

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive disease with the lowest survival rate among all solid tumors. The lethality of PDAC arises from late detection and propensity of the tumor to metastasize and develop resistance against chemo and radiation therapy. A highly complex tumor microenvironment composed of dense stroma, immune cells, fibroblast, and disorganized blood vessels, is the main obstacle to current PDAC therapy. Despite the tremendous success of immune checkpoint inhibitors (ICIs) in cancers, PDAC remains one of the poorest responders of ICIs therapy. The immunologically “cold” phenotype of PDAC is attributed to the low mutational burden, high infiltration of myeloid-derived suppressor cells and T-regs, contributing to a significant immunotherapy resistance mechanism. Thus, the development of innovative strategies for turning immunologically “cold” tumor into “hot” ones is an unmet need to improve the outcome of PDAC ICIs therapies. Other smart strategies, such as nanomedicines, sonic Hedgehog inhibitor, or smoothened inhibitor, are discussed to enhance chemotherapeutic agents’ efficiency by disrupting the PDAC stroma. This review highlights the current challenges and various preclinical and clinical strategies to overcome current PDAC therapy difficulties, thus significantly advancing PDAC research knowledge.
Keywords: Pancreatic cancer, Tumor microenvironments, Nanomedicine, Stroma, Immune checkpoints inhibitors
Graphical Abstract
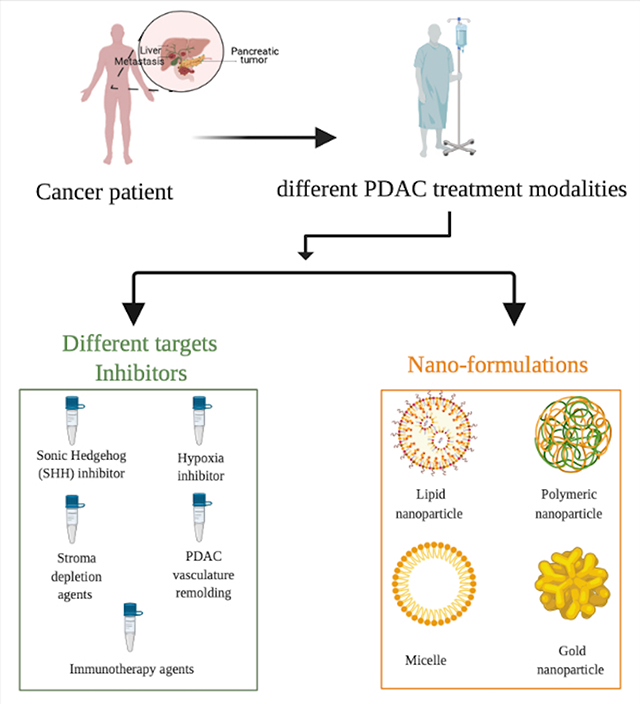
The current work highlights the challenges that play a crucial role in minimizing the therapy efficiencies of pancreatic ductal adenocarcinomas. The potential strategies that can mitigate the challenges in therapy and diagnosis such as utilizing novel targets and nanomedicines are discussed. Together, these methods have potential to improve the overall treatment efficacy and survival rates of patients with pancreatic ductal adenocarcinoma.
1. Introduction
In 2020, pancreatic cancer (PC) was the fourth leading cause of cancer-related death in the US. The 5-year survival rate of pancreatic cancer is the lowest among other solid cancers, 7% [1]. Within PCs, 90% accounts for pancreatic ductal adenocarcinoma (PDAC) [2]. Many risk factors are associated with PDAC, such as smoking, family history, and genetic diseases. In PDAC, poor prognosis is the main challenge due to the tumor’s propensity to metastasize and develop resistance against chemo and radiation therapy [3]. One of the main challenges is dense stroma produced by the mucin-producing gland [4]. Stroma is one of the major tumor microenvironment components that draw attention to researchers due to its role in patients’ poor prognosis [5]. Thus, the presence of stromal components in PDAC elevates the interstitial fluid pressure (IFP), leading to vasoconstriction that in turn impedes the chemotherapeutic delivery efficiency [6,7]. In recent years, researchers have begun to focus on finding new ways to better understanding PDAC’s genetics and biology to improve therapeutic outcomes. Biologically, desmoplastic reaction (DR) is one of the hallmarks of PDAC that contributes to PDAC’s poor prognosis. The dense desmoplasia plays a major role in distorting the normal architecture of pancreatic tissue and blood perfusion; thus, drug penetration decreases, and resistance increases [8]. Tumor hypoxia or low oxygen levels and hypovascular environment is another obstacle that reduces drug delivery efficiency to the tumor site [9]. The vasculature within the PDAC tumor microenvironment (TME) is obscured or obstructed by the tumor-associated fibroblast or pericytes [10]. Therefore, cells that grow within these harsh conditions are resistant to most chemotherapeutic agents and radiotherapy [11]. Within TME, the absence of immune surveillance and inflammatory cells supports aggressiveness and tumorigenesis of PDAC [12]. The signs of genetic alteration in PDAC have been studied extensively; Kristen rat sarcoma viral oncogene homolog (KRAS) mutation and CDKN2A alteration are found to be one of the early events in low-grade pancreatic intraepithelial neoplasia (Figure 1A) [13,14]. On the other hand, alteration of P53 and loss of Smad4 was found to be one of the late events that occur in intraepithelial neoplasia grade 3 (Figure 1A) [13].
Figure 1:
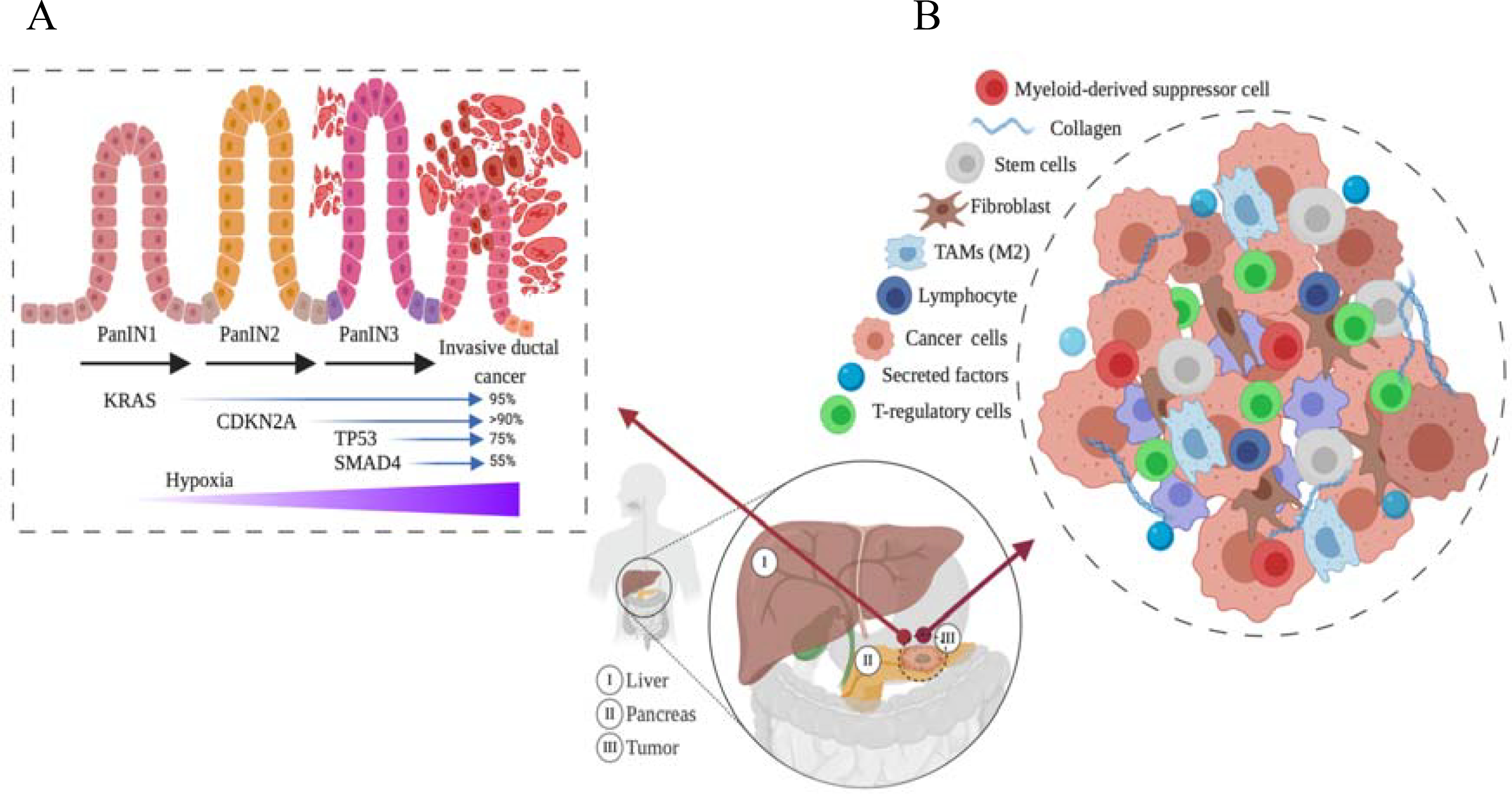
Pancreatic ductal adenocarcinoma (PDAC) stages and tumor microenvironments (TME). (A) different stages of PDAC, and the expression of oncogenes at each stage is shown. (B) The complexity of TME components that attenuate cytotoxic drug penetration is shown.
Several approaches, including immunotherapy, have been studied in patients with pancreatic cancers; nevertheless, most of them have not shown any clinically meaningful outcome noted in other malignancies [15]. Moreover, the complexity of PDAC tumor stroma composition attenuates most PDAC therapies due to poor drug penetration through the stroma barrier. Thus, new immunotherapy approaches need to be established to trigger a potent immune response against tumors. Also, nanoparticles approach is another way to overcome different PDAC barriers through its ability to accumulate in the tumor site [16,17]. Nanomedicine such as liposome and polymeric nanoparticles can overcome additional biological obstacles via protecting encapsulated payloads from degradation in blood circulation, and lowering their accumulation in non-target organs and tissues, thus minimizing the toxicity to normal cells and tissues. Nanoparticles can be designed to improve the delivery efficiency to target tissues, which lowers the needed therapeutic dose. Moreover, nanomedicines can minimize immunogenicity and prolong circulation time [18]. Therefore, nanoparticles provide a viable solution to improve current PDAC therapy’s efficiency. So far, in vitro and in vivo data of nanoparticles that have been tested on PDAC tumors showed promising results. In this review, the current PDAC challenges will be covered. The different ways that clinicians and researchers currently use varying approaches to overcome these challenges and improve the overall disease response will be discussed.
2. Current PDAC therapy
One of the most critical challenges of the PDAC treatment is the tumor microenvironment characterized by constantly changing morphology and genetics that contribute to its aggressiveness [19]. The PDAC tumor microenvironment is also characterized by desmoplasia (dense stromal cells) that adds to the resistance to conventional therapies. However, significant improvements have been made in the treatment strategies of the PDAC, which include monotherapy such as gemcitabine, multidrug regimen like the FOLFIRINOX, and combination therapies (gemcitabine plus adjuvant/neoadjuvant therapies/immune checkpoint inhibitors) (Table 1,2). The following are some of the novel therapeutic strategies for PDAC that have been developed based on the stage of cancer and the resistance mechanisms involved at that stage of the disease.
Table 1:
Clinical trials of Drug delivery in PDAC (Gemcitabine based therapies)
| Study Title | Phase | NCT number |
|---|---|---|
| Gene Therapy of Pancreatic Ductal Adenocarcinoma | Phase 1 | NCT01274455 |
| Study to Investigate the Preliminary Efficacy and Safety of INNO-206 in Advanced Pancreatic Cancer | Phase 2 | NCT01580397 |
| Study of siG12D LODER in Combination with Chemotherapy in Patients with Locally Advanced Pancreatic Cancer | Phase 2 | NCT01676259 |
| Atu027 Plus Gemcitabine in Advanced or Metastatic Pancreatic Cancer (Atu027-I-02) | Phase 1/2 | NCT01808638 |
| Study of Gemcitabine, Abraxane® Plus Placebo Versus Gemcitabine, Abraxane® Plus 1 or 2 Truncated Courses of Demcizumab in Subjects with 1st-Line Metastatic Pancreatic Ductal Adenocarcinoma | Phase 2 | NCT02289898 |
| Modified FOLFIRINOX for Gemcitabine Refractory Pancreatic Cancer: A Phase II Multicenter Trial | Phase 2 | NCT02440958 |
| Phase Ib/II Study of MEDI4736 Evaluated in Different Combinations in Metastatic Pancreatic Ductal Carcinoma | Phase 1/2 | NCT02583477 |
| A Study of Abemaciclib (LY2835219) Alone or in Combination with Other Agents in Participants with Previously Treated Pancreatic Ductal Adenocarcinoma | Phase 2 | NCT02981342 |
| A Study of SLC-0111 and Gemcitabine for Metastatic Pancreatic Ductal Cancer in Subjects Positive for CAIX | Phase 1/2 | NCT03450018 |
| Scheduling Nab-paclitaxel With Gemcitabine | Phase 2 | NCT03529175 |
| Nab-Paclitaxel + Cisplatin + Gemcitabine in Untreated Metastatic Pancreatic Adenocarcinoma | Phase 2 | NCT03915444 |
| Retrospective Analysis of 2nd-line Nab-Paclitaxel + gemcitabine After 1st-line FOLFIRINOX in Pancreatic Cancer | N/A | NCT04133155 |
Table 2:
Clinical trials of Drug delivery in PDAC (Immunotherapy for PDAC)
| Study Title | Phase | NCT number |
|---|---|---|
| A Study of Epacadostat in Combination with Pembrolizumab and Chemotherapy in Subjects with Advanced or Metastatic Solid Tumors (ECHO-207/KEYNOTE-723) | Phase 1/2 | NCT03085914 |
| Paclitaxel Protein Bound Plus Cisplatin Plus Gemcitabine and Paricalcitol for Pancreatic Adenocarcinoma (NABPLAGEMD) | Phase 2 | NCT03415854 |
| Second-line Study of PEGPH20 and Pembro for HA High Metastatic PDAC | Phase 2 | NCT03634332 |
| Study of Pembrolizumab With or Without Defactinib Following Chemotherapy as a Neoadjuvant and Adjuvant Treatment for Resectable Pancreatic Ductal Adenocarcinoma | Phase 2 | NCT03727880 |
| Trial of Neoadjuvant and Adjuvant Nivolumab and BMS-813160 With or Without GVAX for Locally Advanced Pancreatic Ductal Adenocarcinomas. | Phase 1/2 | NCT03767582 |
| A Multiple Ascending Dose Study of MEDI7247 in Advanced or Metastatic Solid Tumors | Phase 1 | NCT03811652 |
| Nivolumab in Combination with Chemotherapy Before Surgery in Treating Patients with Borderline Resectable Pancreatic Cancer | Phase 1/2 | NCT03970252 |
| Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined with Nivolumab and Ipilimumab for Patients with Resected MMR-p Colorectal and Pancreatic Cancer | Phase 1 | NCT04117087 |
| Mutant KRAS G12V-specific TCR Transduced T Cell Therapy for Advanced Pancreatic Cancer | Phase 1/2 | NCT04146298 |
| A Multi-Cancer, Multi-State, Platform Study of Durvalumab (MEDI4736) and Oleclumab (MEDI9447) in Pancreatic Adenocarcinoma, Non-Small Cell Lung Cancer and Squamous Cell Carcinoma of the Head and Neck to Correlate Clinical, Molecular and Immunologic Parameters with DNA Methylation | Phase 2 | NCT04262388 |
Most of the reasonably successful treatments applied so far for PDAC include a combination of chemotherapy, immunotherapy, and radiotherapy. Currently, most clinical trials consist of these combinations (Table 1, 2).
2.1. First-line therapy:
The main PDAC treatment is surgical resection followed by chemotherapy, radiotherapy, and targeted therapy [20]. In resectable tumors, gemcitabine has so far been the most successful and preferred the first-line drug because of its lower toxicity and reasonable response rate [21]. This drug is used as a single therapy and in combination with other drugs such as 5-fluorouracil (5-FU) [22,23]. Using gemcitabine alone for six months after tumor resection has increased the 5-year overall survival (OS) to 20.7% compared to the observation arm ( OS 10.4%) [21]. Other clinical trials had shown no major difference in OS and quality of life after using 5-FU/ leucovorin (23.6 months) or gemcitabine (23 months) in resectable PDAC patients [24]. Moreover, a recent clinical trial (phase III NCT03610100) has designed a modified version of gemcitabine (Acelarin) to overcome the current resistance to gemcitabine and enhance its efficiency via delivering a high intracellular level of dFdCTP (gemcitabine active agent). Due to the pancreatic relapse after 6 months of gemcitabine treatment, a combination regimen should be considered as adjuvant therapy [24]. Neoptoloms et al. have investigated the efficiency of another combination of resectable PDAC using gemcitabine and capecitabine. The team found that this regimen increased OS to 28 months compared to 25.5 months using gemcitabine alone. Interestingly, the adverse effects of using gemcitabine and capecitabine were tolerable [24].
For locally advanced pancreatic cancer, treatment is solely based on combination therapy. One of the more explored combinations with some success in the metastatic PDAC is the FOLFIRINOX (fluorouracil, leucovorin, irinotecan, and oxaliplatin) [25]. Clinical trial PRODIGE 4/ACCORD 11 has shown that FLOFIRINOX increased the OS rate up to 11.1 months compared to 6.8 months of using gemcitabine alone. FLOFIRINOX showed some adverse effects such as thrombocytopenia, neutropenia, febrile neutropenia, diarrhea, and alopecia.; however, the patients’ quality of life was better than gemcitabine alone [26]. In this regard, several retrospective studies have suggested reducing the initial chemotherapy doses of FLOFIRINOX, which could reduce side effects and maintain the regimen efficiency [27]. A phase II trial has shown that using a modified FOLFIRINOX regimen enhanced treatment efficacy and reduced toxicity [28]. Also, PEFG (cisplatin, epirubicin, fluorouracil, and gemcitabine) was one of the first combinations taken to the clinical trials that showed fewer adverse effects and higher therapeutic efficacy [29]. Another combination studied used gemcitabine with the EGFR inhibitor (erlotinib) approved by the FDA for PDAC [30]. Gemcitabine also has been paired with Abraxane, capecitabine alone, and gemcitabine/docetaxel/capecitabine (GTX) to give a good response and better survival rates in PDAC patients [31]. Gemcitabine with Abraxane was a successful combination [32] tried in phase III trials and was approved by the FDA, although it showed severe adverse reactions (such as neutropenia, leukopenia, neuropathy, febrile neutropenia, or fatigue). The reason for approval was because of the advantages that outweighed the side effects of treatment.
Other combination therapies that have shown promise in clinical trial results are listed (Table 1 and 2). Several different combinations are put to the test for PDAC constantly so that we could increase the success rate of therapy and make personalized medicine achievable. The main criteria taken into account in choosing the appropriate combination therapy are efficacy/survival, adverse effects, and patient compliance [33].
2.2. Second-line therapy:
Second-line therapy is mainly depending on the chosen first-line therapy. Thus, healthcare providers should take clinical trials into account for selecting the appropriate regimen that does not impact patients’ quality of life and has minimal side effects. For instance, for patients who received FOLFIRINOX as first-line therapy, a gemcitabine-based regimen should be considered. For patients who failed to respond to FOLFIRINOX, gem-based therapy (gem-nap) played a significant role in improving OS (8.8 months) [34] with manageable side effects. On the other hand, if gem-based therapy is used as first-line therapy, 5-FU can be used either as a single agent or with other agents such as oxaliplatin as the second-line therapy.
2.3. Current ongoing clinical trials for PDAC therapy:
Currently, there are many new pathways and targets for PDAC therapy that are being explored, such as the Hedgehog pathway, KRAS pathways, JAK/STAT pathways, hyaluronidase/hyaluronic acid, angiogenesis, tyrosine kinase inhibitors, and growth factor receptors. Most of these trials are in phase I and phase II and are initially evaluated for their safety and toxicity profiles [35]. CYL-02 is a non-viral gene therapy that is used to sensitize the chemotherapy of PDAC (NCT01274455). Patients with locally advanced PDAC remained free of metastasis after receiving CYL-02. CA 19–9 cancer biomarkers in locally advanced PDAC patients significantly reduced after receiving gene therapy combined with gemcitabine. [36]. Another clinical trial (NCT03450018) combined SLC-0111 (CAIX inhibitor) with gemcitabine for treating metastatic PDAC. SLC-0111 was found to play a role in increasing intratumor acidosis and slowing tumor growth [37]. Some other clinical trials are listed in Tables 1 and 2.
3. Challenges of PDAC treatment
PDAC prognosis is the poorest among all solid tumors. Even though there are new advancements in finding the initial sign of PDAC, inadequate response to current therapies is still prevalent [38]. One of the hypotheses that have emerged in the last few years is that the PDAC microenvironment is responsible for increasing both carcinogenesis and drug resistance [39]. The tumor microenvironment of PDAC is enriched by the stromal barrier, which is composed of fibroblast, immune cells, blood vessels, neural cells, and cellular proteins such as growth factors and cytokines (Figure 1B) [40]. Because of the stromal barrier, most combination therapies failed to increase the survival rate significantly[41]. the overall survival (OS) achieved in resectable tumors aged from 25.5 months using gemcitabine alone or 28 months when combined with capecitabine [24]. While in locally advanced PDAC tumor, the OS ranged from 6.8 using gemcitabine alone or 11.1 months after using FOLFIRINOX (fluorouracil (5-FU), irinotecan, and oxaliplatin) [26]. The following factors are believed to play a role in attenuating current therapeutic efficiency, as discussed below.
3.1. Tumor microenvironments (TME)
One of the main challenges in PDAC therapy is heterogeneous TME [42]. The genetically engineered mouse model (GEMM) of pancreatic cancer has enabled researchers to mimic human pancreatic cancers in many asp,ects such as resistance to gemcitabine, which opens new avenues for understanding pancreatic cancer microenvironments [43–45]. It has been discovered that PDAC has a stroma enriched TME, which keeps changing its composition, especially during progression from preneoplastic pancreatic intraepithelial neoplasms (PanINs) to invasive pancreatic cancer. Pancreatic intraepithelial neoplasms is a PDAC precursor lesion that is classified into a low grade (PanIN-1 and PanIN 2) and high grade (PanIn-3) [46]. The progression from PanIN low grade to PanIN high grade is associated with alteration in cancer-associated genes such as KRAS, P53, CDKN2A, and SMAD4 (figure 1, A) [46]. Stroma, a component of TME, has a major role in increased proliferation, metastasis, immune escape, and drug resistance [40]. Desmoplastic reaction (DR) is one of the PDAC hallmarks that contributes to PDAC’s poor prognosis. The dense desmoplasia plays a major role in distorting the normal architecture of pancreatic tissue and creating a mechanical barrier around the PDAC tumor, that limits tumor vascularization. Thus, drug penetration into tumor site is diminished [47]. Olive, K. et al. have reported that accumulation of active gemcitabine metabolites (2’,2-difluorodeox-cytidine triphosphate (dFdCTP) was high in poor stromal cancer in subcutaneous and orthotopic mice model while it was barely detectable in high dense stroma cancer such as PDAC [48]. One of the pathways that enhances PDAC desmoplastic reaction is sonic hedgehog (SHH) signaling. Importantly, DR consists of multiple components such as fibroblasts, pancreatic stellate cells, and extracellular matrix (collagen I, collagen II, and fibronectin) that all react together, and worsen the clinical outcomes of PDAC patients [49]. The major two components of DR are pancreatic stellate cells (PSCs) and fibroblasts. It has been found that PSCs are involved in secreting many cytokines such as IL6, CLL2, CLL5, and CLL8 [50,51]that are contributing to proliferation, migration, and producing extracellular matrix (ECM) protein [52]. Several studies showed PSCs’ role in reorganizing the collagen fibers to parallel alignments that enhance cancer cells’ migration and invasion [53]. Cross talk between the pancreatic cell and PSCs is a trigger for PDAC cells to grow and migrate via releasing some growth factors such as insulin-like growth factor, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) and cytokines [54]. Several pathways are believed to play a significant role in the PSCs process, such as transforming growth factor-beta (TGF-β), hepatocyte growth factor (HGF), fibroblast growth factor, and epidermal growth factor (EGF). Lohr et al. found that the co-culture of TGF-β expressing Panc-1 is more proliferative and induces more collagen I and fibronectin [52]. Armstrong et al. have shown that PSCs incubated with TGF-β enhanced [3H] thymidine uptake, induce collagen production, promote the malignancy of PDAC tumor, and increase cancer resistance toward chemo and radiation therapies [55,56]. It has been found that fibroblasts play a role in producing secreted protein acidic and rich in cysteine (SPARC) that enhances cell migration and proliferation and worse prognosis in PDAC patients [3,57]. The activated factors of pancreatic cells promote activation of stromal cells, which consequently affect other epithelial tumor components of pancreatic cancer [3]. Therefore, PSCs, fibroblast, and epithelial cells play a significant role in controlling ECM through the proteolytic enzyme of metalloproteinase (MMPs) which expresses in pancreatic cancer cells. It has been found that several enzymes of MMPs such as (MMP-1, MMp-2, and MMp-9) overexpressed when coming in contact with specific ECM proteins [58,59]. ECM is one of the major factors that enhance tumor aggressiveness and invasiveness. Further understanding of PSCs’ activated pathway- PDAC cross-talk is needed to develop PSCs targeted therapy [60,61].
3.2. Cancer Stem Cells (CSCs) in PDAC
It is well known that TME is characterized by its heterogenicity, and its pathological and physiological effects on tumor therapy are not well understood. Epithelial to mesenchymal effect (EMT) is a stem cell characteristic that is believed to have a significant role in promoting tumor heterogenicity and cell metastasis [62]. Within TME, there is a subpopulation of the cells called cancer stem cells (CSCs) that are capable of self-renewal [63], and that can justify why many tumors regenerate after being mostly eradicated during chemotherapeutic treatment [64]. CSCs were firstly found in the hematopoietic system; however, researchers found CSCs to be presented in solid tumors such as breast [65], colon [66], brain [67], and pancreatic cancer [64]. Breast CSCs were characterized by CD44high/CD24low antigenic phenotypes that promote tumor initiation compared to other carcinomas with CD44low/CD24high [65]. However, in the xenograft animal model of pancreatic cancer, it has been identified that a subpopulation of cells has CSC properties and has CD44+, CD24+, and ESA+. In this study, the authors found that CD44+, CD24+, and ESA+ pancreatic cells have a high potential to form a tumor when injected as low as 100 cells per mouse compared with CD44-, CD24-, and ESA- [64].
CD44 is a non-kinase, transmembrane glycoprotein (P-glycoprotein) expressed on several cells and tissues such as embryonic stem cells and bone marrow. It has been found that CD44 is overexpressed in various tumor cells, and it is known as a biomarker of CSCs [68]. Furthermore, CD44 has a significant role in cancer stemness and promoting cancer tumorigenicity [69]. Hyaluronic acid binds to CD44; thus, many drug delivery researchers have reported the efficiency of using hyaluronic acid as CD44 binding ligand to improve the efficiency of PDAC first-line therapy [70–72].
In addition, hepatocyte growth factor receptor (c-Met), present in both normal and tumor cells, is essential for embryonic development and tissue repair [73]. c-Met is a unique receptor with only one ligand that can bind to it, namely, HGF [73]. Upon HGF binding to c-Met, a series of downstream signaling pathway events are mediated, leading to enhancement of normal cell growth, cell motility, and protection of normal cells from apoptosis [74]. In cancer cells, c-Met functions differently than normal cells due to c-Met being overexpressed or mutated, which ultimately enhances pancreatic carcinoma [75]. It has been found that exposing cells to chemotherapeutic agents such as gemcitabine enhanced EMT, increase c-Met phosphorylation that promotes expression of CSCs biomarkers such as CD44 and CD24 [76]. Interestingly enough, CD44 plays a role in regulating the HGF/c-Met signaling pathway, which maintains CSCs function. Overexpression of c-Met in pancreatic cancer is a sign of poor prognosis, which elevates EMT phenotype. Li et al. have studied the role of c-Met in forming tumor spheres. They found that c-Met+ cell lines could form spheres while c-Met– cell lines could not.
Moreover, treating pancreatic xenografts in NOD-SCID mice with a combination of c-Met inhibitor (XL184) and gemcitabine showed significant tumor growth regression even after 32 days of therapy cessation [77]. Also, Alex et al. successfully designed an antibody-drug conjugate (TR1801-ADC) that enhanced the tumor inhibition (in vitro and in vivo) in Met overexpressed pancreatic cancer and worked in synergy with gemcitabine [78]. CSCs are one of the leading players of PDAC distal metastasis. Hermann et al. showed that PDAC distal metastasis is correlated with the presence of CD133+ of pancreatic CSCs and CXCR4+ expression. [79].
Based on the findings mentioned above, PDAC has a subpopulation of cells (CSCs) which plays a crucial role in developing metastasis and fortifying chemotherapeutic resistance. Moreover, c-Met is another PDAC biomarker that enhances the CSCs subpopulation’s tumorigenicity. Together, these studies suggest that CSCs biomarkers (CD44+ and CD133+ cells) and c-Met+ cells can be used as a new target for therapy to minimize tumor growth and enhance the tumor response to current treatment.
3.3. Hypoxia
Hypoxia is an essential feature of PDAC tumor microenvironment, which plays a major role in activating several molecular and signaling pathways contributing to PDAC aggressiveness [80]. Chang Q et al. performed a study to measure the oxygen levels within pancreatic cancer. In this experiment, which was conducted on seven patients, they found a dramatic reduction of oxygen in pancreatic tumors than normal tissue [81]. The ability of cancer cells to survive in the hypoxic condition is attributed to the activation of hypoxia-inducible factor (HIF) pathways. HIF can activate several genes such as STIM1, PKM2, MiR21, and MTA2 [82] that help cancer cells controlling metabolism, survival, pH, migration, as well as some angiogenic growth factors [83–85]. In PDAC, many studies have shown that pancreatic cancer cells induce the angiogenesis process via secreting vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) [86,87].
Furthermore, HIF mediates several pathways, such as c-Met and Hedgehog pathways, that enhance cancer invasiveness and drug resistance [3,88]. Moreover, hypoxia is known to activate the notch signaling pathway, which plays a role in cancer cell proliferation and differentiation [89]. Yoshiharu et al. found almost 21 of 34 examined specimens (62%) showed moderate to high expression of at least one notch in pancreatic cancer as well as in PanIN lesions. [90]. Notch signaling pathway is an early sign of PDAC that acts as a mediator to activate EGF receptors and PDAC precursors [90]. Furthermore, notch pathway activates transforming growth factor-alpha (TGF-α), which functions as an activator of acinar-to-ductal metaplasia [91]. All these factors mediate cancer invasiveness, drug resistance and increase the challenge of reaching drugs to TME of pancreatic cancer. Therefore, new novel targeted therapies or delivery systems that could target hypoxic regions or inhibit notch signaling pathways are needed to improve the overall therapeutic efficiency.
3.4. Inflammation and Immune cells
One of the major characteristics of TME is an absence of immune surveillance and the presence of inflammatory cells that support aggressiveness and tumorigenesis of PDAC [12]. Inflammatory and immune cells are crucial components of TME that play a pivotal role in PDAC aggressiveness. Within TME, at the early stage, many immune cells such as anti-tumor Th1, CD4+, CD8+, and natural killers (NKs) are recruited to eliminate the cancer cells. However, the tumor cells start to develop an escape mechanism via recruiting monocyte and neutrophil, acquiring the anti-inflammatory phenotype (M2 and N2, respectively). Furthermore, tumor cells recruit or polarize T regulatory cells (Treg), shift anti-tumor Th1 to Th2, and recruit myeloid-derived suppressive cells (MDSCs). All these events result in the deactivation of CD4+, CD8+, NKs and increase the tumor progression [92,93].
3.5. Role of RAS in PDAC
The Ras family belongs to a small GTPase composed of HRAS, NRAS, and KRAS [94]. KRAS (KRASG12D and KRASG12V), CDKN2A, TP53, and SMAD4 are the most common genetic mutations found in pancreatic cancer. However, until now, none of them are druggable. The absence of specific inhibitors for all of these genes limits the therapeutic options for PDAC patients [95]. Recent studies found that almost 95% of the patients have a mutational activation of oncogenic KRAS at codon 12 [96] that is crucial in activating and maintaining PDAC. Moreover, KRAS mutation is a sign of poor prognosis in resectable and advanced PDAC patients [97]. Normally, KRAS proteins have a crucial role in cell survival, differentiation, and proliferation [98]. However, the mutated KRAS compromises oncogenic KRAS’s normal function; thus, the pancreatic cells’ growth becomes uncontrollable. In genetically engineered mice models, functional studies have found that KRAS switching off led to dramatic tumor regression [99,100]. Therefore, extensive preclinical and clinical ongoing studies explore KRAS to design an effective targeted therapy.
In OA02.02 phase 1 clinical trials, it has been found that AMG510 can inactivate KRAS by irreversibly occupying His95 groove near the cysteine pocket of KRASG12C. Interestingly, AMG510 is potent against KRASG12C mutation of non-small-cell lung cancer tumor while it is not active against wild type [101]. Moreover, AMG510 showed good anti-tumor activity either alone or with PD-1 checkpoint inhibitors. The adverse effects of AMG510 were tolerable such as nausea and vomiting [101]. However, the use of AMG510 is limited in PDAC due to KRASG12C mutation that accounts for only 2% in PDAC[102]. This study will stimulate researchers to develop new drugs that can inactivate KRASG12D and KRASG12V that is accounting for 80% of PDAC. Due to the undruggable nature of KRASG12D and KRASG12V, targeting the KRAS downstream (MEK-ERK and/or PI3K) is the putative way to manage the KRAS related cancers. Inhibition of a single downstream pathway (RAF, MEK1/2, ERK1/2, and PI3K) of RAS did not show significant clinical effect, and drug resistance may be induced via several compensatory activations mechanisms such as PI3K, which activates several pathways such as Akt and mTOR [103].
Several inhibitors of RAS downstream pathway are clinically tested, as shown in Figure 2. The role of KRAS in enhancing TME is not well understood. Mills, L et al. have demonstrated the role of KRAS in TME. KRAS activates SHH expression, which further induces GLI family zinc finger 1 (transcriptional factor) in fibroblasts. The binding of GLI1 to IL-6 promotor regulates the activation of cancer STAT3. In this study, loss of GLI family zinc finger 1 impaired the carcinogenic of KRAS in PDAC animal model [104]. Thus, targeting the cross-talk between cancer cells and TME components is a viable way for promoting PDAC tumor regression.
Figure 2:
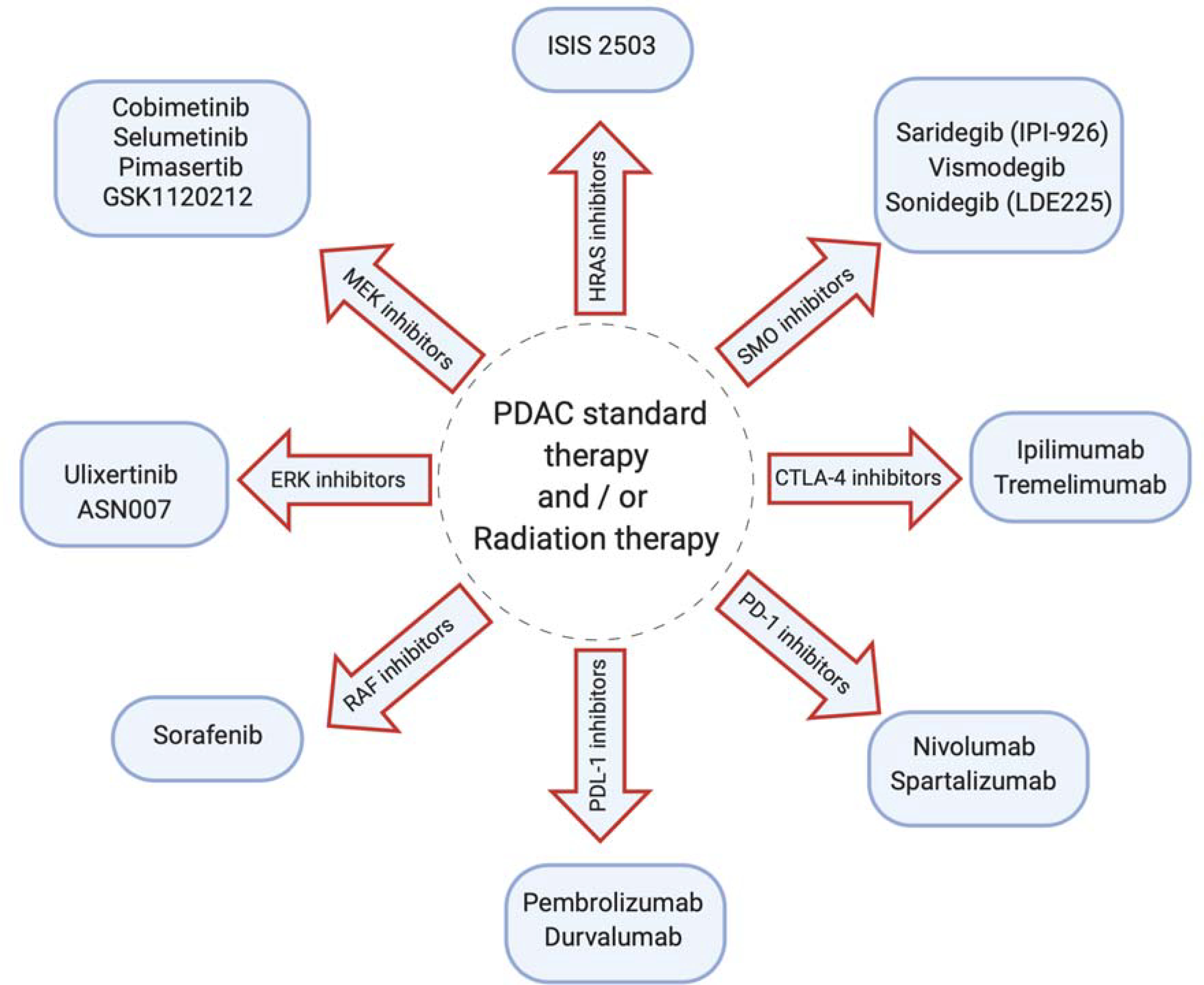
Various classes of drugs that are being explored in the clinical trials in combination with PDAC standard therapy and/or radiation therapy to enhance the efficiency and the overall PDAC response are shown.
4. Strategies to overcome PDAC barriers
4.1. Stroma-Targeting Therapy in PDAC
PDAC tumor is characterized by one of the densest stroma among several tumors types that arise from pancreatic stellate cells (PSCs). Activation of PSCs leads to the formation of an extracellular matrix that enhances the strength and resistance of PDAC cells to chemotherapeutic and radiation therapy [105,106]. Currently, several agents are being tested in preclinical and clinical studies to enhance delivering of cytotoxic drugs to PDAC microenvironments (Figure 2).
4.1.1. Utilizing hyaluronidase enzyme
A variety of methods to enhance the effectiveness of chemical therapeutic agents have been developed to address the physical and biological obstacles to successful PDAC therapies and reverse the effects of stroma on tumor growth. One of these strategies is to use Hyaluronic acid (HA) or hyaluronan, which is one of the major components of PDAC stroma. HA interacts with cellular receptors to ensure tumor cell survival and to activate downstream signaling pathways relevant to tumor progression [107,108]. Accumulating evidence found an abundance of hyaluronan (HA) in PDAC tumors, which significantly enhances tumor proliferation. A high level of HA is a sign of poor prognosis compared to those who have a low level of expression [109]. Therefore, one of the strategies in enhancing PDAC drug delivery is depleting HA using hyaluronidase [110]. In HALO202 clinical trial that recruited 279 patients, PEGylated hyaluronidase (PEHPH20) combined with gem-nap in one arm and gem-nap alone on the other arm. In this study, it has been found that patients who had a high level of HA responded much better than low HA with overall response rate (ORR) (45% vs. 31%) and OS ( 11.5 vs. 8.5 months), respectively [111]. Despite the promising results in phases 1 and 2, the phase 3 clinical study (HALO109–301) failed to achieve the primary endpoint of OS, and further development of PEHPH20 was halted. The authors concluded that targeting desmoplasia alone is not sufficient; other factors such as tumor stroma, epithelial to mesenchymal transition, low tumor mutational burden should be considered to enhance the overall PDAC response [112].
4.1.2. Sonic Hedgehog (SHH) signaling
One of the pathways that participate in improving PDAC desmoplastic reaction is Sonic Hedgehog (SHH) signaling. The Hedgehog pathway is abnormally activated in PDAC [113]. It is evident that Hedgehog signaling pathway stimulates pancreatic stellate cells (PSCs) through direct effects on non-pancreatic tissues and controls stromal deposition [114]. A variety of strategies are being considered to inhibit the Hedgehog pathway, which is the pro-inflammatory stage of PDAC, in order to remove tumor-stroma [115]. The Hedgehog Signal Pathway is blocked by cyclopamine as a natural steroid alkaloid [116]. It reduced the fibronectin content of a PDAC xenograft mouse model and increased tumor vascularization. Cyclopamine improves tumor growth inhibits by 63.3% in combination with nanomaterials loaded with paclitaxel [117]. One research on induced pluripotent stem cells demonstrated that IPI-926, a small molecule that inhibited the Hedgehog pathway, gave favorable effects of inducing blood vessel density and drug concentration in pancreatic ductal adenocarcinoma (PDAC) [118]. In phase, I clinical trial of IPI-926, patients who were previously affected by PDAC did not have any significant side effects [118]. Sadly, despite the promising findings in phase I trials, phase II clinical trials of IPI-926 showed no major benefits to prevent PDAC in pancreatic cancer patients [119]. In similar conditions, in patients with metastatic PDAC relative to gemcitabine alone, Vismodegib, another Hedgehog inhibiter in conjunction with gemcitabine, improved median total survival and progression-free survival significantly [120]. Although multiple therapies that target the Hedgehog pathway showed positive effects in preclinical models, few improved survival rates and their use was followed by side effects and toxicity [121]. Overall, Using SSH signaling inhibitors is an effective way to deplete the stromal barrier and enhance gemcitabine delivery [122,123]. Feig et al. have combined smoothened inhibitor (IPI-926) with gemcitabine which majorly reduced tumor stroma, promoted micro-vessel density, and significantly increase intra-tumor gemcitabine active metabolite (dFdCTP); as a result, the overall survival increased [40]. Olive et al. have found that using Hedgehog inhibitors allows the chemotherapeutic agent to be delivered to the tumor site [48].
4.1.3. Targeting Inflammation and Immune cells in TME
PDAC stroma is enriched in many inflammatory cells, such as mast cells, which are considered the cornerstone of angiogenesis, tumor growth, and lymph node involvement [124]. Therefore, inhibition of mast cell activity is one of the current strategies to limit tumor progression. Ibrutinib, a small molecule that permanently inhibits Bruton’s tyrosine kinase (BTK) protein, is used to decrease fibrosis and inhibit mast cell cytokines release (IL-8, TNFα, and MPC-1) within PDAC TME. Ibrutinib has shown a reduction in tumor fibrosis in a mouse model and improve the mouse response to standard therapy [124,125].
4.1.4. Targeting Hypoxia in TME
Many methods have been utilized by targeting hypoxia, including prodrugs that are activated in hypoxic conditions, drugs that target HIF-1α active cells, and nanoparticles with active hypoxia targeting, which are being developed [126,127]. Evofosfamide (TH-302), a cytotoxic prodrug, comprises a mustard derivative converted in hypoxic conditions to an active metabolite [128]. Clinically, Evofosfamide has been indicated to minimize radiotherapy resistance in PDAC. However, another clinical trial for NSCLC using Evofosfamide with tarloxotinib (a hypoxia-activated tyrosine kinase inhibitor) was terminated early because the patients did not meet the minimum response rate to this combination [128,129]. Another anticancer agent that was identified years ago is POP33. POP33 is a prodrug with the potential to elevate caspase-3 activity and induce apoptosis in HIF-1–active/hypoxic cells [130]. This drug, which is a fusion protein, consists of a cleaved caspase pro-enzyme and a transduction HIF-1α dependent stabilization domain to deliver the drug into cells [130]. Although POP33 exhibited a promise in an animal model of PDAC, it has yet to make its way for human application. While no direct HIF protein inhibitors have reached the clinical studies for PDAC, treatment strategies that target heat shock protein (HSP) 90 have led to HIF degradation [131,132].
4.1.5. Remodeling Tumor Vasculature
The thick fibrotic stroma covering the blood vessels and the proliferation of cancer-associated fibroblasts (CAFs) destroy intratumoral blood vessels, creating a hypoxic microenvironment, which supports pancreatic adenocarcinoma metastasis and the development of chemoresistance [133,134]. Remodeling PDAC vessels improves drug delivery and exposes living cells to better environments which improves drug effectiveness and its disposition in the body. Many agents with varying aims have been employed to target the tumor microenvironment [135]. In 2015, a Phase II study tested the effectiveness of the new medication, FOLFIRINOX, in conjunction with lisinopril in patients with locally advanced pancreatic cancer. The approach was found to have a high proportion of patients achieving R0 resection [136]. It is also noteworthy thatthe lack of the Angiotensin II Type II receptor (AT2R) in pancreatic fibroblasts contributes to tumor cell proliferation [137]. In the future, Ang II signaling pathway will need to be analyzed in more depth before its use in the clinic.
4.2. Other Important strategies
One of the target protein that facilitates PDAC stromal depletion is secreted protein acidic rich in cysteine (SPARC). Abraxane (albumin conjugated paclitaxel) has been hypothesized for its ability to deplete stroma and accumulate in overexpressed SPARC pancreatic TME. There are contradictory results in this regard. One clinical trial has shown that combining gemcitabine with nab-paclitaxel increased the overall survival of high expressed SPARC patients (17.8 months) compared to those who had low SPARC expression (8.1 months) [138,139]. However, stromal depletion was not seen in a preclinical study of the patient-derived xenograft mice model. The author observed that gemcitabine activity was impaired due to activation of reactive oxygen species (ROS). The interesting observation was after using gemcitabine combined with nab-paclitaxel [140]. Thus, the exact role of SPARC is unknown and in-depth investigations are needed to reveal the particular role of this biomarker and its prognostic impact after treating it with nab-paclitaxel or other targeted therapy.
Another target is cancer stem cells (CSC) which have a significant role in tumor proliferation and metastasis [141]. One of the signaling pathways that promote CSC growth is STAT3. It is well documented that STAT3 is activated in PDAC tumor and functions as a tumor promotor. Therefore, inhibition of STAT3 would enhance PDAC response to chemotherapeutic agents and promote tumor inhibition [142].
5. Immunotherapy as a promising approach for PDAC therapy
Since FDA approval of different immunotherapeutic agents such as ipilimumab (2011) and nivolumab (2014), immunotherapy became a candidate therapy for multiple tumors such as melanoma, renal cancer, lung cancer, and others [143]. Using immunotherapies have shown a drastic improvement in OS and enhance the therapeutic response of many solid tumors such as (melanoma and renal cancer). Hence, immune checkpoints in designing a treatment regimen for PDAC is a promising avenue [144]. The most widely used immune checkpoint inhibitors are against the programmed cell-death-1 (PD-1), and the cytotoxic T lymphocyte antigen-4 (CTLA-4) receptors and programmed cell-death ligand-1 (PD-L1) [145].
5.1. Reasons for checkpoint inhibitors failure in PDAC
Immune checkpoints refer to immunogenic regulatory mechanisms pertaining to T-cell immune response. Immune checkpoint blockades (ICB) such as PD-1, PD-L1, and CTLA-4 have shown promising results and varied responses in solid tumors [146]. Cancer immunotherapy has a variable patient outcome that is dependent on the leukocyte population in the tumor microenvironment [147]. Not every patient of either the same or different tumor subtype will elicit a similar anti-tumor immune response to ICB. For highly resectable tumors, chemotherapy may be surpassed by immunotherapy owing to fewer side effects and irreversible tumor regression. However, clinical trials with ICB for PDAC have been disappointing [148].
A major reason for the failure of immunotherapy in PDAC is the dense tumor microenvironment (TME) that is the limiting factor for a number of PDAC chemotherapies. PDAC TME consists of a dense fibrous stroma consisting of tumor cells, immune cells, growth factors for tumor metastasis, extracellular matrix, fibroblasts making it highly complex and heterogenous [3]. Tumor-infiltrating lymphocytes are present at a lower population in the PDAC TME, driving factors for effective clinical response to ICB.
Single-agent clinical trials for employing ICB for PDAC have not shown promising results. Single-agent anti-CTLA-4 (Ipilimumab) dosed intravenously at 3mg/kg/dose (4 doses/course, every 3 weeks, for maximum 2 courses) was practically ineffective for the treatment of advanced pancreatic cancer [149]. Monotherapy with PD-1/PD-L1 ICB durvalumab showed partial response in 2 out of 29 patients who received the intervention and had evaluable data [150]. In essence, single-agent immunotherapies for PDAC have not garnered an effective patient outcome.
Antigenicity and immunogenicity are two major factors responsible for the failure of ICB in PDAC [151]. Reduced antigenicity in PDAC refers to tumor cells’ inability to produce and present tumor-associated antigens to the effector cells of the immune system. This, in turn, negatively affects T-cells’ ability to mount an immune attack in response to antigen producing cells [152]. Immunogenicity, which is the ability to induce an immune response in cancer, is dependent on multiple factors like composition of the stroma, infiltration of CD8+ and CD4+ T-cells, B cells, antigen presentation [152].
PDAC tumor can be classified into subtypes based on RNA expression analysis, and it reveals prominent immune cell signatures specific to the PDAC subtype. Since each subtype has a different signature that characterizes the tumor microenvironment components, they will show different outcomes in response to ICB therapies. Tumors are classified into inflamed (hot) and non-inflamed (cold) based on T-cell infiltration [153]. Most of the preclinical and clinical studies have concluded that only patients who have T-cell inflamed tumors would have a high chance of responding to ICB. However, non-inflamed tumors respond poorly to monotherapy of ICB due to low level of tumor infiltrating lymphocytes (TIL) and PDL-1[153]. Therefore, combining ICB with other agents is a valid strategy to (i) enhance tumor immunogenicity, (ii) recruit more TIL to the tumor site, and (iii) minimize tumor microenvironments immunosuppression [154,155]. PDAC is the lowest mutational tumor among other solid tumors, where the average of mutation is 1 mutation per megabase (Mb) compared to 11 mutations per Mb in melanoma [156]. Thus, immune system recognition of neoantigen is solely based on neoantigen quality, where it can be recognized if it is high-quality neoantigen such as microbial-like sequence [157,158].
5.2. Possible approaches to improve tumor immunogenicity
Various reports have shown the ineffectiveness of monotherapies for PDAC. Hence, it is worthwhile to explore the effect of combinations of ICB or chemotherapy and ICB to tackle PDAC. Patients with poor prognosis and rapidly advancing cases of metastatic PDAC were dosed with a combination of Durvalumab and Tremelimumab. No patients responded well to Durvalumab monotherapy, a response rate of 3.1% was seen in patients receiving combination therapy, although with adverse toxicity in 22% of the cohort of patients [159].
Gemcitabine, frontline chemotherapy for PDAC, has been tested in combination with ICB to disengage PD-1/PD-L1 interaction. Attempts have been made to deliver gemcitabine and anti-PD-L1 in a controlled manner to the tumor [160]. Daniel et al. found that prolonged exposure of PDAC cancer to gemcitabine promotes the expression of several immune markers such as MHC class 1, PD-L1, and PD-L2. The authors also found that gemcitabine plays a role in increasing the secretion of several cytokines and TGFβ [160].
The activity of gemcitabine and anti-PD-1 was tested in vivo on transgenic mice; the current combination failed to inhibit the tumor growth unless the mice had genetic ablation of TGFβ. In TGFβ deficient mice, gemcitabine and anti-PD-1 activity in inhibiting tumor regression were improved by enhancing CTL infiltration into the TME [160]. These observations suggest that gemcitabine primes the PDAC tumor for enhanced antigen presentation, making anti-PD-1 therapy more effective in eliciting a robust CD8+ T-cell response and reducing tumor load [161]. Other drugs such as Cisplatin, albumin-bound paclitaxel, nivolumab are currently under clinical trials to expand on the promising outcomes of the combination of ICB with chemotherapy. So far, it is likely that moving ahead, immunotherapy for PDAC shall shift clinical focus from monotherapy to combination therapy.
Radio and chemotherapy, which have immunogenic cell death effects, have actively reduced tumor burden and enhanced T-cell infiltration to tumor microenvironments. Chemotherapeutic agents that induce immunogenic cell death are platinum-based drugs and taxanes combined with ICB to improve the immune system against cancer cells [162,163].
CCL2-CCR2 leads to recruiting Tumor-associated macrophages (TAM) that play as immunosuppressive within tumor microenvironments [164]. Many clinical trials tried to modulate the immune system to recognize and fight cancer cells. One of the clinical trials is using chemokines receptor 2 antagonist (PF-04136309), which inhibits the binding of chemokines ligand 2 to its receptor CCR2, resulting in inhibition. Immunosuppressive TAM [165].
Another way of inducing immune cells against cancer cells is via using the vaccine. GVAX is one of the vaccines composed of two human pancreatic cells engineered to secret immune cytokines (Granulocyte-Macrophage Colony Stimulating Factor (GM- CSF)) [166]. GVAX efficiency was evaluated as adjuvant therapy in resected PDAC and as a treatment regimen combined with ipilimumab in metastasis PDAC. In clinical trials phase I/II, GVAX was found to induce mesothelin-specific CD8+ T cells in the tumor section and improved the overall survival rate in PDAC patients. Thus, using GVAX, which functions as a trigger for dendritic cells that phagocytes released GM-CSF and migrate to lymph node to activate T cells, is a promising way to enhance tumor immunogenicity in PDAC patients [167,168].
All these methods have been either well established or are being rapidly developed to treat this severe disease. These have a lot of prospects to be explored and challenges to be overcome.
5.3. Other promising targets and strategies
Many teams have used groundbreaking bioinformatics, biochemistry, and cell biology approaches to identify specific mKRAS protein sequences that can be recognized by T cells. These teams have uncovered a set of molecular receptors that enable T cells to be home to mKRAS-expressing cancer cells. On the basis of these findings, Vonderheide RH and his group is undertaking two different clinical trials of new technologies designed to trigger mKRAS immune activity in patients with resected pancreatic cancer. The team is planning to use the most effective T - cell receptor identified and to perform a clinical trial of optimized T -cell therapy for the treatment of metastatic pancreatic cancer [169]. Furthermore, there many other targets and promising strategies are still under development for better therapeutic outcomes as shown in Table 3
Table 3.
Potential Targets in Pancreatic Cancer.
| Molecular targets | Molecular pathways | Reference |
|---|---|---|
| a. Transmembrane Receptor Proteins and downstream signaling cascade | The receptor tyrosine kinases (RTKs): Ras/MAPK, PI3K/AKT, PLCγ/PKC, and JAK/STATs. | [190,191] |
| The phosphoinositide 3 kinases/AKT/ mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway |
[191] | |
| EGFR: detected in up to 90% of PDAC | [192] | |
| FGF/FGFR: The fibroblast growth factor (FGF)/FGFR pathway plays a key role in PDAC development and progression. |
[193] | |
| IGF/IGFR: high expression of Insulin-like Growth Factor-1 (IGF-1) and IGF1R | [192] | |
| TRK: TRK gene fusions | [194,195] | |
| Vascular endothelial growth factor (VEGF)/vegfr): Angiogenesis pathway | [192] | |
| b. Other pathways | WNT/β-CATENIN: | [196,197] |
| NOTCH: | [197] | |
| ROUNDABOUT (ROBO) RECEPTORS/ SLIT GLYCOPROTEIN LIGANDS (SLIT): | [198,199] | |
| TRANSFORMING GROWTH FACTOR BETA (TGF-β): | [192,197] | |
| HEDGEHOG (Hh): | [35,200,201] | |
| NEUREGULIN-1 (NRG1): | [202] | |
| c. Tumor-Suppressor Genes | TP53: | [197] |
| SMAD4 (DPC4): | [203] | |
| BRCA: | [204–206] | |
| MMR DEFICIENCY | [145] | |
| EMT | [193,197,207] | |
| Extracellular Matrix (ECM) | [208,209] | |
| Cancer Stem Cells | [210] |
6. Advances in nanoparticle diagnosis and treatment
The currently available treatment models have limited clinical response, and continuous efforts are being made to increase the treatment efficacy. Nanomedicine and gene therapy have been found to give promising preclinical outcomes in treating aggressive pancreatic tumors, a few of which also went into the clinical trial. Due to the complex tumor stroma composition in PDAC, many drugs failed to penetrate the stroma barrier that negatively impacted their efficiency. Nanomedicines and gene therapy have potential use in enhancing PDAC diagnosis and treatment [18]. Moreover, the exact mechanism of how the nanoparticles accumulate at the PDAC tumor site is unknown. Most of nanoparticles relay on leaky vasculature (EPR effect) to be accumulated in the tumor sites, however, hypovasculature of PDAC tumor compromises the EPR effect and attenuates the nano-medicines delivery [170]. Therefore, using targeted nanoparticles provide a viable solution to improve the diagnostic test’s sensitivity and help treat PDAC as shown in Figure 3. Preclinically, several nanoparticles have been synthesized to enhance PDAC therapy.
Figure 3:
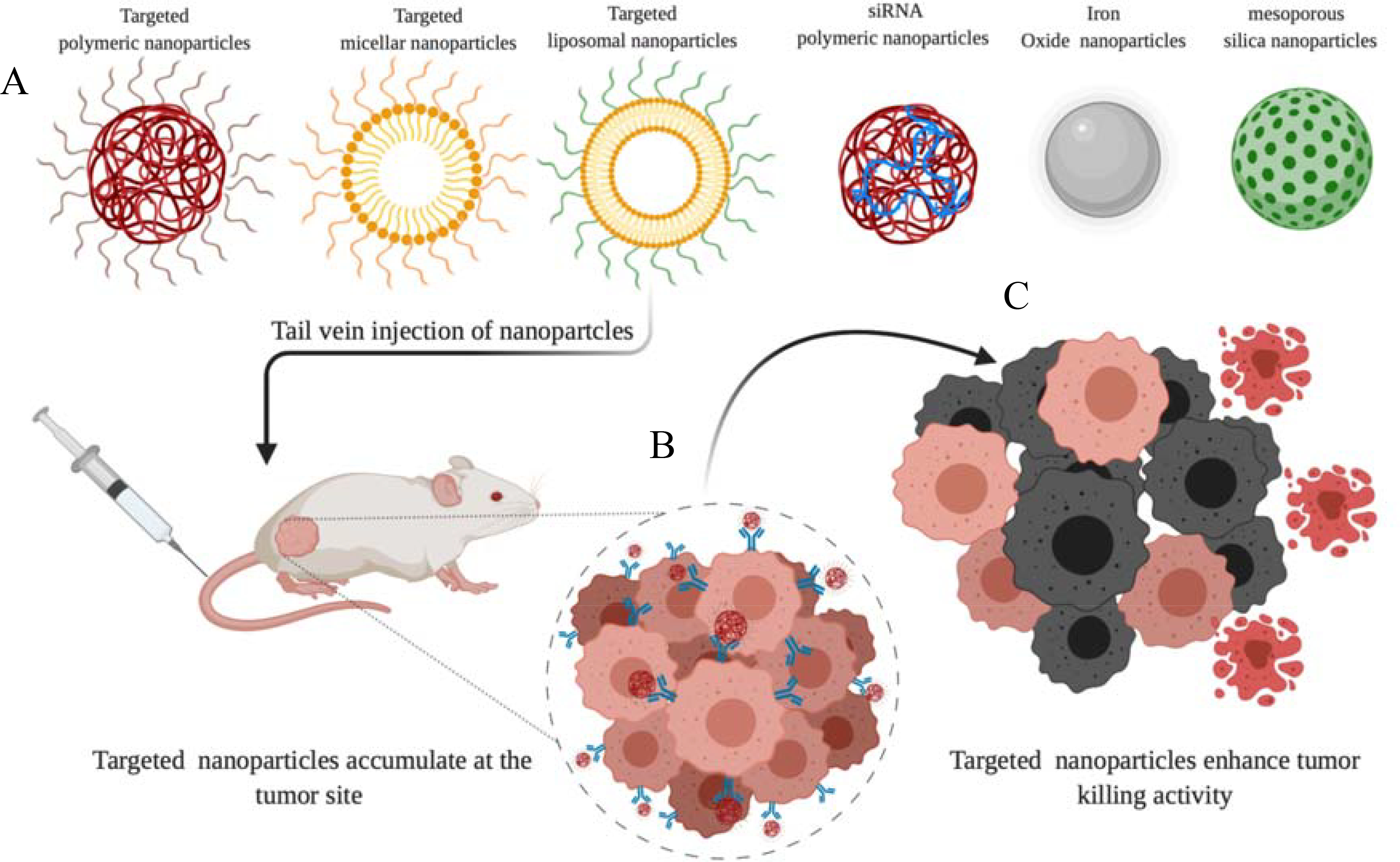
Various types of nanoparticles for PDAC therapy. (A) surface decorated nanoparticles to actively enhance the tumor selectivity is shown. (B) Specific nanoparticles that actively target tumor cells via receptor recognition are shown. (C) The uptake of nanoparticles via the endocytosis process results in cancer cell eradication. (Modified from Brachi et al. “Nanomedicine for imaging and therapy of pancreatic adenocarcinoma.” Frontiers in bioengineering and biotechnology 7 (2019)
Biodegradable nanoparticles characterize by its ability to escape the immune system and improve in vivo circulatory time. [171]. Albumin NPs have been used widely to deliver the cytotoxic gemcitabine to PDAC. Albumin NPs enhance the absorption of the drug in the circulatory system and be renewable and economical [172]. Many studies have revealed that gem-albumin nanoparticles enhanced the tumor regression activity compared to free gem [173–175].
Liposomal formulation characterizes by its biocompatibility, longer circulation time, and most importantly, the potential of surface modulation by ligands for active targeting. Recently, NAPOLI-1 trial showed the role of using irinotecan liposomal formulation combined with 5-FU/folinic acid as second-line therapy for patients who received gem-based therapy as first line therapy. NAPOLI-1 trial confirmed that receiving irinotecan liposomal formulation in combined with 5-FU/folinic acid improved OS (6.1 months) compared to 5-FU/folinic acid OS (4.2 months) [176].
Polymeric nanoparticles have several advantages: size uniformity, good release kinetics, malleability for customization, pH responding properties, and hydrophilic shell; all these properties make it a perfect PDAC delivery system that can deliver different cytotoxic drugs, DNA, proteins, and siRNA [177]. For example, using paclitaxel in its free form does not show significant activity against PDAC, but when administered via poly-lactic-co-glycolic acid (PLGA) nanoparticles, it penetrates the tumor and inhibits protein synthesis. The PDAC tumor uptake of paclitaxel PLGA nanoparticles was 5 folds higher than conventional therapy [178]. Furthermore, Couvreur et al. as shown in Figure 4. have enhanced Gem’s stability and efficiency via using self-assembled nanoparticles composed of gemcitabine and the natural lipid squalene (Gem-SQ) [179]. In mice bearing Panc-1 orthotopic model, Gem-SQ inhibited the tumor regression and enhanced the mice survivability compared to free [180,181].
Figure 4. Anticancer activity of SQgem in nanoparticles platform and gem (5 mg/kg equivalent doses) following intravenous treatment (on 0, 4, 8, and 13 days) of mice bearing P388 subcutaneous tumors.
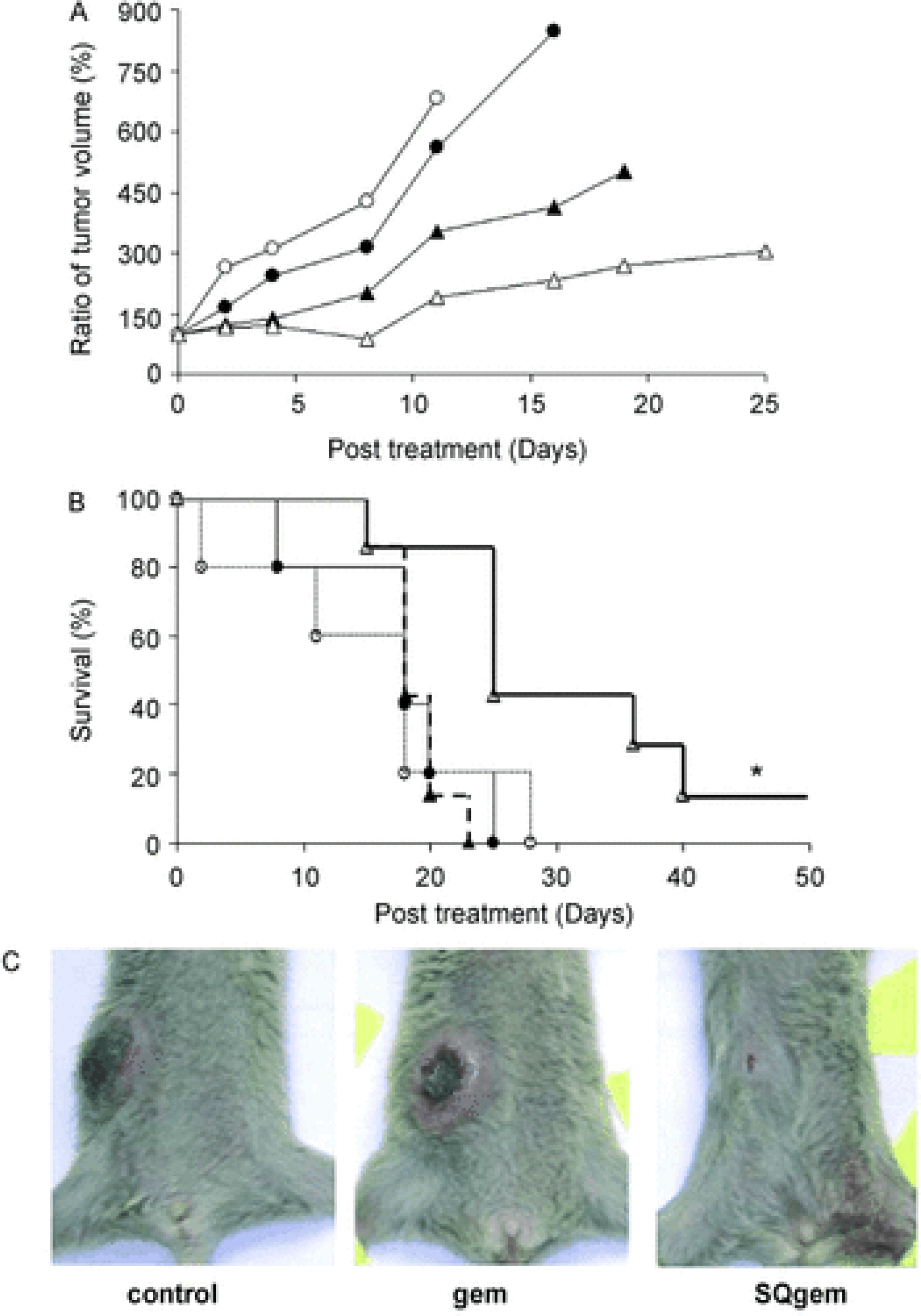
A) Tumor progression: Control (black spheres), saline (white spheres), gem (black diamonds), SQgem nano assemblies (white diamonds). B) Survival curve of mice: Control (solid line), saline (dotted line), gem (dashed line), SQgem nano assemblies (heavy solid line). * indicates P<0.05, as assessed by Kaplan–Meier test. C) Photograph showing the difference in tumor growth in mice following the completion of indicated treatment. Reproduced from [179]
Gene therapy is a promising way to enhance the overall PDAC therapy, but its delivery challenges limit its usage. For example, the main siRNA challenge is their stability issue within the biological systems; therefore, many researchers are trying to overcome this issue using drug delivery systems [182]. Khvalevsky et al. have used a biodegradable polymer matrix loaded with anti-KRASG12D siRNA. siG12D loader improved siRNA stability, overcame renal clearance, and, most importantly, knockdown KRASG12D expression, which improved overall tumor inhibition in the xenograft animal model [183]. Zhao et al. have constructed a hybrid lipid polymer nanoparticle to co-deliver siRNA (si-HIF1α) and gemcitabine (Gem) to target the HIF1α in PDAC cells. They have successfully shown a synergistic killing effect with siRNA and Gem combination therapy in vitro and in vivo and can inhibit tumor metastasis in the orthotopic tumor model [184]. Frederico P. et al. have designed pegylated polymeric nanoparticles conjugated with calcium phosphate to deliver VEGF siRNA. In this work, hybrid nanoparticles enhanced the serum stability and gene silencing efficiency [185].
The hypoxic TME in PDAC produces more HIFs, which are responsible for the activation of genes that control invasion, angiogenesis, chemoresistance, and proliferation [40]. The polymeric lipids coated NPs that loaded with GEM and siRNA, which was complexed to positively charged polylysine residues on the NPs surface, significantly inhibited the growth of subcutaneous PANC-1 tumor xenografts. Thus, it indicated a synergistic effect between HIF-1alpha inhibition and the chemotherapy (Gem). Not only that the combination treatment significantly shrunk the tumor size in an orthotopic PDAC model in comparison with un-encapsulated siRNA and GEM, or with NPs loaded with GEM only, but also, no peritoneal metastases were observed in the combination treatment group as shown in Figure 5. Also, because PDAC TME becomes resistant to chemo and radiotherapy, Wason et al. used nanoparticles to deliver drugs using cerium oxide nanoparticles (CONPs) to regulate production of ROS that might sensitize PDAC cells to radiotherapy (RT) [186,187]. CONPs-based pretreatment limited tumor growth in an orthotopic model nude mice, leading towards significant shrinkage in tumor weight and volume as compared to radiotherapy alone. Several nanomedicine-based strategies have been designed and tested for the treatment of PDAC. There is a need for more smart strategies to overcome these barriers and maximize treatment accumulation in the pancreatic tumor site [188].
Figure 5. In vivo antitumor effect of miRNA–siRNA combination.
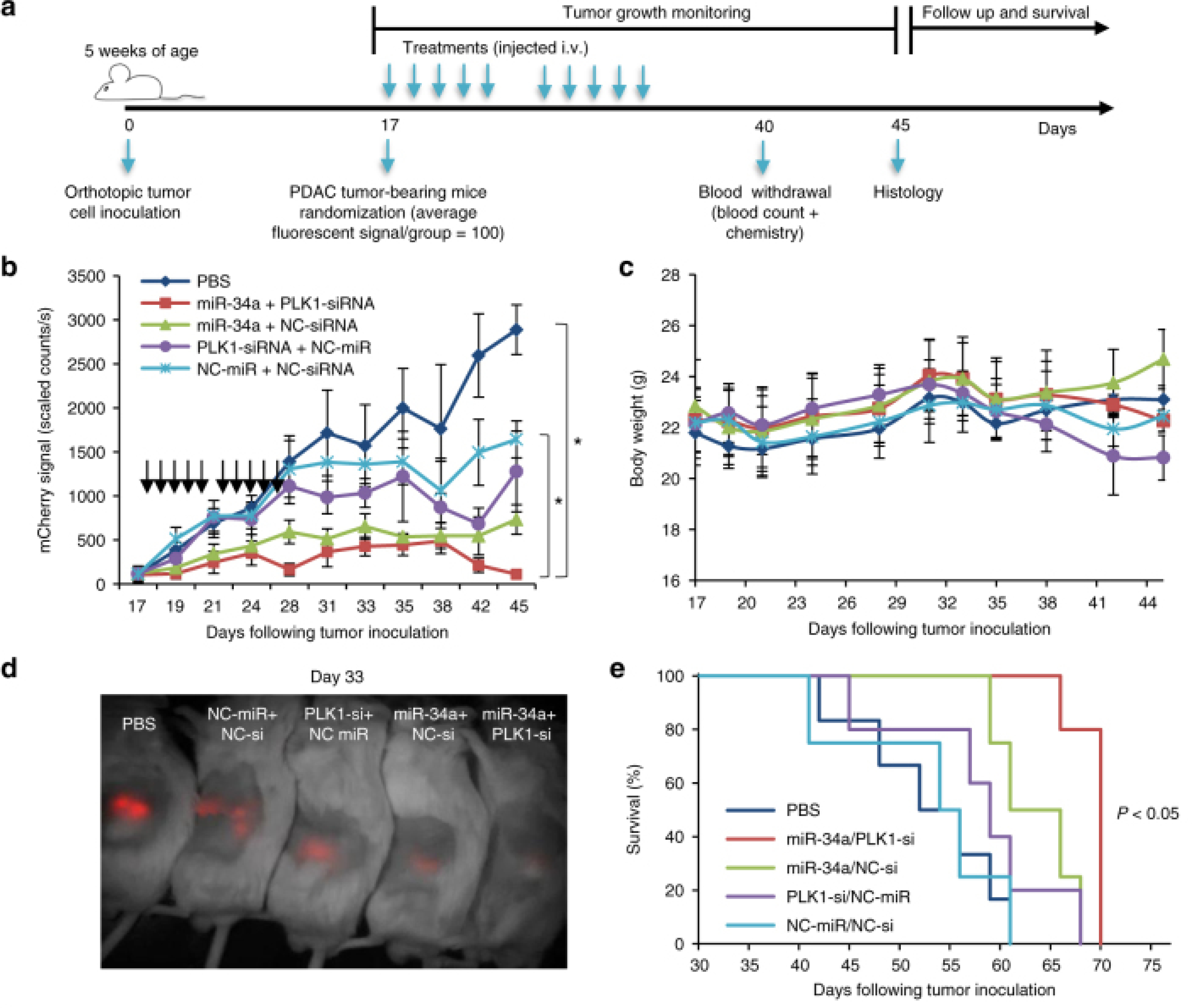
A study design for testing miRNA–siRNA combination efficacy in the orthotopic PDAC model. b Tumor growth curves from twice a week fluorescent measurement of tumor-bearing mice treated with APA complexed with miR-34a/PLK1-siRNA, miR-34a/NC-siRNA, PLK1-siRNA/NC-miR, NC-miR/NC-siRNA or PBS (treatments are marked with arrows). (n = 6, 7). Data represent mean ± SEM. One-way ANOVA. c In vivo toxicity via mouse body weight evaluation. Data represent mean ± SEM. d An image of a representative mouse from each treatment group 33 days post tumor inoculation showing the difference in tumor fluorescent signal. e Kaplan–Meier survival graph. Log-Rank test, P < 0.05 for the combination miR-34a/PLK1-siRNA compared to all other treatment groups. Reproduced from [188].
7. Conclusion and Future Direction
PDAC is an extraordinarily high malignancy cancer entity, particularly characterized by poor prognosis and constantly increasing patient numbers. The aggressive biology and the fact that most patients participate in advanced or disseminated disorder stages make the development of new PDAC therapeutic approaches one of the superordinate modern oncological science tasks. Research over the last 20 years led to a systematic multi-step model of PDAC growth and progress. While this has certainly changed our understanding of PDAC as a disease, so far, neither of these findings could be successfully converted into a medical breakthrough. It is becoming increasingly obvious that the clinical effectiveness of single-agent treatments tends to lag below acceptable estimates, and smart combinations seem to be required instead. In addition, PDAC anti-CSC therapies of the next generation should be produced to attack active stroma cells and target cells such as Wnt-cell components, PSC, MSC, and/or TAMs that are extremely penetrable by small molecules, nanoparticles, or oligonucleotides and perhaps immunotherapy.
Furthermore, nanomedicines’ ability to aggregate and target tumors could be leveraged to enhance early tumor identification, significantly improve survival, and increase the extent of surgical resection. Novel compounds and nanoparticles are continuing to be developed in non-viral vector technology [189]. And a combination of gene therapy with these, along with conventional drug therapy would also prove to be beneficial.
Despite substantial advances in cancer research over the last era, PDAC seems to have very poor survival rates. The present failure to diagnose early-stage prevents the use of effective treatments. Additionally, drug resistance growth is a key factor for recognizing current therapy failure in both the tumor and metastatic tissues. Hence, the improvement in survival of PDAC patients will occur not only through the identification of early serum markers but also through therapeutic approaches directed towards reducing pancreatic CSCs and decreasing drug resistance. Importantly, genetic analyses have strengthened our mechanistic and translational understanding of pancreatic cancer. Genetic principles and techniques are eventually applied to clinical practice, especially for precision medicine initiatives. However,, Epigenomics is evolving rapidly as a promising scientific and computational model for advancing the comprehension of this disease. More significantly, recent studies have identified possible actionable mechanisms supporting the assumption that prospective pancreatic cancer trials will include rigorous epigenomic therapy research. Therefore, epigenomics aims to produce a large amount of new biological as well as scientific relevant information.
In this respect, all the above strategies, and especially modern techniques, represent attractive strategies for both biologically inspired utilizing PDAC TME and immunotherapy strategies that might be the future for finding a new cure to manage PDAC disease.
Acknowledgments:
RA and HA would like to acknowledge the scholarship support from the College of Pharmacy at Taif University and Saudi Arabian Cultural Mission (SACM). SS acknowledges the support of Burroughs Wellcome Fund Collaborative Research Travel Grant (BWFCRTG). AKI acknowledges US Department of Defense CDMRP KCRP Idea Development Award #W81XWH1810471, The American Cancer Society grant #14-238-04-IRG and, The US National Institutes of Health, National Cancer Institute (NIH/NCI) grant R21CA179652 for funding support.
Footnotes
Conflict of Interest:
The authors declare that there are no conflicts of interest.
Reference:
- 1.Siegel RL; Miller KD; Jemal A Cancer statistics, 2020. CA. Cancer J. Clin 2020, 70, 7–30, doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Johnson SI Pancreatic adenocarcinoma. Pancreat. Imaging A Pattern-Based Approach to Radiol. Diagnosis with Pathol. Correl 2017, 371, 73–75, doi: 10.1007/978-3-319-52680-5_18. [DOI] [Google Scholar]
- 3.Rasheed ZA; Matsui W; Maitra A Pathology of pancreatic stroma in PDAC; 2012; ISBN 9788178955483. [PubMed]
- 4.Bortesi L; Pesci A; Bogina G; Castelli P; Zamboni G Ductal Adenocarcinoma of the Pancreas. Surg. Pathol. Clin 2011, 4, 487–521, doi: 10.1016/j.path.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Puleo F; Nicolle R; Blum Y; Cros J; Marisa L; Demetter P; Quertinmont E; Svrcek M; Elarouci N; Iovanna J; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3, doi: 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 6.Dougan SK The pancreatic cancer microenvironment. Cancer J. (United States) 2017, 23, 321–325, doi: 10.1097/PPO.0000000000000288. [DOI] [PubMed] [Google Scholar]
- 7.Erkan M; Hausmann S; Michalski CW; Fingerle AA; Dobritz M; Kleeff J; Friess H The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol 2012, 9, 454–467, doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 8.Kota J; Hancock J; Kwon J; Korc M Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [DOI] [PubMed] [Google Scholar]
- 9.Brown JM; Wilson WR Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [DOI] [PubMed] [Google Scholar]
- 10.Liu X; Jiang J; Meng H Transcytosis - An effective targeting strategy that is complementary to “EPR effect” for pancreatic cancer nano drug delivery. Theranostics 2019, 9, 8018–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown JM; Giaccia AJ The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [PubMed] [Google Scholar]
- 12.Vonderheide RH; Bayne LJ Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol 2013, 25, 200–205, doi: 10.1016/j.coi.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maitra A; Adsay NV; Argani P; Iacobuzio-Donahue C; De Marzo A; Cameron JL; Yeo CJ; Hruban RH Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod. Pathol 2003, 16, 902–912, doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 14.Kanda M; Matthaei H; Wu J; Hong SM; Yu J; Borges M; Hruban RH; Maitra A; Kinzler K; Vogelstein B; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsaab HO; Sau S; Alzhrani R; Tatiparti K; Bhise K; Kashaw SK; Iyer AK PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front. Pharmacol 2017, 8, 561, doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda H The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul 2001, 41, 189–207, doi: 10.1016/S0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 17.Yu B; Zhao X; Lee JL; Lee RJ Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009, 11, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manji GA; Olive KP; Saenger YM; Oberstein P Current and emerging therapies in metastatic pancreatic cancer. Clin. Cancer Res 2017, 23, 1670–1678, doi: 10.1158/1078-0432.CCR-16-2319. [DOI] [PubMed] [Google Scholar]
- 19.Adamska A; Domenichini A; Falasca M Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci 2017, 18, doi: 10.3390/ijms18071338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Zahaby SA; Elnaggar YSR; Abdallah OY Reviewing two decades of nanomedicine implementations in targeted treatment and diagnosis of pancreatic cancer: An emphasis on state of art. J. Control. Release 2019, 293, 21–35. [DOI] [PubMed] [Google Scholar]
- 21.Oettle H; Neuhaus P; Hochhaus A; Hartmann JT; Gellert K; Ridwelski K; Niedergethmann M; Zülke C; Fahlke J; Arning MB; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA - J. Am. Med. Assoc 2013, 310, 1473–1481, doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 22.Di Costanzo F; Carlini P; Doni L; Massidda B; Mattioli R; Iop A; Barletta E; Moscetti L; Recchia F; Tralongo P; et al. Gemcitabine with or without continuous infusion 5-FU in advanced pancreatic cancer: A randomised phase II trial of the Italian oncology group for clinical research (GOIRC). Br. J. Cancer 2005, 93, 185–189, doi: 10.1038/sj.bjc.6602640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neoptolemos JP; Cunningham D; Friess H; Bassi C; Stocken DD; Tait DM; Dunn JA; Dervenis C; Lacaine F; Hickey H; et al. Adjuvant therapy in pancreatic cancer: Historical and current perspectives. Ann. Oncol 2003, 14, 675–692, doi: 10.1093/annonc/mdg207. [DOI] [PubMed] [Google Scholar]
- 24.Neoptolemos JP; Moore MJ; Cox TF; Valle JW; Palmer DH; McDonald AC; Carter R; Tebbutt NC; Dervenis C; Smith D; et al. Effect of adjuvant chemotherapy with fluorouracil plus folinic acid or gemcitabine vs observation on survival in patients with resected periampullary adenocarcinoma: The ESPAC-3 periampullary cancer randomized trial; 2012; Vol. 308;. [DOI] [PubMed] [Google Scholar]
- 25.Vaccaro V; Sperduti I; Milella M FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med 2011, 365, 768–769, doi: 10.1056/nejmc1107627. [DOI] [PubMed] [Google Scholar]
- 26.Ueno M; Ozaka M; Ishii H; Sato T; Ikeda M; Uesugi K; Sata N; Miyashita K; Mizuno N; Tsuji K; et al. Phase II study of modified FOLFIRINOX for chemotherapy-naïve patients with metastatic pancreatic cancer. J. Clin. Oncol 2016, 34, 4111–4111, doi: 10.1200/jco.2016.34.15_suppl.4111. [DOI] [PubMed] [Google Scholar]
- 27.Mahaseth H; Brutcher E; Kauh J; Hawk N; Kim S; Chen Z; Kooby DA; Maithel SK; Landry J; El-Rayes BF Modified FOLFIRINOX regimen with improved safety and maintained efficacy in pancreatic adenocarcinoma. Pancreas 2013, 42, 1311–1315, doi: 10.1097/MPA.0b013e31829e2006. [DOI] [PubMed] [Google Scholar]
- 28.Ohba A; Ueno H; Sakamoto Y; Kondo S; Morizane C; Okusaka T Retrospective comparison of modified FOLFIRINOX with full-dose FOLFIRINOX for advanced pancreatic cancer: A Japanese cancer center experience. J. Clin. Oncol 2018, 36, 469–469, doi: 10.1200/jco.2018.36.4_suppl.469. [DOI] [Google Scholar]
- 29.Reni M; Cordio S; Milandri C; Passoni P; Bonetto E; Oliani C; Luppi G; Nicoletti R; Galli L; Bordonaro R; et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: A randomised controlled multicentre phase III trial. Lancet Oncol 2005, 6, 369–376, doi: 10.1016/S1470-2045(05)70175-3. [DOI] [PubMed] [Google Scholar]
- 30.Ottaiano A; Capozzi M; De Divitiis C; De Stefano A; Botti G; Avallone A; Tafuto S Gemcitabine mono-therapy versus gemcitabine plus targeted therapy in advanced pancreatic cancer: a meta-analysis of randomized phase III trials. Acta Oncol. (Madr) 2017, 56, 377–383, doi: 10.1080/0284186X.2017.1288922. [DOI] [PubMed] [Google Scholar]
- 31.Singh V; Sharma VR Gemcitabine, docetaxel, and capecitabine (GTX) as a first-line regimen in metastatic pancreatic adenocarcinomas (mPAC): A single institution experience. J. Clin. Oncol 2019, 37, 385–385, doi: 10.1200/jco.2019.37.4_suppl.385. [DOI] [Google Scholar]
- 32.Goldstein D; El-Maraghi RH; Hammel P; Heinemann V; Kunzmann V; Sastre J; Scheithauer W; Siena S; Tabernero J; Teixeira L; et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst 2015, 107, 1–10, doi: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- 33.Higuera O; Ghanem I; Nasimi R; Prieto I; Koren L; Feliu J Management of pancreatic cancer in the elderly. World J. Gastroenterol 2016, 22, 764–775, doi: 10.3748/wjg.v22.i2.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pernot S; Bachet JB; Portal A; Taieb J Reply to the comment on “Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: An AGEO prospective multicentre cohort.” Br. J. Cancer 2016, 114, e9, doi: 10.1038/bjc.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Mackelenbergh MG; Stroes CI; Spijker R; Van Eijck CHJ; Wilmink JW; Bijlsma MF; Van Laarhoven HWM Clinical trials targeting the stroma in pancreatic cancer: A systematic review and meta-analysis. Cancers (Basel). 2019, 11, 1–23, doi: 10.3390/cancers11050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buscail L; Bournet B; Vernejoul F; Cambois G; Lulka H; Hanoun N; Dufresne M; Meulle A; Vignolle-Vidoni A; Ligat L; et al. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: Safety, biodistribution, and preliminary clinical findings. Mol. Ther 2015, 23, 779–789, doi: 10.1038/mt.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald PC; Chafe SC; Brown WS; Saberi S; Swayampakula M; Venkateswaran G; Nemirovsky O; Gillespie JA; Karasinska JM; Kalloger SE; et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157, 823–837, doi: 10.1053/j.gastro.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Grossberg AJ; Chu LC; Deig CR; Fishman EK; Hwang WL; Maitra A; Marks DL; Mehta A; Nabavizadeh N; Simeone DM; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA. Cancer J. Clin 2020, 70, 375–403, doi: 10.3322/caac.21626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X; Wang X; Sun W; Cheng K; Qin H; Han X; Lin Y; Wang Y; Lang J; Zhao R; et al. Precision design of nanomedicines to restore gemcitabine chemosensitivity for personalized pancreatic ductal adenocarcinoma treatment. Biomaterials 2018, 158, 44–55, doi: 10.1016/j.biomaterials.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 40.Feig C; Gopinathan A; Neesse A; Chan DS; Cook N; Tuveson DA The pancreas cancer microenvironment. Clin. Cancer Res 2012, 18, 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore MJ; Goldstein D; Hamm J; Figer A; Hecht JR; Gallinger S; Au HJ; Murawa P; Walde D; Wolff RA; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol 2007, 25, 1960–1966, doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 42.Hadden M; Mittal A; Samra J; Zreiqat H; Sahni S; Ramaswamy Y Mechanically stressed cancer microenvironment: Role in pancreatic cancer progression. Biochim. Biophys. Acta - Rev. Cancer 2020, 1874, 188418. [DOI] [PubMed] [Google Scholar]
- 43.Gopinathan A; Tuveson DA The use of GEM models for experimental cancer therapeutics. DMM Dis. Model. Mech 2008, 1, 83–86, doi: 10.1242/dmm.000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh M; Lima A; Molina R; Hamilton P; Clermont AC; Devasthali V; Thompson JD; Cheng JH; Bou Reslan H; Ho CCK; et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol 2010, 28, 585–593, doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 45.Burris HA; Moore MJ; Andersen J; Green MR; Rothenberg ML; Modiano MR; Cripps MC; Portenoy RK; Storniolo AM; Tarassoff P; et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: A randomized trial; 1997; Vol. 15, pp. 2403–2413;. [DOI] [PubMed] [Google Scholar]
- 46.Das KK; Brown JW; Fernandez del-Castillo C; Huynh T; Mills JC; Matsuda Y; Das KM; Mino-Kenudson M mAb Das-1 Identifies Pancreatic Ductal Adenocarcinoma and High-grade Pancreatic Intraepithelial Neoplasia with High Accuracy. Hum. Pathol 2021, doi: 10.1016/j.humpath.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ho WJ; Jaffee EM; Zheng L The tumour microenvironment in pancreatic cancer — clinical challenges and opportunities. Nat. Rev. Clin. Oncol 2020, 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olive KP; Jacobetz MA; Davidson CJ; Gopinathan A; McIntyre D; Honess D; Madhu B; Goldgraben MA; Caldwell ME; Allard D; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (80-. ). 2009, 324, 1457–1461, doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chantrill L; Goldstein D Pancreatic cancer. Cancer Forum 2016, 40, 3–5, doi: 10.5005/jp/books/13056_47. [DOI] [Google Scholar]
- 50.Fu Y; Liu S; Zeng S; Shen H The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das S; Shapiro B; Vucic EA; Vogt S; Bar-Sagi D Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 2020, 80, 1088–1101, doi: 10.1158/0008-5472.CAN-19-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Löhr M; Schmidt C; Ringel J; Kluth M; Müller P; Nizze H; Jesnowski R Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [PubMed] [Google Scholar]
- 53.Pothula SP; Pirola RC; Wilson JS; Apte MV Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418. [DOI] [PubMed] [Google Scholar]
- 54.Erkan M; Adler G; Apte MV; Bachem MG; Buchholz M; Detlefsen S; Esposito I; Friess H; Gress TM; Habisch HJ; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178, doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hwang RF; Moore T; Arumugam T; Ramachandran V; Amos KD; Rivera A; Ji B; Evans DB; Logsdon CD Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926, doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Armstrong T; Packham G; Murphy LB; Bateman AC; Conti JA; Fine DR; Johnson CD; Benyon RC; Iredale JP Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res 2004, 10, 7427–7437, doi: 10.1158/1078-0432.CCR-03-0825. [DOI] [PubMed] [Google Scholar]
- 57.Seux M; Peuget S; Montero MP; Siret C; Rigot V; Clerc P; Gigoux V; Pellegrino E; Pouyet L; N’Guessan P; et al. TP53INP1 decreases pancreatic cancer cell migration by regulating SPARC expression. Oncogene 2011, 30, 3049–3061, doi: 10.1038/onc.2011.25. [DOI] [PubMed] [Google Scholar]
- 58.Mahadevan D; Von Hoff DD Tumor-stroma interactions in pancreatic ductal adenocarcinoma; 2007; Vol. 6, pp. 1186–1197;. [DOI] [PubMed] [Google Scholar]
- 59.Ottaviano AJ; Sun L; Ananthanarayanan V; Munshi HG Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-β1. Cancer Res. 2006, 66, 7032–7040, doi: 10.1158/0008-5472.CAN-05-4421. [DOI] [PubMed] [Google Scholar]
- 60.Jaster R; Sparmann G; Emmrich J; Liebe S Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut 2002, 51, 579–584, doi: 10.1136/gut.51.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider E; Schmid-Kotsas A; Zhao J; Weidenbach H; Schmid RM; Menke A; Adler G; Waltenberger J; Grünert A; Bachem MG Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol. - Cell Physiol 2001, 281, doi: 10.1152/ajpcell.2001.281.2.c532. [DOI] [PubMed] [Google Scholar]
- 62.Mani SA; Guo W; Liao MJ; Eaton EN; Ayyanan A; Zhou AY; Brooks M; Reinhard F; Zhang CC; Shipitsin M; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715, doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reya T; Morrison SJ; Clarke MF; Weissman IL Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [DOI] [PubMed] [Google Scholar]
- 64.Li C; Heidt DG; Dalerba P; Burant CF; Zhang L; Adsay V; Wicha M; Clarke MF; Simeone DM Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037, doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 65.Al-Hajj M; Wicha MS; Benito-Hernandez A; Morrison SJ; Clarke MF Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3983–3988, doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Brien CA; Pollett A; Gallinger S; Dick JE A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110, doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 67.Singh SK; Hawkins C; Clarke ID; Squire JA; Bayani J; Hide T; Henkelman RM; Cusimano MD; Dirks PB Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401, doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 68.Chen C; Zhao S; Karnad A; Freeman JW The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol 2018, 11, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao S; Chen C; Chang K; Karnad A; Jagirdar J; Kumar AP; Freeman JW CD44 expression level and isoform contributes to pancreatic cancer cell plasticity, invasiveness, and response to therapy. Clin. Cancer Res 2016, 22, 5592–5604, doi: 10.1158/1078-0432.CCR-15-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dalla Pozza E; Lerda C; Costanzo C; Donadelli M; Dando I; Zoratti E; Scupoli MT; Beghelli S; Scarpa A; Fattal E; et al. Targeting gemcitabine containing liposomes to CD44 expressing pancreatic adenocarcinoma cells causes an increase in the antitumoral activity. Biochim. Biophys. Acta - Biomembr 2013, 1828, 1396–1404, doi: 10.1016/j.bbamem.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 71.Wickens JM; Alsaab HO; Kesharwani P; Bhise K; Amin MCIM; Tekade RK; Gupta U; Iyer AK Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov. Today 2017, 22, 665–680, doi: 10.1016/j.drudis.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z; Sau S; Alsaab HO; Iyer AK CD44 directed nanomicellar payload delivery platform for selective anticancer effect and tumor specific imaging of triple negative breast cancer. Nanomedicine Nanotechnology, Biol. Med 2018, 14, 1441–1454, doi: 10.1016/j.nano.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sierra JR; Tsao MS c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol 2011, 3, S21–S35, doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsumoto K; Nakamura T Emerging multipotent aspects of hepatocyte growth factor; 1996; Vol. 119;. [DOI] [PubMed] [Google Scholar]
- 75.Corso S; Comoglio PM; Giordano S Cancer therapy: Can the challenge be MET? Trends Mol. Med. 2005, 11, 284–292, doi: 10.1016/j.molmed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 76.Shah AN; Summy JM; Zhang J; Park SI; Parikh NU; Gallick GE Development and Characterization of Gemcitabine-Resistant Pancreatic Tumor Cells. Ann. Surg. Oncol 2007, 14, 3629–3637, doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 77.Li C; Wu J; Hynes M; Dosch J; Sarkar B; Welling TH; Pasca Di Magliano M; Simeone DM C-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, doi: 10.1053/j.gastro.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 78.Cazes A; Betancourt O; Esparza E; Mose ES; Jaquish D; Wong E; Wascher AA; Tiriac H; Gymnopoulos M; Lowy AM A MET Targeting Antibody-Drug Conjugate Overcomes Gemcitabine Resistance in Pancreatic Cancer. Clin. Cancer Res 2021, clincanres.3210.2020, doi: 10.1158/1078-0432.ccr-20-3210. [DOI] [PubMed] [Google Scholar]
- 79.Hermann PC; Huber SL; Herrler T; Aicher A; Ellwart JW; Guba M; Bruns CJ; Heeschen C Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323, doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 80.Yamasaki A; Yanai K; Onishi H Hypoxia and pancreatic ductal adenocarcinoma. Cancer Lett. 2020, 484, 9–15. [DOI] [PubMed] [Google Scholar]
- 81.Chang Q; Jurisica I; Do T; Hedley DW Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011, 71, 3110–3120, doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 82.Jin X; Dai L; Ma Y; Wang J; Liu Z Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pouysségur J; Dayan F; Mazure NM Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443, doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 84.Brihimi-Horn MC; Pouysségur J HIF at a glance. J. Cell Sci 2009, 122, 1055–1057, doi: 10.1242/jcs.035022. [DOI] [PubMed] [Google Scholar]
- 85.Lu X; Kang Y Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res 2010, 16, 5928–5935, doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Erkan M; Reiser-Erkan C; Michalski CW; Deucker S; Sauliunaite D; Streit S; Esposito I; Friess H; Kleeff J Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508, doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koong AC; Mehta VK; Le QT; Fisher GA; Terris DJ; Brown JM; Bastidas AJ; Vierra M Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys 2000, 48, 919–922, doi: 10.1016/S0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 88.Zhong H; De Marzo AM; Laughner E; Lim M; Hilton DA; Zagzag D; Buechler P; Isaacs WB; Semenza GL; Simons JW Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [PubMed] [Google Scholar]
- 89.Cook N; Frese KK; Bapiro TE; Jacobetz MA; Gopinathan A; Miller JL; Rao SS; Demuth T; Howat WJ; Jodrell DI; et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J. Exp. Med 2012, 209, 437–444, doi: 10.1084/jem.20111923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyamoto Y; Maitra A; Ghosh B; Zechner U; Argani P; Iacobuzio-Donahue CA; Sriuranpong V; Iso T; Meszoely IM; Wolfe MS; et al. Notch mediates TGFα-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576, doi: 10.1016/S1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 91.Wagner M; Luhrs H; Kloppel G; Adler G; Schmid RM Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology 1998, 115, 1254–1262, doi: 10.1016/S0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- 92.Chang JH; Jiang Y; Pillarisetty VG Role of immune cells in pancreatic cancer from bench to clinical application An updated review. Med. (United States) 2016, 95, e5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Protti MP; De Monte L Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol 2013, 4 August, doi: 10.3389/fphys.2013.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsai F; Lopes M; Zhou M; … of the OP-P; 2015, undefined K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Natl. Acad Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kleeff J; Korc M; Apte M; La Vecchia C; Johnson CD; Biankin AV; Neale RE; Tempero M; Tuveson DA; Hruban RH; et al. Pancreatic cancer. Nat. Rev. Dis. Prim 2016, 2, 1–22, doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 96.Zeitouni D; Pylayeva-Gupta Y; Der C; Cancers KB-; 2016, undefined KRAS mutant pancreatic cancer: no lone path to an effective treatment. [DOI] [PMC free article] [PubMed]
- 97.Ogura T; Yamao K; Hara K; Mizuno N; Hijioka S; Imaoka H; Sawaki A; Niwa Y; Tajika M; Kondo S; et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J. Gastroenterol 2013, 48, 640–646, doi: 10.1007/s00535-012-0664-2. [DOI] [PubMed] [Google Scholar]
- 98.Cox A; GTPases CD-S; 2010, undefined Ras history: The saga continues. Taylor Fr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh A; Greninger P; Rhodes D; Koopman L; Violette S; Bardeesy N; Settleman J A Gene Expression Signature Associated with “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell 2009, 15, 489–500, doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ying H; Kimmelman AC; Lyssiotis CA; Hua S; Chu GC; Fletcher-Sananikone E; Locasale JW; Son J; Zhang H; Coloff JL; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670, doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Govindan R; Fakih M; Price T; Falchook G; Desai J; Kuo J; Strickler J; Krauss J; Li B; Denlinger C; et al. OA02.02 Phase 1 Study of Safety, Tolerability, PK and Efficacy of AMG 510, a Novel KRASG12C Inhibitor, Evaluated in NSCLC. J. Thorac. Oncol 2019, 14, S208, doi: 10.1016/j.jtho.2019.08.412. [DOI] [Google Scholar]
- 102.Bryant KL; Mancias JD; Kimmelman AC; Der CJ KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci 2014, 39, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cox AD; Fesik SW; Kimmelman AC; Luo J; Der CJ Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov 2014, 13, 828–851, doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mills LD; Zhang Y; Marler RJ; Herreros-Villanueva M; Zhang L; Almada LL; Couch F; Wetmore C; Di Magliano MP; Fernandez-Zapico ME Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem 2013, 288, 11786–11794, doi: 10.1074/jbc.M112.438846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Golan T; Javle M DNA repair dysfunction in pancreatic cancer: A clinically relevant subtype for drug development. JNCCN J. Natl. Compr. Cancer Netw 2017, 15, 1063–1069, doi: 10.6004/jnccn.2017.0133. [DOI] [PubMed] [Google Scholar]
- 106.Kota J; Hancock J; Kwon J; Korc M Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49, doi: 10.1016/j.canlet.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 107.Wickens JM; Alsaab HO; Kesharwani P; Bhise K; Amin MCIM; Tekade RK; Gupta U; Iyer AK Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov. Today 2017, 22, doi: 10.1016/j.drudis.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Minchinton AI; Tannock IF Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [DOI] [PubMed] [Google Scholar]
- 109.Chen IM; Willumsen N; Dehlendorff C; Johansen AZ; Jensen BV; Hansen CP; Hasselby JP; Bojesen SE; Pfeiffer P; Nielsen SE; et al. Clinical value of serum hyaluronan and propeptide of type III collagen in patients with pancreatic cancer. Int. J. Cancer 2020, 146, 2913–2922, doi: 10.1002/ijc.32751. [DOI] [PubMed] [Google Scholar]
- 110.Sato N; Kohi S; Hirata K; Goggins M Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. 2016, 107, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Groot HJ; Lubberts S; de Wit R; Witjes JA; Martijn Kerst J; de Jong IJ; Groenewegen G; van den Eertwegh AJ; Poortmans PM; Klümpen H-J; et al. JOURNAL OF CLINICAL ONCOLOGY Risk of Solid Cancer After Treatment of Testicular Germ Cell Cancer in the Platinum Era. J Clin Oncol 2018, 36, 359–366, doi: 10.1200/JCO. [DOI] [PubMed] [Google Scholar]
- 112.Hakim N; Patel R; Devoe C; Saif MW Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas – Open J. 2019, 3, e1–e4, doi: 10.17140/poj-3-e010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Steele NG; Biffi G; Kemp SB; Zhang Y; Drouillard D; Syu L-J; Hao Y; Oni TE; Brosnan E; Elyada E; et al. Inhibition of Hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin. Cancer Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yauch RL; Gould SE; Scales SJ; Tang T; Tian H; Ahn CP; Marshall D; Fu L; Januario T; Kallop D; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [DOI] [PubMed] [Google Scholar]
- 115.Neesse A Therapeutic Targeting of Stromal Components. In Translational Pancreatic Cancer Research; Springer, 2020; pp. 157–168. [Google Scholar]
- 116.Heretsch P; Tzagkaroulaki L; Giannis A Cyclopamine and hedgehog signaling: chemistry, biology, medical perspectives. Angew. Chemie Int. Ed 2010, 49, 3418–3427. [DOI] [PubMed] [Google Scholar]
- 117.Zhang B; Jiang T; Shen S; She X; Tuo Y; Hu Y; Pang Z; Jiang X Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016, 103, 12–21. [DOI] [PubMed] [Google Scholar]
- 118.Sun J; Russell CC; Scarlett CJ; McCluskey A Small molecule inhibitors in pancreatic cancer. RSC Med. Chem 2020, 11, 164–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bahrami A; Khazaei M; Bagherieh F; Ghayour-Mobarhan M; Maftouh M; Hassanian SM; Avan A Targeting stroma in pancreatic cancer: Promises and failures of targeted therapies. J. Cell. Physiol 2017, 232, 2931–2937. [DOI] [PubMed] [Google Scholar]
- 120.Curran T Reproducibility of academic preclinical translational research: lessons from the development of Hedgehog pathway inhibitors to treat cancer. Open Biol. 2018, 8, 180098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ko AH; LoConte N; Tempero MA; Walker EJ; Kelley RK; Lewis S; Chang W-C; Kantoff E; Vannier MW; Catenacci D V; et al. A phase I study of FOLFIRINOX plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 2016, 45, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bailey JM; Swanson BJ; Hamada T; Eggers JP; Singh PK; Caffery T; Ouellette MM; Hollingsworth MA Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res 2008, 14, 5995–6004, doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tian H; Callahan CA; Dupree KJ; Darbonne WC; Ahn CP; Scales SJ; De Sauvage FJ Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4254–4259, doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Masso-Valles D; Jauset T; Serrano E; Sodir NM; Pedersen K; Affara NI; Whitfield JR; Beaulieu ME; Evan GI; Elias L; et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015, 75, 1675–1681, doi: 10.1158/0008-5472.CAN-14-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Draper A Updates in pancreatic cancer: Modest gains and hopeful targets. J. Oncol. Pharm. Pract 2019, 25, 101–109, doi: 10.1177/1078155218763035. [DOI] [PubMed] [Google Scholar]
- 126.Kizaka-Kondoh S; Itasaka S; Zeng L; Tanaka S; Zhao T; Takahashi Y; Shibuya K; Hirota K; Semenza GL; Hiraoka M Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin. Cancer Res. 2009, 15, 3433–3441, doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]
- 127.Conway JRW; Warren SC; Herrmann D; Murphy KJ; Cazet AS; Vennin C; Shearer RF; Killen MJ; Magenau A; Mélénec P; et al. Intravital Imaging to Monitor Therapeutic Response in Moving Hypoxic Regions Resistant to PI3K Pathway Targeting in Pancreatic Cancer. Cell Rep. 2018, 23, 3312–3326, doi: 10.1016/j.celrep.2018.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hajj C; Russell J; Hart CP; Goodman KA; Lowery MA; Haimovitz-Friedman A; Deasy JO; Humm JL A Combination of Radiation and the Hypoxia-Activated Prodrug Evofosfamide (TH-302) is Efficacious against a Human Orthotopic Pancreatic Tumor Model. Transl. Oncol 2017, 10, 760–765, doi: 10.1016/j.tranon.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Salem A; Asselin MC; Reymen B; Jackson A; Lambin P; West CML; O’Connor JPB; Faivre-Finn C Targeting hypoxia to improve non–small cell lung cancer outcome. J. Natl. Cancer Inst 2018, 110, doi: 10.1093/jnci/djx160. [DOI] [PubMed] [Google Scholar]
- 130.Kizaka-Kondoh S; Itasaka S; Zeng L; Tanaka S; Zhao T; Takahashi Y; Shibuya K; Hirota K; Semenza GL; Hiraoka M Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin. Cancer Res. 2009, 15, 3433–3441, doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]
- 131.Belalcazar A; Shaib WL; Farren MR; Zhang C; Chen Z; Yang L; Lesinski GB; El-Rayes BF; Nagaraju GP Inhibiting heat shock protein 90 and the ubiquitin-proteasome pathway impairs metabolic homeostasis and leads to cell death in human pancreatic cancer cells. Cancer 2017, 123, 4924–4933, doi: 10.1002/cncr.30944. [DOI] [PubMed] [Google Scholar]
- 132.Bobrov E; Skobeleva N; Restifo D; Beglyarova N; Cai KQ; Handorf E; Campbell K; Proia DA; Khazak V; Golemis EA; et al. Targeted delivery of chemotherapy using HSP90 inhibitor drug conjugates is highly active against pancreatic cancer models. Oncotarget 2017, 8, 4399–4409, doi: 10.18632/oncotarget.12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bailey KM; Cornnell HH; Ibrahim-Hashim A; Wojtkowiak JW; Hart CP; Zhang X; Leos R; Martinez GV; Baker AF; Gillies RJ Evaluation of the “steal” phenomenon on the efficacy of hypoxia activated prodrug TH-302 in pancreatic cancer. PLoS One 2014, 9, e113586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guillaumond F; Iovanna JL; Vasseur S Pancreatic tumor cell metabolism: focus on glycolysis and its connected metabolic pathways. Arch. Biochem. Biophys 2014, 545, 69–73. [DOI] [PubMed] [Google Scholar]
- 135.Masamune A; Hamada S; Kikuta K; Takikawa T; Miura S; Nakano E; Shimosegawa T The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand. J. Gastroenterol 2013, 48, 602–609. [DOI] [PubMed] [Google Scholar]
- 136.Murphy JE; Wo JY; Ryan DP; Clark JW; Jiang W; Yeap BY; Drapek LC; Ly L; Baglini CV; Blaszkowsky LS; et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol. 2019, 5, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Doi C; Egashira N; Kawabata A; Maurya DK; Ohta N; Uppalapati D; Ayuzawa R; Pickel L; Isayama Y; Troyer D; et al. Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer 2010, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Infante JR; Matsubayashi H; Sato N; Tonascia J; Klein AP; Riall TA; Yeo C; Iacobuzio-Donahue C; Goggins M Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol 2007, 25, 319–325, doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 139.Mantoni TS; Schendel RRE; Rödel F; Niedobitek G; Al-Assar O; Masamune A; Brunner TB Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol. Ther 2008, 7, 1806–1815, doi: 10.4161/cbt.7.11.6846. [DOI] [PubMed] [Google Scholar]
- 140.Frese KK; Neesse A; Cook N; Bapiro TE; Lolkema MP; Jodrell DI; Tuveson DA Nab-paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269, doi: 10.1158/2159-8290.CD-11-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Razi E; Radak M; Mahjoubin-Tehran M; Talebi S; Shafiee A; Hajighadimi S; Moradizarmehri S; Sharifi H; Mousavi N; Sarvizadeh M; et al. Cancer stem cells as therapeutic targets of pancreatic cancer. Fundam. Clin. Pharmacol. 2020, 34, 202–212, doi: 10.1111/fcp.12521. [DOI] [PubMed] [Google Scholar]
- 142.Venkatasubbarao K; Peterson L; Zhao S; Hill P; Cao L; Zhou Q; Nawrocki ST; Freeman JW Inhibiting signal transducer and activator of transcription-3 increases response to gemcitabine and delays progression of pancreatic cancer. Mol. Cancer 2013, 12, doi: 10.1186/1476-4598-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schizas D; Charalampakis N; Kole C; Economopoulou P; Koustas E; Gkotsis E; Ziogas D; Psyrri A; Karamouzis MV Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [DOI] [PubMed] [Google Scholar]
- 144.Upadhrasta S; Zheng L Strategies in Developing Immunotherapy for Pancreatic Cancer: Recognizing and Correcting Multiple Immune “Defects” in the Tumor Microenvironment. J. Clin. Med 2019, 8, 1472, doi: 10.3390/jcm8091472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Macherla S; Laks S; Naqash AR; Bulumulle A; Zervos E; Muzaffar M Emerging role of immune checkpoint blockade in pancreatic cancer. Int. J. Mol. Sci 2018, 19, 1–12, doi: 10.3390/ijms19113505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koustas E; Sarantis P; Papavassiliou AG; Karamouzis MV The resistance mechanisms of checkpoint inhibitors in solid tumors. Biomolecules 2020, 10, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chattopadhyay PK; Gierahn TM; Roederer M; Love JC Single-cell technologies for monitoring immune systems. Nat. Immunol 2014, 15, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Young K; Hughes DJ; Cunningham D; Starling N Immunotherapy and pancreatic cancer: unique challenges and potential opportunities. Ther. Adv. Med. Oncol 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Royal RE; Levy C; Turner K; Mathur A; Hughes M; Kammula US; Sherry RM; Topalian SL; Yang JC; Lowy I; et al. Phase 2 trial of single agent ipilimumab (Anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother 2010, 33, 828–833, doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Segal NH; Hamid O; Hwu W; Massard C; Butler M; Antonia S; Blake-Haskins A; Robbins PB; Li X; Vasselli J; et al. A Phase I Multi-Arm Dose-Expansion Study of the Anti-Programmed Cell Death-Ligand-1 (Pd-L1) Antibody Medi4736: Preliminary Data. Ann. Oncol 2014, 25, iv365, doi: 10.1093/annonc/mdu342.11. [DOI] [Google Scholar]
- 151.Kabacaoglu D; Ciecielski KJ; Ruess DA; Algül H Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: Current limitations and future options. Front. Immunol 2018, 9, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Amedei A; Niccolai E; Prisco D Pancreatic cancer: Role of the immune system in cancer progression and vaccine-based immunotherapy. Hum. Vaccines Immunother 2014, 10, 3354–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li J; Byrne KT; Yan F; Yamazoe T; Chen Z; Baslan T; Richman LP; Lin JH; Sun YH; Rech AJ; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7, doi: 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Delitto D; Wallet SM; Hughes SJ Targeting tumor tolerance: A new hope for pancreatic cancer therapy? Pharmacol. Ther 2016, 166, 9–29, doi: 10.1016/j.pharmthera.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 155.Smyth MJ; Ngiow SF; Ribas A; Teng MWL Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol 2016, 13, 143–158. [DOI] [PubMed] [Google Scholar]
- 156.Hilmi M; Bartholin L; Neuzillet C Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol 2018, 24, 2137–2151, doi: 10.3748/wjg.v24.i20.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Balachandran VP; Łuksza M; Zhao JN; Makarov V; Moral JA; Remark R; Herbst B; Askan G; Bhanot U; Senbabaoglu Y; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, S12–S16, doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yarchoan M; Johnson BA; Lutz ER; Laheru DA; Jaffee EM Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.O’Reilly EM; Oh DY; Dhani N; Renouf DJ; Lee MA; Sun W; Fisher G; Hezel A; Chang SC; Vlahovic G; et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial JAMA Oncol. 2019, 5, 1431–1438, doi: 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Principe DR; Narbutis M; Kumar S; Park A; Viswakarma N; Dorman MJ; Kamath SD; Grippo PJ; Fishel ML; Hwang RF; et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer Res. 2020, 80, 3101–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Principe DR; Narbutis M; Kumar S; Park A; Viswakarma N; Dorman MJ; Kamath SD; Munshi HG; Rana A Abstract A20: Gemcitabine primes the pancreatic tumor microenvironment for second-line immunotherapy. In Proceedings of the Tumor Microenvironment; American Association for Cancer Research, 2019; Vol. 79, pp. A20–A20. [Google Scholar]
- 162.Duffy AG; Greten TF Immunological off-target effects of standard treatments in gastrointestinal cancers. Ann. Oncol 2014, 25, 24–32, doi: 10.1093/annonc/mdt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bracci L; Schiavoni G; Sistigu A; Belardelli F Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hou P; Kapoor A; Zhang Q; Li J; Wu CJ; Li J; Lan Z; Tang M; Ma X; Ackroyd JJ; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077, doi: 10.1158/2159-8290.CD-19-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nywening TM; Wang-Gillam A; Sanford DE; Belt BA; Panni RZ; Cusworth BM; Toriola AT; Nieman RK; Worley LA; Yano M; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662, doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eric L; Yeo CJ; Lillemoe KD; Biedrzycki B; Kobrin B; Herman J; Sugar E; Piantadosi S; Cameron JL; Solt S; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma: A phase II trial of safety, efficacy, and immune activation. Ann. Surg 2011, 253, 328–335, doi: 10.1097/SLA.0b013e3181fd271c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Le DT; Lutz E; Uram JN; Sugar EA; Onners B; Solt S; Zheng L; Diaz LA; Donehower RC; Jaffee EM; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother 2013, 36, 382–389, doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jaffee EM; Hruban RH; Biedrzycki B; Laheru D; Schepers K; Sauter PR; Goemann M; Coleman J; Grochow L; Donehower RC; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol 2001, 19, 145–156, doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 169.Maoz A; Rennert G; Gruber SB T-Cell Transfer Therapy Targeting Mutant KRAS. N. Engl. J. Med 2017, 376, e11, doi: 10.1056/nejmc1616637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xie Y; Hang Y; Wang Y; Sleightholm R; Prajapati DR; Bader J; Yu A; Tang W; Jaramillo L; Li J; et al. Stromal Modulation and Treatment of Metastatic Pancreatic Cancer with Local Intraperitoneal Triple miRNA/siRNA Nanotherapy. ACS Nano 2020, 14, 255–271, doi: 10.1021/acsnano.9b03978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Murahari M; Yergeri M Identification and Usage of Fluorescent Probes as Nanoparticle Contrast Agents in Detecting Cancer. Curr. Pharm. Des 2013, 19, 4622–4640, doi: 10.2174/1381612811319250009. [DOI] [PubMed] [Google Scholar]
- 172.Birhanu G; Javar HA; Seyedjafari E; Zandi-Karimi A Nanotechnology for delivery of gemcitabine to treat pancreatic cancer. Biomed. Pharmacother. 2017, 88, 635–643. [DOI] [PubMed] [Google Scholar]
- 173.Wang L; An Y; Yuan C; Zhang H; Liang C; Ding F; Gao Q; Zhang D GEM-loaded magnetic albumin nanospheres modified with cetuximab for simultaneous targeting, magnetic resonance imaging, and double-targeted thermochemotherapy of pancreatic cancer cells. Int. J. Nanomedicine 2015, 10, 2507–2519, doi: 10.2147/IJN.S77642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yu X; Di Y; Xie C; Song Y; He H; Li H; Pu X; Lu W; Fu D; Jin C An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line. Int. J. Nanomedicine 2015, 10, 6825–6834, doi: 10.2147/IJN.S93835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li JM; Chen W; Wang H; Jin C; Yu XJ; Lu WY; Cui L; Fu DL; Ni QX; Hou HM Preparation of albumin nanospheres loaded with gemcitabine and their cytotoxicity against BXPC-3 cells in vitro. Acta Pharmacol. Sin 2009, 30, 1337–1343, doi: 10.1038/aps.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang-Gillam A; Li CP; Bodoky G; Dean A; Shan YS; Jameson G; MacArulla T; Lee KH; Cunningham D; Blanc JF; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557, doi: 10.1016/S0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- 177.Kataoka K Smart targeted therapy by self-assembled supramolecular nanosystems. Nanomedicine Nanotechnology, Biol. Med 2016, 12, 451, doi: 10.1016/j.nano.2015.12.010. [DOI] [Google Scholar]
- 178.Sudha T; Bharali DJ; Yalcin M; Darwish NHE; Coskun MD; Keating KA; Lin HY; Davis PJ; Mousa SA Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int. J. Nanomedicine 2017, 12, 1305–1315, doi: 10.2147/IJN.S123742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Couvreur P; Reddy LH; Mangenot S; Poupaert JH; Desmaële D; Lepêtre-Mouelhi S; Pili B; Bourgaux C; Amenitsch H; Ollivon M Discovery of new hexagonal supramolecular nanostructures formed by squalenoylation of an anticancer nucleoside analogue. Small 2008, 4, 247–253, doi: 10.1002/smll.200700731. [DOI] [PubMed] [Google Scholar]
- 180.Réjiba S; Reddy LH; Bigand C; Parmentier C; Couvreur P; Hajri A Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine Nanotechnology, Biol. Med 2011, 7, 841–849, doi: 10.1016/j.nano.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 181.Adiseshaiah PP; Crist RM; Hook SS; McNeil SE Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat. Rev. Clin. Oncol 2016, 13, 750–765, doi: 10.1038/nrclinonc.2016.119. [DOI] [PubMed] [Google Scholar]
- 182.Alzhrani R; Alsaab HO; Petrovici A; Bhise K; Vanamala K; Sau S; Krinock MJ; Iyer AK Improving the therapeutic efficiency of noncoding RNAs in cancers using targeted drug delivery systems. Drug Discov. Today 2020, 25, 718–730, doi: 10.1016/j.drudis.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zorde Khvalevsky E; Gabai R; Rachmut IH; Horwitz E; Brunschwig Z; Orbach A; Shemi AA; Golan T; Domb AJ; Yavin E; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 20723–20728, doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhao X; Li F; Li Y; Wang H; Ren H; Chen J; Nie G; Hao J Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 2015, 46, 13–25, doi: 10.1016/j.biomaterials.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 185.Pittella F; Miyata K; Maeda Y; Suma T; Watanabe S; Chen Q; Christie RJ; Osada K; Nishiyama N; Kataoka K Pancreatic cancer therapy by systemic administration of VEGF siRNA contained in calcium phosphate/charge-conversional polymer hybrid nanoparticles. J. Control. Release 2012, 161, 868–874, doi: 10.1016/j.jconrel.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 186.Vassie JA; Whitelock JM; Lord MS Endocytosis of cerium oxide nanoparticles and modulation of reactive oxygen species in human ovarian and colon cancer cells. Acta Biomater. 2017, 50, 127–141, doi: 10.1016/j.actbio.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 187.Wason MS; Colon J; Das S; Seal S; Turkson J; Zhao J; Baker CH Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine Nanotechnology, Biol. Med 2013, 9, 558–569, doi: 10.1016/j.nano.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gibori H; Eliyahu S; Krivitsky A; Ben-Shushan D; Epshtein Y; Tiram G; Blau R; Ofek P; Lee JS; Ruppin E; et al. Amphiphilic nanocarrier-induced modulation of PLK1 and MIR-34a leads to improved therapeutic response in pancreatic cancer. Nat. Commun 2018, 9, 16, doi: 10.1038/s41467-017-02283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sato-Dahlman M; Wirth K; Yamamoto M Role of gene therapy in pancreatic cancer—A review. Cancers (Basel). 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Conway JR; Herrmann D; Evans TJ; Morton JP; Timpson P Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758, doi: 10.1136/gutjnl-2018-316822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ebrahimi S; Hosseini M; Shahidsales S; Maftouh M; Ferns GA; Ghayour-Mobarhan M; Hassanian SM; Avan A Targeting the Akt/PI3K Signaling Pathway as a Potential Therapeutic Strategy for the Treatment of Pancreatic Cancer. Curr. Med. Chem. 2017, 24, doi: 10.2174/0929867324666170206142658. [DOI] [PubMed] [Google Scholar]
- 192.Polireddy K; Chen Q Cancer of the pancreas: Molecular pathways and current advancement in treatment. J. Cancer 2016, 7, 1497–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kang X; Lin Z; Xu M; Pan J; Wang Z wei Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 2019, 52, e12605, doi: 10.1111/cpr.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Miknyoczki SJ; Lang D; Huang L; Klein-Szanto AJP; Dionne CA; Ruggeri BA Neurotrophins and Trk receptors in human pancreatic ductal adenocarcinoma: Expression patterns and effects on in vitro invasive behavior. Int. J. Cancer 1999, 81, 417–427, doi:. [DOI] [PubMed] [Google Scholar]
- 195.Holmer N; Artola E; Christianson E; Lynn AM; Whitlock KB; Norton S Feasibility of acupuncture to induce sleep for brainstem auditory evoked response testing. Am. J. Audiol 2019, 28, 895–907, doi: 10.1044/2019_AJA-19-0069. [DOI] [PubMed] [Google Scholar]
- 196.Sano M; Driscoll DR; DeJesus-Monge WE; Quattrochi B; Appleman VA; Ou J; Zhu LJ; Yoshida N; Yamazaki S; Takayama T; et al. Activation of WNT/β-Catenin Signaling Enhances Pancreatic Cancer Development and the Malignant Potential Via Up-regulation of Cyr61. Neoplasia (United States) 2016, 18, 785–794, doi: 10.1016/j.neo.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sahin IH; Iacobuzio-Donahue CA; Oreilly EM Molecular signature of pancreatic adenocarcinoma: An insight from genotype to phenotype and challenges for targeted therapy. Expert Opin. Ther. Targets 2016, 20, 341–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pinho AV; Van Bulck M; Chantrill L; Arshi M; Sklyarova T; Herrmann D; Vennin C; Gallego-Ortega D; Mawson A; Giry-Laterriere M; et al. ROBO2 is a stroma suppressor gene in the pancreas and acts via TGF-β signalling. Nat. Commun 2018, 9, 1–14, doi: 10.1038/s41467-018-07497-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Escot S; Willnow D; Naumann H; Di Francescantonio S; Spagnoli FM Robo signalling controls pancreatic progenitor identity by regulating Tead transcription factors. Nat. Commun 2018, 9, 1–12, doi: 10.1038/s41467-018-07474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ma Y; Yu W; Shrivastava A; Srivastava RK; Shankar S Inhibition of pancreatic cancer stem cell characteristics by α-Mangostin: Molecular mechanisms involving Sonic hedgehog and Nanog. J. Cell. Mol. Med 2019, 23, 2719–2730, doi: 10.1111/jcmm.14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Niyaz M; Khan MS; Wani RA; Shah OJ; Mudassar S Sonic Hedgehog Protein is Frequently Up-Regulated in Pancreatic Cancer Compared to Colorectal Cancer. Pathol. Oncol. Res 2020, 26, 551–557, doi: 10.1007/s12253-018-00564-2. [DOI] [PubMed] [Google Scholar]
- 202.Heining C; Horak P; Uhrig S; Codo PL; Klink B; Hutter B; Fröhlich M; Bonekamp D; Richter D; Steiger K; et al. NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 2018, 8, 1087–1095, doi: 10.1158/2159-8290.CD-18-0036. [DOI] [PubMed] [Google Scholar]
- 203.Ahmed S; Bradshaw A-D; Gera S; Dewan M; Xu R The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med 2017, 6, 5, doi: 10.3390/jcm6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Iqbal J; Ragone A; Lubinski J; Lynch HT; Moller P; Ghadirian P; Foulkes WD; Armel S; Eisen A; Neuhausen SL; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009, doi: 10.1038/bjc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lal G; Liu G; Schmocker B; Kaurah P; Ozcelik H; Narod SA; Redston M; Gallinger S Inherited predisposition to pancreatic adenocarcinoma: Role of family history and germ-line p16, BRCA1, and BRCA2 mutations; 2000; Vol. 60;. [PubMed] [Google Scholar]
- 206.Pihlak R; Valle JW; McNamara MG Germline mutations in pancreatic cancer and potential new therapeutic options. Oncotarget 2017, 8, 73240–73257, doi: 10.18632/oncotarget.17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Huang H; Brekken RA The next wave of stroma-targeting therapy in pancreatic cancer. Cancer Res. 2019, 79, 328–330, doi: 10.1158/0008-5472.CAN-18-3751. [DOI] [PubMed] [Google Scholar]
- 208.Awaji M; Singh RK Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers (Basel). 2019, 11, 290, doi: 10.3390/cancers11030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Erdogan B; Webb DJ Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans 2017, 45, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Deshmukh A; Deshpande K; Arfuso F; Newsholme P; Dharmarajan A Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]