

Abstract
Purpose of review
Rapidly emerging evidence implicates an important role of gut-brain-bone marrow axis involving gut microbiota (GM), gut epithelial wall permeability, increased production of proinflammatory bone marrow (BM) cells and neuroinflammation in hypertension (HTN). However, the precise sequence of events involving these organs remains to be established. Furthermore, whether an impaired gut-brain-BM axis is a cause or consequence of HTN is actively under investigation. This will be extremely important for translation of this fundamental knowledge to novel, innovative approaches for the control and management of HTN. Therefore, our objectives are to summarize the latest hypothesis, provide evidence for and against the impaired gut, BM and brain interactions in HTN and discuss perspectives and future directions.
Recent findings
Hypertensive stimuli activate autonomic neural pathways resulting in increased sympathetic and decreased parasympathetic cardiovascular modulation. This directly affects the functions of cardiovascular-relevant organs to increase blood pressure (BP). Increases in sympathetic drive to the gut and BM also trigger sequences of signaling events that ultimately contribute to altered GM, increased gut permeability, enhanced gut- and brain-targeted proinflammatory cells from the BM in perpetuation and establishment of HTN.
Summary
In this review, we present the mechanisms involving the brain, gut, and BM, whose dysfunctional interactions may be critical in persistent neuroinflammation and key in the development and establishment of HTN.
Keywords: Neuroinflammation, Gut Microbiota, Bone Marrow, Autonomic Nervous System, Microglia
Introduction
HTN is the most preventable modifiable risk factor for cardiovascular disease, stroke and chronic kidney disease. American Heart Association/American College of Cardiology estimates that ~48% of Americans have high BP and importantly, ~17% of these hypertensive patients are resistant to all interventions [1]. These numbers are even greater in the African American population who have earlier onset, higher prevalence and more severe pathophysiology than non-Hispanic White Americans. Despite extensive research and development of innovative treatment strategies in the last several decades, HTN-related mortality is increasing. Therefore, there is a crucial need to discover novel and innovative mechanism-based therapies for controlling HTN. This need has added urgency during the current novel coronavirus pandemic because HTN is one of the most common comorbidities with unfavorable outcomes accounting for ~30% of all hospitalized COVID-19 patients [2].
Overwhelming evidence of the last decade has implicated dysregulated commensal microbiota in a variety of chronic diseases including diabetes, obesity, chronic kidney disease, heart failure, cardiovascular diseases, etc. Interestingly, all these diseases impact BP regulation and are risk factors for HTN. Therefore, it is reasonable to infer that GM and functions could have a major impact on BP control and cardiovascular homeostasis. Evidence supporting this view was first provided by our group in 2015 [3■]. The study demonstrated gut microbial dysbiosis in both spontaneously hypertensive rats (SHR) and patients with high BP. Selective decreases in butyrate-producing bacterial communities were observed. This basic finding has been validated by other investigators using both multiple animal models of HTN and patients with HTN [4]. Together, these studies led us to propose the hypothesis of impaired gut-brain-BM communication in HTN.
Hypothesis
We propose that hypertensive stimuli, like stress, salt, diet, impact the brain and the gut. In the brain, particularly autonomic brain regions such as the paraventricular nucleus of the hypothalamus (PVN), microglia become activated, altering their interactions with neurons and neuroinflammation results (Fig. 1). Neuroinflammation increases sympathetic and decreases parasympathetic activity to organs such as the gastrointestinal (GI) tract and BM, and raises BP.
FIGURE 1.
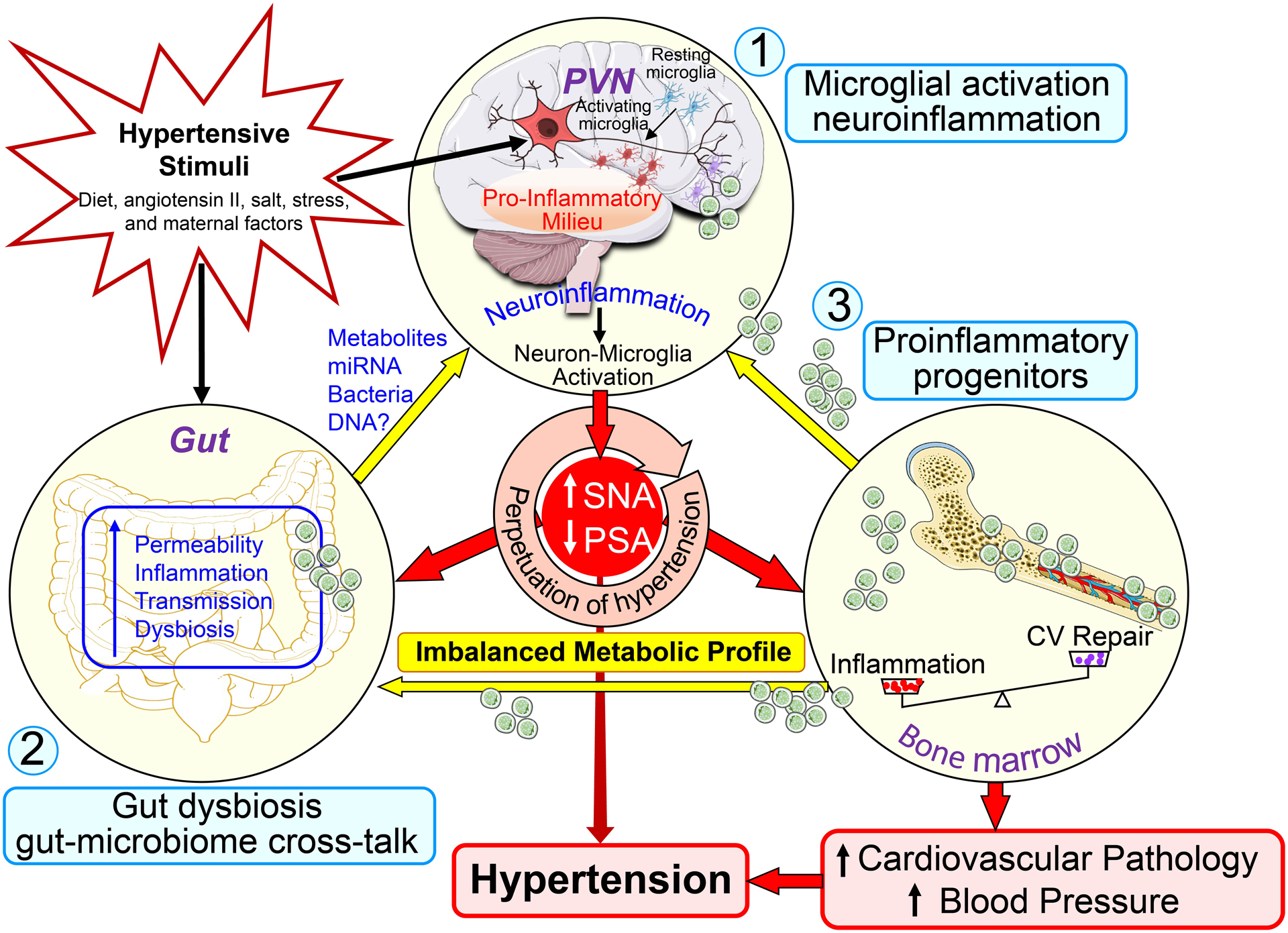
Brain–gut-bone marrow axis. Increases in hypertensive stimuli, such as diet, angiotensin II, salt, stress, and maternal factors, enhance neuronal activity and trigger neuroinflammatory pathways in cardioregulatory brain centers to result in sympathoexcitation. Sympathetic activity to the BM induces mobilization of hematopoietic stem cells, and stimulates their differentiation into inflammatory cells. These cells may then migrate to the brain to become microglia/macrophages and propagate neuroinflammation, as well as to the gut to contribute to intestinal inflammation. Sympathetic activity to the gut may modulate motility and local immune responses. Finally, low-grade inflammation of the gut coupled with alterations in GM may result in bacterial metabolites entering circulation, where they could negatively affect both neuronal activity and immune cells like microglia. This triangular interaction may play an important role in perpetuating the progression of HTN and may be critical in the establishment of HTN. CV, cardiovascular repair; PSA, parasympathetic nerve activity; PVN, paraventricular nucleus; and SNA, sympathetic nerve activity.
In the gut, hypertensive stimuli cause gut microbial dysbiosis, gut barrier weakening and inflammation. Gut dysbiosis results in an imbalanced GI milieu, causing inflammation. Weakened barrier function allows previously excluded gut contents, such as bacteria and bacterial metabolites, to contact the immune system generating inflammation. This exacerbates leakiness and inflammation and recruits pro-inflammatory BM cell progenitors to the gut, that further increase gut inflammation. The factors crossing the weakened gut barrier, and inflammatory mediators generated in the gut, reach the brain via the circulation and contribute to neuroinflammation and mild systemic inflammation (Fig. 1).
Increased sympathetic drive to BM stimulates release of pro-inflammatory progenitors that enter brain and gut to promote inflammation. Decreased release of progenitors capable of vascular repair also results from increased sympathetic stimulation. The interactions of these three organs reinforce their individual responses resulting in a vicious cycle that sustains and increases HTN (Fig. 1).
1. Microglia and Neuroinflammation
Microglia are resident immune cells of the brain arising from the yolk sac [5–7]. They are self-renewing, long-lived cells with a low replication rate [8]. Microglia are extremely dynamic cells. In the adult brain, one of their roles is to surveil the brain parenchyma. During brain development they remove unneeded synapses, promote formation of new synapses and myelination [9–11]. In adulthood, contact between a synapse and microglia process increases activity of the synapse and reinforces the circuit, but this is disrupted during inflammation [12]. Microglia are important in the neuronal stem cell niche [13,14] and blood brain barrier (BBB) [15]. Finally, their development is compromised in germ-free mice [16], being spaced further apart, bearing shortened and less branched projections and surveilling smaller areas, suggesting exposure to microbes or microbial products is important for their proper function. The microglial effects of GM are sex-specific during development and adulthood [17].
Neuroinflammation is observed in animal models of HTN in areas controlling the autonomic nervous system. We hypothesize that hypertensive stimuli impact the brain to cause neuroinflammation, either directly or secondarily following transmission from peripheral tissues e.g. Gl tract (Fig. 1). Neuroinflammation is particularly evident in the PVN [18], but also occurs in brainstem nuclei such as nucleus tractus solitarius and rostroventrolateral medulla during HTN, that is associated with increased sympathetic drive controlled through these brain regions [19].
When rodents are hypertensive, the PVN contains numerous activated microglia producing pro-inflammatory cytokines [18], that with the astrogliosis of established HTN produces a pro-inflammatory milieu [20,21]. BM-derived monocytes are recruited by Ccl2 released from microglia [22], differentiating into cells anatomically indistinguishable from activated microglia [23■■], and contribute to the pro-inflammatory environment [23■■]. Reactive oxygen species (ROS) are generated by pro-inflammatory cytokines binding to their receptors on immune cells, by angiotensin signaling in neurons [24] and the cerebrovasculature via activation of the NADPH-oxidase pathway. ROS increases neuronal firing rate in the PVN [24,25] further increasing ROS. If sustained, this overcomes antioxidant capacity. All these factors can stimulate neuronal firing rates in the PVN, neuroinflammation and increase sympathetic outflow.
PVN neuroinflammation promotes closer interaction of microglia with neurons. The phenotypes of PVN neurons impacted by microglial activation is unclear at present, but worthwhile candidates, but not the only ones, for investigation include pre-autonomic, vasopressinergic, oxytocinergic and corticosterone releasing hormone containing neurons, which all modulate BP [26–28]. These are influenced by activity of interneurons whose responses to pro-inflammatory mediators, and ultimate effect on BP, are becoming better understood [19].
Neuroinflammation changes interactions between microglia and cerebral blood vessels to affect BBB integrity. Astrocytic end feet wrap around capillary endothelial cells sealing them. Upon inflammation resident microglia support the BBB by secreting the tight junction protein, claudin 5, but with prolonged inflammation microglia phagocytose astrocyte end feet, increasing permeability [29]. This allows blood borne substances into the brain; some, like lipopolysaccharides (LPS), stimulate further microglial activation. BBB permeability is affected by the gut microbiome. Germ-free mice have a leaky BBB due to poor production of tight junction proteins, a defect corrected by populating their gut with pathogen-free microbiota [30]. This defect does not result in hypertension because there are no circulating gut microbial products such as LPS to stimulate neuroinflammation and the immune system is less inflammatory in germ-free mice. But in conventionally-raised mice this may be a mechanism connecting GM and HTN, see section 3. Cerebral autoregulation and vascular regulation of brain blood flow are mediated by sympathetic and parasympathetic inputs known to be dysfunctional in essential HTN [31]. This is likely another mechanism by which neuroinflammation, via increased sympathetic outflow, contributes to HTN.
The respective contributions of resident microglia and recruited BM-derived “microglia” to neuroinflammation need clarification. It is likely that initial responses to hypertensive stimuli are mediated by resident microglia, but their fate following BM-derived precursor recruitment is unclear. Another unanswered question is whether BM-derived “microglia” attempt the role of homeostatic microglia but are corrupted by the pro-inflammatory environment or immediately secrete pro-inflammatory cytokines and contribute to neuroinflammation.
In two animal models of HTN, sympathetic drive is increased to the gut and BM before BP increases [32■■–34]. This is discussed in the next sections.
2. Bone marrow.
BM is richly innervated by the sympathetic nervous system [35], but receives no direct parasympathetic input. Sympathetic activity contributes to proliferation, differentiation, maturation, exit and intake of BM cells to the compartment. Norepinephrine (NE) is found in sympathetic fibers [36] that run along the vasculature [37], site of the stem cell niche, and BM cells [38]. The immune BM compartment expresses α- and β-adrenergic receptors. α1-adrenergic receptors affect circadian rhythms of cell proliferation rate [39]; daily rhythm of BM cell release is mediated by β2- or β3-adrenergic receptors via endothelial nitric oxide synthase [40,41]. Sympathetic neurons in BM promote neutrophil egress into circulation by stimulating endothelial cells to produce C-X-C motif chemokine ligand 1 (Cxcl1), inflammation, e.g. by LPS enhance Cxcl1 production and egress; denervation or beta receptor blockade inhibits both processes [42]. Neutrophils interact with microglia to promote neuroinflammation in various brain disorders [43,44] but their role in HTN is unclear.
In animal models of HTN, we observed decreased circulating immune cells capable of vascular repair, but increased BM derived pro-inflammatory cells in the gut and autonomic regions of brain [23■■,33,45]. Likewise, in hypertensive patients, there are increased gut–homing pro-inflammatory immune cells compared to normotensive subjects [46■]. Finally, in mice, HTN stimuli-responsive enhanced memory T-cells are found in BM and are enhanced by sympathetic activity [47].
3.1. Gastrointestinal Tract
Our studies have demonstrated gut pathology and dysbiosis in animal models of HTN. This includes decreased blood flow, increased norepinephrine synthesizing enzyme in small intestine[32■■, 48] thickening and fibrosis of gut muscle layers[32■■], increased gut stiffness [49], shorter villi in small intestine and fewer mucus-producing goblet cells [32■■], increased gut barrier leakiness[32■■,46■] (corrected by butyrate treatment), lower expression of intestinal alkaline phosphatase [50] (an enzyme that by dephosphorylating a variety of substrates including LPS and extracellular ATP, renders them less inflammatory [51]), altered gut immune system [50–52], increased GALT (gastrointestinal associated-lymphoid tissue) activation [50] and increased BM-derived pro-inflammatory cells in the gut [23■■] and most significantly gut microbial dysbiosis[3■,46■]. These are all affected by increased sympathetic drive, and serve to reinforce the interlinked gut pathologies and increase neuroinflammation in a self-perpetuating fashion. Gut dysbiosis and its association with HTN is discussed in more detail below.
Epidemiological studies have consistently implicated the gut in BP regulation. This includes studies documenting the influence of diet, dietary supplements, probiotics and antibiotics on BP. Our studies were among the first to provide experimental evidence supporting this link in animal models of HTN [3■]. They demonstrated dysbiosis, altered alpha diversity and decreased butyrate-producing bacteria in the SHR, an animal model of human HTN. A plethora of publications followed, essentially confirming gut dysbiosis in animal models of HTN including DOCA-salt, Dahl-salt, stroke-prone SHR, chronic angiotensin II-infusion (rats and mice), obstructive sleep apnea (OSA) on high fat diet, and the L-NAME model although differing characteristics of gut dysbiosis were reported among the models [53]. The importance of GM in HTN is supported by studies of Karbach et. al. who demonstrated attenuated vascular dysfunction due to reduced inflammatory immune cell recruitment to the vessels and lower BP in the hypertensive response to chronic angiotensin II-infusion in germ-free mice [54]. Unequivocally, these studies associate gut dysbiosis and HTN. However, they do not address whether it causes an increased BP and establishes HTN.
Fecal microbiota transplantation (FMT) strategies have addressed cause or effect. Durgan’s group was the first to demonstrate that FMT from OSA-HTN rats into normotensive rats resulted in significantly increased BP and altered microbiota [55]. Similarly, our studies showed that FMT from WKY attenuated high BP in SHR and abolished HTN-related vascular dysfunction [56,57]. Additionally, attenuations of enhanced sympathetic activity and neuroinflammation were observed in SHR recipients of WKY FMT. Conversely, FMT from SHR into WKY impaired vascular function and increased neuroinflammation and BP [56,57]. Recently, Joe et. al. demonstrated that germ-free rats had lower BP and reduced vascular contractility; both were reversed by FMT from conventionally-reared rats [58■]. Cumulatively, these observations provide strong support for the presence and contribution of gut dysbiosis to HTN in animal models.
Is there evidence to support this concept in human HTN? Our study with a small cohort of subjects was the first to demonstrate gut dysbiosis in subjects with high BP [46■]. Multiple later studies essentially supported this and described essentially similar bacterial phylogeny and characteristics [53,59–61]. Shotgun metagenomics analyses demonstrated depletion of butyrate-producing bacteria in patients with high BP and circulating butyrate was decreased in a small cohort [46■]. Increases in biomarkers of gut leakiness (fatty acid binding protein 5, zonulin and LPS) and gut targeting pro-inflammatory TH17 cells associated gut dysbiosis with increased permeability and inflammation [46■]. In fact, zonulin tightly correlated with systolic BP (SBP). In addition, a stepwise linear regression model of butyrate-producing bacteria and zonulin predicted SBP with ~55% accuracy and found various bacteria positively and negatively linked to SBP [46■]. Interestingly, some relevant to gut inflammation (Eubacterium siraeum and Alistipes finegoldii) and gut barrier function (Bacteroides thetaiotaomicron) have also been reported elsewhere [62].
Li et. al [63■]. directly addressed cause versus effect of microbiota on BP. First, by demonstrating similar characteristics of microbiota of pre-hypertensive and hypertensive subjects. Further, classifiers based on GM and metabolites discriminated hypertensive and control subjects. Finally, FMT from hypertensive subjects into germ-free mice increased their BP. These data are consistent with animal data and support the conclusion that altered GM, at least in part, is the cause of high BP.
3.2. Dietary sodium, microbiota and hypertension
Sodium consumption is an independent risk factor for HTN. Since the GI tract is among the first organs encountering sodium, it is unsurprising that sodium is increasingly found to influence GM. The first evidence from studies of the Dahl-salt rat model of HTN showed that FMT from salt-resistant rats into salt-sensitive rats exacerbated HTN [64]. Our recent study advanced the concept and demonstrated that a probiotic, Bifidobacterium breve, prevented the increase in BP, cardiac and renal pathology, improved colonic integrity, and restored TH17 and Treg contents in mesenteric lymph nodes and aorta of the DOCA-salt rat model of HTN [65]. Sodium depletes Lactobacillus species, increasing TH17 cells, intestinal inflammation, and high BP. Mice treated with Lactobacillus murinus displayed decreased TH17cells and attenuated high BP. Similarly, challenging a small cohort of humans with moderately high salt reduced GI Lactobacillus species, and increased both TH17 cells and BP [66,67]. These studies suggest the influence of salt on GM-induced inflammation could be the mechanism for salt’s hypertensive potential. Additionally, a recent study implicates intestinally-derived corticosterone in HTN by showing that a diet supplemented with 8% sodium significantly shifted the GM and increased BP [68]. This appeared causative since FMT from high salt diet donor rats increased BP in normal rats. The altered microbiota produced more corticosterone and contained decreased inhibitory enzymes of the aldosterone pathway. This suggests that high salt-induced HTN may be initiated by GM and involves immunomodulation and metabolites derived from bacteria.
3.3. Maternal microbiota and BP control in offspring
Poor maternal diet and disease have been implicated in detrimental fetal programming that permanently alters the physiology of offspring and induces diseases in later life, including HTN. Evidence suggests that GM play an important role in maternal-fetal cross-talk. For example, influencing the GM by maternal administration of probiotics was protective against HTN-associated developmental programming [69]. Similarly, diet-induced maternal influences affect interplay between microbiota and hypertensive programming [70–73]. Our studies investigated a direct involvement of maternal microbiota in HTN by using an antihypertensive drug, captopril, in the SHR model. Captopril treatment altered maternal GM, attenuated gut pathology and neuroinflammation [74]. Their male offspring demonstrated persistently decreased SBP and neuroinflammation and improvements in gut inflammation and pathology [75■]. These observations demonstrate that lowering maternal high BP rebalances the GM, improves the dysregulated gut-brain axis, and transmits this to their offspring. Thus, targeting the maternal gut-brain axis may be a viable strategy for control of HTN in subsequent generations. One caveat is the use of captopril, that as an ACE (angiotensin converting enzyme) inhibitor, is contraindicated in pregnancy. Therefore, this concept requires validation with other classes of anti-hypertensive drugs and strategies like probiotics and high-fiber diet. The translational and therapeutic implications are profound for breaking transgenerational transmission of HTN.
Conclusion
Experimental and clinical studies strongly implicate altered GM, gut leakiness and gut pathology with HTN. Furthermore, evidence for a dysfunctional gut-brain-BM axis is mounting. Interventional strategies correcting GM have generally lowered high BP and rebalanced the gut-brain axis. However, many questions remain to be answered before establishing full clinical and translational impacts of the GM in HTN. The following deserve consideration for moving this concept towards possible therapeutics.
Mechanisms of cross-talk between gut epithelium and microbiota must be investigated; organoid cultures from animal models [76] and human subjects would be invaluable here.
Comprehensive cohort studies with different ethnic groups and both sexes are needed to address environmental influences such as diet, salt sensitivity, genetic background, etc.
Impacts of antihypertensive drugs, particularly, ACE inhibitors, angiotensin receptor blockers and calcium channel antagonists on GM need to be correlated to their effectiveness in controlling BP, focusing on ethnic and gender disparities.
Are there unique ethnic/gender gut microbiomes and gut bacterial metabolite signatures in HTN?
HTN is a risk factor for COVID-19 infection. HTN and COVID-19 both reflect altered GM and inflammation. It would be interesting to investigate whether unique host-GM communication in hypertensive patients renders them more susceptible to COVID-19.
Key points.
Neuroinflammation is particularly evident during HTN and contributes to HTN via increased sympathetic outflow.
BM pro-inflammatory progenitors extravasate to the brain, and contribute to neuroinflammation.
Human studies and animal models strongly support the presence and contribution of gut dysbiosis to HTN.
Hypertensive stimuli (diet, angiotensin II, salt, stress, and maternal factors) dysregulate the gut-brain-BM axis, leading to high BP.
Financial support and sponsorship
This work was supported by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) grants HL033610, HL110170, and HL132448.
Footnotes
Conflicts of interest
None.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Whelton P, Carey R, Aronow W, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: A report of the American College of Cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol 2017. November7; pii: S0735–1097(17):41519–1. [Google Scholar]
- 2.Tadic M, Cuspidi C, Mancia G, Dell’Oro R, Grassi G. COVID-19, hypertension and cardiovascular diseases: Should we change the therapy? Pharmacol Res 2020; 158:104906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.■.Yang T, Santisteban MM, Rodriguez V, et al. Gut dysbiosis is linked to hypertension. Hypertension 2015; 65(6):1331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the link between gut dysbiosis and hypertension.
- 4.Vallianou N, Geladari E, Kounatidis D. Microbiome and hypertension, where are we now? J Cardiovasc Med (Hagerstown) 2020; 21(2):83–88. [DOI] [PubMed] [Google Scholar]
- 5.Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 1999; 117:145–52. [DOI] [PubMed] [Google Scholar]
- 6.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330(6005):841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015; 518(7540):547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Réu P, Khosravi A, Bernard S, et al. The lifespan and turnover of microglia in the human brain. Cell Rep 2017; 20(4):779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paolicelli R, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333:1456–8. [DOI] [PubMed] [Google Scholar]
- 10.Parkhurst C, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013; 155:1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wlodarczyk A, Holtman I, Krueger M, et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. Embo j 2017; 36:3292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyoshi R, Wake H, Kato D, et al. Microglia enhance synapse activity to promote local network synchronization. Eneuro 2018; 5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribeiro Xavier A, Kress B, Goldman S, Lacerda de Menezes J, Nedergaard M. A distinct population of microglia supports adult neurogenesis in the subventricular zone. J Neurosci 2015; 35(34):11848–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shigemoto-Mogami Y, Hoshikawa K, Goldman J, Sekino Y, Sato K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci 2014; 34(6):2231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grossmann R, Stence N, Carr J, et al. Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia 2002; 37(3):229–40. [PubMed] [Google Scholar]
- 16.Erny D, Hrabě de Angelis AL, Jaitin D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nature Neurosci 2015; 18(7): 965–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thion M, Low D, Silvin A, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 2018; 172(3):500–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi P, Diez-Freire C, Jun J, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension 2010; 56(2):297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dampney R, Michelini L, Li D, Pan H. Regulation of sympathetic vasomotor activity by the hypothalamic paraventricular nucleus in normotensive and hypertensive states. Am J Physiol Heart Circ Physiol 2018; 315(5):H1200–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang T, Rodriguez V, Malphurs W, et al. Butyrate regulates inflammatory cytokine expression without affecting oxidative respiration in primary astrocytes from spontaneously hypertensive rats. Physiol Rep 2018; 6(14): e13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sumners C, Alleyne A, Rodríguez V et al. Brain angiotensin type-1 and type-2 receptors: Cellular locations under normal and hypertensive conditions. Hypertens Res 2020; 43 (4)281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hickman S, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol Disord Drug Targets 2010; 9(2):168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.■■.Santisteban MM, Ahmari N, Carvajal JM, et al. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ Res 2015; 117(2):178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper discloses that BM contributes to hypertension by increasing peripheral inflammatory cells and their extravasation into the brain.
- 24.Sun C, Sellers K, Sumners C, Raizada M. NAD(P)H oxidase inhibition attenuates neuronal chronotropic actions of angiotensin II. Circ Res 2005; 6(6):659–66. [DOI] [PubMed] [Google Scholar]
- 25.Peterson J, Sharma R, Davisson R. Reactive oxygen species in the neuropathogenesis of hypertension. Curr Hypertens Rep 2006; 8(3):232–41. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Nguyen D, Mifflin S. Corticotropin-releasing hormone projections from the paraventricular nucleus of the hypothalamus to the nucleus of the solitary tract increase blood pressure. J Neurophysiol 2019; 121(2):602–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lozić M, Šarenac O, Murphy D, Japundžić-Žigon N. Vasopressin, central autonomic control and blood pressure regulation. Curr Hypertens Rep 2018; 20(2):11. [DOI] [PubMed] [Google Scholar]
- 28.De Melo V, Saldanha R, Dos Santos C, et al. Ovarian hormone deprivation reduces oxytocin expression in paraventricular nucleus preautonomic neurons and correlates with baroreflex impairment in rats. Front Physiol 2016; 7:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haruwaka K, Ikegami A, Tachibana Y, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun 2019; 10(1):5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braniste V, Al-Asmakh M, Kowal C, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 2014; 6(263):263ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marmarelis V, Shin D, Oesterreich M, Mueller M. Quantification of dynamic cerebral autoregulation and co2 dynamic vasomotor reactivity impairment in essential hypertension. J Appl Physiol (1985) 2020; 128(2):397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.■■.Santisteban MM, Qi Y, Zubcevic J, et al. Hypertension-linked pathophysiological alterations in the gut. Circ Res 2017; 120(2):312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes gut pathology associated with hypertension.
- 33.Zubcevic J, Jun J, Kim S, et al. Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension 2014; 63(3):542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmari N, Santisteban M, Miller D, et al. Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. Am J Physiol Heart Circ Physiol 2019; 317(2):H279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wee N, Lorenz M, Bekirov Y, Jacquin M, and Scheller E. Shared autonomic pathways connect bone marrow and peripheral adipose tissues across the central neuraxis. Front Endocrinol (Lausanne) 2019; 10:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marino F, Cosentino M, Bombelli R, et al. Measurement of catecholamines in mouse bone marrow by means of HPLC with electrochemical detection. Haematologica 1997; 82(4):392–4. [PubMed] [Google Scholar]
- 37.Xiao L, do Carmo L, Foss J, Chen W, Harrison D. Sympathetic enhancement of memory T-cell homing and hypertension sensitization. Circ Res 2020; 126(6):708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maestroni G, Conti A. Modulation of hematopoiesis via alpha 1-adrenergic receptors on bone marrow cells. Exp Hematol 1994; 22(3):313–20. [PubMed] [Google Scholar]
- 39.Maestroni G, Cosentino M, Marino F, et al. Neural and endogenous catecholamines in the bone marrow. circadian association of norepinephrine with hematopoiesis? Exp Hematol 1998; 26(12):1172–7. [PubMed] [Google Scholar]
- 40.Récalde A, Richart A, Guérin C, et al. Sympathetic nervous system regulates bone marrow-derived cell egress through endothelial nitric oxide synthase activation: Role in postischemic tissue remodeling. Arterioscler Thromb Vasc Biol 2012; 32(3):643–53. [DOI] [PubMed] [Google Scholar]
- 41.Scheiermann C, Kunisaki Y, Lucas D, et al. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 2012; 37(2):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ao T, Kikuta J, Sudo T, et al. Local sympathetic neurons promote neutrophil egress from the bone marrow at the onset of acute inflammation. Int Immunol 2020; 32(11):727–36. [DOI] [PubMed] [Google Scholar]
- 43.Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol 2015; 129:239–57. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Lee H, Lee H, Kim I, Lee J. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain, Acta Neuropathol Commun 2019; 7(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zubcevic J, Waki H, Raizada M, Paton J. Autonomic-immune-vascular interaction: An emerging concept for neurogenic hypertension. Hypertension 2011; 57(6):1026–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.■.Kim S, Goel R, Kumar A, et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci (Lond) 2018;132(6):701–18. [DOI] [PMC free article] [PubMed] [Google Scholar]; This demonstrates that intestinal barrier dysfunction and microbiome function are linked to hypertension in humans
- 47.Nicholls A, Wen S, Hall P, Hickey M, Wong C. Activation of the sympathetic nervous system modulates neutrophil function. J Leukoc Biol 2018; 103(2):295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toral M, Robles-Vera I, de la Visitación N, et al. Critical role of the interaction gut microbiota - sympathetic nervous system in the regulation of blood pressure. Front Physiol 2019; 10:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart D, Rubiano A, Santisteban M, et al. Hypertension-linked mechanical changes of rat gut. Acta Biomater 2016; 45:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang T, Li H, Oliveira A, et al. Transcriptomic signature of gut microbiome-contacting cells in colon of spontaneously hypertensive rats. Physiol Genomics 2019; 52(3):121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lalles J. Intestinal alkaline phosphatase: Novel functions and protective effects. Nutr Rev 2014; 72(2):82–94. [DOI] [PubMed] [Google Scholar]
- 52.Yang T, Ahmari N, Schmidt J, et al. Shifts in the gut microbiota composition due to depleted bone marrow beta adrenergic signaling are associated with suppressed inflammatory transcriptional networks in the mouse colon. Front Physiol 2017; 8:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang T, Richards E, Pepine C, Raizada M. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol 2018; 14(7):442–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karbach S, Schönfelder T, Brandão I, et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 2016; 30;5(9):e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durgan DJ, Ganesh BP, Cope JL, et al. Role of the gut microbiome in obstructive sleep Apnea–Induced hypertension. Hypertension 2016; 67:469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toral M, Robles-Vera I, de la Visitación N, et al. Role of the immune system in vascular function and blood pressure control induced by fecal microbiota transplantation in rats. Acta Physiol (Oxf) 2019; 227(1):e13285. [DOI] [PubMed] [Google Scholar]
- 57.Toral M, Robles-Vera I, de la Visitación N, et al. Critical role of the interaction gut microbiota sympathetic nervous system in the regulation of blood pressure. Acta Physiol (Oxf) 2019; 10:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.■.Joe B, McCarthy C, Edwards J, et al. Microbiota introduced to germ-free rats restores vascular contractility and blood pressure. Hypertension 2020; 76(6):1847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that vascular system senses and responds to the presence of microbiota with a notable impairment in vascular contractility via enhanced actin depolymerization.
- 59.Richards EM, Pepine CJ, Raizada MK, Kim S. The gut, its microbiome, and hypertension. Curr Hypertens Rep 2017; 19(4):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marques F, Mackay C, Kaye D. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat Rev Cardiol 2018; 15:20–32. [DOI] [PubMed] [Google Scholar]
- 61.Verhaar B, Prodan A, Nieuwdorp M, Muller M. Gut microbiota in hypertension and atherosclerosis: A review. Nutrients 2020; 12(10):2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robles-Vera I, Marta Toral M, Duarte J. Microbiota and hypertension: Role of the sympathetic nervous system and the immune system. Am J Hypertens 2020; 33(10):890–901. [DOI] [PubMed] [Google Scholar]
- 63.■.Li J, Zhao F, Wang Y, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017; 5(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates gut dysbiosis before the onset of hypertension in an Asian population.
- 64.Mell B, Jala V, Mathew A, et al. Evidence for a link between gut microbiota and hypertension in the dahl rat. Physiol Genomics 2015;47(6):187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robles-Vera I, de la Visitación N, Toral M, et al. Probiotic bifidobacterium breve prevents DOCA-salt hypertension. Faseb j 2020; August 11: doi: 10.1096/fj.202001532R. [DOI] [PubMed] [Google Scholar]
- 66.Wilck N, Matus M, Kearney S, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017; 551(7682):585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haase S, Wilck N, Kleinewietfeld M, Müller D, Linker R. Sodium chloride triggers Th17 mediated autoimmunity. J Neuroimmunol 2019; 329:9–13. [DOI] [PubMed] [Google Scholar]
- 68.Yan X, Jin J, Su X, et al. Intestinal flora modulates blood pressure by regulating the synthesis of intestinal-derived corticosterone in high salt-induced hypertension. Circ Res 2020; 126(7):839–53. [DOI] [PubMed] [Google Scholar]
- 69.Hsu C, Lin Y, Hou C, Tain Y. Maternal administration of probiotic or prebiotic prevents male adult rat offspring against developmental programming of hypertension induced by high fructose consumption in pregnancy and lactation. Nutrients 2018; 10(9):1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen H, Lin Y, Lin I, et al. Resveratrol prevents combined prenatal NG-nitro-L-arginine-methyl ester (L-NAME) treatment plus postnatal high-fat diet induced programmed hypertension in adult rat offspring: Interplay between nutrient-sensing signals, oxidative stress and gut microbiota. J Nutr Biochem 2019; 70:28–37. [DOI] [PubMed] [Google Scholar]
- 71.Hsu C, Hou C, Lee C, Chan J, Tain Y. The interplay between maternal and post-weaning high-fat diet and gut microbiota in the developmental programming of hypertension. Nutrients 2019; 11(9):1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu C, Chan J, Yu H, et al. Targeting on gut microbiota-derived metabolite trimethylamine to protect adult male rat offspring against hypertension programmed by combined maternal high-fructose intake and dioxin exposure. Int J Mol Sci 2020; 21(15):5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rubaye H, Adamson C, Jadavji N. The role of maternal diet on offspring gut microbiota development: A review. J Neurosci Res 2020; February 28: doi: 10.1002/jnr.24605. [DOI] [PubMed] [Google Scholar]
- 74.Yang T, Aquino V, Lobaton G, et al. Sustained captopril-induced reduction in blood pressure is associated with alterations in gut-brain axis in the spontaneously hypertensive rat. J Am Heart Assoc 2019;8(4): e010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.■.Li H, Yang T, Richards E, Pepine C, Raizada M. Maternal treatment with captopril persistently alters gut-brain communication and attenuates hypertension of male offspring. Hypertension 2020; 75(5): 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that maternal captopril treatment persistently alters the gut-brain axis and attenuates hypertension of male offspring.
- 76.Li J, Stevens BR, Richards EM, Raizada MK. SARS-CoV-2 Receptor ACE2 (Angiotensin-Converting Enzyme 2) Is Upregulated in Colonic Organoids from Hypertensive Rats. Hypertension 2020; 76: E26–E28. [DOI] [PMC free article] [PubMed] [Google Scholar]