increase in senescent cells within the heart and vasculature of older adults (relative to young adults)(94). Senescent cells are permanently arrested in the G1 stage of the cell cycle in response to various stressors, such as telomere attrition, stem cell exhaustion, DNA damage and genotoxic stress(93). It is important to note, however, that cellular senescence is implicated in the physiological processes that can also be highly beneficial for the organism(93), such as wound healing, fibrosis, tissue remodeling and embryonic development. Similar to chronological aging, anthracycline chemotherapy promotes cellular senescence in a variety of cell and tissue types, including cardiac tissue(95).
Cancer is defined in the simplest form as an uncontrolled division of abnormal cells. Accordingly, the cellular senescence response (cell cycle arrest) induced by anthracycline chemotherapy is thought to be a key mechanism by which anthracyclines elicits their antitumorigenic effects(96). However, if we consider the role of cellular senescence in CVD and the senescence-inducing property of anthracycline chemotherapy, it is possible that cellular senescence also may serve as a therapeutic target for the prevention and/or treatment of anthracycline chemotherapy-mediated vascular dysfunction.
7.1. Influence of cellular senescence on vascular health.
Vascular cells (endothelial and vascular smooth muscle cells) undergo cellular senescence(97, 98). Considering a key component of the SASP is secretion of pro-inflammatory cytokines, the SASP could directly remodel the vasculature. Thus, targeting the pathophysiological increase in senescent cells and the associated SASP could mitigate anthracycline-associated vascular dysfunction. Cellular senescence can be quantified in a variety of ways, as there is a unique “signature” of up- and down-regulated genes and proteins in senescent cells(99). A key aspect of this “signature” is the upregulation of the tumor suppressor proteins p16 and p21(93, 99).
We have shown that primary arterial endothelial cells isolated from healthy sedentary middle-aged/older adults have greater p16 and p21 abundance relative to young sedentary adults, which was inversely related to brachial artery FMD(100). The results of the same study demonstrated that habitually exercising middle-aged/older adults have lower endothelial cell p16 and p21 abundance as well as higher FMD, relative to their sedentary counterparts(100). Together, these findings suggest that lowering vascular cell senescence may hold promise for improving vascular endothelial function in a wide variety of pathological settings.
To explore this possibility, we recently used a reverse translation approach to determine the role of cellular senescence in regulating age-related arterial dysfunction in mice. Our preliminary results indicate that treatment of old mice with the senolytic ABT-263 (aka Navitoclax) reverses the age-related increase in aortic PWV and increases vascular endothelial function to levels observed in young adult mice(101). Our observations are consistent with previous findings that treatment of old mice with the senolytic cocktail Dasatanib plus Quercetin can increase vascular endothelial function in old mice(102).
As previously mentioned, DOXO increases cellular senescence in a variety of cell and tissue types, including cardiac tissue, but the role of cellular senescence in mediating DOXO-induced vascular dysfunction remains to be determined. Although senolytic-based therapeutic strategies have been used to determine the influence of cellular senescence on chronological aging-related vascular dysfunction, it has yet to be determined whether clearance of senescent cells following DOXO chemotherapy can prevent arterial stiffening and vascular endothelial dysfunction. By using a mouse model in which p16+ cells can be readily cleared (p16-3MR mouse), Demaria et al.(95) showed that clearance of p16+ cells following DOXO administration could prevent DOXO-mediated cardiac dysfunction. Given that vascular dysfunction is a key antecedent to cardiac dysfunction, we hypothesized that clearance of senescent cells following DOXO administration would prevent DOXO-mediated arterial stiffness and vascular endothelial dysfunction. Our preliminary results support this concept. Specifically, using the p16-3MR mouse model, we found that clearing senescent cells following DOXO in young adult mice prevented arterial stiffening and vascular endothelial dysfunction induced by DOXO treatment(103). This effect appeared to be mediated in part by the preservation of NO bioavailability and prevention of increased aortic intrinsic mechanical wall stiffness. Together, these preliminary data suggest that cellular senescence may be a novel therapeutic target for preventing and/or treating DOXO-driven vascular dysfunction. Unfortunately, there are currently no approved therapeutic strategies for reducing cellular senescence-associated vascular dysfunction in the setting of DOXO chemotherapy. Thus, a logical next step will be to develop safe and effective translational interventions that can reduce cellular senescence-associated vascular dysfunction in patients treated with anthracycline chemotherapeutic agents. This topic will be discussed further in the “Research Gaps” section.
8. NAD+
8.1. Linking mitochondrial dysfunction to reduced NAD+ bioavailability.
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor for the maintenance of cellular homeostasis. NAD+ is produced via oxidation of nicotinamide adenine dinucleotide + hydrogen (NADH; reduced form of NAD+) in the mitochondrial electron transport system(104). Reduced bioavailability of NAD+ is a common manifestation of advancing age(105) and impaired NAD+ bioavailability in vascular cells has been linked to age-related vascular dysfunction(106) (reviewed in detail elsewhere(107)). This observation has led many to consider boosting NAD+ bioavailability as a viable therapeutic strategy to prevent and/or treat common age-related diseases, including vascular disorders. Reduced NAD+ bioavailability is a key component of the aging hallmark “reduced energy sensing”(61) and there is evidence to suggest that this mechanism interacts with other hallmarks such as mitochondrial dysfunction and cellular senescence. Interestingly, like aging, anthracycline chemotherapy has shown to reduce NAD+ levels in a variety of cell types. Accordingly, we will discuss: 1) how NAD+ interacts with mitochondrial dysfunction and cellular senescence; 2) results of NAD+-targeted interventions for improving age-related vascular function; and 3) targeting NAD+ bioavailability for preventing and/or treating anthracycline-induced vascular dysfunction.
8.2. Mitochondrial dysfunction and cellular senescence: interactive effects of reduced NAD+.
It is well established that dysfunctional mitochondria can drive cellular senescence(108). As previously stated, mitochondria oxidize NADH to NAD+; therefore, mitochondrial dysfunction can directly decrease the bioavailability of NAD+. Furthermore, reduced NAD+ bioavailability has been shown to directly evoke mitochondrial dysfunction and cellular senescence in fibroblasts in vitro(108). As such, there seems to be an interactive effect within the common aging hallmarks of mitochondrial dysfunction, cellular senescence and impaired energy sensing.
Much of the focus on NAD+ bioavailability is due to the ability of the molecule to activate a family of NAD+-dependent energy sensing enzymes called sirtuins(109). In the mammalian genome, there are seven sirtuins (SIRT 1-7), and SIRT1 is the most highly studied among them(110). The abundance and activity of SIRT1 is reduced in many tissue types in the setting of chronological aging, including the vasculature(111). Most importantly, SIRT1 activity is reduced in a variety of cell and tissue types following administration of DOXO(112-115). As it relates to mitochondria, loss of mitochondrial sirtuin activity directly causes mitochondrial dysfunction and cellular senescence(108). Accordingly, targeting NAD+ bioavailability and/or sirtuin activity may represent a viable alternative approach to circumvent reduced mitochondrial function and prevent cellular senescence. However, the interaction between these processes and anthracycline chemotherapy, and how that interaction influences vascular function are incompletely understood.
8.3. Targeting NAD+ and sirtuins: potential therapeutic opportunities to improve vascular function in anthracycline-treated cancer survivors.
Caloric restriction (CR) is a lifestyle intervention used to increase bioavailability of NAD+. Our laboratory has shown that short-term CR initiated in late life reverses vascular dysfunction with aging(116) and that lifelong CR prevents the development of age-related vascular dysfunction(117). However, due to the increased risks of sarcopenia and frailty associated with CR in the setting of aging, this approach is not suitable for translation to older adults, which is likely also the case with anthracycline-treated cancer survivors, given the potential (relative to age-matched healthy controls) for cachexia and frailty in this patient population(118-120). Alternatively, CR-mimetics (NAD+ precursors/ NAD+ boosting compounds) such as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) can be used to increase NAD+ bioavailability with aging(121). We have shown that short-term NMN supplementation in late life can reverse age-related vascular dysfunction(122). We also have demonstrated that NR supplementation in middle-aged and older adults, free of overt CVD, is well tolerated, increases circulating NAD+ levels, and may reduce arterial stiffness(123). Activation of SIRT1 via the SIRT-specific activator SRT1720(124) and the non-specific activator resveratrol(125) also have shown to improve vascular function with aging. Of note, DOXO exposure to cardiomyocytes reduces SIRT1 activity, which is prevented by co-exposure of DOXO with resveratrol(126), and resveratrol supplementation prevents the DOXO-induced reduction in aortic EDD of young adult rats(127). Therefore, NAD+ boosting compounds may represent still another intriguing therapeutic strategy for improving vascular dysfunction following anthracycline chemotherapy.
8.4. NAD+ bioavailability: the balance between salvage and consumption.
NAD+ bioavailability is driven by a balance between the NAD+ salvage pathway (e.g., NMN and NR) and NAD+ consuming enzymes, such as poly(ADP-ribose) polymerase 1 (PARP1) and CD38(105). With advancing age, CD38 is increased in a variety of tissue types, and old CD38 knockout mice are void of a variety of age-related physical dysfunctions(128); however, the effect of CD38 on vascular function with aging is unknown. Via increased ROS production, DOXO can activate PARP1 in cardiomyocytes, ultimately reducing NAD+ bioavailability(129). Furthermore, treatment of macrophages with DOXO in vitro increases CD38 activity(130). Yet, it has not been established how anthracyclines influence vascular PARP1 or CD38.
Currently, there are natural and pharmaceutical inhibitors of PARP1 (e.g., nicotinamide(131) and Rucaparib(132)) and CD38 (e.g., apigenin(133, 134) and daratumumab(135)), which may show promise for preventing and/or treating anthracycline-related vascular dysfunction. Preliminary results from our laboratory suggest that short-term oral supplementation (in drinking water) with apigenin can reverse age-related vascular dysfunction and reduce foam cell formation (initiating step in the progression of atherosclerosis) in vitro(136). Given that DOXO reduces NAD+ bioavailability via a reduction in NMN/NR(137) and an increase in CD38 activity in cell types outside of the vasculature, a viable therapeutic option may be to simultaneously supplement a NAD+ boosting compound combined with a CD38 inhibitor. However, more work is needed to establish efficacy of dose, timing and potential toxicity of this approach.
9. AMPK and mTOR
Other key components of the aging hallmark “deregulated nutrient sensing” include reduced adenosine monophosphate protein-activated kinase (AMPK) and an increase in the mammalian target of rapamycin (mTOR)(61). During low states of cellular energy availability (high ADP: AMP ratio), AMPK is activated, which serves to increase glucose and lipid oxidation and stimulates mitochondrial oxidative metabolism(138). Contrary to AMPK, mTOR is activated during times of energy surplus to activate protein translation/synthesis and stimulate cellular growth pathways(139). With advanced age, there is reduced AMPK(140-142) and increased mTOR(143, 144) across a variety of cell and tissue types, which have been associated with a myriad of age-related diseases. Importantly, the age-related reduction in AMPK(145) and increase in mTOR(146) have been associated with vascular dysfunction.
In endothelial cells in vitro, pharmacological stimulation of AMPK induces antioxidant effects and activates endothelial NO synthase (enzyme responsible for NO production)(147). Our laboratory has shown that old mice have reduced vascular AMPK, which was associated with impaired vascular endothelial function(145). Furthermore, we have shown that short-term (2 weeks) activation of AMPK by aminoimidazole carboxamide ribonucleotide (AICAR) increases vascular AMPK and improves vascular endothelial function of old mice via suppression of tonic superoxide production(145). Furthermore, AICAR increases resistance artery vasodilation in a spontaneously hypertensive rat model(148). AMPK also is a direct inhibitor of mTOR(61), so it is plausible that activation of AMPK by AICAR could be improving vascular function via suppression of mTOR.
Inhibition of mTOR by dietary rapamycin has shown to delay age-associated diseases and extend lifespan in mice(149-151). There also is evidence to suggest mTOR inhibition can reduce oxidative stress in a variety of tissues and cell types(152, 153), including the heart(154). However, until recently, it was unknown how mTOR inhibition influenced the aging vasculature. Our laboratory demonstrated that feeding old mice a rapamycin (mTOR inhibitor)-enriched diet enhanced vascular AMPK activation, reversed age-related arterial stiffness, and increased vascular endothelial function via suppression of tonic oxidative stress (146).
As mentioned throughout this review, the hallmarks of aging do not occur in a vacuum and are remarkably interactive. A study by Laberge et al.(155) demonstrated that activation of the SASP was dependent upon mTOR activation. As such, an additional mechanism by which rapamycin improves vascular function could be via suppression of the SASP. As previously mentioned, we have shown that DOXO-mediated arterial dysfunction occurs via cellular senescence(103). Additionally, we have recently demonstrated that suppression of cellular senescence following DOXO prevents arterial stiffness via suppression of mTOR(103). However, the role of AMPK activation and mTOR inhibition as potential therapeutic targets for the prevention and/or treatment of anthracycline-mediated arterial dysfunction are not currently understood. Considering AMPK activation (via AICAR) and mTOR inhibition (via rapamycin) improve natural vascular aging, a reasonable next step could be to determine whether these therapeutic approaches can improve vascular function in the setting of anthracycline chemotherapy.
10. Aerobic exercise as an “all-inclusive” therapeutic approach for enhancing mitochondrial function, reducing cellular senescence and improving energy sensing pathways.
Aerobic exercise has remarkable positive effects on the aging vasculature (reviewed in detail elsewhere(57)), whether that is in the setting of chronological aging or accelerated aging via consumption of a Western-style diet(28, 156). Specifically, we have shown that voluntary aerobic exercise improves vascular function with aging via suppression of tonic oxidative stress and a reduction in vascular inflammation(28). However, as described throughout this review, other mechanisms such as reduced mitochondrial function, increased cellular senescence and impaired energy sensing may be key upstream mechanisms by which oxidative stress and inflammation are regulated. (Figure 8).
Figure 8. The influence of aerobic exercise on mitochondrial dysfunction, cellular senescence and dysregulated energy sensing.

Chronological aging leads to mitochondrial dysfunction, cellular senescence and dysregulated energy sensing, all of which are key hallmarks of aging. Aerobic exercise is an effective therapeutic strategy for preventing and treating these hallmarks in the setting of chronological aging. Anthracycline chemotherapies (e.g., doxorubicin [DOXO]) induce similar effects to aging. Thus, aerobic exercise may improve vascular function in anthracycline-treated cancer survivors by targeting these key mechanistic pathways.
10.1. Aerobic exercise and mitochondrial function.
Data from our laboratory suggest that late-life voluntary aerobic exercise in mice improves vascular endothelial function via suppression of excess vascular mitochondrial ROS production (oxidative stress) and enhanced vascular mitochondrial “fitness”, including increased stress resistance and mitochondrial biogenesis(157). We also recently demonstrated that lifelong aerobic exercise in mice can prevent vascular dysfunction in the settings of chronological aging and accelerated aging (via a Western-style diet) via suppression of tonic vascular mitochondrial oxidative stress(28). Taken together these observations strongly suggest that aerobic exercise may be a valuable therapeutic approach for targeting vascular mitochondrial oxidative stress with aging.
10.2. Aerobic exercise and vascular senescence.
Our laboratory has shown in a cross-sectional study in humans that sedentary healthy middle-aged and older adults have greater endothelial cell senescence relative to young sedentary adults, which was associated with impaired vascular endothelial function(100). In this study, there was an absence of age-related increases in endothelial cell senescence in older adults who had habitually participated in aerobic exercise, which was associated with preserved vascular endothelial function(100). Furthermore, a recent systematic review and meta-analysis on the topic suggests that aerobic exercise may be used as a senolytic therapy(158). However, to date, there has been no comprehensive investigation of the role of cellular senescence in regulating the protective effects of aerobic exercise on the aging vasculature.
10.3. Aerobic exercise and energy sensing.
Aerobic exercise increases NAD+ bioavailability, in part due to enhanced mitochondrial function, which in turn enhances SIRT activity (reviewed in detail elsewhere(159)). Furthermore, old mice administered with the CD38 inhibitor thiazologuin(az)olin(on)e have enhanced NAD+ bioavailability and aerobic exercise capacity relative to sham-treated old controls, suggesting there is an interactive effect of aging, NAD+ bioavailability and aerobic exercise(160). In terms of the influence of aerobic exercise on AMPK and mTOR, AICAR is commonly referred to as an “exercise mimetic”, as aerobic exercise is well known for activating AMPK and suppressing mTOR. As such, enhanced energy sensing may be a mechanism by which aerobic exercise maintains vascular function, but more work is needed to establish that possibility.
10.4. Therapeutic effect of aerobic exercise on vascular function with anthracycline chemotherapy.
Considering that aerobic exercise enhances cellular resilience/stress resistance of arteries, such that vascular function is preserved upon exposure to a stressor that would otherwise cause dysfunction(157), Gibson et al.(161) hypothesized that voluntary aerobic exercise pre-conditioning in rats would prevent the DOXO-induced impairment in aortic EDD. Contrary to their hypothesis, aortas from exercise trained rats had similar EDD to sedentary controls. In contrast, Lee et al.(162) demonstrated that 8 weeks of high-intensity interval training on a cycle ergometer (7x: 1 minute at 90% peak power output, followed by 2 minutes at 10% peak power output; 3 sessions/week), performed throughout anthracycline chemotherapy preserved vascular endothelial function in patients undergoing treatment for breast cancer. Consistent with these findings, Long et al.(163) have shown that 24 weeks of exercise, following the guidelines for exercise in cancer survivors put forth by the American College of Sports Medicine, could increase vascular endothelial function in young adult childhood cancer survivors.
Taken together, exercise training may be a favorable lifestyle intervention/strategy for preserving and improving vascular function during or following anthracycline chemotherapy, respectively. However, the mechanisms by which exercise training improves vascular function in anthracycline-treated cancer survivors remain to be elucidated. Furthermore, the influence of exercise training on arterial stiffness during or following anthracycline chemotherapy remains to be determined.
11. Anthracycline chemotherapy in older adults — a “double-hit” to the vasculature.
The total number of older adults is projected to increase significantly over the next 50 years worldwide(164, 165). Advancing age is the primary risk factor for the development of both cancer(166) and vascular dysfunction(167). Thus, as a result, there will be an increased number of older adults undergoing chemotherapy in the coming years. Unfortunately, elderly patients have a reduced ability to tolerate treatment and are more vulnerable (relative to children and young adults) to the toxic effects of chemotherapy(168) (Figure 9).
Figure 9. Aging and cancer – a “double hit” to vascular dysfunction and cardiovascular disease (CVD) risk.

Chronological aging is the primary risk factor for both cancer and vascular dysfunction. Cancer is most commonly treated with chemotherapeutic agents (e.g., anthracyclines like doxorubicin [DOXO]) that can independently drive vascular dysfunction. Vascular dysfunction is a key antecedent to CVD and elevated CVD risk places an individual at increased risk for developing future cancers. Thus, cancer treatment in the setting of advanced age markedly increases the risk for developing CVD (relative to aging alone) due to being a “second-hit” to the vasculature.
Anthracyclines are also first-line chemotherapeutic agents to treat multiple cancers most commonly observed in older adults – breast and prostate cancer and Non-Hodgkin’s lymphoma(169, 170). Furthermore, The American Society of Clinical Oncology estimates ~2- to 7-fold increased risk of CVD in older adults (≥ 60 years of age) undergoing anthracycline chemotherapy when compared to young adults undergoing similar treatment(171). Accordingly, the combination of advancing age and chemotherapy treatment may be a “double-hit” to the vasculature. Moreover, the overall dose of chemotherapy is often reduced in older adults (relative to children and young adults), especially those with pre-existing CVD, so as to not place undue stress on the cardiovascular system(169). This all points to the biomedical importance of developing therapeutic strategies for preventing and/or treating anthracycline-mediated vascular dysfunction to mitigate the accelerated rate of CVD progression in young and older adults. not just in young adults.
12. Research gaps and potential future directions.
Vascular dysfunction is a key antecedent to CVD, in particular large elastic artery stiffening and endothelial dysfunction. In this review we have discussed the central roles impaired mitochondrial function, cellular senescence and reduced energy sensing as key pathophysiological mechanisms mediating anthracycline-induced vascular dysfunction. We have also reviewed established therapies that could target these pathways (Table 1).
Table 1.
Potential therapeutic targets and treatment options for anthracycline-mediated vascular dysfunction.
| Therapeutics | Description |
|---|---|
| Mitochondria-targeted compounds | |
| MitoQ | Mitochondria-targeted antioxidant |
| SS-31 | Mitochondria-targeted peptide |
| Urolithin-A | Gut microbiome-derived mitophagy activator |
| Dexrazoxane | Mitochondria DNA damage inhibitor |
| Senolytics | |
| ABT-263 (Navitoclax) | BCL pathway (anti-apoptosis) inhibitor |
| Dasatanib + Quercetin | BCL (anti-apoptosis) pathway inhibitor |
| Natural compounds | Food-derived compounds (e.g., flavonoids) |
| Galacto-conjugated compounds | Target only cells expressing β-Galactosidase |
| NAD+ boosting compounds | |
| Nicotinamide Mononucleotide | NAD+ salvage pathway activator |
| Nicotinamide Riboside | NAD+ salvage pathway activator |
| CD-38 inhibitors | |
| Apigenin | Food-derived (flavonoid) CD-38 inhibitor |
| Daratumumab | Synthetic CD-38 inhibitor |
| Thiazologuin(az)olin(on)e | Synthetic CD-38 inhibitor |
| Sirtuin activators | |
| Resveratrol | Food-derived (plant polyphenol) surtuin activator |
| SRT1720 | Synthetic sirtuin activator |
| PARP inhibitors | |
| Nicotinamide | Inhibits PARP and increases NAD+ bioavailability |
| Rucaparib | Inhibits PARP and increases NAD+ bioavailability |
| AMPK activator | |
| AICAR | AMP analog (increases circulating AMP) |
| mTOR inhibitor | |
| Rapamycin | Immunosuppressive compounds which inhibits mTOR |
MitoQ, Mitoquinol mesylate; SS-31, Szeto-Schiller peptide; BCL, B-cell lymphoma; NAD+, nicotinamide adenine dinucleotide; PARP, Poly (ADP-ribose) polymerase; AMPK, adenine monophosphate protein kinase; AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide; mTOR, mammalian target of rapamycin
There remain several important knowledge gaps in the field; the following represent some potential future, biomedically significant directions for research related to anthracycline-mediated vascular dysfunction (Figure 10).
Figure 10. Research gaps and future directions.

MitoQ, mitoquinone; Szeto-Schiller peptide, SS-31; Nicotinamide adenine dinucleotide, NAD+; Nicotinamide mononucleotide, NMN; Nicotinamide riboside, NR; Sirtuins, SIRT; Inspiratory muscle strength training, IMST.
12.1. Mitochondrial-targeted therapies.
MitoQ.
As described above, mitochondrial dysfunction may be a therapeutic target for preventing and/or treating anthracycline-mediated vascular dysfunction. In our preclinical study, 4 weeks of MitoQ supplementation (in drinking water) following DOXO administration prevented DOXO-mediated vascular dysfunction(27). Importantly regarding clinical translation, MitoQ has shown to be safe(85, 86) and effective for improving vascular function in middle-aged/older adults(81). As such, MitoQ supplementation trials in anthracycline-treated cancer survivors are warranted.
Dexrazoxane.
Dexrazoxane, a compound which is commonly administered with anthracyclines to mitigate excess mitochondrial oxidative stress (via reduced mitochondrial DNA damage), is proven to reduce the cardiac toxicity associated with anthracyclines(172, 173). However, the effect of Dexrazoxane on vascular function in the setting of anthracycline chemotherapy is currently unknown. Considering vascular dysfunction is a common antecedent to cardiac dysfunction, it is plausible that the protective effect of Dexrazoxane on the heart could be a result of improved vascular function.
SS-31.
There are also other emerging mitochondrial-targeted therapies that could be considered. Specifically, Szeto-Schiller peptide (SS-31; aka Elamipretide), a mitochondrial-targeted peptide, improves physiological function in a variety of pre-clinical settings(174-176). Regarding vascular aging, SS-31 treatment improved cerebrovascular EDD by increasing NO bioavailability in old mice(176). SS-31 also normalized mitochondrial ROS production and restored basal mitochondrial respiration in cerebrovascular endothelial cells from old rats to levels observed in cells from young animals(176). Early phase clinical trials in humans have shown mixed results on various physiological functions, but SS-31 has yet to be assessed for improving vascular function in older adults or in patients receiving or who have received anthracycline chemotherapy.
Urolithin A.
Another mitochondrial-targeted therapy that has recently gained significant attention is ellagitannin-derived Urolithin A, a gut microbiome-derived metabolite that activates mitophagy(177). Urolithin A treatment preserves mitochondrial function with aging and extends lifespan of Caenorhabditis elegans and improves skeletal muscle function in rodents(177). Moreover, acute and chronic administration of the compound appears to be safe and well-tolerated in sedentary older adults(178). However, it is currently unknown whether Urolithin A can improve vascular function. Considering it is safe and well-tolerated, Urolithin A may be an effective therapeutic approach for preventing and/or treating anthracycline-induced vascular dysfunction.
12.2. Senolytics.
Future work is needed to determine how senolytic therapies affect vascular function in anthracycline-treated cancer survivors. Given the many interactions described above, future studies should investigate how senolytic therapies influence other hallmarks of aging(61). Below, we discuss the various senolytic strategies that could be used to help address current research gaps.
Food-derived compounds.
There are a variety of naturally derived compounds that have gained attention as potential senolytic therapies. A study by Yousefzadeh et al.(179) screened a variety of flavonoid compounds that have senolytic properties. Results of the screening demonstrated that fisetin (found primarily in strawberries and cucumbers) had the greatest senolytic effect in mouse primary embryonic fibroblasts and IMR90 cells. To translate these findings, the same study demonstrated that fisetin supplementation could reduce senescent cell burden in a variety of tissues in progeroid mice (a model of accelerated aging) and chronologically aged wild-type mice, as well as extend lifespan in wild-type mice. Currently, there is an ongoing open-label clinical trial (NCT04733534) assessing the effect of fisetin on reducing senescence and frailty in childhood cancer survivors Presently, however, the influence of fisetin on vascular function in cancer survivors has not been investigated.
Dasatanib + Quercetin.
Numerous preclinical studies have assessed the combination of Dasatanib + Quercetin for reducing cellular senescence in the setting of advanced age in a variety of cell and tissue types (reviewed elsewhere(180)). Specifically, one study has shown that Dasatanib + Quercetin can improve aortic EDD in old mice, which was associated with reduced senescent cell burden in the aorta(102); however, the influence of D+Q on arterial stiffness remains to be determined. Furthermore, nothing is known regarding D+Q as a potential therapeutic approach to improve vascular function with anthracycline chemotherapy. Importantly, D+Q has shown to be safe for human use(181) and is being administered to childhood cancer survivors to determine its influence on peripheral T-cell senescence and frailty (NCT04733534).
ABT-263 (Navitoclax).
ABT-263 is a senolytic compound designed to inhibit the anti-apoptotic BCL-2 pathway in senescent cells(182). Administration of ABT-263 rejuvenates the hematopoetic stem cell niche in old mice(183) and preserves cognitive function following whole brain irradiation in young adult mice(184). Our preliminary results suggest that ABT-263 administration to old mice reverses aortic stiffening and improves EDD with aging in mice(101). However, the influence of ABT-263 on vascular senescence and function in the setting of anthracycline chemotherapy has yet to be determined.
Galactosidase-conjugated senolytics.
As described above, senolytics are designed to have preferential cytotoxic activity for senescent cells, yet translation of senolytic therapies to human disease is hindered by their suboptimal specificity for senescent cells and notable toxicities. It is well established that senescent cells highly express the senescence marker β-galactosidase (SA-β-gal)(185), which can be used to specifically release tracers and cytotoxic cargoes from galactose-encapsulated nanoparticles within these cells. Recently, Gonzalez-Gualda et al.(186) demonstrated that galacto-conjugated ABT-263 can be preferentially activated by SA-β-gal activity in a variety of cell types. This approach revealed greater senolytic activity relative to standard ABT-263 administration, as a result of reduced activation in nonsenescent cells, and resulted in lower platelet toxicity relative to standard ABT-263. Accordingly, future studies aimed at targeting senescent cells to improve vascular function, without off-target toxic effects, should consider using galacto-conjugated senolytics.
12.3. NAD+ Boosting Compounds/CR mimetics/SIRT activators.
As described previously, several therapeutic strategies aimed at mimicking the effects of CR (e.g., NMN, NR, SIRT activators, resveratrol, CD38ases) are effective for reversing chronological aging-related vascular dysfunction and/or other diseases of aging. However, the influence of these therapies on vascular function in the setting of anthracycline chemotherapy are not well understood. DOXO reduces NAD+ bioavailability and SIRT activity, both of which are associated with vascular dysfunction; as such, future studies are warranted to better understand the interaction of NAD+ bioavailability and SIRT activity with anthracycline chemotherapy and vascular dysfunction.
12.4. Lifestyle interventions and interaction with aging hallmarks.
Above we described the potential efficacy of regular aerobic exercise for improving EDD with anthracycline chemotherapy. But it should be noted that long-term adherence to aerobic exercise is generally poor, particularly in groups of lower education and/or income status(187). Future studies are warranted to determine the extent to which healthy lifestyle interventions, including novel modes of more time-efficient exercise training (188), may interact with the hallmarks of aging to improve vascular function in settings of both natural aging and anthracycline chemotherapy.
12.5. Sex differences.
Estrogen has demonstrated to be protective against DOXO-mediated cardiac dysfunction in preclinical studies, such that cardiac function is worse in male mice relative to female mice following an equivalent dose of DOXO(189-191). Furthermore, female anthracycline-treated cancer survivors have greater endothelial function relative to their male counterparts(50). However, the mechanisms by which estrogen protects against anthracycline-induced vascular dysfunction are incompletely understood and deserving of additional investigation, including in estrogen-deficient postmenopausal cancer survivors. Novel preclinical models that better recapitulate sex hormone regulation throughout the lifespan represent a potentially useful experimental mechanistic approach in this regard(192-195).
12.6. Interactions between different prevention and treatment strategies.
It remains to be determined whether there are interactions among the aforementioned therapeutic strategies. Future work should consider experimental opportunities for determining the independent and interactive effects of therapies targeting the so-called hallmark of aging cellular processes for preventing/treating vascular dysfunction induced by anthracycline-based chemotherapies.
Acknowledgements
Work from the authors’ research was supported by U.S. National Institute of Health Awards: R01 AG055822 (DRS); 3R01 AG055822-03S1 (SM); T32 DK007135 (ZSC); and F32 HL151022 (ZSC). MitoQ Limited provided MitoQ for preclinical studies. Authors have no conflicts of interest to disclose. Data from the original research presented in this review will be available upon reasonable request to the corresponding author.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Data Availability Statement
All data will be made available following a reasonable request to the corresponding author.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137(12):e67–e492. Epub 2018/01/31. doi: 10.1161/CIR.0000000000000558. PubMed PMID: 29386200. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. Epub 2019/01/08. doi: 10.3322/caac.21551. PubMed PMID: 30620402. [DOI] [PubMed] [Google Scholar]
- 3.Halpern MT, Yabroff KR. Prevalence of outpatient cancer treatment in the United States: estimates from the Medical Panel Expenditures Survey (MEPS). Cancer Invest. 2008;26(6):647–51. doi: 10.1080/07357900801905519. PubMed PMID: 18584358. [DOI] [PubMed] [Google Scholar]
- 4.Truong J, Yan AT, Cramarossa G, Chan KK. Chemotherapy-induced cardiotoxicity: detection, prevention, and management. Can J Cardiol. 2014;30(8):869–78. Epub 2014/05/04. doi: 10.1016/j.cjca.2014.04.029. PubMed PMID: 25064580. [DOI] [PubMed] [Google Scholar]
- 5.Meinardi MT, Gietema JA, van Veldhuisen DJ, van der Graaf WT, de Vries EG, Sleijfer DT. Long-term chemotherapy-related cardiovascular morbidity. Cancer Treat Rev. 2000;26(6):429–47. doi: 10.1053/ctrv.2000.0175. PubMed PMID: 11139373. [DOI] [PubMed] [Google Scholar]
- 6.Lau ES, Paniagua SM, Liu E, Jovani M, Li SX, Takvorian K, et al. Cardiovascular Risk Factors are Associated with Future Cancer. JACC: CardioOncology 2021. p. 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipshultz SE, Karnik R, Sambatakos P, Franco VI, Ross SW, Miller TL. Anthracycline-related cardiotoxicity in childhood cancer survivors. Curr Opin Cardiol. 2014;29(1):103–12. doi: 10.1097/HCO.0000000000000034. PubMed PMID: 24284979. [DOI] [PubMed] [Google Scholar]
- 8.Lipshultz SE, Franco VI, Miller TL, Colan SD, Sallan SE. Cardiovascular disease in adult survivors of childhood cancer. Annu Rev Med. 2015;66:161–76. doi: 10.1146/annurev-med-070213-054849. PubMed PMID: 25587648; PubMed Central PMCID: PMCPMC5057395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaklamani VG, Gradishar WJ. Epirubicin versus doxorubicin: which is the anthracycline of choice for the treatment of breast cancer? Clin Breast Cancer. 2003;4 Suppl 1:S26–33. PubMed PMID: 12756076. [DOI] [PubMed] [Google Scholar]
- 10.Khanna A, Pequeno P, Gupta S, Thavendiranathan P, Lee DS, Abdel-Qadir H, et al. Increased Risk of All Cardiovascular Disease Subtypes Among Childhood Cancer Survivors: Population-Based Matched Cohort Study. Circulation. 2019;140(12):1041–3. Epub 2019/08/26. doi: 10.1161/CIRCULATIONAHA.119.041403. PubMed PMID: 31446776. [DOI] [PubMed] [Google Scholar]
- 11.Luu AZ, Chowdhury B, Al-Omran M, Teoh H, Hess DA, Verma S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl Sci. 2018;3(6):861–70. Epub 2018/12/31. doi: 10.1016/j.jacbts.2018.06.005. PubMed PMID: 30623145; PubMed Central PMCID: PMCPMC6314956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a "set up" for vascular disease. Circulation. 2003;107(1):139–46. PubMed PMID: 12515756. [DOI] [PubMed] [Google Scholar]
- 13.Adji A, O'Rourke MF, Namasivayam M. Arterial stiffness, its assessment, prognostic value, and implications for treatment. Am J Hypertens. 2011;24(1):5–17. Epub 2010/09/09. doi: 10.1038/ajh.2010.192. PubMed PMID: 20940710. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121(4):505–11. Epub 2010/01/18. doi: 10.1161/CIRCULATIONAHA.109.886655. PubMed PMID: 20083680; PubMed Central PMCID: PMCPMC2836717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chirinos JA, Segers P, Hughes T, Townsend R. Large-Artery Stiffness in Health and Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74(9):1237–63. doi: 10.1016/j.jacc.2019.07.012. PubMed PMID: 31466622; PubMed Central PMCID: PMCPMC6719727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension. 2008;51(1):99–104. Epub 2007/11/19. doi: 10.1161/HYPERTENSIONAHA.107.093674. PubMed PMID: 18025297. [DOI] [PubMed] [Google Scholar]
- 17.Pase MP, Beiser A, Himali JJ, Tsao C, Satizabal CL, Vasan RS, et al. Aortic Stiffness and the Risk of Incident Mild Cognitive Impairment and Dementia. Stroke. 2016;47(9):2256–61. Epub 2016/08/04. doi: 10.1161/STROKEAHA.116.013508. PubMed PMID: 27491735; PubMed Central PMCID: PMCPMC4995162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang MC, Tsai WC, Chen JY, Huang JJ. Stepwise increase in arterial stiffness corresponding with the stages of chronic kidney disease. Am J Kidney Dis. 2005;45(3):494–501. doi: 10.1053/j.ajkd.2004.11.011. PubMed PMID: 15754271. [DOI] [PubMed] [Google Scholar]
- 19.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107(3):490–7. doi: 10.1161/01.cir.0000048894.99865.02. PubMed PMID: 12551876. [DOI] [PubMed] [Google Scholar]
- 20.Fleenor BS. Large elastic artery stiffness with aging: novel translational mechanisms and interventions. Aging Dis. 2013;4(2):76–83. Epub 2012/12/11. PubMed PMID: 23696949; PubMed Central PMCID: PMCPMC3659253. [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107(5):615–9. Epub 2010/07/15. doi: 10.1161/CIRCRESAHA.110.221846. PubMed PMID: 20634486; PubMed Central PMCID: PMCPMC2936100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinderliter AL, Caughey M. Assessing endothelial function as a risk factor for cardiovascular disease. Curr Atheroscler Rep. 2003;5(6):506–13. PubMed PMID: 14525685. [DOI] [PubMed] [Google Scholar]
- 23.Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis. 2016;248:97–109. Epub 2016/03/09. doi: 10.1016/j.atherosclerosis.2016.03.007. PubMed PMID: 26994427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond). 2011;120(9):357–75. doi: 10.1042/CS20100476. PubMed PMID: 21244363; PubMed Central PMCID: PMCPMC3482987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreau KL, Donato AJ, Seals DR, DeSouza CA, Tanaka H. Regular exercise, hormone replacement therapy and the age-related decline in carotid arterial compliance in healthy women. Cardiovasc Res. 2003;57(3):861–8. doi: 10.1016/s0008-6363(02)00777-0. PubMed PMID: 12618248. [DOI] [PubMed] [Google Scholar]
- 26.Butlin M, Tan I, Spronck B, Avolio AP. Measuring Arterial Stiffness in Animal Experimental Studies. Arterioscler Thromb Vasc Biol. 2020;40(5):1068–77. Epub 2020/04/09. doi: 10.1161/ATVBAHA.119.313861. PubMed PMID: 32268787; PubMed Central PMCID: PMCPMC7176337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clayton ZS, Brunt VE, Hutton DA, VanDongen NS, D'Alessandro A, Reisz JA, et al. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC CardioOncol. 2020;2(3):475–88. Epub 2020/09/15. doi: 10.1016/j.jaccao.2020.06.010. PubMed PMID: 33073250; PubMed Central PMCID: PMCPMC7561020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gioscia-Ryan RA, Clayton ZS, Zigler MC, Richey JJ, Cuevas LM, Rossman MJ, et al. Lifelong voluntary aerobic exercise prevents age- and Western diet- induced vascular dysfunction, mitochondrial oxidative stress and inflammation in mice. J Physiol. 2020. Epub 2020/10/25. doi: 10.1113/JP280607. PubMed PMID: 33103241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossman MJ, Gioscia-Ryan RA, Santos-Parker JR, Ziemba BP, Lubieniecki KL, Johnson LC, et al. Inorganic Nitrite Supplementation Improves Endothelial Function With Aging: Translational Evidence for Suppression of Mitochondria-Derived Oxidative Stress. Hypertension. 2021;77(4):1212–22. Epub 2021/03/01. doi: 10.1161/HYPERTENSIONAHA.120.16175. PubMed PMID: 33641356; PubMed Central PMCID: PMCPMC7946806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunt VE, Gioscia-Ryan RA, Casso AG, VanDongen NS, Ziemba BP, Sapinsley ZJ, et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension. 2020;76(1):101–12. Epub 2020/06/10. doi: 10.1161/HYPERTENSIONAHA.120.14759. PubMed PMID: 32520619; PubMed Central PMCID: PMCPMC7295014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green D Point: Flow-mediated dilation does reflect nitric oxide-mediated endothelial function. J Appl Physiol (1985). 2005;99(3):1233–4; discussion 7-8. doi: 10.1152/japplphysiol.00601.2005. PubMed PMID: 16103524. [DOI] [PubMed] [Google Scholar]
- 32.Kaplon RE, Hill SD, Bispham NZ, Santos-Parker JR, Nowlan MJ, Snyder LL, et al. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging (Albany NY). 2016;8(6):1167–83. doi: 10.18632/aging.100962. PubMed PMID: 27208415; PubMed Central PMCID: PMCPMC4931825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209(1):13–22. doi: 10.1083/jcb.201412052. PubMed PMID: 25869663; PubMed Central PMCID: PMCPMC4395483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nowak KL, Rossman MJ, Chonchol M, Seals DR. Strategies for Achieving Healthy Vascular Aging. Hypertension. 2018;71(3):389–402. Epub 2018/01/08. doi: 10.1161/HYPERTENSIONAHA.117.10439. PubMed PMID: 29311256; PubMed Central PMCID: PMCPMC5812814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunt VE, Gioscia-Ryan RA, Richey JJ, Zigler MC, Cuevas LM, Gonzalez A, et al. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J Physiol. 2019. Epub 2019/02/04. doi: 10.1113/JP277336. PubMed PMID: 30714619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gioscia-Ryan RA, Battson ML, Cuevas LM, Eng JS, Murphy MP, Seals DR. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J Appl Physiol (1985). 2018;124(5):1194–202. Epub 2017/10/26. doi: 10.1152/japplphysiol.00670.2017. PubMed PMID: 29074712; PubMed Central PMCID: PMCPMC6008077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka H, Dinenno FA, Monahan KD, Clevenger CM, DeSouza CA, Seals DR. Aging, habitual exercise, and dynamic arterial compliance. Circulation. 2000;102(11):1270–5. doi: 10.1161/01.cir.102.11.1270. PubMed PMID: 10982542. [DOI] [PubMed] [Google Scholar]
- 38.Parr SK, Liang J, Schadler KL, Gilchrist SC, Steele CC, Ade CJ. Anticancer Therapy-Related Increases in Arterial Stiffness: A Systematic Review and Meta-Analysis. J Am Heart Assoc. 2020:e015598. Epub 2020/07/10. doi: 10.1161/JAHA.119.015598. PubMed PMID: 32648507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budinskaya K, Puchnerová V, Svačinová J, Novák J, Hrstková H, Nováková M, et al. Non-invasive assessment of vascular system function and damage induced by anthracycline treatment in the pediatric cancer survivors. Physiol Res. 2017;66(Suppl 4):S553–S60. doi: 10.33549/physiolres.933794. PubMed PMID: 29355384. [DOI] [PubMed] [Google Scholar]
- 40.Herceg-Cavrak V, Ahel V, Batinica M, Matec L, Kardos D. Increased arterial stiffness in children treated with anthracyclines for malignant disease. Coll Antropol. 2011;35(2):389–95. PubMed PMID: 21755708. [PubMed] [Google Scholar]
- 41.Krystal JI, Reppucci M, Mayr T, Fish JD, Sethna C. Arterial stiffness in childhood cancer survivors. Pediatr Blood Cancer. 2015;62(10):1832–7. Epub 2015/04/20. doi: 10.1002/pbc.25547. PubMed PMID: 25895119. [DOI] [PubMed] [Google Scholar]
- 42.Jenei Z, Bárdi E, Magyar MT, Horváth A, Paragh G, Kiss C. Anthracycline causes impaired vascular endothelial function and aortic stiffness in long term survivors of childhood cancer. Pathol Oncol Res. 2013;19(3):375–83. Epub 2012/12/16. doi: 10.1007/s12253-012-9589-6. PubMed PMID: 23242567. [DOI] [PubMed] [Google Scholar]
- 43.Arnold N, Merzenich H, Wingerter A, Schulz A, Schneider A, Prochaska JH, et al. Promotion of Arterial Stiffness by Childhood Cancer and Its Characteristics in Adult Long-Term Survivors. J Am Heart Assoc. 2021;10(5):e015609. Epub 2021/02/24. doi: 10.1161/JAHA.119.015609. PubMed PMID: 33624513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frye JN, Sutterfield SL, Caldwell JT, Behnke BJ, Copp SW, Banister HR, et al. Vascular and autonomic changes in adult cancer patients receiving anticancer chemotherapy. J Appl Physiol (1985). 2018;125(1):198–204. Epub 2018/03/22. doi: 10.1152/japplphysiol.00005.2018. PubMed PMID: 29565770. [DOI] [PubMed] [Google Scholar]
- 45.Chaosuwannakit N, D'Agostino R, Hamilton CA, Lane KS, Ntim WO, Lawrence J, et al. Aortic stiffness increases upon receipt of anthracycline chemotherapy. J Clin Oncol. 2010;28(1):166–72. Epub 2009/11/09. doi: 10.1200/JCO.2009.23.8527. PubMed PMID: 19901105; PubMed Central PMCID: PMCPMC2799231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drafts BC, Twomley KM, D'Agostino R, Lawrence J, Avis N, Ellis LR, et al. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc Imaging. 2013;6(8):877–85. Epub 2013/05/01. doi: 10.1016/j.jcmg.2012.11.017. PubMed PMID: 23643285; PubMed Central PMCID: PMCPMC3745801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mizia-Stec K, Gościńska A, Mizia M, Haberka M, Chmiel A, Poborski W, et al. Anthracycline chemotherapy impairs the structure and diastolic function of the left ventricle and induces negative arterial remodelling. Kardiol Pol. 2013;71(7):681–90. doi: 10.5603/KP.2013.0154. PubMed PMID: 23907900. [DOI] [PubMed] [Google Scholar]
- 48.Jang WJ, Choi DY, Jeon IS. Vascular endothelial dysfunction after anthracyclines treatment in children with acute lymphoblastic leukemia. Korean J Pediatr. 2013;56(3):130–4. Epub 2013/03/18. doi: 10.3345/kjp.2013.56.3.130. PubMed PMID: 23559975; PubMed Central PMCID: PMCPMC3611047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chow AY, Chin C, Dahl G, Rosenthal DN. Anthracyclines cause endothelial injury in pediatric cancer patients: a pilot study. J Clin Oncol. 2006;24(6):925–8. doi: 10.1200/JCO.2005.03.5956. PubMed PMID: 16484703. [DOI] [PubMed] [Google Scholar]
- 50.Dengel DR, Ness KK, Glasser SP, Williamson EB, Baker KS, Gurney JG. Endothelial function in young adult survivors of childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30(1):20–5. doi: 10.1097/MPH.0b013e318159a593. PubMed PMID: 18176175. [DOI] [PubMed] [Google Scholar]
- 51.Hader SN, Zinkevich N, Norwood Toro LE, Kriegel AJ, Kong A, Freed JK, et al. Detrimental effects of chemotherapy on human coronary microvascular function. Am J Physiol Heart Circ Physiol. 2019;317(4):H705–H10. Epub 2019/08/09. doi: 10.1152/ajpheart.00370.2019. PubMed PMID: 31397169; PubMed Central PMCID: PMCPMC6843017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seals DR, Kaplon RE, Gioscia-Ryan RA, LaRocca TJ. You're only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology (Bethesda). 2014;29(4):250–64. doi: 10.1152/physiol.00059.2013. PubMed PMID: 24985329; PubMed Central PMCID: PMCPMC4103060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ Res. 2018;123(7):849–67. doi: 10.1161/CIRCRESAHA.118.311378. PubMed PMID: 30355080; PubMed Central PMCID: PMCPMC6248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clayton ZS, Brunt VE, Hutton DA, Casso AG, Ziemba BP, Melov S, et al. Tumor Necrosis Factor Alpha-Mediated Inflammation and Remodeling of the Extracellular Matrix Underlies Aortic Stiffening Induced by the Common Chemotherapeutic Agent Doxorubicin. Hypertension. 2021:HYPERTENSIONAHA12016759. Epub 2021/03/15. doi: 10.1161/HYPERTENSIONAHA.120.16759. PubMed PMID: 33719511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–37. doi: 10.1152/ajpcell.1996.271.5.C1424. PubMed PMID: 8944624. [DOI] [PubMed] [Google Scholar]
- 56.Crabtree MJ, Channon KM. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide. 2011;25(2):81–8. Epub 2011/04/22. doi: 10.1016/j.niox.2011.04.004. PubMed PMID: 21550412; PubMed Central PMCID: PMCPMC5357050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seals DR. Edward F. Adolph Distinguished Lecture: The remarkable anti-aging effects of aerobic exercise on systemic arteries. J Appl Physiol (1985). 2014;117(5):425–39. Epub 2014/05/22. doi: 10.1152/japplphysiol.00362.2014. PubMed PMID: 24855137; PubMed Central PMCID: PMCPMC4157159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mitchell GF, Parise H, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, et al. Changes in arterial stiffness and wave reflection with advancing age in healthy men and women: the Framingham Heart Study. Hypertension. 2004;43(6):1239–45. Epub 2004/05/03. doi: 10.1161/01.HYP.0000128420.01881.aa. PubMed PMID: 15123572. [DOI] [PubMed] [Google Scholar]
- 59.Shi R, Gao S, Smith AH, Li H, Shao M, Shangguan J, et al. Superoxide-induced Type I collagen secretion depends on prolyl 4-hydroxylases. Biochem Biophys Res Commun. 2020;529(4):1011–7. Epub 2020/07/30. doi: 10.1016/j.bbrc.2020.07.002. PubMed PMID: 32819558; PubMed Central PMCID: PMCPMC7442856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, et al. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2012;32(3):745–55. Epub 2011/12/22. doi: 10.1161/ATVBAHA.111.243121. PubMed PMID: 22199367; PubMed Central PMCID: PMCPMC3288772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. PubMed PMID: 23746838; PubMed Central PMCID: PMCPMC3836174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977;72(2):441–55. doi: 10.1083/jcb.72.2.441. PubMed PMID: 833203; PubMed Central PMCID: PMCPMC2110997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park SY, Gifford JR, Andtbacka RH, Trinity JD, Hyngstrom JR, Garten RS, et al. Cardiac, skeletal, and smooth muscle mitochondrial respiration: are all mitochondria created equal? Am J Physiol Heart Circ Physiol. 2014;307(3):H346–52. Epub 2014/06/06. doi: 10.1152/ajpheart.00227.2014. PubMed PMID: 24906913; PubMed Central PMCID: PMCPMC4121645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112(8):1171–88. doi: 10.1161/CIRCRESAHA.111.300233. PubMed PMID: 23580773; PubMed Central PMCID: PMCPMC3700369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100(8):1128–41. doi: 10.1161/01.RES.0000261970.18328.1d. PubMed PMID: 17463328. [DOI] [PubMed] [Google Scholar]
- 66.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res. 2006;99(9):924–32. doi: 10.1161/01.RES.0000248212.86638.e9. PubMed PMID: 17068300. [DOI] [PubMed] [Google Scholar]
- 67.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100(4):460–73. doi: 10.1161/01.RES.0000258450.44413.96. PubMed PMID: 17332437. [DOI] [PubMed] [Google Scholar]
- 68.Ungvari Z, Sonntag WE, Csiszar A. Mitochondria and aging in the vascular system. J Mol Med (Berl). 2010;88(10):1021–7. Epub 2010/08/17. doi: 10.1007/s00109-010-0667-5. PubMed PMID: 20714704; PubMed Central PMCID: PMCPMC3045746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadlec AO, Beyer AM, Ait-Aissa K, Gutterman DD. Mitochondrial signaling in the vascular endothelium: beyond reactive oxygen species. Basic Res Cardiol. 2016;111(3):26. Epub 2016/03/18. doi: 10.1007/s00395-016-0546-5. PubMed PMID: 26992928; PubMed Central PMCID: PMCPMC6749826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu RM, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565–77. Epub 2015/10/10. doi: 10.1016/j.redox.2015.09.009. PubMed PMID: 26496488; PubMed Central PMCID: PMCPMC4625010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med. 2004;37(6):768–84. doi: 10.1016/j.freeradbiomed.2004.06.008. PubMed PMID: 15304253. [DOI] [PubMed] [Google Scholar]
- 72.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5. Epub 2010/12/01. doi: 10.1038/nature09663. PubMed PMID: 21124315. [DOI] [PubMed] [Google Scholar]
- 73.López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13(2):106–18. Epub 2013/01/16. doi: 10.1016/j.mito.2013.01.003. PubMed PMID: 23333405. [DOI] [PubMed] [Google Scholar]
- 74.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208(3):417–20. Epub 2011/02/28. doi: 10.1084/jem.20110367. PubMed PMID: 21357740; PubMed Central PMCID: PMCPMC3058577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–5. Epub 2010/02/19. doi: 10.1038/nri2725. PubMed PMID: 20168318. [DOI] [PubMed] [Google Scholar]
- 76.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261(7):3060–7. PubMed PMID: 3456345. [PubMed] [Google Scholar]
- 77.Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440–6. doi: 10.1097/FPC.0b013e32833ffb56. PubMed PMID: 21048526; PubMed Central PMCID: PMCPMC3116111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chaiswing L, Cole MP, Ittarat W, Szweda LI, St Clair DK, Oberley TD. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol Cancer Ther. 2005;4(7):1056–64. doi: 10.1158/1535-7163.MCT-04-0322. PubMed PMID: 16020663. [DOI] [PubMed] [Google Scholar]
- 79.Yen HC, Oberley TD, Gairola CG, Szweda LI, St Clair DK. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch Biochem Biophys. 1999;362(1):59–66. doi: 10.1006/abbi.1998.1011. PubMed PMID: 9917329. [DOI] [PubMed] [Google Scholar]
- 80.Fleenor BS, Seals DR, Zigler ML, Sindler AL. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell. 2012;11(2):269–76. Epub 2012/01/19. doi: 10.1111/j.1474-9726.2011.00783.x. PubMed PMID: 22168264; PubMed Central PMCID: PMCPMC3409251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension. 2018;71(6):1056–63. Epub 2018/04/16. doi: 10.1161/HYPERTENSIONAHA.117.10787. PubMed PMID: 29661838; PubMed Central PMCID: PMCPMC5945293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rossman MJ, Gioscia-Ryan RA, Clayton ZS, Murphy MP, Seals DR. Targeting mitochondrial fitness as a strategy for healthy vascular aging. Clin Sci (Lond). 2020;134(12):1491–519. doi: 10.1042/CS20190559. PubMed PMID: 32584404. [DOI] [PubMed] [Google Scholar]
- 83.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126–67. Epub 2013/10/22. doi: 10.1089/ars.2012.5149. PubMed PMID: 23991888; PubMed Central PMCID: PMCPMC3929010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hutton D, Brunt V, Casso A, Ziemba B, Seals D, Clayton ZS, editors. Increased Large Elastic Artery Stiffening with The Anthracycline Chemotherapy Drug Doxorubicin: Potential Role of Excess Mitochondrial Superoxide. Experimental Biology; 2020. [Google Scholar]
- 85.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, et al. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30(7):1019–26. Epub 2010/05/18. doi: 10.1111/j.1478-3231.2010.02250.x. PubMed PMID: 20492507. [DOI] [PubMed] [Google Scholar]
- 86.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord. 2010;25(11):1670–4. doi: 10.1002/mds.23148. PubMed PMID: 20568096. [DOI] [PubMed] [Google Scholar]
- 87.Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006;20(11):1889–91. Epub 2006/08/04. doi: 10.1096/fj.05-5189fje. PubMed PMID: 16891621. [DOI] [PubMed] [Google Scholar]
- 88.Sarti P, Arese M, Bacchi A, Barone MC, Forte E, Mastronicola D, et al. Nitric oxide and mitochondrial complex IV. IUBMB Life. 2003;55(10-11):605–11. doi: 10.1080/15216540310001628726. PubMed PMID: 14711006. [DOI] [PubMed] [Google Scholar]
- 89.Ballinger SW, Patterson C, Yan CN, Doan R, Burow DL, Young CG, et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. 2000;86(9):960–6. doi: 10.1161/01.res.86.9.960. PubMed PMID: 10807868. [DOI] [PubMed] [Google Scholar]
- 90.Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54(4):469–87. PubMed PMID: 14726604. [PubMed] [Google Scholar]
- 91.Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim Biophys Acta. 2006;1760(2):267–71. Epub 2005/11/28. doi: 10.1016/j.bbagen.2005.10.012. PubMed PMID: 16337743. [DOI] [PubMed] [Google Scholar]
- 92.Wojcik T, Buczek E, Majzner K, Kolodziejczyk A, Miszczyk J, Kaczara P, et al. Comparative endothelial profiling of doxorubicin and daunorubicin in cultured endothelial cells. Toxicol In Vitro. 2015;29(3):512–21. Epub 2014/12/18. doi: 10.1016/j.tiv.2014.12.009. PubMed PMID: 25529801. [DOI] [PubMed] [Google Scholar]
- 93.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40. doi: 10.1038/nrm2233. PubMed PMID: 17667954. [DOI] [PubMed] [Google Scholar]
- 94.Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40(8-9):634–42. doi: 10.1016/j.exger.2005.04.010. PubMed PMID: 15970413. [DOI] [PubMed] [Google Scholar]
- 95.Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7(2):165–76. Epub 2016/12/15. doi: 10.1158/2159-8290.CD-16-0241. PubMed PMID: 27979832; PubMed Central PMCID: PMCPMC5296251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515(7525):134–7. Epub 2014/08/24. doi: 10.1038/nature13638. PubMed PMID: 25156255. [DOI] [PubMed] [Google Scholar]
- 97.Tian XL, Li Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics. 2014;41(9):485–95. Epub 2014/08/14. doi: 10.1016/j.jgg.2014.08.001. PubMed PMID: 25269674. [DOI] [PubMed] [Google Scholar]
- 98.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72(1):9–17. Epub 2006/06/06. doi: 10.1016/j.cardiores.2006.06.004. PubMed PMID: 16824498. [DOI] [PubMed] [Google Scholar]
- 99.Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1):e3000599. Epub 2020/01/16. doi: 10.1371/journal.pbio.3000599. PubMed PMID: 31945054; PubMed Central PMCID: PMCPMC6964821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313(5):H890–H5. Epub 2017/09/29. doi: 10.1152/ajpheart.00416.2017. PubMed PMID: 28971843; PubMed Central PMCID: PMCPMC5792201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mahoney SA, Hutton DA, Rossman MJ, Brunt VE, VanDongen NS, Casso AG, et al. Late-life treatment with the senolytic ABT-263 reverses aortic stiffening and improves endothelial function with aging. FASEB J 2021. 35:S1. [Google Scholar]
- 102.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–7. Epub 2016/08/05. doi: 10.1111/acel.12458. PubMed PMID: 26864908; PubMed Central PMCID: PMCPMC5013022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hutton DA, Brunt VE, Mahoney SA, Casso AG, Greenberg NT, VanDongen NS, et al. Cellular senescence mediates doxorubicin-induced arterial dysfunction via activation of mitochondrial oxidative stress and the mammalian target of rapamycin. FASEB J 2021. 35:S1. [Google Scholar]
- 104.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130(6):1095–107. doi: 10.1016/j.cell.2007.07.035. PubMed PMID: 17889652; PubMed Central PMCID: PMCPMC3366687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schultz MB, Sinclair DA. Why NAD(+) Declines during Aging: It's Destroyed. Cell Metab. 2016;23(6):965–6. doi: 10.1016/j.cmet.2016.05.022. PubMed PMID: 27304496; PubMed Central PMCID: PMCPMC5088772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Das A, Huang GX, Bonkowski MS, Longchamp A, Li C, Schultz MB, et al. Impairment of an Endothelial NAD. Cell. 2018;173(1):74–89.e20. doi: 10.1016/j.cell.2018.02.008. PubMed PMID: 29570999; PubMed Central PMCID: PMCPMC5884172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Csiszar A, Tarantini S, Yabluchanskiy A, Balasubramanian P, Kiss T, Farkas E, et al. Role of endothelial NAD. Am J Physiol Heart Circ Physiol. 2019;316(6):H1253–H66. Epub 2019/03/15. doi: 10.1152/ajpheart.00039.2019. PubMed PMID: 30875255; PubMed Central PMCID: PMCPMC6620681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016;23(2):303–14. Epub 2015/12/10. doi: 10.1016/j.cmet.2015.11.011. PubMed PMID: 26686024; PubMed Central PMCID: PMCPMC4749409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amjad S, Nisar S, Bhat AA, Shah AR, Frenneaux MP, Fakhro K, et al. Role of NAD. Mol Metab. 2021:101195. Epub 2021/02/17. doi: 10.1016/j.molmet.2021.101195. PubMed PMID: 33609766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. PubMed PMID: 20078221; PubMed Central PMCID: PMCPMC2866163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA, Seals DR. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol. 2011;589(Pt 18):4545–54. Epub 2011/07/11. doi: 10.1113/jphysiol.2011.211219. PubMed PMID: 21746786; PubMed Central PMCID: PMCPMC3208223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009;46(12):1589–97. Epub 2009/03/19. doi: 10.1016/j.freeradbiomed.2009.03.011. PubMed PMID: 19303434. [DOI] [PubMed] [Google Scholar]
- 113.Ruan Y, Dong C, Patel J, Duan C, Wang X, Wu X, et al. SIRT1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell Physiol Biochem. 2015;35(3):1116–24. Epub 2015/02/06. doi: 10.1159/000373937. PubMed PMID: 25766524. [DOI] [PubMed] [Google Scholar]
- 114.Wu YZ, Zhang L, Wu ZX, Shan TT, Xiong C. Berberine Ameliorates Doxorubicin-Induced Cardiotoxicity via a SIRT1/p66Shc-Mediated Pathway. Oxid Med Cell Longev. 2019;2019:2150394. Epub 2019/12/06. doi: 10.1155/2019/2150394. PubMed PMID: 31885776; PubMed Central PMCID: PMCPMC6918936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Angelis A, Piegari E, Cappetta D, Russo R, Esposito G, Ciuffreda LP, et al. SIRT1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int J Cardiol. 2015;189:30–44. Epub 2015/04/02. doi: 10.1016/j.ijcard.2015.03.438. PubMed PMID: 25889431. [DOI] [PubMed] [Google Scholar]
- 116.Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010;9(3):304–12. Epub 2010/03/13. doi: 10.1111/j.1474-9726.2010.00557.x. PubMed PMID: 20121721; PubMed Central PMCID: PMCPMC2894368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Donato AJ, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, et al. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. 2013;12(5):772–83. Epub 2013/07/02. doi: 10.1111/acel.12103. PubMed PMID: 23714110; PubMed Central PMCID: PMCPMC3772986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ness KK, Armstrong GT, Kundu M, Wilson CL, Tchkonia T, Kirkland JL. Frailty in childhood cancer survivors. Cancer. 2015;121(10):1540–7. Epub 2014/12/19. doi: 10.1002/cncr.29211. PubMed PMID: 25529481; PubMed Central PMCID: PMCPMC4424063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Krnavek NJ, Ajasin S, Arreola EC, Zahiri M, Noun M, Lupo PJ, et al. Sensor-Based Frailty Assessment in Survivors of Childhood Cancer: A Pilot Study. J Frailty Aging. 2021;10(2):176–81. doi: 10.14283/jfa.2020.71. PubMed PMID: 33575708. [DOI] [PubMed] [Google Scholar]
- 120.Hayek S, Gibson TM, Leisenring WM, Guida JL, Gramatges MM, Lupo PJ, et al. Prevalence and Predictors of Frailty in Childhood Cancer Survivors and Siblings: A Report From the Childhood Cancer Survivor Study. J Clin Oncol. 2020;38(3):232–47. Epub 2019/12/04. doi: 10.1200/JCO.19.01226. PubMed PMID: 31800343; PubMed Central PMCID: PMCPMC6968796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martens CR, Seals DR. Practical alternatives to chronic caloric restriction for optimizing vascular function with ageing. J Physiol. 2016;594(24):7177–95. Epub 2016/11/29. doi: 10.1113/JP272348. PubMed PMID: 27641062; PubMed Central PMCID: PMCPMC5157076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15(3):522–30. Epub 2016/03/11. doi: 10.1111/acel.12461. PubMed PMID: 26970090; PubMed Central PMCID: PMCPMC4854911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD. Nat Commun. 2018;9(1):1286. Epub 2018/03/29. doi: 10.1038/s41467-018-03421-7. PubMed PMID: 29599478; PubMed Central PMCID: PMCPMC5876407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gano LB, Donato AJ, Pasha HM, Hearon CM, Sindler AL, Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307(12):H1754–63. Epub 2014/10/17. doi: 10.1152/ajpheart.00377.2014. PubMed PMID: 25326534; PubMed Central PMCID: PMCPMC4269699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim EN, Kim MY, Lim JH, Kim Y, Shin SJ, Park CW, et al. The protective effect of resveratrol on vascular aging by modulation of the renin-angiotensin system. Atherosclerosis. 2018;270:123–31. Epub 2018/02/02. doi: 10.1016/j.atherosclerosis.2018.01.043. PubMed PMID: 29407880. [DOI] [PubMed] [Google Scholar]
- 126.Liu MH, Shan J, Li J, Zhang Y, Lin XL. Resveratrol inhibits doxorubicin-induced cardiotoxicity via sirtuin 1 activation in H9c2 cardiomyocytes. Exp Ther Med. 2016;12(2):1113–8. Epub 2016/06/08. doi: 10.3892/etm.2016.3437. PubMed PMID: 27446329; PubMed Central PMCID: PMCPMC4950526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Olukman M, Can C, Erol A, Oktem G, Oral O, Cinar MG. Reversal of doxorubicin-induced vascular dysfunction by resveratrol in rat thoracic aorta: Is there a possible role of nitric oxide synthase inhibition? Anadolu Kardiyol Derg. 2009;9(4):260–6. PubMed PMID: 19666426. [PubMed] [Google Scholar]
- 128.Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23(6):1127–39. doi: 10.1016/j.cmet.2016.05.006. PubMed PMID: 27304511; PubMed Central PMCID: PMCPMC4911708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Damiani RM, Moura DJ, Viau CM, Brito V, Morás AM, Henriques JAP, et al. Influence of PARP-1 inhibition in the cardiotoxicity of the topoisomerase 2 inhibitors doxorubicin and mitoxantrone. Toxicol In Vitro. 2018;52:203–13. Epub 2018/06/15. doi: 10.1016/j.tiv.2018.06.013. PubMed PMID: 29913208. [DOI] [PubMed] [Google Scholar]
- 130.Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG, et al. Senescent cells promote tissue NAD. Nat Metab. 2020;2(11):1265–83. Epub 2020/11/16. doi: 10.1038/s42255-020-00305-3. PubMed PMID: 33199924; PubMed Central PMCID: PMCPMC7908681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nikas IP, Paschou SA, Ryu HS. The Role of Nicotinamide in Cancer Chemoprevention and Therapy. Biomolecules. 2020;10(3). Epub 2020/03/20. doi: 10.3390/biom10030477. PubMed PMID: 32245130; PubMed Central PMCID: PMCPMC7175378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang S, Han L, Han J, Li P, Ding Q, Zhang QJ, et al. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat Chem Biol. 2019;15(12):1223–31. Epub 2019/10/28. doi: 10.1038/s41589-019-0379-2. PubMed PMID: 31659317; PubMed Central PMCID: PMCPMC6864272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, et al. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21(13):3629–39. Epub 2007/06/21. doi: 10.1096/fj.07-8290com. PubMed PMID: 17585054. [DOI] [PubMed] [Google Scholar]
- 134.Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62(4):1084–93. Epub 2012/11/19. doi: 10.2337/db12-1139. PubMed PMID: 23172919; PubMed Central PMCID: PMCPMC3609577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Oberle A, Brandt A, Alawi M, Langebrake C, Janjetovic S, Wolschke C, et al. Long-term CD38 saturation by daratumumab interferes with diagnostic myeloma cell detection. Haematologica. 2017;102(9):e368–e70. Epub 2017/05/18. doi: 10.3324/haematol.2017.169235. PubMed PMID: 28522580; PubMed Central PMCID: PMCPMC5685239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Clayton ZS, Hutton DA, Brunt VE, VanDongen NS, Ziemba BP, Casso AG, et al. Apigenin restores endothelial function by ameliorating oxidative stress, prevents foam cell formation, reverses aortic stiffening, and mitigates vascular inflammation with aging. FASEB J 2021. 35:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158(6):1324–34. doi: 10.1016/j.cell.2014.07.040. PubMed PMID: 25215490; PubMed Central PMCID: PMCPMC4163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–35. Epub 2017/10/04. doi: 10.1038/nrm.2017.95. PubMed PMID: 28974774; PubMed Central PMCID: PMCPMC5780224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–76. doi: 10.1016/j.cell.2017.02.004. PubMed PMID: 28283069; PubMed Central PMCID: PMCPMC5394987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li T, Mu N, Yin Y, Yu L, Ma H. Targeting AMP-Activated Protein Kinase in Aging-Related Cardiovascular Diseases. Aging Dis. 2020;11(4):967–77. Epub 2020/07/23. doi: 10.14336/AD.2019.0901. PubMed PMID: 32765957; PubMed Central PMCID: PMCPMC7390518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang BZ, Yang JJ, Zhang H, Smith CA, Jin K. AMPK Signaling Regulates the Age-Related Decline of Hippocampal Neurogenesis. Aging Dis. 2019;10(5):1058–74. Epub 2019/10/01. doi: 10.14336/AD.2019.0102. PubMed PMID: 31595203; PubMed Central PMCID: PMCPMC6764723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–41. Epub 2011/12/15. doi: 10.1016/j.arr.2011.12.005. PubMed PMID: 22186033. [DOI] [PubMed] [Google Scholar]
- 143.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–45. doi: 10.1038/nature11861. PubMed PMID: 23325216; PubMed Central PMCID: PMCPMC3687363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stallone G, Infante B, Prisciandaro C, Grandaliano G. mTOR and Aging: An Old Fashioned Dress. Int J Mol Sci. 2019;20(11). Epub 2019/06/06. doi: 10.3390/ijms20112774. PubMed PMID: 31174250; PubMed Central PMCID: PMCPMC6600378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lesniewski LA, Zigler MC, Durrant JR, Donato AJ, Seals DR. Sustained activation of AMPK ameliorates age-associated vascular endothelial dysfunction via a nitric oxide-independent mechanism. Mech Ageing Dev. 2012;133(5):368–71. Epub 2012/03/28. doi: 10.1016/j.mad.2012.03.011. PubMed PMID: 22484146; PubMed Central PMCID: PMCPMC3359767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lesniewski LA, Seals DR, Walker AE, Henson GD, Blimline MW, Trott DW, et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell. 2017;16(1):17–26. Epub 2016/09/22. doi: 10.1111/acel.12524. PubMed PMID: 27660040; PubMed Central PMCID: PMCPMC5242306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, et al. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006;26(6):1281–7. Epub 2006/04/06. doi: 10.1161/01.ATV.0000221230.08596.98. PubMed PMID: 16601232. [DOI] [PubMed] [Google Scholar]
- 148.Ford RJ, Teschke SR, Reid EB, Durham KK, Kroetsch JT, Rush JW. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. J Hypertens. 2012;30(4):725–33. doi: 10.1097/HJH.0b013e32835050ca. PubMed PMID: 22306847. [DOI] [PubMed] [Google Scholar]
- 149.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–5. Epub 2009/07/08. doi: 10.1038/nature08221. PubMed PMID: 19587680; PubMed Central PMCID: PMCPMC2786175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014;69(2):119–30. Epub 2013/05/16. doi: 10.1093/gerona/glt056. PubMed PMID: 23682161; PubMed Central PMCID: PMCPMC4038246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016;5. Epub 2016/08/23. doi: 10.7554/eLife.16351. PubMed PMID: 27549339; PubMed Central PMCID: PMCPMC4996648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Singh AK, Singh S, Garg G, Rizvi SI. Rapamycin alleviates oxidative stress-induced damage in rat erythrocytes. Biochem Cell Biol. 2016;94(5):471–9. Epub 2016/06/06. doi: 10.1139/bcb-2016-0048. PubMed PMID: 27633009. [DOI] [PubMed] [Google Scholar]
- 153.Jiang J, Zuo Y, Gu Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson's disease. Int J Mol Med. 2013;31(4):825–32. Epub 2013/02/20. doi: 10.3892/ijmm.2013.1280. PubMed PMID: 23426728. [DOI] [PubMed] [Google Scholar]
- 154.Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem. 2014;289(7):4145–60. Epub 2013/12/26. doi: 10.1074/jbc.M113.521062. PubMed PMID: 24371138; PubMed Central PMCID: PMCPMC3924280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–61. Epub 2015/07/06. doi: 10.1038/ncb3195. PubMed PMID: 26147250; PubMed Central PMCID: PMCPMC4691706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lesniewski LA, Zigler ML, Durrant JR, Nowlan MJ, Folian BJ, Donato AJ, et al. Aging compounds western diet-associated large artery endothelial dysfunction in mice: prevention by voluntary aerobic exercise. Exp Gerontol. 2013;48(11):1218–25. Epub 2013/08/13. doi: 10.1016/j.exger.2013.08.001. PubMed PMID: 23954368; PubMed Central PMCID: PMCPMC3840721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gioscia-Ryan RA, Battson ML, Cuevas LM, Zigler MC, Sindler AL, Seals DR. Voluntary aerobic exercise increases arterial resilience and mitochondrial health with aging in mice. Aging (Albany NY). 2016;8(11):2897–914. doi: 10.18632/aging.101099. PubMed PMID: 27875805; PubMed Central PMCID: PMCPMC5191877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen XK, Yi ZN, Wong GT, Hasan KMM, Kwan JS, Ma AC, et al. Is exercise a senolytic medicine? A systematic review. Aging Cell. 2021;20(1):e13294. Epub 2020/12/30. doi: 10.1111/acel.13294. PubMed PMID: 33378138; PubMed Central PMCID: PMCPMC7811843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.White AT, Schenk S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am J Physiol Endocrinol Metab. 2012;303(3):E308–21. Epub 2012/03/20. doi: 10.1152/ajpendo.00054.2012. PubMed PMID: 22436696; PubMed Central PMCID: PMCPMC3423123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tarragó MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD. Cell Metab. 2018;27(5):1081–95.e10. doi: 10.1016/j.cmet.2018.03.016. PubMed PMID: 29719225; PubMed Central PMCID: PMCPMC5935140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gibson NM, Greufe SE, Hydock DS, Hayward R. Doxorubicin-induced vascular dysfunction and its attenuation by exercise preconditioning. J Cardiovasc Pharmacol. 2013;62(4):355–60. doi: 10.1097/FJC.0b013e31829c9993. PubMed PMID: 23719093. [DOI] [PubMed] [Google Scholar]
- 162.Lee K, Kang I, Mack WJ, Mortimer J, Sattler F, Salem G, et al. Effects of high-intensity interval training on vascular endothelial function and vascular wall thickness in breast cancer patients receiving anthracycline-based chemotherapy: a randomized pilot study. Breast Cancer Res Treat. 2019;177(2):477–85. Epub 2019/06/24. doi: 10.1007/s10549-019-05332-7. PubMed PMID: 31236810; PubMed Central PMCID: PMCPMC6661195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Long TM, Rath SR, Wallman KE, Howie EK, Straker LM, Bullock A, et al. Exercise training improves vascular function and secondary health measures in survivors of pediatric oncology related cerebral insult. PLoS One. 2018;13(8):e0201449. Epub 2018/08/09. doi: 10.1371/journal.pone.0201449. PubMed PMID: 30092052; PubMed Central PMCID: PMCPMC6084859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Petsko GA. A seat at the table. Genome Biol. 2008;9(12):113. doi: 10.1186/gb-2008-9-12-113. PubMed PMID: 19144208; PubMed Central PMCID: PMCPMC2646288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Harper S. Economic and social implications of aging societies. Science. 2014;346(6209):587–91. doi: 10.1126/science.1254405. PubMed PMID: 25359967. [DOI] [PubMed] [Google Scholar]
- 166.DePinho RA. The age of cancer. Nature. 2000;408(6809):248–54. doi: 10.1038/35041694. PubMed PMID: 11089982. [DOI] [PubMed] [Google Scholar]
- 167.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933–44. Epub 2011/01/24. doi: 10.1161/CIR.0b013e31820a55f5. PubMed PMID: 21262990. [DOI] [PubMed] [Google Scholar]
- 168.Ha H, Keam B, Kim TM, Jeon YK, Lee SH, Kim DW, et al. Reduced Dose Intensities of Doxorubicin in Elderly Patients with DLBCL in Rituximab Era. Cancer Res Treat. 2016;48(1):304–11. Epub 2015/04/09. doi: 10.4143/crt.2014.339. PubMed PMID: 25865654; PubMed Central PMCID: PMCPMC4720063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reddy P, Shenoy C, Blaes AH. Cardio-oncology in the older adult. J Geriatr Oncol. 2017;8(4):308–14. Epub 2017/05/09. doi: 10.1016/j.jgo.2017.04.001. PubMed PMID: 28499724; PubMed Central PMCID: PMCPMC5776715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carver JR, Schuster SJ, Glick JH. Doxorubicin cardiotoxicity in the elderly: old drugs and new opportunities. J Clin Oncol. 2008;26(19):3122–4. doi: 10.1200/JCO.2008.16.5274. PubMed PMID: 18591552. [DOI] [PubMed] [Google Scholar]
- 171.Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2017;35(8):893–911. Epub 2016/12/05. doi: 10.1200/JCO.2016.70.5400. PubMed PMID: 27918725. [DOI] [PubMed] [Google Scholar]
- 172.Bansal N, Adams MJ, Ganatra S, Colan SD, Aggarwal S, Steiner R, et al. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology. 2019;5:18. Epub 2019/12/02. doi: 10.1186/s40959-019-0054-5. PubMed PMID: 32154024; PubMed Central PMCID: PMCPMC7048046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zheng H, Dimayuga C, Hudaihed A, Katz SD. Effect of dexrazoxane on homocysteine-induced endothelial dysfunction in normal subjects. Arterioscler Thromb Vasc Biol. 2002;22(7):E15–8. doi: 10.1161/01.atv.0000023187.25914.5b. PubMed PMID: 12117747. [DOI] [PubMed] [Google Scholar]
- 174.Chiao YA, Zhang H, Sweetwyne M, Whitson J, Ting YS, Basisty N, et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife. 2020;9. Epub 2020/07/10. doi: 10.7554/eLife.55513. PubMed PMID: 32648542; PubMed Central PMCID: PMCPMC7377906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hou Y, Li S, Wu M, Wei J, Ren Y, Du C, et al. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am J Physiol Renal Physiol. 2016;310(6):F547–59. Epub 2015/12/30. doi: 10.1152/ajprenal.00574.2014. PubMed PMID: 26719366. [DOI] [PubMed] [Google Scholar]
- 176.Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Fulop GA, Hertelendy P, Gautam T, et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17(2). Epub 2018/02/06. doi: 10.1111/acel.12731. PubMed PMID: 29405550; PubMed Central PMCID: PMCPMC5847870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879–88. Epub 2016/07/11. doi: 10.1038/nm.4132. PubMed PMID: 27400265. [DOI] [PubMed] [Google Scholar]
- 178.Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1(6):595–603. Epub 2019/06/14. doi: 10.1038/s42255-019-0073-4. PubMed PMID: 32694802. [DOI] [PubMed] [Google Scholar]
- 179.Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. Epub 2018/09/29. doi: 10.1016/j.ebiom.2018.09.015. PubMed PMID: 30279143; PubMed Central PMCID: PMCPMC6197652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc. 2017;65(10):2297–301. Epub 2017/09/04. doi: 10.1111/jgs.14969. PubMed PMID: 28869295; PubMed Central PMCID: PMCPMC5641223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–63. Epub 2019/01/05. doi: 10.1016/j.ebiom.2018.12.052. PubMed PMID: 30616998; PubMed Central PMCID: PMCPMC6412088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–35. Epub 2016/03/18. doi: 10.1111/acel.12445. PubMed PMID: 26711051; PubMed Central PMCID: PMCPMC4854923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83. Epub 2015/12/14. doi: 10.1038/nm.4010. PubMed PMID: 26657143; PubMed Central PMCID: PMCPMC4762215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yabluchanskiy A, Tarantini S, Balasubramanian P, Kiss T, Csipo T, Fülöp GA, et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience. 2020;42(2):409–28. Epub 2020/01/20. doi: 10.1007/s11357-020-00154-8. PubMed PMID: 31960269; PubMed Central PMCID: PMCPMC7205933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798–806. doi: 10.1038/nprot.2009.191. PubMed PMID: 20010931. [DOI] [PubMed] [Google Scholar]
- 186.González-Gualda E, Pàez-Ribes M, Lozano-Torres B, Macias D, Wilson JR, González-López C, et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell. 2020;19(4):e13142. Epub 2020/03/31. doi: 10.1111/acel.13142. PubMed PMID: 32233024; PubMed Central PMCID: PMCPMC7189993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Troiano RP, Berrigan D, Dodd KW, Mâsse LC, Tilert T, McDowell M. Physical activity in the United States measured by accelerometer. Med Sci Sports Exerc. 2008;40(1):181–8. doi: 10.1249/mss.0b013e31815a51b3. PubMed PMID: 18091006. [DOI] [PubMed] [Google Scholar]
- 188.Craighead DH, Heinbockel TC, Rossman MJ, Jankowski LR, Jackman RA, Bailey EF, et al. Inspiratory muscle strength training lowers resting systolic blood pressure and improves vascular endothelial function in middle-aged and older adults. FASEB J 2019. p. 541.4. [Google Scholar]
- 189.Zhang XJ, Cao XQ, Zhang CS, Zhao Z. 17β-estradiol protects against doxorubicin-induced cardiotoxicity in male Sprague-Dawley rats by regulating NADPH oxidase and apoptosis genes. Mol Med Rep. 2017;15(5):2695–702. Epub 2017/03/16. doi: 10.3892/mmr.2017.6332. PubMed PMID: 28447737. [DOI] [PubMed] [Google Scholar]
- 190.Pokrzywinski KL, Biel TG, Rosen ET, Bonanno JL, Aryal B, Mascia F, et al. Doxorubicin-induced cardiotoxicity is suppressed by estrous-staged treatment and exogenous 17β-estradiol in female tumor-bearing spontaneously hypertensive rats. Biol Sex Differ. 2018;9(1):25. Epub 2018/06/15. doi: 10.1186/s13293-018-0183-9. PubMed PMID: 29907135; PubMed Central PMCID: PMCPMC6003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cadeddu Dessalvi C, Pepe A, Penna C, Gimelli A, Madonna R, Mele D, et al. Sex differences in anthracycline-induced cardiotoxicity: the benefits of estrogens. Heart Fail Rev. 2019;24(6):915–25. doi: 10.1007/s10741-019-09820-2. PubMed PMID: 31256318. [DOI] [PubMed] [Google Scholar]
- 192.Kappeler CJ, Hoyer PB. 4-vinylcyclohexene diepoxide: a model chemical for ovotoxicity. Syst Biol Reprod Med. 2012;58(1):57–62. doi: 10.3109/19396368.2011.648820. PubMed PMID: 22239082; PubMed Central PMCID: PMCPMC3307534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Brooks HL, Pollow DP, Hoyer PB. The VCD Mouse Model of Menopause and Perimenopause for the Study of Sex Differences in Cardiovascular Disease and the Metabolic Syndrome. Physiology (Bethesda). 2016;31(4):250–7. doi: 10.1152/physiol.00057.2014. PubMed PMID: 27252160; PubMed Central PMCID: PMCPMC5504385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pollow DP, Romero-Aleshire MJ, Sanchez JN, Konhilas JP, Brooks HL. ANG II-induced hypertension in the VCD mouse model of menopause is prevented by estrogen replacement during perimenopause. Am J Physiol Regul Integr Comp Physiol. 2015;309(12):R1546–52. Epub 2015/10/21. doi: 10.1152/ajpregu.00170.2015. PubMed PMID: 26491098; PubMed Central PMCID: PMCPMC4698416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Perez JN, Chen H, Regan JA, Emert A, Constantopoulos E, Lynn M, et al. Effects of chemically induced ovarian failure on voluntary wheel-running exercise and cardiac adaptation in mice. Comp Med. 2013;63(3):233–43. PubMed PMID: 23759526; PubMed Central PMCID: PMCPMC3690429. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data will be made available following a reasonable request to the corresponding author.